

Abstract
The Smith–Lemli–Opitz syndrome (SLOS) is an autosomal recessive multiple congenital anomaly/mental retardation disorder caused by an inborn error of post-squalene cholesterol biosynthesis. Deficient cholesterol synthesis in SLOS is caused by inherited mutations of 3β-hydroxysterol-Δ7 reductase gene (DHCR7). DHCR7 deficiency impairs both cholesterol and desmosterol production, resulting in elevated 7DHC/8DHC levels, typically decreased cholesterol levels and, importantly, developmental dysmorphology. The discovery of SLOS has led to new questions regarding the role of the cholesterol biosynthesis pathway in human development. To date, a total of 121 different mutations have been identified in over 250 patients with SLOS who represent a continuum of clinical severity. Two genetic mouse models have been generated which recapitulate some of the developmental abnormalities of SLOS and have been useful in elucidating the pathogenesis. This mini review summarizes the recent insights into SLOS genetics, pathophysiology and potential therapeutic approaches for the treatment of SLOS.
Keywords: brain, cholesterol, development, neural tissue, sterol precursors
Genetic defects in enzymes responsible for post-squalene cholesterol biosynthesis have recently emerged as important causes of congenital dysmorphology syndromes. Identification of the genetic defect in the Smith–Lemli–Opitz syndrome (SLOS, MIM270400) (1–3) led to the discovery that other similar dysmorphology syndromes were caused by inborn errors of cholesterol biosynthesis in human and mouse: desmosterolosis (4), lathosterolosis (5), CHILD syndrome (6, 7), CDPX2 (a form of chondrodysplasia punctata) and hydrops-ectopic calcification-moth-eaten skeletal (HEM, also known as Greenberg) dysplasia (8–11). All of these syndromes have been linked to deficiency of the specific enzymes required for the production of cholesterol from lanosterol, as summarized in Table 1. Patients with these disorders present with complex malformation syndromes involving different organs and systems. These disorders have led to a new appreciation for the essential role that cholesterol plays in development. Given the multiple and critical roles that sterols play, it is not surprising that mutations in genes encoding sterol biosynthetic enzymes can have a dramatic effect on eucaryotic development. Antley–Bixler syndrome (ABS) was previously classified as a sterol synthesis disorder. However, a recent large survey of patients with ABS shows that individuals with an ABS-like phenotype and normal steroidogenesis have fibroblast growth factor receptor (FGFR) mutations, whereas those with ambiguous genitalia are caused by genetic defects of P450 oxidoreductase (POR), which is responsible for steroidogenesis (12, 13). Therefore, ABS with POR mutations belongs to a disorder related to steroid metabolism instead of sterol synthesis (14). The detailed descriptions of these malformation syndromes have been extensively reviewed (15–17). In the present review, some recent insights into SLOS genetics, pathophysiology and potential therapeutic approaches are highlighted.
Table 1.
Post-squalene cholesterol biosynthesis disorders in human and mouse models
| Chromosome location
|
|||||||
|---|---|---|---|---|---|---|---|
| Disease | MIM | Inheritance | Enzyme | Gene | Human | Mouse | Mouse model |
| Smith–Lemli–Opitz syndrome | 270400 | AR | 3β-hydroxysterol-Δ7 reductase | DHCR7 | 11q13 | 7F5 | Dhcr7 −/− |
| Desmosterolosis | 602398 | AR | 3β-hydroxysterol-Δ24 reductase | DHCR24 | 1p33-31 | 4C7 | Dhcr24 −/− |
| Lathosterolosis | 607330 | AR | 3β-hydroxysterol-Δ5 reductase | SC5D | 11q23 | 9B | Sc5d −/− |
| CDPX2a | 302960 | X-linked | 3β-hydroxysterol-Δ8, 7 reductase | EBP | Xp11 | XA1.1 | Bpa and Str miced |
| CHILD syndromeb | 308050 | X-linked | 3β-hydroxysterol dehydrogenase | NSDHL | Xq28 | XA7.1 | Tdho mousee |
| HEM skeletal dysplasiac | 215140 | AR | 3β-hydroxysterol-Δ14 reductase | LBR | 1q32 | 1H5 | Ichthyosis mouse |
CDPX2, chondrodysplasia punctata type 2.
CHILD, congenital hemidysplasia, ichthyosis and limb defects.
HEM, hydrops-ectopic calcification-moth-eaten.
Bare patches and striated mice.
Tdho mouse, tattered-Hokkaido mouse.
