

Abstract
A genome-scale metabolic model of Helicobacter pylori 26695 was constructed from genome sequence annotation, biochemical, and physiological data. This represents an in silico model largely derived from genomic information for an organism for which there is substantially less biochemical information available relative to previously modeled organisms such as Escherichia coli. The reconstructed metabolic network contains 388 enzymatic and transport reactions and accounts for 291 open reading frames. Within the paradigm of constraint-based modeling, extreme-pathway analysis and flux balance analysis were used to explore the metabolic capabilities of the in silico model. General network properties were analyzed and compared to similar results previously generated for Haemophilus influenzae. A minimal medium required by the model to generate required biomass constituents was calculated, indicating the requirement of eight amino acids, six of which correspond to essential human amino acids. In addition a list of potential substrates capable of fulfilling the bulk carbon requirements of H. pylori were identified. A deletion study was performed wherein reactions and associated genes in central metabolism were deleted and their effects were simulated under a variety of substrate availability conditions, yielding a number of reactions that are deemed essential. Deletion results were compared to recently published in vitro essentiality determinations for 17 genes. The in silico model accurately predicted 10 of 17 deletion cases, with partial support for additional cases. Collectively, the results presented herein suggest an effective strategy of combining in silico modeling with experimental technologies to enhance biological discovery for less characterized organisms and their genomes.
Genomics has reshaped many approaches to studying the biology of an organism. It has made possible studies in comparative genomics (1, 2, 17, 34), genome-wide transcriptional profiling (16), and a host of integrated analyses aimed at understanding the systems biology of organisms (14, 25, 68).
For model organisms such as Escherichia coli and Saccharomyces cerevisiae genome sequencing has generated a wealth of insight into the physiology of these microbes. However, the availability of extensive biological information and literature regarding these organisms has undoubtedly made it easier to reconcile data against the genome and interpret many characteristic properties of the genome and its genetic contents.
To date most of the common model organisms have had their genomes sequenced and current sequencing efforts are naturally moving on to less-well characterized organisms. This begs the question: what can be learned about less characterized organisms from their genomic sequence in the relative absence of extensive previous laboratory research? Additionally, how can the genome be utilized to assist in guiding future research and discovery into the particular organism of interest?
Herein we explore one aspect of these questions as it relates to metabolism. In particular we focus on the metabolic physiology and capabilities of a less-well characterized organism, namely, Helicobacter pylori. H. pylori is a human pathogen that colonizes the gastric mucosa, resulting in an acute inflammatory response and damage to epithelial cells and progressing to a number of disease states, including gastritis, peptic ulceration, and gastric cancer (36, 52). This organism serves as a good example of what can be learned primarily from the genome sequence of specialized organisms for which there is only limited supporting literature, particularly as it relates to metabolism.
Due in large part to the availability of the complete genome sequence of this microbe (23, 66), it is now possible to reconstruct its metabolic network and assess the organism's metabolic capabilities and fitness. Previously we have reconstructed the metabolic network of Haemophilus influenzae Rd, based primarily on its genome sequence, and performed extensive constraint-based metabolic modeling and pathway analysis of this organism (18, 55a, 60). In this work we report a comparable analysis for H. pylori 26695 and present these specific results along with comparisons between the metabolic potential and fitness of H. pylori and H. influenzae. The environment of the lungs and that of the stomach have shaped the evolution of both of these bacteria in very different ways. Both provide excellent examples of Darwinian evolution and survival in that they have exploited the resources available to them in creative ways to ensure their survival and fitness. In silico analysis supports this notion and represents a new form of comparative genomics.
Herein, we take a comprehensive look at the metabolic capabilities of H. pylori and present an examination of computed essential genes, with a comparison to experimentally validated essential genes. Taken together, the results aim to demonstrate what can be learned from the genome of less-characterized organisms when analyzed in conjunction with an in silico model.
MATERIALS AND METHODS
The overall procedures discussed below for model development and simulation are summarized in Fig. 1. The detailed computational methods and theory behind much of the work presented herein have been published in a collection of recent primary technical manuscripts and reviews (14, 21, 57-59, 61).
FIG. 1.

Overview of the in silico modeling methods and their conceptual basis. (A) Hypothetical network describing a set of metabolic reactions. Based on genomic, biochemical, and physiological data, such a reaction network can be reconstructed to represent the set of chemical reactions predicted to occur within an organism. (B) Extreme pathways are calculated for the reconstructed reaction network. Reactions that do not occur in any of the extreme pathways constitute unused reactions. Certain reactions always participate together when active in an extreme pathway. These groups of concomitantly occurring reactions constitute reaction subsets, as illustrated. (C) FBA is used to determine what input substrates and balanced reaction fluxes are required to meet the demand of producing metabolites I and J simultaneously in a fixed ratio. In this example only substrate B is available. (D) The reaction from metabolite B to E is eliminated, requiring the use of substrate A to meet the production demands of I and J. This situation also leads to the production of metabolite H as a by-product. Note that this reaction is essential if substrate A is not present.
Metabolic reconstruction.
Beginning from genomic sequence data, relevant genes in primary metabolism are identified to assist in reconstructing the metabolic network of H. pylori. This information is complemented with biochemical information and cell physiology data and is used to construct an organism-specific reaction index (the set of all reactions included in the reconstruction). The result of this process is a metabolic map illustrated in Fig. 1A. The reaction index underlying such a map represents the best determination of the biochemical reactions that the organism is believed to be capable of carrying out based on all available data. The detailed process of metabolic reconstruction and model development has been recently reviewed (14).
The metabolic genotype of H. pylori was determined from its annotated genome sequence (66) as provided by the TIGR Microbial Database as well as from GenBank (4). To begin establishing the metabolic reaction index for H. pylori, the annotation of all the open reading frames (ORFs) was carefully examined, and only gene assignments based on a high level of sequence similarity and pertaining to metabolic enzymes or membrane transporters were selected for inclusion in the metabolic genotype. From general biochemistry the complete set of potential metabolic reactions carried out by the associated gene products was determined. This list of reactions was augmented using additional experimental information from the literature pertaining to the genetic and enzymatic characterization of precise biochemical functions in the cell and general nutrient uptake characteristics (5, 7-9, 11, 13, 15, 17, 22, 24, 26-32, 34, 35, 37, 39-51, 53, 54, 55, 56, 62-65).
Pathway analysis.
