

Summary
Directional cell movement is initiated by the generation of a PtdIns(3,4,5)P3 gradient at the cell membrane, which is followed by Rac GTPase-mediated actin rearrangement. The evolutionarily conserved DOCK180 protein has an indispensable role in cell migration by functioning as an atypical guanine nucleotide exchange factor for Rac via its DOCK Homology Region (DHR)-2 domain. Here, we report that another conserved protein domain, DHR-1, also has an important role in DOCK180 signaling. A form of DOCK180 that lacks the DHR-1 domain is capable of inducing GTP-loading of Rac, but it fails to promote cell elongation and migration. The DHR-1 domain directly and specifically interacts with PtdIns(3,4,5)P3 in vitro and in vivo, and mediates the DOCK180 signaling complex localization at sites of PtdIns(3,4,5)P3 accumulation. A chimeric form of DOCK180 in which the DHR-1 domain has been replaced by a canonical PtdIns(3,4,5)P3-binding PH domain is fully functional at inducing cell elongation and migration, suggesting that the main function of DHR-1 is to bind PtdIns(3,4,5)P3. These results demonstrate that DOCK180, via its DHR-1 and DHR-2 domains, couples PtdIns(3,4,5)P3 signaling to Rac GTP-loading, which is essential for directional cell movement.
The key response of a cell to a migratory attractant is to extend membrane protrusions in the direction of migration. This polarization is triggered by the recruitment of phosphatidylinositol (PtdIns) 3-kinases to the cellular sites that initially sense the attractant 1,2. PtdIns(3,4,5)P3 subsequently accumulates at the leading edge while it is being degraded at the rear of the cell by PTEN-like activity, resulting in a PtdIns(3,4,5)P3 gradient at the membrane 3–6. PtdIns(3,4,5)P3 accumulation is followed by the recruitment of lipid-binding proteins that promote actin-based membrane protrusions at the leading edge 1. Rho GTPases, such as Rac, have a key role in orchestrating the cytoskeletal rearrangements necessary for cell migration 7. When in an active GTP-bound form, Rho GTPases couple to their effectors to induce specific biological outputs. Rac is preferentially activated at the leading edge where it induces membrane extensions, in part, through the WAVE-Arp2/3 complex 8. At present, the signaling mechanisms that facilitate localized Rac activation are not understood.
Mammalian DOCK180 was originally identified as a major binding partner for the Crk adapter protein 9. Subsequent genetic studies demonstrated that the DOCK180 orthologues Myoblast City and Ced-5 in Drosophila and C. elegans, respectively, have a crucial role in regulating Rac signaling in processes such as myoblast fusion, cell migration and phagocytosis 10–14. Studies in C. elegans have further identified Ced-12 (ELMO) as an upstream regulator of Rac that functions genetically at the same step as DOCK180 15–17. In mammalian cells, the Crk-ELMO-DOCK180 complex activates several Rac-dependent pathways, including JNK kinase cascade, actin remodeling, cell migration and engulfment of apoptotic cells 15,17–20. Recent work by us and others has identified an evolutionarily conserved DOCK180-related protein superfamily, several members of which are guanine nucleotide exchange factors (GEFs) of Rho GTPases 21,22. In accordance with the genetic studies, DOCK180 functions as a specific GEF for Rac 21,23. The DOCK180 family members lack a discernable tandem DH-PH domain, which is the hallmark catalytic domain of the canonical Rho-GEFs. Instead, they rely on a DHR-2 (also known as CZH2 or DOCKER) domain that, similar to the DH-PH module, interacts with a nucleotide-free GTPase and promotes its activation 21–23. We have previously identified another evolutionarily conserved domain, termed DHR-1 (also known as CZH1), in all known members of the DOCK180 superfamily 21,22. Here, we report that the DHR-1 domain is a novel PtdIns(3,4,5)P3-binding module, which localizes the DOCK180 signaling complex to the leading edge, and is crucial for DOCK180-mediated cell elongation and cell migration.
Results
DHR-1 domain is required for DOCK180-induced cell elongation and motility
The significance of the DHR-1 domain in DOCK180-mediated signaling was examined by expressing DOCK180 with a deletion of the DHR-1 domain (DOCK180 DHR-1; Supplementary Information, Fig. S1) in LR73 cells (a CHO cell variant commonly used for cell spreading and migration studies) 15,23,24. As shown in Fig. 1a, control-transfected cells spread and assembled actin stress fibers on fibronectin. Cells that had been transfected with wild-type DOCK180 or ELMO1 alone, or DOCK180 and ELMO1 together were morphologically indistinguishable from the control cells (not shown). As reported previously 15, expression of CrkII alone or CrkII together with DOCK180 or ELMO1 resulted in membrane ruffling (not shown). Upon coexpression of DOCK180, ELMO and CrkII, more than 40% of the cells adopted a highly elongated morphology (Fig. 1a, Fig. 7c). Thus, simultaneous expression of CrkII, ELMO and DOCK180 is required for an extended cellular morphology, reflecting the in vivo finding that all three gene products are indispensable in C. elegans for processes such as cell migration. Importantly, cells expressing DOCK180 DHR-1, ELMO1 and CrkII failed to acquire an elongated morphology (Fig. 1a, Fig. 7c). Furthermore, a strong correlation was observed between the elongated phenotype and cell migration. Only when DOCK180, ELMO1 and CrkII were expressed together, a highly significant, 7-fold increase in cell migration was observed (Fig. 1c-d). Cells coexpressing DOCK180 DHR-1, ELMO1 and CrkII failed to exhibit significantly enhanced motility, demonstrating that the DHR-1 domain is essential for DOCK180-mediated directional cell movement.
Fig. 1. The DHR-1 domain is required for DOCK180-mediated cell elongation and motility.
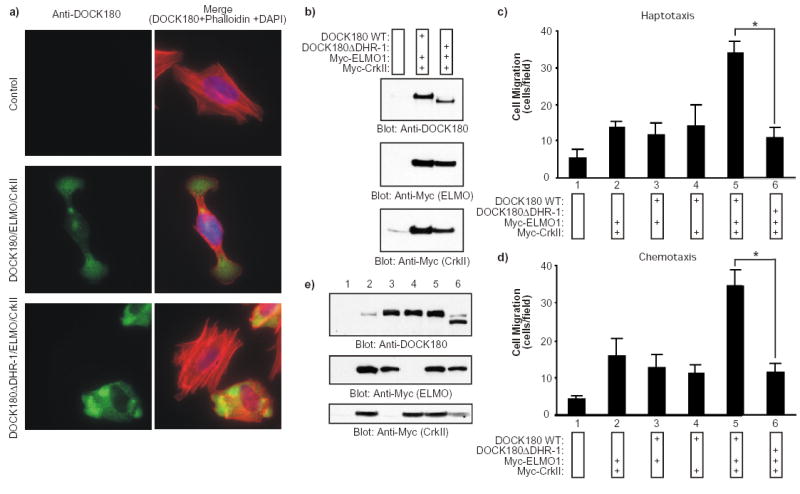
a) Serum-starved LR73 cells that had been transfected with the indicated plasmids were detached and plated on fibronectin-coated cover slips for 2 h in the absence of serum prior to fixing. Cells in the left panels were stained with anti-DOCK180 (H-4), while the panels on the right represent an overlay of the anti-DOCK180, rhodamine-phalloidin and DAPI stains. Cells were photographed at a 60X magnification. b) Expression levels of the transfected proteins were analyzed by immunoblotting of cell lysates with anti-DOCK180 (C-19) and anti-Myc antibodies, as indicated. c, d) Serum-starved LR73 cells that had been transfected with a GFP plasmid together with the indicated plasmids were detached and placed in the upper compartment of a Boyden-chamber. In the haptotactic migration assay (c), the cells were allowed to migrate for 4 h towards fibronectin. In the chemotactic assay (d), the top and bottom sides of the membrane were precoated with fibronectin and the cells were allowed to migrate for 4 h towards 10% serum as a chemoattractant in the lower chamber. Cells were fixed and stained with DAPI. GFP/DAPI double positive cells that had migrated across the membrane were counted from photographs taken at a 40X magnification. The experiments described above were performed independently three times in duplicate. Data are shown as mean +/- SD. *, P < 0.001; one-way ANOVA. e) Expression levels of the transfected proteins were analyzed by immunoblotting of cell lysates with anti-DOCK180 (C-19) and anti-Myc antibodies, as indicated.
Fig. 7. The PH domain of BMX/Etk can functionally replace the DHR-1 domain in DOCK180.