SLOS genetics
Incidence
SLOS is an autosomal recessive disorder; loss-of-function mutations in both copies of DHCR7 on chromosome 11q13 are necessary to have disease. Both male and females are equally affected. DHCR7 reduces the C7-C8 double bond in the sterol B ring to form cholesterol or desmosterol depending upon the precursor. Desmosterol can be converted to cholesterol by 3β-hydroxysterol-Δ24 reductase (DHCR24). Therefore, SLOS patients usually have low plasma cholesterol levels and invariably have elevated levels of cholesterol precursors 7-dehydrocholesterol (and its spontaneous isomer 8-dehydrocholesterol) and absent desmosterol. SLOS is currently recognized as one of the most common autosomal recessive inherited disorders after cystic fibrosis and phenylketonuria in the North American Caucasian population. Based on elevated 7DHC levels, the incidence of SLOS is estimated to range from 1/10,000 to 1/60,000 of live births, depending upon the region. SLOS seems to be more prevalent among peoples of Caucasian descent, but infrequently reported in peoples of Asian or African background. In the US, the incidence of SLOS has been estimated to be approximately 1/50,000 (18, 19). A recent prenatal diagnostic study in the US, using maternal serum unconjugated estriol as a predictor for SLOS, indicated an incidence of 1/60,000 births (20). Another 3-year prospective population surveillance performed in Canada reported that the minimum incidence was 1/70,358 live births with a minimum prevalence of approximately 1/950,000 (21). There is, however, a discrepancy between the incidence of SLOS and the higher prevalence of carrier status of mutant alleles of DHCR7. Population screens for mutant DHCR7 alleles suggest a 3–4% carrier frequency in Caucasian populations, giving a hypothetical birth incidence about 1/2500–1/4500, assuming no foetal loss (17, 22, 23). This twofold discrepancy suggests that the true incidence may be higher due to unrecognized miscarriages or missed diagnoses in mildly affected individuals (e.g. subtle physical stigmata, minor behaviour or mental problems). More milder cases of SLOS have now been reported, due to increased awareness and access to a diagnostic test (24–27). Severe mutations such as homozygosity for null alleles may result in intrauterine or perinatal lethality (28–30). Prenatal mortality for the most severely affected cases of SLOS may be as high as 80% (22). In cases where a woman suffers an early spontaneous abortion, but who subsequently gives birth to a normal child (who may be wild type or heterozygous for a mutant DHRC7 allele) will not be further investigated. As early spontaneous abortion is not uncommon, perhaps this pattern may have masked the true incidence of SLOS. In this context, a true estimate of the incidence of SLOS is required using a more widespread screening.
DHCR7 mutations
Correa-Cerro and Porter recently summarized 105 different mutant alleles in a review of 559 published DHCR7 mutations (17). Sixteen additional novel missense mutations have since been reported (31–33). The mutational spectrum in SLOS appears consistent with that of most other autosomal recessive disorders, with missense mutations accounting for the vast majority of mutations, with less frequent null mutations (e.g. frameshift, nonsense or splice-site mutations). A number of SLOS cases (seven cases in published reports and two cases from our unpublished data) have been identified with only a single mutation (1, 3, 34), screened by bidirectional sequencing of PCR-amplified coding exons and adjacent intronic sequences. To date, a total of 121 mutations (685 alleles) in DHCR7 have been identified (Fig. 1). Mutations are located throughout the coding region (exons 3–9), including 105 missense, five nonsense, three splice-site and eight nucleotide insertions or deletions. The missense mutations account for 87.6% of the total spectrum. Fifty per cent of the missense mutations are located in one of the nine predicted transmembrane domains. In descending order, the commonest DHCR7 mutations, with the relative frequencies over 4%, are IVS8-1G>C (27.3%), T93M (10.4%), W151X (5.7%), V326L (4.8%) and R404C (4.5%). These five mutations account for 50–60% of reported mutant alleles. The other less common mutations (indicated in red typescript, Fig. 1) include R352W (2.8%), E448K (2.8%), G410S (1.9%), R242C (1.6%), S169L (1.6%) and F302 (1.5%).
Fig. 1.
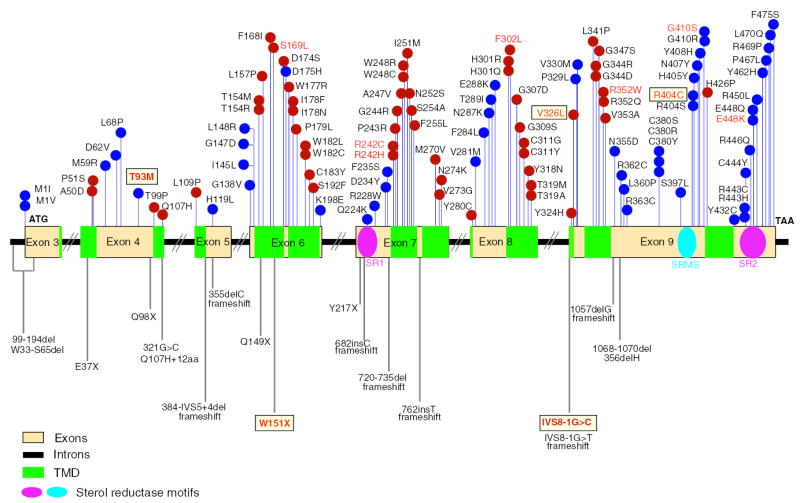
Mutational spectrum in SLOS. A total of 121 mutations have been reported in DHCR7 in SLOS patients as of June 2005. Missense mutations are found in all coding exons as shown in the upper panel of the figure. The red dots represent mutations in predicted transmembrane domains (TMD) and blue dots for those outside of these regions. Mutations that cause frameshift or premature chain termination are shown below the gene structure. Boxes indicate the most frequent mutations (<4%), and those whose frequencies are 1–4% are shown in red typescript. All others have frequencies of <1%.
Pathogenesis
Phenotype heterogeneity and possible affecting factors
The classic paradigm for the pathogenesis of an inborn error of metabolism includes the accumulation of a toxic precursor and/or deficiency of an essential product as a result of an enzyme deficiency. In the case of SLOS, accumulation of sterol precursors, such as 7DHC (7DHC has two conjugated Δ5,7 double bonds in the B-ring, but cholesterol has a unique Δ5 double bond), could be potentially toxic (but has not been convincingly demonstrated) (35, 36), and cholesterol deficiency is almost certainly detrimental (37, 38), but how sterol deficiency leads to dysmorphology has yet to be elucidated. A number of potential popular mechanisms have been championed, such as disruption of sonic hedgehog signalling, yet none of these have led to any mechanistic insight.