The reaction index of H. pylori can be defined mathematically in the form of a stoichiometric matrix, S, which has dimensions of (m by n) where m is the number of metabolites in the reaction network and n is the number of reactions. Therefore, each column represents a reaction and each row represents the stoichiometric participation of a specific metabolite in each of the reactions. A particular flux distribution of the network, v, indicates the flux levels through each of the reactions. Based on principles of conservation of mass and the assumption of a steady state, the flux distribution through a reaction network can be characterized by the following equation:
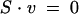 |
(1) |
From this equation it is possible to determine all of the chemically balanced metabolic routes through the network that span the entire range of metabolic capabilities for the network. These pathways are referred to as extreme pathways, and their method of calculation is based on principles of convex analysis (59). Previously an algorithm for extreme-pathway calculation has been published along with a pathway classification scheme (59). Figure 1B illustrates the extreme pathways for the hypothetical reaction network. Note that these extreme pathways can each have multiple input substrates and multiple output products.
For large networks such as for H. pylori or H. influenzae, it is more manageable to subdivide the network into smaller subsystems for extreme-pathway calculation. The strategy for extreme-pathway enumeration in subdivided networks is discussed in the literature (60) and was used in this study to determine the pathway structure of the H. pylori metabolic network as it was previously used to study H. influenzae.
Briefly, the metabolic network of H. pylori was subdivided into six discrete subsystems, and each of the reactions was assigned to one of the six subsystems. These subsystems include amino acid biosynthesis and degradation, nucleotide biosynthesis and degradation, vitamin and cofactor biosynthesis, lipid and cell envelope biosynthesis, central metabolism, and transport and energy-redox metabolism, including the immediate assimilation and dissimilation of various carbon sources. The extreme pathways for each of the subsystems were then calculated. From these extreme pathways a list of reactions that do not appear in any of the pathways can be determined. In addition reaction subsets can be identified as sets of reactions that always occur together in pathways at fixed stoichiometric ratios (see Fig. 1B for examples).
FBA.
Using the extreme pathways and/or the reaction network to describe the connectivity of the system allows for the calculation of network properties through the use of flux balance analysis (FBA). FBA is a method to assess the production capabilities and systemic properties of a metabolic network. The fundamentals of FBA have been reviewed elsewhere (6, 21, 58, 67). Extreme-pathway analysis and FBA share a common theoretical basis which has been described in detail previously (57). Specifically, FBA utilizes the principles of linear programming (LP), which is a subset of convex analysis.
Constraints are placed on individual reactions that state the upper and lower bounds on the range of flux values that each of the reactions can have. This constraint is described in the following form:
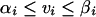 |
(2) |
where αi is the lower bound on flux vi and βi is the upper bound. If no information about flux levels is available, the value of αi is set to zero for irreversible fluxes, and in all other cases αi and βi are left unconstrained, allowing the flux to take on any value, positive or negative. The capabilities of the metabolic system are then explored using LP (12, 67). A reaction is first selected as an objective function that is to be maximized or minimized. A solution is then calculated that satisfies all of the constraints of equations 1 and 2. The result is the optimal flux distribution that will allow the highest flux through the chosen objective reaction. LINDO was used to solve the LP problems (LINDO Systems, Inc., Chicago, Ill.).
While FBA can be used to generate highly quantitative results that have been validated against experimental data (19, 20), here the approach is used primarily to assess the qualitative metabolic capabilities of the H. pylori reaction network. Figure 1C and D provide examples of how FBA was used herein.
Objective function.
To calculate an optimal flux distribution, an objective must be defined. For physiologically meaningful results, we have defined the objective as the ability to produce the required components of cellular biomass (e.g., amino acids, nucleotides, and phospholipids, etc.) that enable the organism to grow and survive. This growth objective is mathematically defined as an output flux utilizing each biomass precursor metabolite as a substrate. Forty-seven (45) metabolites were selected as required biomass constituents (Table 1). An output biomass reaction (exchange flux) was created that utilized these constituents in equal stoichiometric ratios to be used as a means to assess the ability of the network to produce all of the required demands based on particular substrate availability conditions. This reaction was selected as the objective in all of the calculations. The complete ability of the network to produce all of the biomass constituents leads to a positive flux value for this objective reaction.
TABLE 1.
Forty-seven metabolites considered to be required for biomass generation and maintenance in H. pylori
| Classification | Abbreviation | Complete name |
|---|---|---|
| Amino acids | ALA | Alanine |
| ARG | Arginine | |
| ASN | Asparagine | |
| ASP | Aspartate | |
| CYS | Cysteine | |
| GLU | Glutamate | |
| GLN | Glutamine | |
| GLY | Glycine | |
| HIS | Histidine | |
| ILE | Isoleucine | |
| LEU | Leucine | |
| LYS | Lysine | |
| MET | Methionine | |
| PHE | Phenylalanine | |
| PRO | Proline | |
| SER | Serine | |
| THR | Threonine | |
| TRP | Tryptophan | |
| TYR | Tyrosine | |
| VAL | Valine | |
| Polyamines | PTRC | Putrescine |
| SPMD | Spermidine | |
| Ribonucleotides | ATP | Adenosine 5′-triphosphate |
| GTP | Guanine 5′-triphosphate | |
| CTP | Cytidine 5′-triphosphate | |
| UTP | Uridine 5′-triphosphate | |
| Deoxyribonucleotides | DATP | Deoxyadenosine 5′-triphosphate |
| DGTP | Deoxyguanine 5′-triphosphate | |
| DCTP | Deoxycytidine 5′-triphosphate | |
| DTTP | Deoxythymidine 5′-triphosphate | |
| Phospholipids | PS | Phosphatidyl serine |
| PE | Phosphatidyl ethanolamine | |
| PG | Phosphatidyl glycerol | |
| Cell envelope | PEPTIDO | Peptidoglycan |
| LPS | Lipopolysaccharide | |
| Isoprenoids | OPP | Octaprenyl pyrophosphate |
| UDPP | Undecaprenyl pyrophosphate | |
| Vitamins and cofactors | NAD | Nicotinamide adenine dinucleotide |
| NADP | Nicotinamide adenine dinucleotide phosphate | |
| FAD | Flavin adenine dinucleotide | |
| CoA | Coenzyme A | |
| ACP | Acyl carrier protein | |
| PTH | Pantothenate | |
| Thiamine | ||
| MTHF | Methyl-tetrahydrofolate | |
| MK | Menaquinone | |
| DMK | Dimethylmenaquinone |
Minimal medium determination.
Beginning with all of the extracellular metabolites available to the metabolic network, each of the metabolites was removed individually to determine if they were required for producing all of the biomass constituents. This determination was accomplished by constraining the exchange flux or uptake reaction for the metabolite to zero and optimizing for the biomass objective reaction. After thorough examination a set of metabolites was arrived at for which the removal of any one metabolite would render the network unable to produce the biomass demands. This set of metabolites constitutes a defined minimal medium required for the in silico model to support growth of H. pylori.