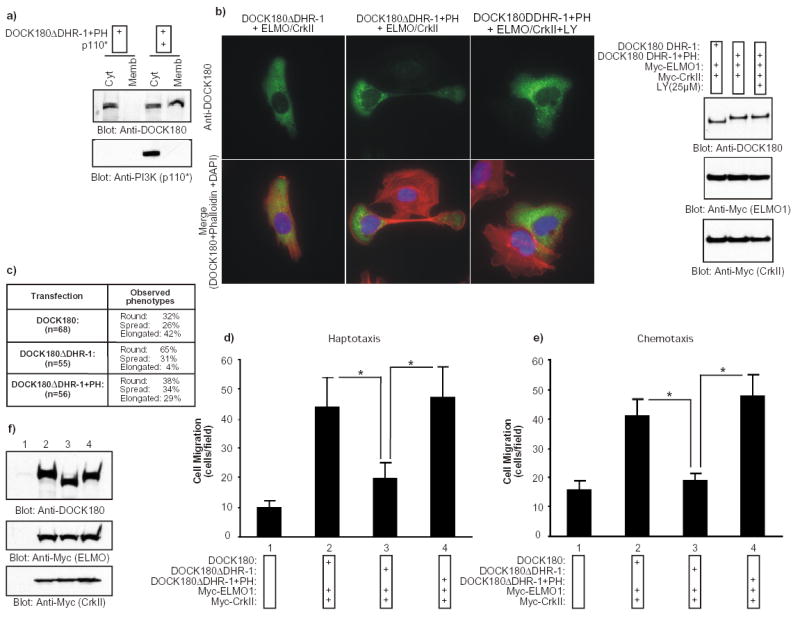
a) A chimeric DOCK180 protein (DOCK180 DHR-1+PH) in which the DHR-1 domain had been replaced with the PH domain of Bmx/Etk translocates to the membrane in response to PtdIns(3,4,5)P3 production. HEK293T cells were transfected with the indicated plasmids. After 24 h, the cytosolic and membrane fractions were biochemically purified and the distribution of DOCK180 DHR-1+PH and p110* was analyzed by immunoblotting with the indicated antibodies. b) The introduction of the PH domain of Bmx/Etk in the DHR-1 mutant rescues the cell polarization defects. Serum-starved LR73 cells expressing the indicated plasmids were detached and allowed to spread on fibronectin for 2 h. Cells were then analyzed as described in Fig 1a. When treated with LY294002, the cells were preincubated with the inhibitor for 30 min prior to plating and the inhibitor was left on the cells throughout the experiment. In the right panel, expression levels of the proteins were analyzed by immunoblotting the cell lysates with anti-DOCK180 (C19) and anti-Myc antibodies. c) Quantification of the effect on cell elongation by the DOCK180 DHR-1+PH chimeric protein. Cells were processed exactly as in (b) and several independent fields were photographed. The cells were visually inspected and scored for three phenotypes: 1- Round (attached and minimally spread cells), 2- Spread (clearly spread and flat cells) and 3- Elongated (elongated cells with a polarity). This is a representative experiment of three independent assays. d–e) The introduction of the PH domain of Bmx/Etk in the DHR-1 mutant rescues the cell migration defects. Serum-starved LR73 expressing the indicated plasmids were detached and subjected to haptotactic and chemotactic migration assays, as described in Fig. 1. Data represent mean +/- SD of a representative experiment performed in duplicate. *, P<0.001; one-way ANOVA. f) Expression levels of the transfected proteins were analyzed by immunoblotting of cell lysates with anti-DOCK180 and anti-Myc antibodies, as indicated.
In a reverse experiment, downregulation of DOCK180 by siRNA resulted in a reduction in the number of fully spread cells (70% of the cells are fully spread on fibronectin upon control siRNA-treatment vs. 54% upon DOCK180 siRNA-treatment; Fig. 2a–c). Importantly, re-expression of the wild-type DOCK180 protein in DOCK180 siRNA-treated cells significantly rescued the spread morphology and assembly of actin stress fibers (71% of the cells are spread; Fig. 2b–c). In contrast, the DOCK180ΔDHR-1 mutant failed to rescue the spread phenotype (47% of the cells are spread; Fig. 2b-c). Since total pools of transfected cells were analyzed in these studies, we also performed immunofluorescence analysis of individual transfected cells and similar results were obtained (Fig. 2d–e). 57% of DOCK180 siRNA-treated cells re-expressing high levels of DOCK180 displayed a fully spread morphology, while 60% of the DOCK180 siRNA-treated cells expressing equally high levels of the DOCK180ΔDHR-1 protein remained round (Fig. 2d–e). Taken together, these studies support the notion that the DHR-1 domain has an essential role in DOCK180 signaling.
Fig. 2. The DHR-1 domain is required for cell spreading.
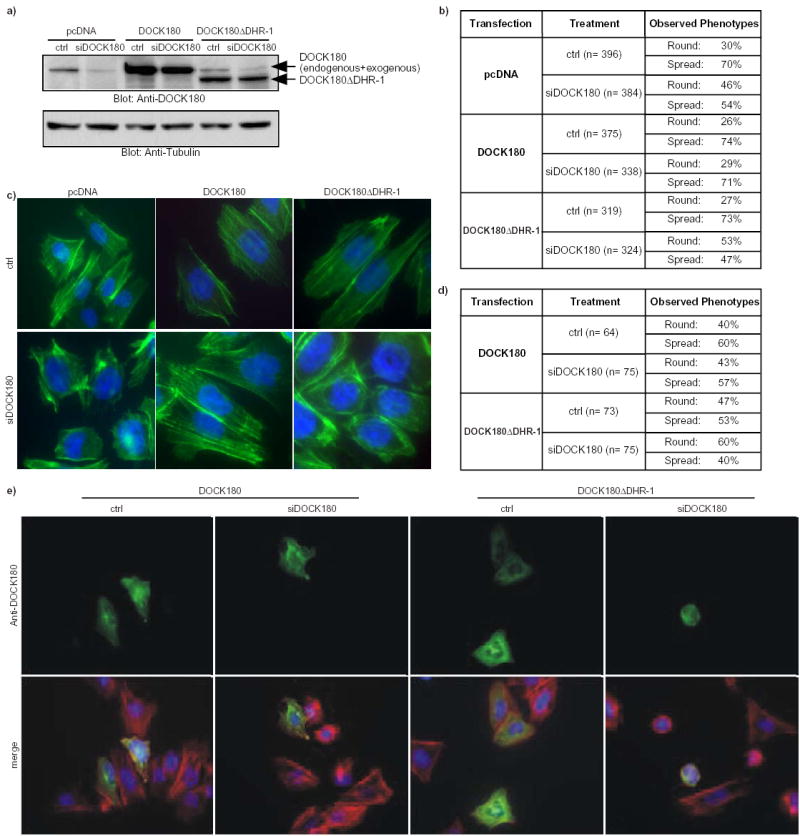
a) Immunoblot analysis with an anti-DOCK180 antibody (C-19) of total LR73 cell lysates that had been treated with either control siRNA (ctrl) or siRNA against DOCK180 (siDOCK180), and simultaneously transfected with an empty vector (pcDNA), or a vector coding for the wild-type DOCK180 or the DOCK180 DHR-1 mutant (top panel). Anti-Tubulin blot was used as a loading control (bottom panel). b–c) siRNA-mediated downregulation of DOCK180 levels leads to an impaired cell spreading which can be rescued by re-expression of wild-type DOCK180 protein but not of DOCK180 DHR-1. Micrographs of cells from three independent experiments were pooled and used to score for two phenotypes: 1-Round and 2-Spread. Quantification of the results is presented in a table format in (b). Representative micrographs in (c) are an overlay of FITC-phalloidin and DAPI stains. The cells were photographed at a 60X magnification. d–e) Cells treated as in (b–c) were stained for DOCK180 (H-4) to allow for identification and specific analysis of those re-expressing the wild-type DOCK180 or the DOCK180 DHR-1 mutant proteins. Cells with equal green fluorescent intensity were scored for the round and spread phenotypes. Quantification of the results in a representative experiment is presented in a table format in (d), while representative micrographs of the same experiment are shown in (e). In (e), upper panels show anti-DOCK180 stains, while the bottom panels are overlays of DOCK180, rhodamine-phalloidin and DAPI stains.