Although the phenotype severity correlates inversely with blood cholesterol levels and with predicted or measured DHCR7 activity, there exists considerable phenotype heterogeneity in patients with identical DHCR7 mutations (34, 39–41). The SLOS phenotypic spectrum is broad and variable, from early embryonic non-viability to varying levels of severity postnatally, including distinctive facial appearance, growth and mental retardation, autistic behaviour, hypotonia, failure to feed, decreased lifespan and variable structural anomalies of the heart, the lungs, the brain, the gastrointestinal tract, the limbs, the genitalia and the kidneys. Other factors in addition to genotype and the cholesterol and/or 7DHC levels at the time of diagnosis have been considered to influence the clinical severity. These factors could involve maternal factors that may increase the supply of cholesterol to the developing foetus, the amounts of cholesterol that can accumulate in the neural tissues before the blood–brain barrier is formed, perhaps the rate at which potentially toxic precursors can accumulate or be removed from the foetal tissues, and undefined other genetic and epigenetic factors (42–44). Recently, the maternal apolipoprotein E genotype (ApoE) was implicated in phenotype heterogeneity (45). Maternal ApoE2 genotypes were associated with a severe SLOS phenotype, whereas ApoE genotypes without the E2 allele were associated with a milder phenotype (45). However, as the numbers of mothers with the ApoE2/ApoE2 genotypes were small, some precaution should be expressed.
Notably, despite the apparently limited mother–foetus cholesterol transport, severely affected SLOS patients with homozygous ‘null’ mutations have detectable levels of cholesterol and do not invariably manifest a severe phenotype (40, 46). A recent study that the DHCR7 transcriptional signal was detectable in cultured fibroblasts from a patient with the ‘null’ mutations (W151X/IVS8-1G>C) suggests that the IVS8-1G>C splice mutation may be ‘leaky’ (46, 47), a phenomenon described in other genetic disorders (48). It is theoretically possible that another enzyme may be able to reduce the double bond at C7-8 of sterol precursors, albeit at a very low level. There is considerable amino acid sequence homology between DHCR7 and other sterol synthesis enzymes or ones that share the putative reductase domain, such as sterol Δ8 isomerase, sterol C14 reductase, the lamin B receptor and TM7SR2. These reductase motifs in DHCR7 are sterol reductase motif 1, SR1, amino acids 213–228, GNFFYNYMMGIEFNPR in a loop between the fourth and fifth transmembrane regions, sterol reductase 2, SR2, amino acids 439–462, LLTHRCLRDEHRCASKYGRDWERY in the carboxy terminus (CT) and a 12 amino acid sterol reductase signature motif, SRSM, amino acids 394–405, LLXSGWWGXXRH in the fourth loop which contains the binding site for NADPH. These motifs remain to be confirmed experimentally, but many of the missense mutations map to these motifs – Q224K and R228W are located in SR1; R443H, R443C, C444Y, E448K, E448Q, R450L and Y462H in SR2; S397L, R404S and H405Y in SRSM.
Knockout and transgenic mice
Two genetically manipulated murine models that mimic the biochemical and phenotypic hallmarks of the SLOS have been generated. Targeted disruption of Dhcr7 results in early postnatal death with severe sterol metabolic alterations and a number of developmental abnormalities (49–51), confirming the need for de novo cholesterol synthesis for normal mammalian embryogenesis.
Although a number of neurophysiological abnormalities, involving glutamate and serotonin pathways, have been reported in Dhcr7 −/− mice (50, 52), it remains unclear whether underlying neurodevelopmental defects contribute significantly to neonatal lethality of Dhcr7 null mice (49, 50). To investigate the consequences of cholesterol deficiency in postnatal central nervous system (CNS) development, we have generated transgenic mice expressing human DHCR7 under the control of a CNS-specific nestin promoter. Minimally expressed transgene in Dhcr7 null background results in a stochastically partial rescue (12%) of neonatal lethality. Rescued Dhcr7 −/− mice (TgRKO) survived 3–17 days postnatally, showing severe growth retardation and impaired movement suggestive of a neurological dysfunction (53). TgRKO mice present with profound cholesterol deficiency in the brain and less severe in peripheral tissues, indicating that the dietary cholesterol was not available to the brains of rescued mice. CNS defects are observed in TgRKO mice, including a dilated ventricular system and a number of structural defects. Although the total sterol level is not reduced in postnatal brains of rescued mice (consisting of approximately 10% cholesterol and approximately 90% of different sterol precursors), a delayed postnatal myelination and defective neurogenesis are clearly evident in TgRKO mice. In this context, conditional knockout of squalene synthase (the committed step for cholesterol synthesis) in neurons or oligodendrocytes causes either early postnatal death due to abnormal neurogenesis or premature death resulting from hypomyelination, indicating that normal CNS cholesterol synthesis is critical for postnatal survival (54, 55). Our study indicates that CNS defects, involving abnormal myelination and neurogenesis, contribute significantly to the early postnatal lethality of Dhcr7 −/− mice and may be important in the severity of SLOS in humans. Another conclusion is that even small and subtle changes in brain sterol metabolism were sufficient to enable partial rescue, suggesting that mechanism(s) involved in the CNS dysfunction may involve mechanisms other than the bulk requirement for cholesterol in membranes. One such pathway may be the disruption of critical neurosteroid synthesis within the CNS, a pathway highlighted by preliminary studies by Shackleton and his colleagues (56).
Another interesting development is the creation of a hypomorph mouse, the Dhcr7T93M. Mice homozygous for Dhcr7T93M or heterozygous for compound mutations (Dhcr7T93M/null) are viable, fertile and appear to have normal growth. Phenotypically Dhcr7T93M/T93M and Dhcr7T93M/null mice have mild dilatation of the third and lateral ventricles, and T93M/null mice have 2–3 toe syndactyly (57). The viability of these mice may allow investigations of therapeutic approaches for management of SLOS.