Alternative carbon source determination.
In the minimal medium alanine and arginine fulfill the bulk carbon requirements for the network (as presented in the Results section). To determine what other major carbon sources can be used by the network the reactions used to assimilate alanine and arginine into central metabolism were simultaneously removed. This was accomplished by removing the d-amino acid dehydrogenase, alanine dehydrogenase, and 1-pyrroline-5-carboxylate dehydrogenase. With these reactions removed, alanine and arginine are still utilized by the network to fulfill certain biomass requirements; however, their broad utilization by the metabolic network is no longer possible with the removal of the above-mentioned reactions. Under these conditions, individual metabolites were included in the minimal medium composition to determine if the network could support the use of the biomass reaction. All of the possible substrates available to the in silico model were screened to determine their ability to serve as a major source of carbon to the organism. These carbon sources are referred to as the alternative carbon sources.
Deletion studies.
All of the reactions in central metabolism were examined to determine the essentiality of the reaction to the network under a variety of medium conditions. To assess the consequences of deleting a reaction from the network, the constraints on the particular reaction are set to zero. A simulation is then run to see if the network can support growth by optimizing for the biomass objective reaction. If the network cannot support growth, then the deleted reaction is deemed essential under the particular environment and medium conditions used in the simulation.
Four different medium conditions are presented in this deletion study. These include (i) the in silico minimal medium (described in Results), (ii) the minimal medium plus glucose, (iii) the minimal medium plus all of the alternative carbon sources (described in Results), and (iv) the minimal medium plus all the alternative carbon sources and all of the amino acids.
RESULTS
H. pylori metabolic reconstruction.
In the present metabolic reconstruction for H. pylori there are 291 genetic loci or ORFs included in the metabolic genotype, corresponding to 272 associated reactions. Based on experimental information from the literature, 116 additional reactions were included, bringing the final number of reactions in the H. pylori reaction index to 388. The 116 additional reactions fall into two categories, (i) reactions for which the associated genetics are unknown and (ii) reactions for which associated genetics are known in other organisms but no evidence for their existence has been found in the H. pylori genome. The complete list of reactions and associated genes is provided online as supplementary material along with a list of all the metabolite abbreviations (http://gcrg.ucsd.edu/supplementary_data/JBACT/Hpylori.xls). Ta-ble 2 provides a comparison of some of the basic characteristic properties of the H. pylori genome and metabolic model versus the model previously developed for H. influenzae.
TABLE 2.
Comparison of genomic characteristics and in silico metabolic model characteristics of H. pylori and H. influenzae (63)
| Properties | Bacterial strain
|
|
|---|---|---|
| H. pylori 26695 | H. influenzae Rd | |
| Genome characteristics | ||
| Genome length (bp) | 1,667,867 | 1,830,135 |
| G+C content (%) | 39 | 38 |
| No. of ORFs | 1,590 | 1,748 |
| No. with identified database match | 1,091 | 1,011 |
| No. with no database match | 499 | 732 |
| In silico metabolic networks | ||
| No. of genes included (% of known ORF) | 291 (≈27) | 400 (≈40) |
| No. of associated reactions | 272 | 412 |
| No. of other reactions | 116 | 49 |
| No. of metabolites (internal/external) | 339/64 | 367/84 |
| No. of internal fluxes | 534 | 632 |
| No. of exchange fluxes | 115 | 135 |
| Dimensions of S (metabolites by reactions) | 403 by 649 | 451 by 767 |
Figure 2 provides a graphical illustration of the central metabolism subsystem in the reconstructed network. H. pylori contains many of the glycolytic reactions; however, several key enzymes, including phosphofructokinase and pyruvate kinase, have not been identified. The absence of these two enzymes affects the organism's ability to extract energy from hexoses. The pentose phosphate reactions are all present in H. pylori with the exception of 6-phosphogluconate dehydrogenase, which makes the oxidative branch of the pathway incomplete. The first reaction in the oxidative branch, glucose-6-phosphate (G6P) dehydrogenase (G6PD), utilizes either one of two redox carriers (NAD or NADP). H. pylori has the reactions of the Entner-Doudoroff pathway allowing for carbon to be shunted away from the pentose phosphate pathway and into glyceraldehyde 3-phosphate and pyruvate. Furthermore, there were no carboxylases identified to convert phosphoenolpyruvate (PEP) into oxaloacetate, and we did not identify an anaplerotic reaction to convert PEP into a tricarboxylic acid (TCA) cycle intermediate. However, we included a malic enzyme in the metabolic reconstruction (28). The TCA cycle in the metabolic reconstruction is complete, but not in a traditional sense as in E. coli. There are reactions included such as α-ketoglutarate oxidase (AKO) (55), succinyl coenzyme A (CoA):acetoacetate CoA transferase (SCOT) (13), and malate:quinone oxidoreductase (MQO) (31), which allow complete connectivity between the metabolites of the TCA cycle, although these reactions are not typically considered to be part of a TCA cycle. Notably, the fumarate reductase included in the reconstruction is reversible, as evidenced from experimental literature, indicating the oxidation of succinate (11, 31) and as supported by a number of modeling calculations in agreement with physiological findings.
FIG. 2.

Reconstructed central metabolic subsystem for H. pylori. The different metabolite colors indicate the ability of the metabolite to leave the subsystem for participation in reactions in other subsystems. (Supplementary data are available at http://gcrg.ucsd.edu/supplementary_data/JBACT/Hpylori.xls.)
Extreme-pathway calculations.
Based on the metabolic reconstruction the extreme pathways were calculated for the six subsystems. The total number of pathways and the ratios of pathways to reactions are provided in Table 3. The ratio of pathways to reactions in most of the subsystems is very similar to the results for H. influenzae with the exception of the nucleotide subsystem, which contained nearly 10 times fewer pathways than those found in H. influenzae. This difference indicates that there is significantly less flexibility in the H. pylori metabolic network with respect to the processing of nucleotides due to the lack of catabolic pathways available to provide different nucleotide substrates to the cell from the environmental resources.
TABLE 3.