Impaired signaling by the DHR-1 mutant of DOCK180 is not due to reduced Rac-GTP loading
The failure of DOCK180 DHR-1 to promote cell elongation and cell migration raised the possibility that ablating the DHR-1 domain could impair the ability of DOCK180 to induce Rac GTP-loading. This was not found to be the case, as DOCK180 DHR-1 was able to promote Rac GTP-loading to the same extent as wild-type DOCK180 both in the presence and absence of ELMO1 and CrkII (Fig. 3a). As previously reported 21, the DHR-2 domain was found to be essential for DOCK180-mediated Rac GTP-loading (Fig. 3a). We found that the inability of DOCK180 DHR-1 to promote cell elongation and cell migration is not a consequence of a failure to interact with its binding partners either, as DOCK180 DHR-1 was found to associate with CrkII and ELMO1 to the same extent as wild-type DOCK180 in coimmunoprecipitation experiments (Fig. 3b).
Fig. 3. Intact GTP-loading of Rac and coupling to ELMO1 and CrkII by the DHR-1 mutant of DOCK180.
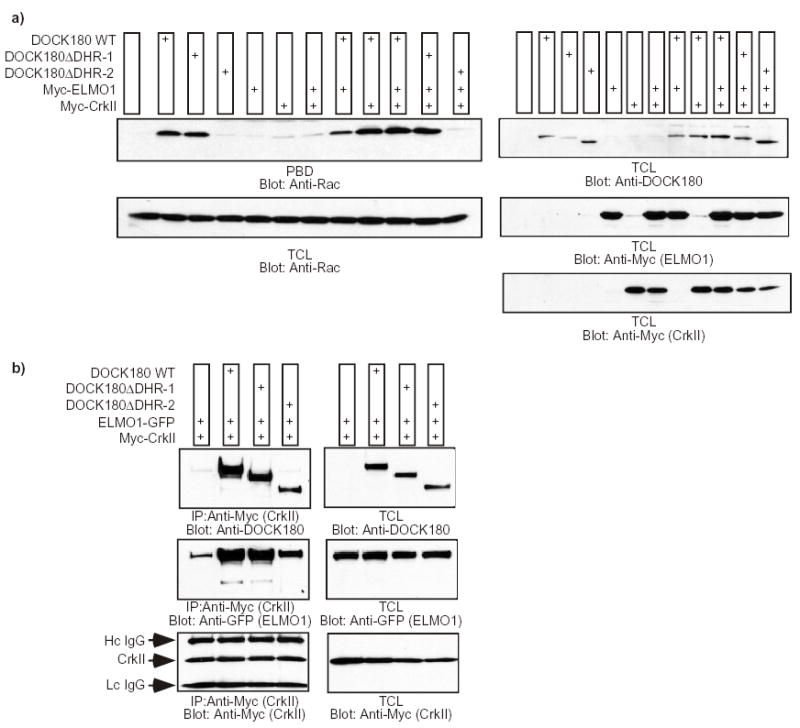
a) LR73 cells were transfected with the indicated plasmids and the GTP-loading status of Rac was analyzed by affinity precipitation with the PBD domain of PAK immobilized to glutathione-sepharose beads (upper panel). As a loading control for Rac and to verify the expression of each protein, 20 μl of total cell lysates (TCL) were analyzed by immunoblotting with antibodies against Rac, DOCK180 and Myc (for ELMO1 and CrkII). The experiment was performed independently three times, and no statistical differences were observed between the capabilities of the wild-type DOCK180 and the DOCK180ΔDHR-1 mutant to induce GTP-loading of Rac (the DOCK180ΔDHR-1 mutant demonstrated, in arbitrary units, a 0.94±0.05-fold Rac activation when co-expressed with ELMO1 and CrkII, compared to 1.0-fold activation by wild-type DOCK180, Crk and ELMO). b) DOCK180 lacking the DHR-1 forms a trimolecular complex with ELMO1 and CrkII. LR73 cells were transfected with the indicated plasmids and Triton X-100 lysates were immunoprecipitated with an antibody against the Myc-epitope. The coprecipitation of the various DOCK180 proteins and ELMO1-GFP, in addition to the verification of equal Myc-tagged CrkII precipitation, was analyzed by immunoblotting with antibodies against DOCK180 (C19), GFP and Myc, respectively (left panels). The expression levels of each protein were analyzed by immunoblotting 10 μg of the total Triton X-100 lysates with antibodies against DOCK180 (C19), GFP and Myc (right panels).
The DHR-1 domain of DOCK180 binds PtdIns(3,5)P2 and PtdIns(3,4,5)P3
We previously noted a putative C2 domain in the DHR-1 region of DOCK180 by SMART analysis 21. C2 domains mediate non-specific binding to anionic phospholipids such as phosphatidylserine in a Ca2+-regulated (type I topology) or non-regulated (type II topology) manner 25. In rare instances, C2 domains can interact with both phosphoinositides and proteins 25–28. The lipid binding activity of C2 domains resides in three loops (CBR1–3) that are enriched in basic amino acids, and we noticed such clusters of conserved basic amino acids in the putative CBR1 and CBR3 loops in the DHR-1 domain (Supplementary Information, Fig. S1). We therefore decided to investigate whether the DHR-1 domain interacts with lipids, and focused on phosphoinositides due to their known role in cell migration. When incubated with beads coated with various phosphoinositides, the DHR-1 domain interacted specifically with PtdIns(3,5)P2 and PtdIns(3,4,5)P3 (Fig. 4a). Similar binding was observed in the presence of either 1.1 mM Ca2+ or 5 mM EDTA, suggesting that the interaction is not Ca2+-regulated (not shown). In a control experiment, the PH domain of Akt was specifically precipitated by PtdIns(3,4,5)P3-beads 29 (Fig. 4a). Sequential truncations in the carboxyl-terminus of the DHR-1 domain demonstrated that the minimal lipid-binding site encompassed residues 422–619, the boundaries of the putative C2 domain being residues 422–587 (Fig. 4b).
Fig. 4. The DHR-1 domain of DOCK180 has lipid-binding activity toward phosphoinositides in vitro.
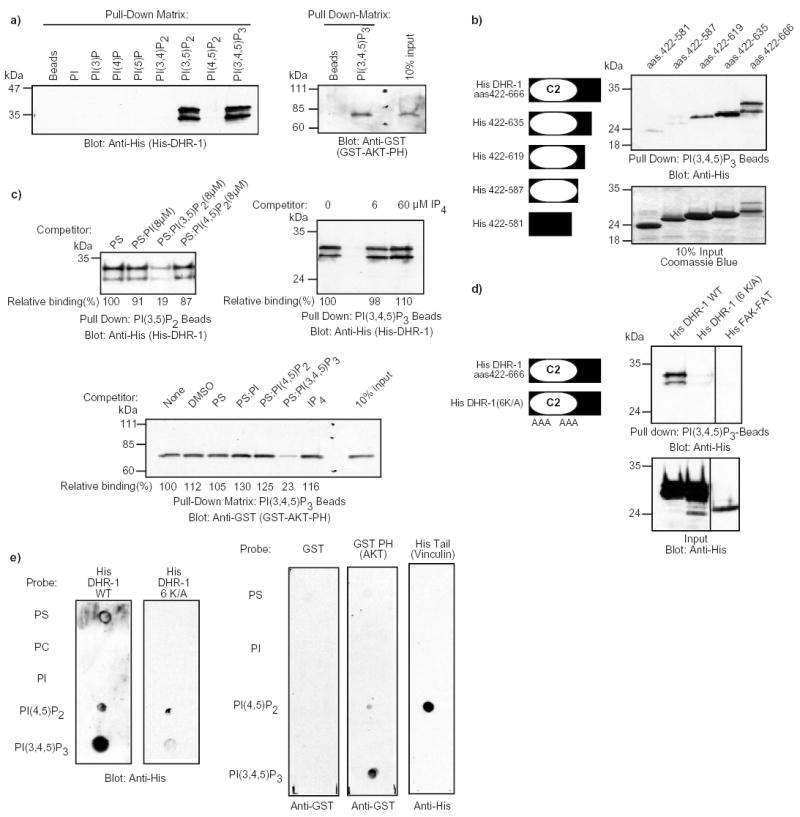
a) The DHR-1 domain of DOCK180 binds to PtdIns(3,5)P2 and PtdIns(3,4,5)P3 in vitro. 5 μg of purified recombinant His-DHR-1 or GST-Akt-PH proteins were incubated with beads coated with the indicated phosphoinositides, or as a control, with beads alone. The bound proteins were detected by immunoblotting with anti-His or anti-GST antibodies. b) Mapping of the minimal PtdIns(3,4,5)P3 binding site in the DHR-1 domain. A graphical representation of the various DHR-1 domain constructs is shown on the left. The carboxyl-terminal truncations of the DHR-1 domain were tested for their ability to interact with beads coated with PtdIns(3,4,5)P3 and the bound material was detected as described in (a). Fractions of the purified proteins were analyzed on SDS-PAGE and stained with Coomassie Blue as a control. c) Liposome competition assay. The DHR-1 domain of DOCK180 and the PH domain of Akt were preincubated with various liposomes, or Ins(1,3,4,5)P4, prior to a pull-down experiment with the indicated lipid-coated beads. The bound material was detected as described in (a). Relative binding is indicated. d) Mutation of six conserved lysines residues in the putative CBR1 and CBR3 loops of the DHR-1 domain abolishes the lipid binding activity. A graphical representation of the DHR-1 mutant is shown on the left. The indicated proteins were purified and subjected to binding to PtdIns(3,4,5)P3-beads, as described in (a). In the lower panel, the purified proteins were analyzed on SDS-PAGE and stained with Coomassie Blue as a control. FAK-FAT, focal adhesion targeting motif of FAK. e) Protein-lipid overlay assay. Lipid-spotted membranes were overlayed with the indicated purified proteins. Bound proteins were detected by immunoblotting with anti-His and anti-GST antibodies, as indicated.