Molecular mechanism
Currently, the pathophysiological mechanisms involved in SLOS have not been fully elucidated. The most popular theory advanced has been that the key morphogen, Sonic hedgehog (and its related proteins Indian and Desert hedgehog), is affected, as this protein needs covalently attached cholesterol for regulated short and long-range signalling processes (58–61). Shh post-translational autoprocessing (51, 62) and expression in CNS and lung are not affected in the Dhcr7 −/− embryos (51), validating the previous conclusions that precursor sterols participate as efficiently as cholesterol in the Shh processing reaction (58) and can also substitute for cholesterol for structural requirements such as constituents of bilayer membranes (63). With regard to the latter, cholesterol is a known important component of membrane lipid rafts, and any subtle defects in raft formation could conceivably alter signalling pathways (64, 65). An appropriate composition of cholesterol in the cell membranes is crucial for optimal enzymatic activity, ion and metabolite transport or channelling, protein–protein and protein–lipid interactions, and signal transduction. The pathogenetic mechanisms of cholesterol deficiency caused by abnormal cholesterol biosynthesis may extend beyond disruption of morphogenic pathways. For example, the role of cholesterol in normal synaptogenesis is independent of the Shh pathway (66).
Prenatal diagnosis and screening
Prenatal diagnosis of SLOS, using elevated 7DHC, has now been performed in many pregnancies (67, 68). An elevated 7DHC/cholesterol ratio in foetal tissues or amniotic fluid (AF) is diagnostic of SLOS, and chorionic villus (CV) sampling at 11–12 weeks of gestation (69) has been reported. A direct correlation between the level of 7DHC in AF and the clinical severity of SLOS has been demonstrated in 20 pregnancies eventually confirmed to have SLOS (70). Interestingly, the levels of cholesterol in the AF of foetuses with SLOS are not significantly different from those of controls (71), supporting the concept that some of the pathophysiology may involve cholesterol precursor molecules and/or their metabolites.
Maternal serum or urine levels of unconjugated estriol (μE3) to screening for SLOS foetal pregnancies is a promising non-invasive new method for early diagnosis (20). Synthesis of μE3, a pregnancy-related steroid hormone, is dependent on cholesterol produced by the foetal tissues for synthesis by the placenta. Low to undetectable levels of μE3 have been observed in the urine, amniotic fluid and serum of pregnant women carrying foetuses affected with SLOS. In pregnancies affected with SLO, the levels of μE3 in maternal serum are abnormally low. Additionally, the utilization of precursor sterols in the foetal adrenals and gonads leads to the synthesis of abnormal equine-type oestrogens, which enter the maternal plasma and urine. These compounds are detectable using gas chromatography-mass spectrometry (72), although this test is not available for routine clinical use. It is also possible that this test may distinguish between affected and unaffected foetuses as early as 12 weeks of gestation, when the foetal production of steroids increases rapidly.
The diagnosis of SLOS by sonographic evaluation alone of the foetus is not reliable, except possibly in cases with severe malformations and a previous history of affected family members.
Prenatal diagnosis of SLOS by molecular analysis on DNA extracted from CV samples in families with known genotype has been reported (73, 74), although the high risk of spontaneous abortion limits its widespread use at this time.
Therapeutic approaches
There is no known cure for SLOS. Surgical repair of physical anomalies such as congenital heart defects, cleft palate, genital anomalies, craniofacial, gastrointestinal and limb defects involves a number of specialities in the care of survivors. Much of the treatment is supportive. The inability to synthesize cholesterol leads to predictable deficiency in adrenal steroid synthesis and may require steroid replacement. As bile acids are also synthesized from cholesterol, deficiency of this leads to inability to absorb dietary cholesterol and fat-soluble vitamin deficiencies. Dietary cholesterol supplementation restores both adrenal and bile salt deficiencies. Once sufficient cholesterol has been converted to bile salts by the liver, the absorption of dietary cholesterol should be restored. On a balanced diet, this should ensure a relatively normal cholesterol delivery to endocrine organs for sufficient steroid hormone synthesis. As the adrenals in SLOS depend upon lipoprotein-delivered means for cortisol synthesis, adrenal insufficiency should be considered when faced with a child with a prolonged illness. Intravenous plasma therapy has been reported for a case diagnosed antenatally, although it is unclear whether this is necessary or effective in reducing morbidity and mortality (75). Treatment strategies of dietary cholesterol supplementation are focused on supplying exogenous crystalline cholesterol by various vehicles (i.e. oil-based or aqueous suspension) in an attempt to increase body cholesterol levels and to secondarily decrease the levels of 7DHC/8DHC, through feedback inhibition of HMG-CoA reductase. Dietary cholesterol supplementation is now routine, although there are no controlled studies to validate their efficacy, and this issue remains unresolved. Not all children show a positive response to cholesterol supplementation (76). The response to therapy has varied between patients and protocols. Merkens et al. (77) reported that total sterols in plasma, LDL and HDL increased with dietary cholesterol supplementation. LDL and HDL cholesterol increased significantly, while the concentrations of 7DHC or 8DHC in LDL did not change. While plasma cholesterol levels and the ratio of cholesterol to total sterols increased, a significant decrease in 7DHC was not seen. A significant negative correlation was noted between cholesterol and 7DHC levels. The data indicate that those with the best response to dietary cholesterol in terms of cholesterol levels also had the lowest 7DHC levels, which would support the current practice of cholesterol supplementation as a treatment modality for SLOS in terms of improving the biochemical phenotype and providing a pool of cholesterol for transport to the tissues.