Subsystem characteristics and pathway calculation results for H. pylori indicating the total number of extreme pathways in each subsystema
| Subsystem | No. of reactions | No. of fluxes
|
No. of metabolites | No. of extreme pathways of type:
|
Total no. of pathways | Pathway/flux ratio | Total no. of H. influenzaee pathways | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Internal | External | I | II | III | ||||||
| A | 75 | 97 | 65 | 114 | 36 | 1 | 21 | 58 | 0.23 | 65 |
| C | 34 | 55 | 38 | 48 | 50 | 1 | 17 | 68 | 0.55 | 83 |
| L | 50 | 62 | 33 | 76 | 11 | 1 | 12 | 24 | 0.13 | 36 |
| N | 68 | 105 | 49 | 86 | 51 | 8 | 36 | 95 | 0.38 | 764 |
| T | 86 | 129 | 122 | 154 | 102 | 3 | 20 | 125 | 0.42 | 210 |
| V | 75 | 80 | 45 | 113 | 16 | 1 | 5 | 22 | 0.14 | 21 |
Abbreviations for subsystems: A, amino acid biosynthesis and degradation; C, central or core metabolism; L, lipid and cell envelope biosynthesis; N, nucleotide biosynthesis and degradation; T, transport and energy-redox metabolism, (V) vitamin and cofactor biosynthesis. Pathways are classified according to the scheme presented in reference,63 wherein the primary metabolic pathways of functional significance are those of type I.
Unused reactions.
As in H. influenzae there are a number of reactions in the metabolic network that do not appear in any of the extreme pathways. These reactions will not be used in any flux distribution representing metabolic activity of the network. The reason for these reactions not being present in any extreme pathway is that supporting reactions to generate substrates or consume the products are not present in the network, and hence the flux can never be active. The list of unused reactions in H. pylori is provided in Table 4 along with the comparison numbers for H. influenzae.
TABLE 4.
Reactions not active in any extreme pathways in each of the subsystems of H. influenzae and H. pyloria
| Sub- systemb | Total no. of unused reactions
|
Unused reactions in H. pylori | |
|---|---|---|---|
| H. influenzae | H. pylori | ||
| A | 28 | 6 | METB1, METB2, ILVC1, ILVC2, ILVE1, ILVE3 |
| C | 2 | 0 | |
| L | 1 | 0 | |
| N | 4 | 10 | ADK2, GMK2, DEOD1, DEOD3, DEOD4, DEOD7, GPT1, DEOB1, ADNUC, GNNUC |
| T | 11 | 11 | DPEPTP, OPEPTP, GALE, GLACTP, PMI, DSERTP, NUPCTP2, NUPCTP3, NUPCTP6, NARK, NMNTP |
| V | 6 | 10 | BIOF, BIOA, BIOD, BIOB, UBIA, THIM, UNKRXNI, THID, THIB, THIF |
The unused reactions listed in Table 4 can be used to direct questions related to the network reconstruction, as well as genome annotation, and biochemical function of particular enzymes. As an example consider the METB1 and METB2 reactions in the amino acid subsystem that are coded for by the metB gene (locus HP0106). These reactions are unused in their typical role of methionine biosynthesis (a required amino acid), which indicates that further experimentation may be necessary to determine the role of the metB gene product. ILVC1 and ILVC2 are unused; however, ILVC3 is used, which would suggest that the role of the ilvC gene (locus HP0330) is solely related to pantothenate biosynthesis rather than having a dual function in branched-chain amino acid biosynthesis as is the case in both the H. influenzae and E. coli in silico models. Other interesting results include the GALE reaction (galE, locus HP0360), which would be used in the conversion of UDP-galactose to and from UDP-glucose, but there are no reactions and genes that appear to be related to UDP-galactose production or utilization.
As in the H. influenzae metabolic network the reactions for biotin biosynthesis and a number of thiamine biosynthetic reactions are not used. The reason for biotin reactions not being used is the fact that the initial steps are unknown; therefore, no substrate is supplied to the reactions. For thiamine the reactions are not used because of missing steps in the biosynthetic pathway, which make thiamine an auxotrophic requirement for H. pylori similar to H. influenzae.
Reaction subsets.
The reactions that always occur together in the same ratio in each of the extreme pathways in which they are active can be calculated for each of the subsystems. These reaction subsets would be expected to contain reactions whose associated genes might be coregulated and possibly exist within the same regulon. The list of calculated reaction subsets in H. pylori is provided in Table 5.
TABLE 5.
Reaction subsets calculated from extreme pathways of each subsystem of the H. pylori metabolic networka
| Metabolism group and reaction subsets |
|---|
| Amino acid |
| DHS1, AROB, AROQ, AROE, AROK, AROA, AROC |
| TRPD, TRPC1, TRPC2, TRPAB |
| TYRA1, TYRA2, ASPB2 |
| METL2, THRB, THRC |
| DAPA, DAPB, DAPD, DAPC, DAPE, DAPF |
| ADCSASE_r, METH |
| SERA, SERC, SERB |
| SPEA, SPEB |
| SPED, SPEE, MTHAKN, MTHRKN, MTHIPIS, NE1PH, NE3UNK, TNSUNK |
| CYSDN, CYSC, CYSH, CYSU, CYSE, CYSK |
| Central |
| FBP, FBA_r |
| GAP, PGK |
| PGM, ENO |
| PGL, EDD, EDA |
| GLTA, ACNB, ICD |
| SCOT, ATOB |
| Lipid and cell envelope |
| ACCABCD, FABD |
| FABH1, FABF |
| C120SN, DGKA, LPXA, ENVA, LPXD, USHA12, LPXB, LPXK, KDTAI, KDOLIPH, ASPISO, KDSA, KDOPH, KDSB, PAPHTSE, GMHA, LPSSYN |
| C141SY, C160SN, C161SY, C181SY, PLS |
| PGSA2, PGPP |
| GLMS, GLMM, GLMU |
| MURZ, MURB, MURC, MURD, MURE, MURF, GLR, DDLA, MRAY, MURG |
| Nucleotide |
| PYRA, PYRB, PYRC, PYRD |
| PYRE, PYRF |
| PURF, PURD, PURL, PURM, PURK, PURE, PURC, PURB1, PURH1, PURH2 |
| PURA, PURB2 |
| GUAB, GUAA |
| NDK4, NRDAB4 |
| NDK6, NRDAB1 |
| NDK7, NRDAB3 |
| NDK8, TMK1 |
| DEOD2, DEOD8_r |
| Transport |
| ADHE2, ETHTP_r |
| PTA, ACKA |
| GALU, ALGC1 |
| GLCTP, GLK1 |
| PROTPI, NATP_r |
| LACTP, DLD |
| BCRBTP_r, ICFA |
| GLCD, GLLDHR, KATA |
| Vitamin and cofactor |
| FOLE, DNTPH, DHPPH, FOLB, FOLK, PABB, PABC, FOLP, FOLC |
| FOLD1, FOLD2 |
| GLTX, HEMA, HEML, HEMB, HEMC, HEMD, HEME, HEMF, HEMG, HEMH |
| RIBA, RIBD1, RIBD2, PMDPHT, RIBB, RIBE, RIBC, RIBF1, RIBF2 |
| PANB, ILVC3, PAND, PANC, COAA, PCLIG, PCDCL, PATRAN, DPHCOAK |
| IPPPISO, ISPA1, ISPA2 |
| NADB, NADA, NADC, NADD, NADE |
| MENF, MEND1, MEND2, MENC, MENE, MENB, MENA |
Each set of reactions always occurs together in the same flux ratios in all type I and type II pathways in which the particular fluxes are active. Reverse enzymatic activity is indicated by the suffix “_r” at the end of a flux name.