A liposome competition assay demonstrated that the binding between the DHR-1 domain and the PtdIns(3,4,5)P3-beads is specific. Preincubation of DHR-1 with phosphatidylserine (PS) liposomes containing 8 μM PtdIns(3,5)P2 inhibited the binding of DHR-1 to the PtdIns(3,5)P2- beads by approximately 80% (Fig. 4c), and this effect was dose-dependent (not shown). Liposomes of PS alone, PS containing either 8 μM PtdIns or PtdIns(4,5)P2, or Ins(1,3,4,5)P4 did not significantly compete the DHR-1 binding. In a control experiment, PS:PtdIns(3,4,5)P3-liposomes specifically disrupted the binding of the PH domain of Akt to the PtdIns(3,4,5)P3-beads (Fig. 4c).
A mutant form of DHR-1 with a predicted loss of function in the PtdIns(3,4,5)P3-binding activity was generated. The mutated residues were selected based on sequence alignment between the DHR-1 domains of the DOCK180 superfamily (Supplementary Information, Fig. S1), and between the DOCK180 DHR-1 domain and the C2 domain of PtdIns 3-Kinase γ (not shown). In the construct DHR-1(6K/A), six lysines, three each in the putative CBR1 and CBR3 loops, were mutated to alanine (see Methods). We hypothesized that these positively charged residues were directly involved in coordinating PtdIns(3,4,5)P3. When subjected to a pull-down with the PtdIns(3,4,5)P3-beads, DHR-1(6K/A) exhibited little lipid binding activity (Fig. 4d). A negative control, the FAT domain of FAK 30 similarly failed to interact with the PtdIns(3,4,5)P3- beads. Importantly, cells expressing a full-length form of DOCK180 in which the same six lysines had been mutated to alanine [DOCK180(6K/A)], together with ELMO1 and CrkII, failed to display an elongated morphology (Supplementary Information, Fig. S2).
Lipid-blot overlay assays 31 were performed to confirm the PtdIns(3,4,5)P3-binding activity of the DHR-1 domain. In agreement with the bead data, the wild-type form of the DHR-1 domain readily interacted with PtdIns(3,4,5)P3, while the DHR-1(6 K/A) failed to do so. In control experiments, the PH domain of Akt 29 and the tail-domain of Vinculin 32 interacted with PtdIns(3,4,5)P3 and PtdIns(4,5)P2, respectively (Fig. 4e). Taken together, these data suggest that the DHR-1 domain is a lipid binding module that is necessary for the DOCK180 function.
The DHR-1 domain mediates DOCK180 membrane translocation in response to PtdIns(3,4,5)P3 production
Several lines of investigation were undertaken to examine the role of the DHR-1 domain in lipid binding in cells, as well as the significance of PtdIns(3,4,5)P3 in DOCK180 signaling. First, clarified lysates of cells expressing either the wild-type DOCK180 or the DOCK180 DHR-1 mutant were subjected to a pull-down by the PtdIns(3,4,5)P3-beads. While the wild-type DOCK180 protein readily interacted with the beads, DOCK180 DHR-1 was only weakly precipitated in this assay (Fig. 5a). These data suggest that the majority of the PtdIns(3,4,5)P3-binding activity of DOCK180 can be attributed to the DHR-1 domain.
Fig. 5. The DHR-1 domain displays lipid binding activity in vivo.
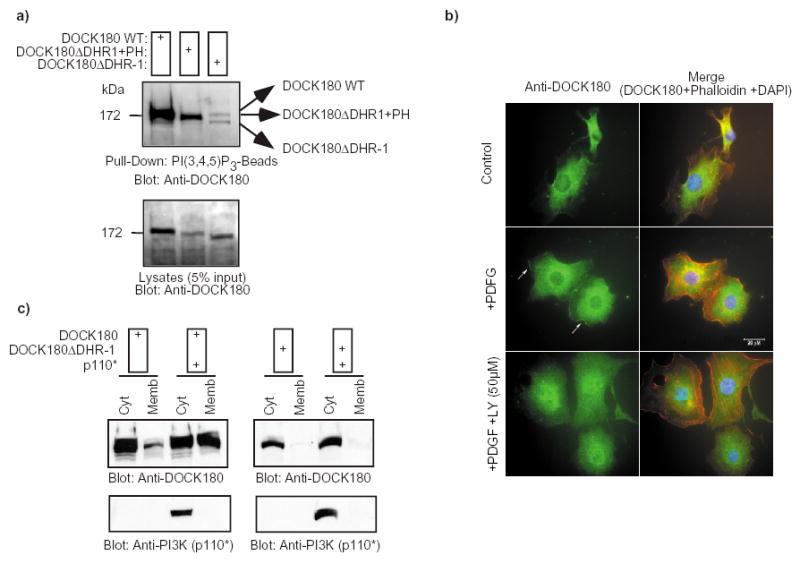
a) The DOCK180 DHR-1 mutant fails to interact with PtdIns(3,4,5)P3. DOCK180 WT, DOCK180 DHR-1 or DOCK180 DHR-1+PH (see Fig. 7 and supplemental information, Fig. S1) were expressed in LR73 cells. Cell lysates were subjected to a pull-down by PtdIns(3,4,5)P3-beads. Bound proteins were detected by immunoblotting with an anti-DOCK180 antibody (C19). The lower panels demonstrate the expression levels of the exogenous DOCK180 proteins. b) Endogenous DOCK180 localizes to the plasma membrane in response to PDGF stimulation in a PtdIns 3-kinase dependent manner. Serum-starved NIH3T3 cells were treated with either DMSO (top and middle panels) or 50 μM LY294002 for 30 min (bottom panels). Cells were subsequently left untreated (top panels) or stimulated with 10 ng/ml PDGF (middle and bottom panels) for 10 min prior to fixing. Cells in the left panels were stained with an anti-DOCK180 rabbit polyclonal antibody, while the panels on the right represent an overlay of the anti-DOCK180, rhodamine-phalloidin and DAPI stains. Cells were photographed at a 60X magnification. c) DOCK180 translocates to the membrane in response to PtdIns(3,4,5)P3 production in a DHR-1-dependent manner. HEK293T cells were transfected with the indicated plasmids. After 24 h, the cytosolic and membrane fractions were biochemically purified and the distribution of DOCK180, DOCK180 DHR-1 and p110* was analyzed by immunoblotting with the indicated antibodies.
We next investigated whether the endogenous DOCK180 protein would translocate to the membrane in response to PtdIns 3-kinase activation. DOCK180 was readily recruited to the membrane in NIH 3T3 cells that had been treated with PDGF to activate PtdIns 3-kinase 33. Importantly, this membrane recruitment was blocked by a pre-treatment of the cells with the PtdIns 3-kinase inhibitor LY294002 (Fig. 5b). The importance of the DHR-1 domain for DOCK180 membrane translocation was subsequently addressed. As an experimental system, an active form of PtdIns 3-kinase, p110* 34, was used to induce PtdIns(3,4,5)P3 production. The wild-type DOCK180 protein was primarily localized in the cytosol when expressed alone (Fig. 5c). When coexpressed with p110*, it readily localized to the membrane fraction. This translocation was found to be dependent on the DHR-1 domain, as the DOCK180 DHR-1 protein remained cytosolic both in the presence and absence of p110*. As a control for fractionation, p110* was found exclusively in the cytosolic fraction, in agreement with the reported localization of this mutant protein 34 (Fig. 5c). These results demonstrate that the DHR-1 domain is critical in mediating PtdIns 3-kinase-dependent translocation of DOCK180 to the cell membrane.