Another novel therapy proposed is the use of a ‘statin’ drug, simvastatin, an HMG-CoA reductase inhibitor, aimed at blocking the cholesterol synthesis pathway to avoid the formation of large amounts of precursor sterols, such as 7DHC/8DHC, thereby limiting exposure to potentially toxic metabolites. Simvastatin can also cross the blood–brain barrier and may provide a means to treat the biochemical defect present in the CNS of SLOS patients (78). A major effect of statin therapy is the transcriptional upregulation of genes controlled by the transcriptional factor SREBP, one of which is DHCR7. Thus, if any residual activity is present in the mutant DHCR7, its upregulation could increase intracellular cholesterol synthesis. Simvastatin use in SLOS patients resulted in a paradoxical increase in serum and cerebral spinal fluid cholesterol levels (79). Treatment of mutant and control fibroblasts with simvastatin significantly increased DHCR7 expression, whether cells were cultured in either cholesterol-containing or cholesterol-deficient media (41). To date, reports of statin therapy in SLOS have been confined to case reports, and further carefully constructed trials in larger series of cases are needed. Determination of residual DHCR7 enzymatic activity may be helpful in selecting SLOS patients being considered for a beneficial response of statins, as this treatment could theoretically be detrimental in subjects with little or no DHCR7 activity (33, 80).
Finally, we propose another therapy, that of direct cholesterol delivery to the CNS by low-pressure catheter infusion for the treatment of the one clinical manifestation in SLOS that has no direct treatment. Mental retardation, behavioural disturbance and movement disorders are major causes of morbidity. Therapy aimed specifically at this organ is likely to have a greater benefit than the reversal of non-CNS biochemical parameters. Restoration of adequate cholesterol stores by direct delivery is feasible, and as the turnover of cholesterol in the CNS is very slow, its effects may last for months, perhaps years, before re-treatment may be required. Toxicity of overdosing with cholesterol should be limited, as the normal CNS mechanisms regulating sterol exit out of the brain remain intact. Additionally, a slight overreplacement of cholesterol should lead to a suppression of the sterol synthesis pathway and allow for clearance of precursors. Finally, repletion of cholesterol in the brain may allow the brain to remodel and develop normally, especially if this can be carried out as early as possible after diagnosis.
Acknowledgments
Funding for this work has been provided by a grant from the National Institutes of Health, NHLBI, HL689660 (SBP).
References
- 1.Wassif CA, Maslen C, Kachilele-Linjewile S, et al. Mutations in the human sterol delta7-reductase gene at 11q12-13 cause Smith–Lemli–Opitz syndrome. Am J Hum Genet. 1998;63:55–62. doi: 10.1086/301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waterham HR, Wijburg FA, Hennekam RC, et al. Smith–Lemli–Opitz syndrome is caused by mutations in the 7-dehydrocholesterol reductase gene. Am J Hum Genet. 1998;63:329–338. doi: 10.1086/301982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitzky BU, Witsch-Baumgartner M, Erdel M, et al. Mutations in the Delta7-sterol reductase gene in patients with the Smith–Lemli–Opitz syndrome. Proc Natl Acad Sci USA. 1998;95:8181–8186. doi: 10.1073/pnas.95.14.8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waterham HR, Koster J, Romeijn GJ, et al. Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet. 2001;69:685–694. doi: 10.1086/323473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunetti-Pierri N, Corso G, Rossi M, et al. Lathosterolosis, a novel multiple-malformation/mental retardation syndrome due to deficiency of 3beta-hydroxysteroid-delta5-de-saturase. Am J Hum Genet. 2002;71:952–958. doi: 10.1086/342668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bornholdt D, Konig A, Happle R, et al. Mutational spectrum of NSDHL in CHILD syndrome. J Med Genet. 2005;42:e17. doi: 10.1136/jmg.2004.024448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grange DK, Kratz LE, Braverman NE, Kelley RI. CHILD syndrome caused by deficiency of 3beta-hydroxysteroid-delta8, delta7-isomerase. Am J Med Genet. 2000;90:328–335. doi: 10.1002/(sici)1096-8628(20000214)90:4<328::aid-ajmg13>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 8.Braverman N, Lin P, Moebius FF, et al. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat Genet. 1999;22:291–294. doi: 10.1038/10357. [DOI] [PubMed] [Google Scholar]
- 9.Derry JM, Gormally E, Means GD, et al. Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet. 1999;22:286–290. doi: 10.1038/10350. [DOI] [PubMed] [Google Scholar]
- 10.Seo KW, Kelley RI, Okano S, Watanabe T. Mouse Tdho abnormality results from double point mutations of the emopamil binding protein gene (Ebp) Mamm Genome. 2001;12:602–605. doi: 10.1007/s00335-001-3010-1. [DOI] [PubMed] [Google Scholar]