If many of these reactions are to be coregulated, then a good place to begin examining their regulation would be at the genetic level, in particular their location within the genome relative to one another. There are a few subsets that have reactions whose associated genes are located adjacent to each other within the genome in what appear to be the same operon. These include subset T2, A2, C2, C6, and portions of C4 and C5. However, the majority of the genes in most subsets are not located adjacent to one another, but rather they are scattered far across the genome. Examples include the following subsets: A1, A4, A5, A7, C5, V1, V3, V4, N1, N2, N4, and L6. The implications of these results are explored further in the discussion section.
Growth requirements.
For H. pylori the calculated set of required substrates includes eight amino acids: alanine, arginine, histidine, isoleucine, leucine, methionine, phenylalanine, and valine, as well as thiamine, oxygen, a phosphate source (inorganic phosphate), and sulfate or cysteine as a sulfur source. No purine source is required as the network is capable of performing de novo purine biosynthesis (38). Figure 3 illustrates the connectivity of these substrates to each of the biomass constituents for which their biosynthesis is associated.
FIG. 3.

Minimal substrate requirements for H. pylori to generate the 47 required biomass constituents which can be synthesized. Lines connecting substrates to biomass constituents indicate that the network is unable to generate the product without the substrate. The dashed lines indicate that either alanine or arginine is required for the production of the connected biomass requirement. See Table 1 for abbreviations.
The requirement of oxygen for the production of NAD, NADP, the pyrimidine nucleotides (CTP and UTP) and deoxynucleotides (dCTP and dTTP) is based on the need for an electron acceptor for the electrons from the flavin-containing electron carrier, flavin adenine dinucleotide (FAD, oxidized; FADH, reduced). The only reaction in the network capable of oxidizing FADH back to FAD is through the transfer of electrons to the menaquinone pool and subsequent acceptance of the electrons by oxygen. Without oxygen the pathway combinations are unable to balance the generation of FADH, violating the mass balance equations governing the system, and so a number of biomass constituents are seen to have a dependency on oxygen.
Taking a closer look at the connectivity in Fig. 3, it is seen that alanine and arginine are the major sources of carbon for the network responsible for the synthesis of a large number of biomass constituents when considering the minimal substrates. To utilize alanine the organism must use the alanine deaminase reaction converting alanine into pyruvate, liberating ammonia in the process. Once the carbon is fed into central metabolism as pyruvate, the formation of the necessary precursor metabolites for widespread biosynthesis can be accomplished. In the case of arginine, it is first converted to ornithine, then to l-glutamate g-semialdehyde, then to glutamate, and ultimately into α-ketoglutarate, which can be routed throughout the majority of central metabolism.
Other substrates that are capable of fulfilling the bulk carbon requirements of the organism include glucose, pyruvate, lactate, malate, fumarate, succinate, α-ketoglutarate and the amino acids asparagine, aspartate, glutamine, glutamate, and serine, as well as the nucleosides adenosine and guanosine. Collectively, these 14 substrates are referred to as alternative carbon sources. Glucose enters the central metabolic subsystem in Fig. 2 as G6P. Lactate, alanine, and serine are all assimilated into the subsystem as pyruvate. Glutamine and glutamate are assimilated as α-ketoglutarate, while aspartate and asparagine are assimilated as fumarate. The nucleosides are both assimilated as ribose-5-phosphate.
Deletion study.
The effect of the loss in enzymatic function corresponding to a gene deletion was examined to determine if the metabolic network could support the production of the required biomass constituents in the absence of the selected reaction. Each reaction in the central metabolic network of H. pylori (depicted in Fig. 2) was individually eliminated from the network and the effects were studied under four different environmental/media conditions (compositions described in the methods section). The results of this deletion study are presented in Tables 6 and 7.
TABLE 6.
Model results for reactions deleted in central metabolism under four different substrate availability conditionsa
| Deleted reaction | In silico essentiality in medium
|
|||
|---|---|---|---|---|
| Minimal | Minimal + glucose | Minimal + carbon sources | Minimal + carbon sources + amino acids | |
| ACEB | − | − | − | − |
| ACNB | + | + | + | + |
| AKO | + | + | + | + |
| ATOB | + | + | + | + |
| ENO | − | + | + | + |
| FBA | − | + | + | + |
| FRD | + | + | + | + |
| FUMC | + | + | + | + |
| G6PD1 | + | + | + | + |
| G6PD2 | + | + | + | + |
| GAP | − | + | + | + |
| GLTA | + | + | + | + |
| ICD | + | + | + | + |
| MAEB | + | + | + | + |
| PGI | − | + | + | + |
| PGK | − | + | + | + |
| PGL | + | + | + | + |
| PGM | − | + | + | + |
| PPA | − | − | − | − |
| PPSA | − | + | + | + |
| PRSA | − | − | − | − |
| RPE | − | − | + | + |
| RPI | − | − | + | + |
| SCOT | + | + | + | + |
| TKTA1 | + | + | + | + |
| TKTA2 | − | − | + | + |
| TPI | − | − | − | − |
The composition of each of the four media is described in the Results section. Symbols: (+), ability to make all 47 biomass constituents in the absence of the indicated reaction; (−), inability to make the biomass constituents.
TABLE 7.