LY294002 was also used to examine the importance of PtdIns(3,4,5)P3 in DOCK180-induced cell elongation and migration. As shown in Fig. 6a, cells expressing DOCK180, ELMO1 and CrkII that had been treated with LY294002 failed to adopt an elongated phenotype when plated on fibronectin (compare to Fig. 1a). LY294002 also abrogated the ability of the CrkII-ELMO-DOCK180 complex to promote significant cell movement, as shown in Fig. 6b. These findings further support the role of PtdIns(3,4,5)P3 in CrkII-ELMO-DOCK180 signaling.
Fig 6. Generation of PtdIns(3,4,5)P3 is necessary for DOCK180-induced cell elongation and migration.
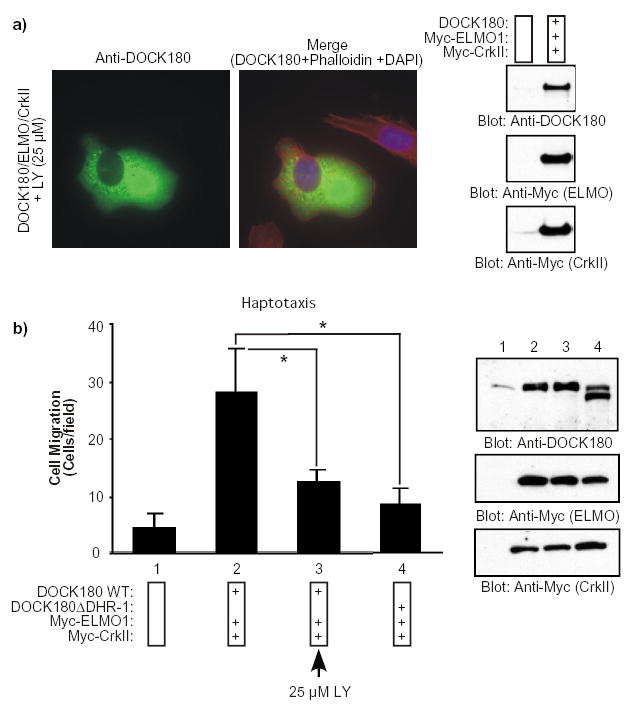
a) CrkII-ELMO1-DOCK180-induced cell elongation is sensitive to the PtdIns 3-kinase inhibitor LY294002. Serum-starved LR73 cells expressing the indicated plasmids were detached and incubated with the LY294002 inhibitor (25 μM) for 30 min in suspension. The cells were then allowed to spread for 2 h on fibronectin-coated slides in the presence of LY294002, fixed and stained with anti-DOCK180 (H-4) antibody, Rhodamine-phalloidin and DAPI. Photographs were taken at a 60X magnification. (Compare to Fig. 1a). The expression levels of the transfected proteins were analyzed by immunoblotting of total cell lysates with anti-DOCK180 (C19) and anti-Myc antibodies, as indicated (right panel). b) CrkII-ELMO1-DOCK180-induced cell migration is sensitive to LY294002. Serum-starved LR73 cells expressing the indicated plasmids, together with a plasmid for GFP, were subjected to a haptotactic migration assay. For the sample treated with 25 μM of LY294002, the inhibitor was added to both the upper and the lower compartment of the modified Boyden chamber. Cell migration was analyzed as described in Fig. 1a. Data represent mean +/− SD of a representative experiment performed in duplicate. *, P<0.001; one-way ANOVA. In the lower panel, expression levels of the transfected proteins were analyzed by immunoblotting of cell lysates with anti-DOCK180 (C19) and anti-Myc antibodies, as indicated.
The CrkII-ELMO1-DOCK180 complex is enriched in membrane extensions during cell elongation and colocalizes with PtdIns(3,4,5)P3
We next investigated whether the CrkII-ELMO-DOCK180 complex localizes to sites of membrane extensions by co-expression of differentially tagged forms of DOCK180, CrkII and ELMO. In sequential coimmunoprecipitation experiments, we first observed that more than 90% of both endogenous and exogenously expressed ELMO and DOCK180 proteins are found in the same complex (Supplementary Information, Fig. S2). Thus, the localization of GFP-ELMO1 or RFP-ELMO1 was subsequently used as a marker for the CrkII-ELMO-DOCK180 complex in cells. As shown in a time-lapse analysis (Supplementary Information, Video S1 and Fig. S3), GFP-ELMO was enriched in membranes undergoing dynamic rearrangements during cell elongation on fibronectin. Once the leading edge had been established (Supplementary Information, Fig S3, t=14’), less membrane ruffling, and localization of ELMO1 at the leading edge, was observed. Similarly, transfected DOCK180 localized at the leading edge in membrane ruffles of cells that had been allowed to spread for 60 min on fibronectin (Supplementary Information, Fig. S2), but once the leading edge had been established, less localization of DOCK180 at the membrane ruffles was observed (Fig. 1a and not shown).
To examine whether the CrkII-ELMO-DOCK180 complex colocalizes with PtdIns(3,4,5)P3 at the leading edge, Flag-tagged DOCK180, Myc-tagged CrkII and RFP-tagged ELMO1 were co-expressed with the GFP-tagged PH domain of BTK, a validated in vivo probe for PtdIns(3,4,5)P3. COS-7 cells were chosen in this experiment to allow the use of EGF as a positive control for the recruitment of the PH domain of BTK to the membrane. As shown in a time-lapse movie (Supplementary Information, Video S2 and Fig. S3), colocalization of the RFP-ELMO1 protein and the GFP-BTK-PH domain was observed in membrane ruffles at the leading edge upon cell elongation on fibronectin for 1 h. When the same cell was subsequently stimulated with EGF for 10 min, membrane ruffling occurred randomly throughout the cell, leading to colocalization of RFP-ELMO1 and GFP-BTK-PH at these sites. These results suggest that the CrkII-ELMO-DOCK180 complex localizes to PtdIns(3,4,5)P3 accumulation sites, which agrees with our biochemical data demonstrating that DOCK180 translocates to the membrane in response to PtdIns(3,4,5)P3 generation.
A PH domain with PtdIns(3,4,5)P3-binding activity can functionally replace the DHR-1 domain in DOCK180
To determine whether the main function of the DHR-1 domain of DOCK180 during cell elongation and migration is to interact with PtdIns(3,4,5)P3, we generated a DOCK180 construct in which the DHR-1 domain was replaced with the PtdIns(3,4,5)P3-binding PH domain of the BMX/Etk tyrosine kinase 35 (Supplementary Information, Fig. S1). In control experiments, we found that both the isolated PH domain of BMX/Etk and the chimeric DOCK180 DHR-1+PH protein bound to beads coated with PtdIns(3,4,5)P3 (Fig. 5a, Supplementary Information, Fig. S4). We also confirmed that DOCK180 DHR-1+PH promoted Rac GTP-loading and CrkII-ELMO-DOCK180 complex formation to the same extent as wild-type DOCK180 (Supplementary Information, Fig. S4). Additionally, coexpression of p110* resulted in translocation of DOCK180 DHR-1+PH, similar to wild-type DOCK180, to the membrane compartment (Fig. 7a).
In subsequent functional studies, we found that when coexpressed with ELMO1 and CrkII, DOCK180 DHR-1+PH was capable of promoting cell elongation (Fig. 7b). Furthermore, cell extension promoted by DOCK180 DHR-1+PH was sensitive to PtdIns 3-kinase inhibition (Fig. 7b). Cells expressing DOCK180 DHR-1+PH were morphologically indistinguishable from the cells expressing wild-type DOCK180. Quantitatively, we found that approximately 42% of the cells expressing wild-type DOCK180 and 29% of the cells expressing the PH domain chimera had an elongated morphology, while only 4% of the DOCK180 DHR-1-transfected cells displayed this phenotype (Fig. 7c). As shown in Fig. 7d–e, the DOCK180 DHR-1+PH protein was found to fully recapitulate the function of the wild-type DOCK180 protein both in haptotactic and chemotactic migration assays. These results demonstrate that a canonical PtdIns(3,4,5)P3-binding domain can functionally replace the DHR-1 domain during DOCK180-mediated cell elongation and cell migration. These results also support our conclusion that the role of the DHR-1 domain, via PtdIns(3,4,5)P3 binding, is to couple PtdIns 3-kinase signaling to the regulation of the CrkII-ELMO-DOCK180 complex in processes that require localized Rac activation at sites of membrane extensions, such as cell motility.
Discussion
DOCK180 and its orthologs are essential activators of various Rac GTPase-dependent biological processes 14–17,21,23. We and others have demonstrated that DOCK180 promotes GTP-loading of Rac through an atypical GEF domain termed the DHR-2 domain 21,23. We report here that another conserved domain in DOCK180-related proteins, DHR-1, is also indispensable for the biological function of DOCK180, as it couples the production of PtdIns(3,4,5)P3 at the membrane to DOCK180-mediated Rac GTP loading.