- 11.Waterham HR, Koster J, Mooyer P, et al. Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol delta 14-reductase deficiency due to mutations in the lamin B receptor gene. Am J Hum Genet. 2003;72:1013–1017. doi: 10.1086/373938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang N, Pandey AV, Agrawal V, et al. Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet. 2005;76:729–749. doi: 10.1086/429417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukami M, Horikawa R, Nagai T, et al. Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: molecular and clinical studies in 10 patients. J Clin Endocrinol Metab. 2005;90:414–426. doi: 10.1210/jc.2004-0810. [DOI] [PubMed] [Google Scholar]
- 14.Tint GS, Xu G, Batta A, Salen G. Birth defect/malformation syndromes caused by inborn errors of cholesterol biosynthesis. Ital J Pediatr. 2004;30:371–376. [Google Scholar]
- 15.Andersson HC. Disorders of post-squalene cholesterol biosynthesis leading to human dysmorphogenesis. Cell Mol Biol (Noisy-le-Grand) 2002;48:173–177. [PubMed] [Google Scholar]
- 16.Herman GE. Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum Mol Genet. 2003;12:R75–R88. doi: 10.1093/hmg/ddg072. [DOI] [PubMed] [Google Scholar]
- 17.Correa-Cerro LS, Porter FD. 3beta-hydroxysterol Delta7-reductase and the Smith–Lemli–Opitz syndrome. Mol Genet Metab. 2005;84:112–126. doi: 10.1016/j.ymgme.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Kelley RI, Hennekam RC. The Smith–Lemli–Opitz syndrome. J Med Genet. 2000;37:321–335. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelley RI. RSH/Smith–Lemli–Opitz syndrome: mutations and metabolic morphogenesis. Am J Hum Genet. 1998;63:322–326. doi: 10.1086/301987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoen E, Norem C, O’Keefe J, Krieger R, Walton D, To TT. Maternal serum unconjugated estriol as a predictor for Smith–Lemli–Opitz syndrome and other fetal conditions. Obstet Gynecol. 2003;102:167–172. doi: 10.1016/s0029-7844(03)00370-3. [DOI] [PubMed] [Google Scholar]
- 21.Nowaczyk MJ, Zeesman S, Waye JS, Douketis JD. Incidence of Smith–Lemli–Opitz syndrome in Canada: results of three-year population surveillance. J Pediatr. 2004;145:530–535. doi: 10.1016/j.jpeds.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 22.Opitz JM, Gilbert-Barness E, Ackerman J, Lowichik A. Cholesterol and development: the RSH (‘Smith–Lemli–Opitz’) syndrome and related conditions. Pediatr Pathol Mol Med. 2002;21:153–181. doi: 10.1080/15227950252852078. [DOI] [PubMed] [Google Scholar]
- 23.Battaile KP, Battaile BC, Merkens LS, Maslen CL, Steiner RD. Carrier frequency of the common mutation IVS8-1G>C in DHCR7 and estimate of the expected incidence of Smith–Lemli–Opitz syndrome. Mol Genet Metab. 2001;72:67–71. doi: 10.1006/mgme.2000.3103. [DOI] [PubMed] [Google Scholar]
- 24.Langius FA, Waterham HR, Romeijn GJ, et al. Identification of three patients with a very mild form of Smith–Lemli–Opitz syndrome. Am J Med Genet A. 2003;122:24–29. doi: 10.1002/ajmg.a.20207. [DOI] [PubMed] [Google Scholar]
- 25.Anderson AJ, Stephan MJ, Walker WO, Kelley RI. Variant RSH/Smith–Lemli–Opitz syndrome with atypical sterol metabolism. Am J Med Genet. 1998;78:413–418. doi: 10.1002/(sici)1096-8628(19980806)78:5<413::aid-ajmg4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 26.Angle B, Tint GS, Yacoub OA, Clark AL. Atypical case of Smith–Lemli–Opitz syndrome: implications for diagnosis. Am J Med Genet. 1998;80:322–326. [PubMed] [Google Scholar]
- 27.Neklason DW, Andrews KM, Kelley RI, Metherall JE. Biochemical variants of Smith–Lemli–Opitz syndrome. Am J Med Genet. 1999;85:517–523. doi: 10.1002/(sici)1096-8628(19990827)85:5<517::aid-ajmg18>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 28.Loffler J, Trojovsky A, Casati B, Kroisel PM, Utermann G. Homozygosity for the W151X stop mutation in the delta7-sterol reductase gene (DHCR7) causing a lethal form of Smith–Lemli–Opitz syndrome: retrospective molecular diagnosis. Am J Med Genet. 2000;95:174–177. doi: 10.1002/1096-8628(20001113)95:2<174::aid-ajmg16>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 29.Linck LM, Hayflick SJ, Lin DS, et al. Fetal demise with Smith–Lemli–Opitz syndrome confirmed by tissue sterol analysis and the absence of measurable 7-dehydrocholesterol Delta (7)-reductase activity in chorionic villi. Prenat Diagn. 2000;20:238–240. [PubMed] [Google Scholar]
- 30.Nowaczyk MJ, Farrell SA, Sirkin WL, et al. Smith-Lemli-Opitz (RHS) syndrome: holoprosencephaly and homozygous IVS8-1G–>C genotype. Am J Med Genet. 2001;103:75–80. doi: 10.1002/1096-8628(20010915)103:1<75::aid-ajmg1502>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 31.Witsch-Baumgartner M, Clayton P, Clusellas N, et al. Identification of 14 novel mutations in DHCR7 causing the Smith–Lemli–Opitz syndrome and delineation of the DHCR7 mutational spectra in Spain and Italy. Hum Mutat. 2005;25:412. doi: 10.1002/humu.9328. [DOI] [PubMed] [Google Scholar]
- 32.Cardoso WL, Balreira A, Martins E, et al. Molecular studies in Portuguese patients with Smith–Lemli–Opitz syndrome and report of three new mutations in DHCR7. Mol Genet Metab. 2005;85:228–235. doi: 10.1016/j.ymgme.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Ginat S, Battaile KP, Battaile BC, Maslen C, Gibson KM, Steiner RD. Lowered DHCR7 activity measured by ergosterol conversion in multiple cell types in Smith–Lemli–Opitz syndrome. Mol Genet Metab. 2004;83:175–183. doi: 10.1016/j.ymgme.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Yu H, Lee MH, Starck L, et al. Spectrum of Delta (7)-dehydrocholesterol reductase mutations in patients with the Smith–Lemli–Opitz (RSH) syndrome. Hum Mol Genet. 2000;9:1385–1391. doi: 10.1093/hmg/9.9.1385. [DOI] [PubMed] [Google Scholar]