In silico results compared to experimental results for the essentiality of 17 genes in the modela
| Deleted reaction | Gene(s) | Locus identification(s) | In vitro essentiality | In silico essentiality in medium
|
Agree- ment | |||
|---|---|---|---|---|---|---|---|---|
| Minimal | Minimal + glucose | Minimal + carbon sources | Minimal + carbon sources + amino acids | |||||
| ACPS | acpS | HP0808 | E | − | − | − | − | Yes |
| AROQ | aroQ | HP1038 | E | − | − | − | − | Yes |
| DCD | dcd | HP0372 | NE | + | + | + | + | Yes |
| EDA | eda | HP1099 | E | + | + | + | + | |
| EDD | edd | HP1100 | E | + | + | + | + | |
| FBP | fbp | HP1385 | E | − | + | + | + | |
| GLCD | glcD | HP0509 | E | − | − | − | − | Yes |
| GPT1/2/3 | gpt | HP0735 | E | + | + | + | + | |
| MQO | mqo | HP0086 | E | − | − | + | + | |
| MURB | murB | HP1418 | E | − | − | − | − | Yes |
| MURF | murF | HP0740 | E | − | − | − | − | Yes |
| OOR | oorA, oorB, oorC, oorD | HP0589, HP0590, HP0591, HP0588 | E | + | + | + | + | |
| POR | porA, porB, porC, porD | HP1110, HP1111, HP1108, HP1109 | E | − | − | − | − | Yes |
| PYRE | pyrE | HP1257 | E | − | − | − | − | Yes |
| SPEE | speE | HP0832 | NE | − | − | − | − | |
| TAL | tal | HP1495 | NE | + | + | + | + | Yes |
| TRXB | trxA, trxB | HP0824, HP0825 | E | − | − | − | − | Yes |
All of the essential in silico predictions are shaded, with a − sign indicating the inability to produce the biomass demands. The in vitro essentiality data is taken from reference 10 (E, essential; NE, not essential).
In Table 6 there are four enzymatic deletions that render the network incapable of generating the required energy and biomass constituents to support growth under all conditions examined. The corresponding genes for these reactions are aceB, ppa, prsA, and tpi. The loss of ppa does not allow the network to convert pyrophosphate (PPi) into inorganic phosphate (Pi); however, this reaction would likely occur spontaneously at a reduced rate. Without the prsA gene the network is not capable of generating phosphoribosyl pyrophosphate and subsequently cannot make any of the nucleotides or deoxynucleotides required for growth. The inability of the tpi in silico mutant to grow is a result of H. pylori lacking the phosphofructokinase enzyme; therefore, without the TPI reaction H. pylori is unable to make dihydroxyacetone-phosphate. We also find that the tktA gene is essential under all conditions if both the TKTA1 and TKTA2, reactions are simultaneously deleted. Without these two reactions the network cannot generate sedoheptulose-7-phosphate and erythrose-4-phosphate (E4P), required for the generation of a number of biomass constituents.
There is biochemical evidence suggesting that malate synthase (ACEB) activity occurs in H. pylori (27, 55). However, there is little evidence in support of isocitrate lyase activity, leading to the conclusion that there is an incomplete glyoxylate bypass in H. pylori. The in silico model predicts that ACEB is essential. This result is due to the necessity of this reaction for the processing of glyoxylate, which is produced as net result of the production of glycolaldehyde. There is consistency between this in silico finding and similar results for the GLCD reaction supported by the experimentally determined essentiality of the glcD gene (HP0509) encoding glycolate oxidase.
Table 7 contains a series of 17 deletion results comparing the in silico-determined essentiality of genes whose in vitro essentiality was recently determined using a vector-free allelic replacement mutagenesis technique (10). In 10 out of the 17 cases the in silico model is in complete agreement with the in vitro determinations. In 2 of the remaining cases (fbp and mqo) there is partial agreement with the in vitro data depending on the conditions considered.
The discrepancies between the in silico deletion study and the experimental study lead to interesting testable hypotheses that can be performed to further probe the organisms' capabilities. As an example, consider the eda and edd genes of the Entner-Doudoroff pathway. These genes are essential as determined in vitro, which is thought to be due to their critical role in the catabolism of glucose. However, they are not required in the model because glucose catabolism is not required. If glucose is used as an alternative carbon source to alanine and arginine, the model predicts edd and eda to be essential; however, there is not enough experimental evidence to convincingly support the requirement of glucose for growth even though it clearly enhances the growth rate. This is an area where further investigation will help to refine the model and improve its predictive capabilities. The gpt gene encoding guanine phosphoribosyl transferase is essential in vitro, which would imply that de novo purine biosynthesis does not occur. If any of the genes for purine biosynthesis are removed from the model, then gpt is calculated to be essential.
DISCUSSION
Here we have initiated the in silico model development process for H. pylori, a less-characterized organism particularly from a metabolic standpoint. With the complete genome sequence available along with additional biochemical and physiological data, the metabolic network for this organism was reconstructed. The constraint-based modeling methodology was then applied to assess the performance capabilities of the organism. This assessment included the use of extreme-pathway analysis and FBA. The overall modeling process can assist in accelerating the pace of biological discovery by generating experimentally testable hypotheses. These hypotheses may represent novel insights into the organism, help to support previous insights, or resolve existing discrepancies between different sets of experimental results which may be inconclusive. The in silico model is then improved through each iteration of modeling and experimentation, so that it may provide the most concise representation of the organism's known functional capabilities.
There are many examples of topics which the model can assist in exploring further. Two such examples include the de novo biosynthesis of alanine by the organism as well as its ability to perform de novo purine biosynthesis. In each case there is evidence in the literature that can be interpreted to support both sides of the argument for the ability of the organism to produce these biomass constituents. Each of the possible cases can be examined with the model and used to design experiments in defined media aimed at determining these capabilities. In combination with experimental determination of whole-genome expression patterns and bioinformatic sequence analysis, it should be possible to determine the location of genes responsible for these metabolic activities.
As another example, exploration into the reactions in central metabolism that would allow for the conversion of α-ketoglutarate into succinate to complete the TCA cycle can be investigated. This is a critical issue in central metabolism, as it appears that fumarate reductase can function in a reversible manner, thereby allowing for the assimilation of a wide range of additional compounds. Growth studies can be designed that can test the ability of the organism to utilize succinate, α-ketoglutarate, and compounds which can be assimilated into α-ketoglutarate as major carbon sources. The function of the SCOT reaction associated with loci HP0691 and HP0692 can be investigated in relation to the proposed AKO reaction through experiments which introduce acetoacetate as an available substrate within the organism. In this way the in silico model can be used to generate testable hypotheses which can be rapidly validated or invalidated through integration with experimentation, leading to improved insight into the TCA cycle and the critical role that it plays in H. pylori. This is another simple example of how system-modeling strategies can be used to drive experimental research on an organism.