We found that forms of DOCK180 which either lack DHR-1 or contain point mutations in that domain fail to induce cell elongation and directional cell movement. The DOCK180 DHR-1 protein was found to induce Rac GTP-loading to the same extent as the wild-type DOCK180, supporting the notion that GTP-loading of Rac alone is not sufficient to induce processes that require cell polarization 36. Instead, it is known that formation of a PtdIns(3,4,5)P3 gradient at the membrane mediates Rac-dependent actin reorganization at the leading edge, which is required for directional cell movement 4–6. What had remained unclear is how PtdIns(3,4,5)P3 integrates with DOCK180 signaling to regulate Rac function upon cell polarization. We report here that the DHR-1 domain directly binds to PtdIns(3,4,5)P3. Furthermore, a form of DOCK180 in which the DHR-1 domain had been replaced by a canonical PtdIns(3,4,5)P3 binding PH domain efficiently mediated cell elongation and cell migration. Thus, the two conserved domains in DOCK180, DHR-1 and DHR-2, function to integrate PtdIns(3,4,5)P3 signaling with Rac GTP-loading, and this integration is essential for membrane polarization and cell migration to occur.
Our finding that DOCK180 translocates to the plasma membrane in response to PtdIns(3,4,5)P3 production is in agreement with a report by Kobayashi et al. 37. These authors suggested that a cluster of basic amino acids in the carboxyl-terminus of DOCK180 could mediate an ionic interaction with phosphate residues of PtdIns(3,4,5)P3. Kobayashi et al. noted, however, that this ionic interaction is not sufficient for the binding of full-length DOCK180 to PtdIns(3,4,5)P3 because Ins(1,3,4,5)P4 (which inhibits an ionic interaction) cannot compete with PtdIns(3,4,5)P3 binding to full-length DOCK180 37. Thus, region(s) other than the basic domain must participate in the recognition of PtdIns(3,4,5)P3 and DOCK180 membrane translocation. Our studies identify the DHR-1 domain as this region since we found that it binds to PtdIns(3,4,5)P3 in a non-ionic manner, and that it is essential for PtdIns 3-kinase-induced membrane translocation of DOCK180.
The structural basis of the DHR-1 domain-mediated PtdIns(3,4,5)P3 binding is presently unknown. Identification of a putative C2 domain within the DHR-1 domain is interesting, but requires experimental verification. C2 domains typically interact with lipids such as phosphatidylserine and phosphatidylcholine: we failed to detect such interactions for DHR-1. Recently, the C2 domains of JFC1 and Rsb5 proteins have been reported to bind rather promiscuously to several phosphoinositides, including PtdIns(3,4,5)P3 26,28. Therefore, subsets of C2 domains are emerging as genuine phosphoinositide-binding modules. Our finding that point mutations in the putative CBR1 and CBR3 loops in the DHR-1 domain abolish both the lipid binding activity and the function of the protein provide supporting evidence for the presence of a C2-like domain within DHR-1. Since the sequences of the DHR-1 domain are conserved across the DOCK180-related proteins, it is plausible that the lipid binding function is also conserved. Indeed, in vitro experiments demonstrate that several other DHR-1 domains [those of DOCK3, DOCK6 and DOCK9 (zizimin1)] also have lipid-binding activity (not shown).
Our results and previous studies by others demonstrate that the presence of all three components of the CrkII-ELMO-DOCK180 complex is required for cell elongation and migration 15,21,23. Our studies here provide a molecular explanation for the role of DOCK180 in this complex, that of coupling PtdIns(3,4,5)P3 signaling with Rac GTP-loading. Going forward, it will be important to identify the biological events regulated by this multiprotein complex that are PtdIns(3,4,5)P3-dependent. Also of note, our studies do not rule out the possibility that ELMO1 and CrkII could provide membrane targeting activity. For example, it has been suggested that the PH domain of Ced-12 (ELMO) is required for membrane targeting and protein function in C. elegans 17. We have also detected lipid binding activity in the PH domain of ELMO (not shown), but further studies will be required to determine the specificity of this domain and the biological function of this activity. Additional work is needed to understand the full biological significance of the CrkII-ELMO-DOCK180 protein complex and its regulation in cellular signaling.
Methods
Constructs, reagents and antibodies
Flag-tagged DOCK180 in pCNX2 and Myc-tagged CrkII in pCAGGS have been described previously 38 and were obtained from Dr. M. Matsuda. DOCK180 DHR-2 in pcDNA3.1(Zeor) and Myc-tagged ELMO1 in pcDNA3.1 have been described in 21. pEGFP-ELMO1 15 was obtained from Dr. K. Ravichandran. pEGFP-BTK PH domain was a gift from Dr. T. Mustelin. The plasmid p110* has been described in 34 and was a gift from Dr. J. Olefsky. The FAT domain of FAK in pET has been described in 30. The various His-DOCK180 DHR-1 constructs for bacterial expression were generated by PCR using the DOCK180 cDNA as a template, and cloned to the NdeI/BamHI-site of pET28a (Novagen). The DOCK180 DHR-1 construct was generated by replacing a NotI/BglII-fragment, coding for residues 1–602, of the wild-type DOCK180 in pcDNA3.1(Zeor) with a PCR-generated NotI-BglII-fragment coding for residues 1–421 of human DOCK180. The result was an in-frame deletion of the sequences coding for residues 422–602 of DOCK180. To generate DOCK180 DHR-1+PH in pcDNA3.1(Zeor), the PH domain-coding sequences of BMX/Etk, residues 1–150 of human Bmx/Etk 39, were amplified by PCR and ligated into the BglII site of DOCK180 DHR-1 (see above). This resulted in a construct coding for a chimeric protein in which the residues 422–602 of DOCK180 were replaced by the PH domain of BMX/Etk. The PH domain of BMX/Etk was also subcloned in the bacterial expression vector pGEX4T1. The pET28 DHR-1 mutant (K439,442,446,522,524,527A) and the full length DOCK180 with the 6K/A mutations were generated by site directed mutagenesis (QuickChange, Stratagene).
Fibronectin was purified from human serum as described in 40 and used at a 10 μg/ml concentration to coat tissue culture surfaces. The following reagents were obtained from commercial sources: LY294002 (Calbiochem); Ins(1,3,4,5)P4 (Sigma); PtdIns(3,5)P2, PtdIns(3,4,5)P3 and PIPn-beads (Echelon Bioscience); Phosphatidylserine, Phosphatidylcholine, PtdIns and PtdIns(4,5)P2 (Avanti Polar Lipids); anti-DOCK180 (C-19 and H-4), anti-Myc (9E10), anti-GST-HRP (Z-5) and anti-GFP (B-2) antibodies (Santa Cruz Biotechnologies); monoclonal antibodies against CrkII and the p85 subunit of PtdIns 3-kinase (Pharmingen), anti-His antibody (Qiagen); polyclonal antibody against ELMO1 (Abcam); Rac monoclonal antibody 23A8 (Upstate); mouse monoclonal anti-Tubulin antibody Ab-1 (Calbiochem); the control and DOCK180 mouse specific siRNAs (Santa Cruz Biotechnologies); purified GST PH domain of AKT (Upstate). The recombinant Vinculin tail has been described in 32 and was obtained from Dr. R. Liddington. The polyclonal antibody against DOCK180 was a gift from Dr. M. Matsuda.
Cell culture and transfections
LR73 cells were maintained in Alpha MEM supplemented with 10% fetal bovine serum, penicillin and streptomycin. HEK293-T, COS-7 and NIH3T3 cells were cultured in DMEM supplemented with 10% fetal serum, penicillin and streptomycin. 750,000 LR73 cells were plated in a 6-well plate 24 h prior to transfection. 2–2.5 μg of plasmids and 8 μl of Lipofectamine2000 (Invitrogen), mixed in OptiMEM media, were used for transfection as per manufacturer’s instructions. Cells were analyzed 24 h later. 1X106 HEK293-T cells were plated in a 6-well plate 24 h prior to transfection. The cells were transfected with up to 4 μg of plasmid DNA and 8 μl of Cytopure reagent (Q-Biogen) in OptiMEM media as per manufacturer’s instructions. Cells were analyzed 24 h later. For siRNA experiments, 2.2X105 LR73 cells were seeded/well in a 12-well plate in an antibiotic-free medium. The following day, cells were co-transfected with plasmids encoding for DOCK180 (1.0 μg), DOCK180 DHR-1 (2.0 μg) or empty vector, and 80–100nM of siRNA against mouse DOCK180 (SC-35208) or control siRNA (SC-45037). Transfections were performed in triplicate, using 2 μl of Lipofectamine2000 (Invitrogen) and 500 μl of OptiMEM media per well, according to manufacturer’s instructions. 18–20 h post-transfection, cells were starved in alpha-MEM containing 0.5% FBS overnight and duplicates were harvested for cell morphology assays (see below). Cells from the third well were lysed in RIPA buffer, and 15–20 μg of the cleared lysates were used for immunoblot analysis.