- 35.Gaoua W, Chevy F, Roux C, Wolf C. Oxidized derivatives of 7-dehydrocholesterol induce growth retardation in cultured rat embryos: a model for antenatal growth retardation in the Smith–Lemli–Opitz syndrome. J Lipid Res. 1999;40:456–463. [PubMed] [Google Scholar]
- 36.Gaoua W, Wolf C, Chevy F, Ilien F, Roux C. Cholesterol deficit but not accumulation of aberrant sterols is the major cause of the teratogenic activity in the Smith–Lemli–Opitz syndrome animal model. J Lipid Res. 2000;41:637–646. [PubMed] [Google Scholar]
- 37.Ohashi K, Osuga J, Tozawa R, et al. Early embryonic lethality caused by targeted disruption of the 3-hydroxy-3-methylglutaryl-CoA reductase gene. J Biol Chem. 2003;278:42936–42941. doi: 10.1074/jbc.M307228200. [DOI] [PubMed] [Google Scholar]
- 38.Tozawa R, Ishibashi S, Osuga J, et al. Embryonic lethality and defective neural tube closure in mice lacking squalene synthase. J Biol Chem. 1999;274:30843–30848. doi: 10.1074/jbc.274.43.30843. [DOI] [PubMed] [Google Scholar]
- 39.Tint GS, Salen G, Batta AK, et al. Correlation of severity and outcome with plasma sterol levels in variants of the Smith–Lemli–Opitz syndrome. J Pediatr. 1995;127:82–87. doi: 10.1016/s0022-3476(95)70261-x. [DOI] [PubMed] [Google Scholar]
- 40.Witsch-Baumgartner M, Fitzky BU, Ogorelkova M, et al. Mutational spectrum in the Delta7-sterol reductase gene and genotype-phenotype correlation in 84 patients with Smith–Lemli–Opitz syndrome. Am J Hum Genet. 2000;66:402–412. doi: 10.1086/302760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wassif CA, Krakowiak P, Wright BS, et al. Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in Smith–Lemli–Opitz syndrome fibroblasts. Mol Genet Metab. 2005;85:46–107. doi: 10.1016/j.ymgme.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 42.Bjorkhem I, Starck L, Andersson U, et al. Oxysterols in the circulation of patients with the Smith–Lemli–Opitz syndrome: abnormal levels of 24S- and 27-hydroxycholesterol. J Lipid Res. 2001;42:366–371. [PubMed] [Google Scholar]
- 43.Wassif CA, Yu J, Cui J, Porter FD, Javitt NB. 27-Hydroxylation of 7- and 8-dehydrocholesterol in Smith–Lemli–Opitz syndrome: a novel metabolic pathway. Steroids. 2003;68:497–502. doi: 10.1016/s0039-128x(03)00090-4. [DOI] [PubMed] [Google Scholar]
- 44.Woollett LA. The origins and roles of cholesterol and fatty acids in the fetus. Curr Opin Lipidol. 2001;12:305–312. doi: 10.1097/00041433-200106000-00010. [DOI] [PubMed] [Google Scholar]
- 45.Witsch-Baumgartner M, Gruber M, Kraft HG, et al. Maternal apo E genotype is a modifier of the Smith–Lemli–Opitz syndrome. J Med Genet. 2004;41:577–584. doi: 10.1136/jmg.2004.018085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Correa-Cerro LS, Wassif CA, Waye JS, et al. DHCR7 nonsense mutations and characterisation of mRNA nonsense mediated decay in Smith–Lemli–Opitz syndrome. J Med Genet. 2005;42:350–357. doi: 10.1136/jmg.2004.022749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nalla VK, Rogan PK. Automated splicing mutation analysis by information theory. Hum Mutat. 2005;25:334–342. doi: 10.1002/humu.20151. [DOI] [PubMed] [Google Scholar]
- 48.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 49.Fitzky BU, Moebius FF, Asaoka H, et al. 7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith–Lemli–Opitz/RSH syndrome. J Clin Invest. 2001;108:905–915. doi: 10.1172/JCI12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wassif CA, Zhu P, Kratz L, et al. Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith–Lemli–Opitz syndrome. Hum Mol Genet. 2001;10:555–564. doi: 10.1093/hmg/10.6.555. [DOI] [PubMed] [Google Scholar]
- 51.Yu H, Wessels A, Chen J, et al. Late gestational lung hypoplasia in a mouse model of the Smith–Lemli–Opitz syndrome. BMC Dev Biol. 2004;4:1. doi: 10.1186/1471-213X-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waage-Baudet H, Lauder JM, Dehart DB, et al. Abnormal serotonergic development in a mouse model for the Smith–Lemli–Opitz syndrome: implications for autism. Int J Dev Neurosci. 2003;21:451–459. doi: 10.1016/j.ijdevneu.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 53.Yu H, Wessels A, Tint GS, Patel SB. Partial rescue of neonatal lethality of Dhcr7 null mice by a nestin promoter-driven DHCR7 transgene expression. Brain Res Dev Brain Res. 2005;156:46–60. doi: 10.1016/j.devbrainres.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 54.Saher G, Goebbels S, Ishibashi S, Nave KA. Impaired cortical layer formation caused by cre-mediated inactivation of cholesterol biosynthesis in neurons. Federation European Neuroscience Soc (FENS) 2004;2:A211–A226. (Abstract). [Google Scholar]
- 55.Saher G, Brugger B, Lappe-Siefke C, et al. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- 56.Marcos J, Guo LW, Wilson WK, Porter FD, Shackleton C. The implications of 7-dehydrosterol-7-reductase deficiency (Smith–Lemli–Opitz syndrome) to neurosteroid production. Steroids. 2004;69:51–60. doi: 10.1016/j.steroids.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 57.Correa-Cerro LS, Wassif CA, Kratz L, Kelly RI, Porter FD. Development, characterization, and treatment of a hypomorphic SLOS mouse model. Society for inherited metabolic disorders (simd) 2004 (Abstract). [Available at http://www.simd.org/meetings/SIMD2004/abstract/abstractviewone.asp?selectedabstract=59].