In H. pylori an examination of the reaction subsets indicates that the genetic order of the genes in a reaction subset is not well conserved in terms of operon structure. This general result may suggest that there is well-coordinated regulation of many of these genes to ensure their simultaneous expression. However, in the original examination of the genome there were only four helix-turn-helix motifs found throughout the entire genome (66), as well as relatively few two-component systems (3), indicating that only a limited amount of regulation occurs at the genetic level in H. pylori. If the organism has only a limited level of transcriptional regulation, then a possible hypothesis is that many of the genes in the organism (particularly those in the calculated reaction subsets) are constitutively expressed in the organism. This could be investigated with the use of whole-genome expression analysis to examine the expression of the genes that comprise a particular subset. An example subset for further investigation would be the A5 set comprising the reactions for lysine biosynthesis, which is associated with five genes widely dispersed in the genome (dapA-HP1013, dapB-HP0510, dapD-HP0626, dapE-HP0212, and dapF-HP0566). Overall the results of the reaction subset gene locations would tend to support a notion that the bacterium contains little regulation and its metabolic network is inflexible, being adapted for a very limited range of environmental conditions.
While it is clear from cell physiology that H. pylori requires oxygen, the precise dependence on oxygen to generate the biomass constituents is most likely not the reason for the true physiological requirement of oxygen (33). Without oxygen the cell cannot effectively accept electrons from the various carriers and subsequently is unable to generate energy through oxidative phosphorylation. In many bacteria this situation is overcome by utilizing other electron acceptors or a shift to fermentative growth with the use of substrate level phosphorylation via glycolysis. From the reconstruction and pathway analysis of the network it is clear that H. pylori has a limited ability to perform substrate level phosphorylation due to the absence of a pyruvate kinase reaction to complete glycolysis.
A particularly interesting conclusion stemming from these studies relates to the amino acid requirements of the organism. The human host that H. pylori colonizes has nutritional requirements for nine amino acids. These are referred to as the nine human essential amino acids and are histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. Upon careful examination of these requirements, it is seen that of the eight amino acids required by the bacterium, six are also amino acids that the human body must acquire from nutritional sources. Alanine and arginine are the two amino acids that H. pylori requires which are not required by the human body (65).
This general strategy for metabolic design would seem ideal for the bacterium. It appears as if it may have evolved to eliminate many of the genes and enzymes of the energetically unfavorable biosynthetic pathways for amino acid production, as it is guaranteed of their presence in the host gastric environment where proteolysis is continuously occurring. For H. influenzae the number of required amino acids is only three (glutamate, arginine, and cysteine), none of which are human essential amino acids. These amino acid requirements provide an example of the possible adaptations of each of these organisms to their particular environmental niche. In fact, further use of the model suggests that the use of amino acids as a major carbon source for H. pylori leads to a significant increase in the generation of ammonia that the organism may use to neutralize the surrounding extracellular environment.
From comparative modeling there are many distinct properties of the metabolism of each of these organisms that further support this notion of environmental adaptation. These include the range of preferred carbon substrates, as well as additional growth requirements. In H. pylori there was much less evidence to support the coordinated regulation of reactions and genes comprising reaction subsets than there was in H. influenzae. This finding may indicate that H. pylori does not witness as many significant changes in the environment that necessitate a reprogramming of its genetic expression. This lack of regulation might also have to do with the extremely limited amount of competition that the organism faces in the lining of the stomach where it is protected from the host's immune response.
Due to its role as a prominent human pathogen there is interest in identifying essential genes in H. pylori. From the deletion analysis it is clear that the essential nature of a gene is dependent on the environmental conditions, as there are many genes essential under some conditions but not others. Seven genes were identified that are predicted to be essential under all of the conditions examined, and these would represent interesting candidates for experimental validation studies. Currently there are limited studies focusing on the identification of essential genes in H. pylori, with the exception of one recent study by Chalker et al. (10). The in silico model contained 24 genes corresponding to 17 reactions that were experimentally examined for essentiality and provide validation for the model. Ten of the 17 case predictions (∼60%) were in accordance with experimental data, while 2 were in agreement under certain medium conditions. The experimental medium conditions were complex, making the direct comparison difficult. In general these results are encouraging. Through integrated modeling and experimentation it would be expected that the accuracy of the model would increase as a result of enhanced biological discovery into the organism's metabolic capabilities. The accuracy of prediction for H. pylori is less than what we have previously observed with similar predictions in E. coli and S. cerevisiae, for which predictions of gene deletions are 80 to 90% accurate (20; I. Famili, J. Foerster, J. Nielsen, and B. O. Palsson, submitted for publication). For those in silico models, improved accuracy is anticipated, as they are model organisms which have been more thoroughly characterized.
At the outset we posed the question of what can be learned from the genomes of less-characterized organisms. Through the use of constraint-based modeling we have demonstrated that there is much that can be derived from the genome when it is examined in parallel with existing experimental data. Integrating genomic, biochemical, and physiological data together in the context of an in silico model serves as a practical tool in the quest to understand the physiology of this organism. In addition to the task of data-driven model development, there appears to be much to be gained from prospective implementation of an in silico model for biological discovery. The success of such model-driven discovery is dependent on the ability to design experimentally testable hypotheses and carry out these studies in support of an iterative model development paradigm. For less-characterized organisms the notion of integrating a model with experimentation should lead to more-efficient discovery, perhaps even at a higher rate than that for common model organisms. This paradigm for efficient discovery is facilitated and enabled by the information contained in the genome, so there is much to be gained from the genome of less-characterized organisms such as H. pylori, provided that there are methods developed to capitalize on this wealth of information. Such genome-enabled science provides additional justification for genome sequencing efforts.
Acknowledgments
We acknowledge the insightful and highly constructive reviews that were received for the manuscript. These reviews led to a number of enhancements to the model in terms of its biological content that improved the predictive capabilities and quality of the results presented. This is exemplary of the true nature of collaborative and iterative model development.
This work was funded in part by the NIH (GM57089), the NSF (MCB 73384 and BES 14092) and the Whitaker Foundation through graduate student fellowships in biomedical engineering (C.S. and I.F.). We also acknowledge funding from the NSF, DOE, and DARPA to the Church Laboratory.