Immunoprecipitations and Rac-GTP assays
Cells were lysed in a buffer consisting of 25 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 5 mM NaF, 0.1 mM Na3VO4 and 1x Complete protease inhibitors (Roche) for 10 min. Clarified cell lysates were incubated with the appropriate antibody and immune complex formation was carried out for 1 h on ice. Protein A-sepharose was added for 30 min to isolate the immune complex. The beads were washed three times with lysis buffer and the bound proteins were analyzed by SDS-PAGE and immunoblotting. In one experiment, we used 0.5% NP-40 and RIPA buffers to isolate the soluble and membrane bound proteins, respectively, as described in 41. The GTP-loading status of Rac was analyzed by GST PAK-PBD affinity precipitation as described previously 21.
Cell morphology analysis and time-lapse microscopy
LR73 cells, treated with various siRNAs or transfected with the indicated plasmids were serum-starved overnight in media supplemented with 0.5% serum. A typical transfection consisted of a total of 1.5 μg of plasmid DNA (0.9 μg of DOCK180, 0.3 μg of ELMO1 and 0.3 μg of CrkII), and, for the various experimental conditions, the amount of total plasmid was always kept constant by adding empty vector. The day of the assay, the cells were detached and harvested in 1 X Hanks balanced salt solution supplemented with 5 mM EDTA and 0.1% trypsin, followed by one wash with Fibroblast Basal Media (FBM, Clonetics) supplemented with 0.5% BSA (Sigma). Cells were resuspended in 600 μl of FBM-BSA, counted and adjusted to 1X106 cells/ml. From each sample, 100,000 cells were plated on 4-well chamber slides (LabTek) that had been pre-coated with fibronectin (10 μg/ml). The cells were allowed to spread for 2 h before fixing with 4% paraformaldehyde. The fixed material was permeabilized by incubation with PBS-0.2% Triton X-100 for 10 min followed by blocking in PBS-1% BSA for 30 min. Cells were stained with anti-DOCK180 antibody (H-4) at a 1:100 dilution in blocking buffer for 1 h. Following several PBS washes, a secondary mouse antibody coupled to Alexa 488 (Molecular Probes) was added at a 1:100 dilution in blocking buffer for 30 min. Actin was stained with Rhodamine-Phalloidin (5 μl/well, Molecular Probes), and the nuclei were stained with DAPI (Molecular Probes) for 30 min. Chamber slides were mounted with a coverslip in the presence of the antifade reagent DABCO (FUKA) dissolved in 30% glycerol. Cells were analyzed and photographed at a 60X magnification using a Nikon inverted microscope. For endogenous DOCK180 localization, NIH3T3 cells were starved for 16 h prior to treatment with either DMSO or 50 μM LY294002, as indicated in the figure legend. The cells were subsequently treated or not with 10 ng/ml of PDGF for 20 min, fixed and stained as described above. The DOCK180 polyclonal antibody was used at a 1:75 dilution in the immunofluorescence analysis. For time-lapse microscopy, images were acquired on an Olympus microscope with an automatic stage performing the objective changes (Nomarski, confocal Green and confocal Red). Cells were photographed at a 40 X magnification. The video reconstitution of the images was created in Metamorph.
Cell migration assays
LR73 cells were transfected and prepared as described above with the exception that a GFP plasmid (0.5–1 μg per well) was included in these experiments as a marker for transfected cells. A typical transfection consisted of a total of 2–2.5 μg of plasmid DNA (0.9 μg of DOCK180, 0.3 μg of ELMO1, 0.3 μg of CrkII and 0.5–1 μg of pEGFP), and the amount of total plasmid was always kept constant by adding empty vector. The migration assays were performed in modified Boyden chambers (COSTAR, 8 μm pore size). For haptotactic assays, the underside of the Boyden chambers were pre-coated with fibronectin (10 μg/ml) for 2 h before being transferred to wells containing FBM-BSA. For chemotactic assays, the Boyden chambers were pre-coated on both sides with fibronectin (10 μg/ml) and were transferred to wells containing FBM-BSA + 10% serum. To initiate the migration assays, 100,000 cells were added in the upper chambers of the Boyden chambers, in duplicate, and allowed to migrate for 4–6 h. Cells were then fixed in 4% paraformaldehyde for 10 min, stained with DAPI and the remaining cells in the upper chambers were mechanically removed using cotton swabs. The membranes were mounted on coverslips and the GFP and DAPI-positive cells that had migrated to the underside of the membranes were photographed (40X, inverted Nikon microscope) and counted. At least 3 independent fields per membrane were analyzed in each sample. One-way analysis of variance (ANOVA) and All Pairwise Multiple Comparison Procedures (Holm-Sidak method) were performed for statistical analysis using the SygmaStat 3.1 software package. Typically, n=6 for each data set, alpha level was 0.05 and a normality test was performed to ensure normal data distribution in each experiment. The transfection efficiency was monitored by replating an aliquot of the cells from each condition on fibronectin-coated slides for 4 h. Aliquots were also analyzed by western blotting to monitor protein expression levels.
Lipid binding assays
His-tagged DHR-1 constructs in pET28a were expressed in the BL21 strain and the induced cell lysates were mixed with a Nickel-Agarose matrix (Sigma). Bound material was washed extensively with 10 column volumes of Tris-HCl pH 8.0/0.5 M KCl/20 mM Imidazole and 2 column volumes of Tris-HCl-pH 8.0/1 M KCl) prior to elution in Tris-HCl pH 8.0/100 mM KCl/300 mM Imidazole. The GST PH domain of BTK was purified on glutathione sepharose (GibcoBRL) beads and eluted in PBS/10 mM glutathione at pH 8.0, as recommended by the manufacturer. Fractions containing the purified proteins were pooled and the amounts of proteins were immediately quantified by SDS-PAGE/Coomassie staining, using BSA as a standard. Approximately 5 μg of the proteins were then diluted in 500 μl of PtdInsP beads binding buffer (10 mM HEPES pH 7.5, 150 mM NaCl, 0.25% NP-40). The diluted proteins were mixed with 20 μl of packed beads coated with the various lipids and incubated at room temperature for 1 h. The beads were then washed three times in binding buffer. The bound material was subjected to SDS-PAGE and detected by immunoblotting using the anti-His or anti-GST antibodies. Alternatively, the proteins were used to probe lipid-spotted Hybond C-Extra membranes, as previously described 31. Bound proteins were detected by immunoblotting with the anti-His and anti-GST specific antibodies.
Cell fractionation
Cell fractionation was performed as described in 42. Briefly, 1x106 HEK293-T cells that had been transfected with the indicated plasmids were harvested by gently suspending in PBS and the suspension was centrifuged at 800g for 5 min to collect the cells. The cells were then suspended in 300 μl of Buffer A (20 mM Tris-HCl pH 7.5/150 mM NaCl/5 mM NaF/1 mM Na3VO4/Complete protease inhibitors), subjected to a single freeze/thaw cycle in liquid nitrogen and 37oC water bath followed by centrifugation at 16,000g for 10 min. The supernatant was collected and termed the “cytosolic” fraction. The pellet was washed with 500 μl of Buffer A before extraction with Buffer A + 1% Triton X-100. The clarified supernatant was termed the “membrane” fraction. Equal amounts of proteins (typically between 15–20 μg) were resolved by SDS-PAGE and the proteins of interest were analyzed by immunoblotting with specific antibodies.
Supplementary Material
Acknowledgments
We thank Drs. M. Hurwitz, H.R. Horvitz, D. Schlaepfer, and H. Katoh for useful discussions. We acknowledge the expert technical help provided by Dr. E. Monosov for microscopy experiments. J.-F. C. is a Research Fellow of The Terry Fox Foundation/National Cancer Institute of Canada. This work was supported by grants from the National Institutes of Health (to K. V.).