- 58.Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 59.Lewis PM, Dunn MP, McMahon JA, et al. Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell. 2001;105:599–612. doi: 10.1016/s0092-8674(01)00369-5. [DOI] [PubMed] [Google Scholar]
- 60.Porter JA, von Kessler DP, Ekker SC, et al. The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature. 1995;374:363–366. doi: 10.1038/374363a0. [DOI] [PubMed] [Google Scholar]
- 61.Porter JA, Ekker SC, Park WJ, et al. Hedgehog patterning activity: role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell. 1996;86:21–34. doi: 10.1016/s0092-8674(00)80074-4. [DOI] [PubMed] [Google Scholar]
- 62.Cooper MK, Wassif CA, Krakowiak PA, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33:508–513. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- 63.Keller RK, Arnold TP, Fliesler SJ. Formation of 7-dehydrocholesterol-containing membrane rafts in vitro and in vivo, with relevance to the Smith–Lemli–Opitz syndrome. J Lipid Res. 2004;45:347–355. doi: 10.1194/jlr.M300232-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simons K, Ikonen E. How cells handle cholesterol. Science. 2000;290:1721–1726. doi: 10.1126/science.290.5497.1721. [DOI] [PubMed] [Google Scholar]
- 65.Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29:190–201. doi: 10.1016/j.mcn.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 67.Kelley RI. Diagnosis of Smith–Lemli–Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts. Clin Chim Acta. 1995;236:45–58. doi: 10.1016/0009-8981(95)06038-4. [DOI] [PubMed] [Google Scholar]
- 68.Rossiter JP, Hofman KJ, Kelley RI. Smith–Lemli–Opitz syndrome: prenatal diagnosis by quantification of cholesterol precursors in amniotic fluid. Am J Med Genet. 1995;56:272–275. doi: 10.1002/ajmg.1320560307. [DOI] [PubMed] [Google Scholar]
- 69.Nowaczyk MJ, Heshka T, Kratz LE, Kelley RE. Difficult prenatal diagnosis in mild Smith–Lemli–Opitz syndrome. Am J Med Genet. 2000;95:396–398. [PubMed] [Google Scholar]
- 70.Dallaire L, Mitchell G, Giguere R, Lefebvre F, Melancon SB, Lambert M. Prenatal diagnosis of Smith–Lemli–Opitz syndrome is possible by measurement of 7-dehydrocholesterol in amniotic fluid. Prenat Diagn. 1995;15:855–858. doi: 10.1002/pd.1970150911. [DOI] [PubMed] [Google Scholar]
- 71.Kratz LE, Kelley RI. Prenatal diagnosis of the RSH/Smith–Lemli–Opitz syndrome. Am J Med Genet. 1999;82:376–381. [PubMed] [Google Scholar]
- 72.Shackleton CH, Roitman E, Kratz LE, Kelley RI. Equine type estrogens produced by a pregnant woman carrying a Smith–Lemli–Opitz syndrome fetus. J Clin Endocrinol Metab. 1999;84:1157–1159. doi: 10.1210/jcem.84.3.5660. [DOI] [PubMed] [Google Scholar]
- 73.Loeffler J, Utermann G, Witsch-Baumgartner M. Molecular prenatal diagnosis of Smith–Lemli–Opitz syndrome is reliable and efficient. Prenat Diagn. 2002;22:827–830. doi: 10.1002/pd.419. [DOI] [PubMed] [Google Scholar]
- 74.Nowaczyk MJ, Garcia DM, Eng B, Waye JS. Rapid molecular prenatal diagnosis of Smith–Lemli–Opitz syndrome. Am J Med Genet. 2001;102:387–388. doi: 10.1002/ajmg.1503. [DOI] [PubMed] [Google Scholar]
- 75.Irons MB, Nores J, Stewart TL, et al. Antenatal therapy of Smith–Lemli–Opitz syndrome. Fetal Diagn Ther. 1999;14:133–137. doi: 10.1159/000020906. [DOI] [PubMed] [Google Scholar]
- 76.Sikora DM, Ruggiero M, Petit-Kekel K, Merkens LS, Connor WE, Steiner RD. Cholesterol supplementation does not improve developmental progress in Smith–Lemli–Opitz syndrome. J Pediatr. 2004;144:783–791. doi: 10.1016/j.jpeds.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 77.Merkens LS, Connor WE, Linck LM, Lin DS, Flavell DP, Steiner RD. Effects of dietary cholesterol on plasma lipoproteins in Smith–Lemli–Opitz syndrome. Pediatr Res. 2004;56:726–732. doi: 10.1203/01.PDR.0000141522.14177.4F. [DOI] [PubMed] [Google Scholar]
- 78.Saheki A, Terasaki T, Tamai I, Tsuji A. In vivo and in vitro blood–brain barrier transport of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors. Pharm Res. 1994;11:305–311. doi: 10.1023/a:1018975928974. [DOI] [PubMed] [Google Scholar]
- 79.Jira PE, Wevers RA, de Jong J, et al. Simvastatin. A new therapeutic approach for Smith–Lemli–Opitz syndrome. J Lipid Res. 2000;41:1339–1346. [PubMed] [Google Scholar]
- 80.Starck L, Lovgren-Sandblom A, Bjorkhem I. Simvastatin treatment in the SLO syndrome: a safe approach? Am J Med Genet. 2002;113:183–189. doi: 10.1002/ajmg.10722. [DOI] [PubMed] [Google Scholar]