References
- 1.Merlot S, Firtel RA. Leading the way: Directional sensing through phosphatidylinositol 3-kinase and other signaling pathways. J Cell Sci. 2003;116:3471–3478. doi: 10.1242/jcs.00703. [DOI] [PubMed] [Google Scholar]
- 2.Postma M, Bosgraaf L, Loovers HM, Van Haastert PJ. Chemotaxis: signalling modules join hands at front and tail. EMBO Rep. 2004;5:35–40. doi: 10.1038/sj.embor.7400051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Funamoto S, Meili R, Lee S, Parry L, Firtel RA. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 2002;109:611–623. doi: 10.1016/s0092-8674(02)00755-9. [DOI] [PubMed] [Google Scholar]
- 4.Wang F, et al. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol. 2002;4:513–518. doi: 10.1038/ncb810. [DOI] [PubMed] [Google Scholar]
- 5.Weiner OD, et al. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol. 2002;4:509–513. doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- 7.Wennerberg K, Der CJ. Rho-family GTPases: it's not only Rac and Rho (and I like it) J Cell Sci. 2004;117:1301–1312. doi: 10.1242/jcs.01118. [DOI] [PubMed] [Google Scholar]
- 8.Takenawa T, Miki H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci. 2001;114:1801–1809. doi: 10.1242/jcs.114.10.1801. [DOI] [PubMed] [Google Scholar]
- 9.Hasegawa H, et al. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16:1770–1776. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. 2001;107:17–26. doi: 10.1016/s0092-8674(01)00502-5. [DOI] [PubMed] [Google Scholar]
- 11.Erickson MR, Galletta BJ, Abmayr SM. Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J Cell Biol. 1997;138:589–603. doi: 10.1083/jcb.138.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nolan KM, et al. Myoblast city, the Drosophila homolog of DOCK180/CED-5, is required in a Rac signaling pathway utilized for multiple developmental processes. Genes Dev. 1998;12:3337–3342. doi: 10.1101/gad.12.21.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rushton E, Drysdale R, Abmayr SM, Michelson AM, Bate M. Mutations in a novel gene, myoblast city, provide evidence in support of the founder cell hypothesis for Drosophila muscle development. Development. 1995;121:1979–1988. doi: 10.1242/dev.121.7.1979. [DOI] [PubMed] [Google Scholar]
- 14.Reddien PW, Horvitz HR. The Engulfment Process of Programmed Cell Death in Caenorhabditis elegans. Annu Rev Cell Dev Biol. 2004;20:193–221. doi: 10.1146/annurev.cellbio.20.022003.114619. [DOI] [PubMed] [Google Scholar]
- 15.Gumienny TL, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001;107:27–41. doi: 10.1016/s0092-8674(01)00520-7. [DOI] [PubMed] [Google Scholar]
- 16.Wu YC, Tsai MC, Cheng LC, Chou CJ, Weng N. Y C elegans CED-12 acts in the conserved crkII/DOCK180/Rac pathway to control cell migration and cell corpse engulfment. Dev Cell. 2001;1:491–502. doi: 10.1016/s1534-5807(01)00056-9. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Z, Caron E, Hartwieg E, Hall A, Horvitz HR. The C. elegans PH domain protein CED-12 regulates cytoskeletal reorganization via a Rho/Rac GTPase signaling pathway. Dev Cell. 2001;1:477–489. doi: 10.1016/s1534-5807(01)00058-2. [DOI] [PubMed] [Google Scholar]
- 18.Cheresh DA, Leng J, Klemke RL. Regulation of cell contraction and membrane ruffling by distinct signals in migratory cells. J Cell Biol. 1999;146:1107–1116. doi: 10.1083/jcb.146.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolfi F, et al. The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc Natl Acad Sci U S A. 1998;95:15394–15399. doi: 10.1073/pnas.95.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiyokawa E, et al. Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev. 1998;12:3331–3336. doi: 10.1101/gad.12.21.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cote JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci. 2002;115:4901–4913. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 22.Meller N, Irani-Tehrani M, Kiosses WB, del Pozo MA, Schwartz MA. Zizimin1, a novel Cdc42 activator, reveals a new GEF domain for Rho proteins. Nat Cell Biol. 2002;4:639–647. doi: 10.1038/ncb835. [DOI] [PubMed] [Google Scholar]
- 23.Brugnera E, et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. 2002;4:574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 24.Grimsley CM, et al. Dock180 and ELMO1 proteins cooperate to promote evolutionarily conserved Rac-dependent cell migration. J Biol Chem. 2004;279:6087–6097. doi: 10.1074/jbc.M307087200. [DOI] [PubMed] [Google Scholar]
- 25.Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Catz SD, Johnson JL, Babior BM. The C2A domain of JFC1 binds to 3'-phosphorylated phosphoinositides and directs plasma membrane association in living cells. Proc Natl Acad Sci U S A. 2002;99:11652–11657. doi: 10.1073/pnas.172382799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.dara Berkowitz JK, et al. JFC1, a novel tandem C2 domain-containing protein associated with the leukocyte NADPH oxidase. J Biol Chem. 2001;276:18855–18862. doi: 10.1074/jbc.M011167200. [DOI] [PubMed] [Google Scholar]
- 28.Dunn R, Klos DA, Adler AS, Hicke L. The C2 domain of the Rsp5 ubiquitin ligase binds membrane phosphoinositides and directs ubiquitination of endosomal cargo. J Cell Biol. 2004;165:135–144. doi: 10.1083/jcb.200309026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirai T, et al. Specific detection of phosphatidylinositol 3,4,5-trisphosphate binding proteins by the PIP3 analogue beads: an application for rapid purification of the PIP3 binding proteins. Biochim Biophys Acta. 1998;1402:292–302. doi: 10.1016/s0167-4889(98)00014-7. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi I, Vuori K, Liddington RC. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat Struct Biol. 2002;9:101–106. doi: 10.1038/nsb755. [DOI] [PubMed] [Google Scholar]
- 31.Dowler S, Kular G, Alessi DR. Protein lipid overlay assay. Sci STKE. 2002;2002:PL6. doi: 10.1126/stke.2002.129.pl6. [DOI] [PubMed] [Google Scholar]
- 32.Bakolitsa C, de Pereda JM, Bagshaw CR, Critchley DR, Liddington RC. Crystal structure of the vinculin tail suggests a pathway for activation. Cell. 1999;99:603–613. doi: 10.1016/s0092-8674(00)81549-4. [DOI] [PubMed] [Google Scholar]
- 33.Kazlauskas A, Cooper JA. Phosphorylation of the PDGF receptor beta subunit creates a tight binding site for phosphatidylinositol 3 kinase. EMBO J. 1990;9:3279–3286. doi: 10.1002/j.1460-2075.1990.tb07527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klippel A, et al. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol Cell Biol. 1996;16:4117–4127. doi: 10.1128/mcb.16.8.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varnai P, Rother KI, Balla T. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in single living cells. J Biol Chem. 1999;274:10983–10989. doi: 10.1074/jbc.274.16.10983. [DOI] [PubMed] [Google Scholar]
- 36.Srinivasan S, et al. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol. 2003;160:375–385. doi: 10.1083/jcb.200208179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi S, et al. Membrane recruitment of DOCK180 by binding to PtdIns(3,4,5)P3. Biochem J. 2001;354:73–78. doi: 10.1042/0264-6021:3540073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kiyokawa E, Hashimoto Y, Kurata T, Sugimura H, Matsuda M. Evidence that DOCK180 up-regulates signals from the CrkII-p130(Cas) complex. J Biol Chem. 1998;273:24479–24484. doi: 10.1074/jbc.273.38.24479. [DOI] [PubMed] [Google Scholar]
- 39.Tamagnone L, et al. BMX, a novel nonreceptor tyrosine kinase gene of the BTK/ITK/TEC/TXK family located in chromosome Xp22.2. Oncogene. 1994;9:3683–3688. [PubMed] [Google Scholar]
- 40.Ruoslahti E, Hayman EG, Pierschbacher M, Engvall E. Fibronectin: purification, immunochemical properties, and biological activities. Methods Enzymol. 1982;82(Pt A):803–831. doi: 10.1016/0076-6879(82)82103-4. [DOI] [PubMed] [Google Scholar]
- 41.Polte TR, Hanks SK. Complexes of focal adhesion kinase (FAK) and Crk-associated substrate (p130(Cas)) are elevated in cytoskeleton-associated fractions following adhesion and Src transformation. Requirements for Src kinase activity and FAK proline-rich motifs. J Biol Chem. 1997;272:5501–5509. doi: 10.1074/jbc.272.9.5501. [DOI] [PubMed] [Google Scholar]
- 42.Katoh H, Negishi M. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature. 2003;424:461–464. doi: 10.1038/nature01817. [DOI] [PubMed] [Google Scholar]
- 43.Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.