REFERENCES
- 1.Alm, R. A., J. Bina, B. M. Andrews, P. Doig, R. E. Hancock, and T. J. Trust. 2000. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect. Immun. 68:4155-4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arigoni, F., F. Talabot, M. Peitsch, M. D. Edgerton, E. Meldrum, E. Allet, R. Fish, T. Jamotte, M. L. Curchod, and H. Loferer. 1998. A genome-based approach for the identification of essential bacterial genes. Nat. Biotechnol. 16:851-856. [DOI] [PubMed] [Google Scholar]
- 3.Beier, D., and R. Frank. 2000. Molecular characterization of two-component systems of Helicobacter pylori. J. Bacteriol. 182:2068-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benson, D. A., M. S. Boguski, D. J. Lipman, J. Ostell, and B. F. Ouellette. 1998. GenBank. Nucleic Acids Res. 26:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birkholz, S., U. Knipp, E. Lemma, A. Kroger, and W. Opferkuch. 1994. Fumarate reductase of Helicobacter pylori—an immunogenic protein. J. Med. Microbiol. 41:56-62. [DOI] [PubMed] [Google Scholar]
- 6.Bonarius, H. P. J., G. Schmid, and J. Tramper. 1997. Flux analysis of underdetermined metabolic networks: the quest for the missing constraints. Trends Biotechnol. 15:308-314. [Google Scholar]
- 7.Burns, B. P., S. L. Hazell, and G. L. Mendz. 1995. Acetyl-CoA carboxylase activity in Helicobacter pylori and the requirement of increased CO2 for growth. Microbiology 141:3113-3118. [DOI] [PubMed] [Google Scholar]
- 8.Burns, B. P., G. L. Mendz, and S. L. Hazell. 1998. A novel mechanism for resistance to the antimetabolite N-phosphonoacetyl-l-aspartate by Helicobacter pylori. J. Bacteriol. 180:5574-5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chalk, P. A., A. D. Roberts, and W. M. Blows. 1994. Metabolism of pyruvate and glucose by intact cells of Helicobacter pylori studied by 13C NMR spectroscopy. Microbiology (Reading) 140:2085-2092. [DOI] [PubMed] [Google Scholar]
- 10.Chalker, A. F., H. W. Minehart, N. J. Hughes, K. K. Koretke, M. A. Lonetto, K. K. Brinkman, P. V. Warren, A. Lupas, M. J. Stanhope, J. R. Brown, and P. S. Hoffman. 2001. Syst. identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J. Bacteriol. 183:1259-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen, M., L. P. Andersen, L. Zhai, and A. Kharazmi. 1999. Characterization of the respiratory chain of Helicobacter pylori. FEMS Immunol. Med. Microbiol. 24:169-174. [DOI] [PubMed] [Google Scholar]
- 12.Chvatal, V. 1983. Linear programming. W. H. Freeman and Company, New York, N.Y.
- 13.Corthesy-Theulaz, I. E., G. E. Bergonzelli, H. Henry, D. Bachmann, D. F. Schorderet, A. L. Blum, and L. N. Ornston. 1997. Cloning and characterization of Helicobacter pylori succinyl CoA:acetoacetate CoA-transferase, a novel prokaryotic member of the CoA-transferase family. J. Biol. Chem. 272:25659-25667. [DOI] [PubMed] [Google Scholar]
- 14.Covert, M. W., C. H. Schilling, I. Famili, J. S. Edwards, I. I. Goryanin, E. Selkov, and B. O. Palsson. 2001. Metabolic modeling of microbial strains in silico. Trends Biochem. Sci. 26:179-186. [DOI] [PubMed] [Google Scholar]
- 15.De Reuse, H., A. Labigne, and D. Mengin-Lecreulx. 1997. The Helicobacter pylori ureC gene codes for a phosphoglucosamine mutase. J. Bacteriol. 179:3488-3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devaux, F., P. Marc, and C. Jacq. 2001. Transcriptomes, transcription activators and microarrays. FEBS Lett. 498:140-144. [DOI] [PubMed] [Google Scholar]
- 17.Doig, P., B. L. de Jonge, R. A. Alm, E. D. Brown, M. Uria-Nickelsen, B. Noonan, S. D. Mills, P. Tummino, G. Carmel, B. C. Guild, D. T. Moir, G. F. Vovis, and T. J. Trust. 1999. Helicobacter pylori physiology predicted from genomic comparison of two strains. Microbiol. Mol. Biol. Rev. 63:675-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards, J., and B. Palsson. 1999. Systems properties of the Haemophilus influenzae Rd metabolic genotype. J. Biol. Chem. 274:17410-17416. [DOI] [PubMed] [Google Scholar]
- 19.Edwards, J. S., R. U. Ibarra, and B. O. Palsson. 2001. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 19:125-130. [DOI] [PubMed] [Google Scholar]
- 20.Edwards, J. S., and B. O. Palsson. 2000. The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. USA 97:5528-5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards, J. S., R. Ramakrishna, C. H. Schilling, and B. O. Palsson. 1999. Metabolic flux balance analysis, p. 13-57. In S. Y. Lee and E. T. Papoutsakis (ed.), Metabolic engineering. Marcel Dekker, New York, N.Y.
- 22.Finel, M. 1998. Does NADH play a central role in energy metabolism in Helicobacter pylori? Trends Biochem. Sci. 23:412-413. [DOI] [PubMed] [Google Scholar]
- 23.Fleischmann, R. D., M. D. Adams, O. White, R. A. Clayton, E. F. Kirkness, A. R. Kerlavage, C. J. Bult, J. F. Tomb, B. A. Dougherty, J. M. Merrick, K. McKenney, G. Sutton, W. Fitzhugh, C. Fields, J. D. Gocayne, J. Scott, R. Shirley, L. I. Liu, A. Glodek, J. M. Kelley, J. F. Weidman, C. A. Phillips, T. Spriggs, E. Hedblom, M. D. Cotton, T. R. Utterback, M. C. Hanna, D. T. Nguyen, D. M. Saudek, R. C. Brandon, L. D. Fine, J. L. Fritchman, J. L. Fuhrmann, N. S. M. Geoghagen, C. L. Gnehm, L. A. McDonald, K. V. Small, C. M. Fraser, H. O. Smith, and J. C. Venter. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496-498, 507-512. [DOI] [PubMed] [Google Scholar]
- 24.Ge, Z., and D. E. Taylor. 1997. The Helicobacter pylori gene encoding phosphatidylserine synthase: sequence, expression, and insertional mutagenesis. J. Bacteriol. 179:4970-4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gombert, A. K., and J. Nielsen. 2000. Mathematical modelling of metabolism. Curr. Opin. Biotechnol. 11:180-186. [DOI] [PubMed] [Google Scholar]
- 26.Hazell, S. L., and G. L. Mendz. 1997. How Helicobacter pylori works: an overview of the metabolism of Helicobacter pylori. Helicobacter 2:1-12. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman, P. S., A. Goodwin, J. Johnsen, K. Magee, and S. J. Veldhuyzen van Zanten. 1996. Metabolic activities of metronidazole-sensitive and -resistant strains of Helicobacter pylori: repression of pyruvate oxidoreductase and expression of isocitrate lyase activity correlate with resistance. J. Bacteriol. 178:4822-4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes, N. J., P. A. Chalk, C. L. Clayton, and D. J. Kelly. 1995. Identification of carboxylation enzymes and characterization of a novel four-subunit pyruvate:flavodoxin oxidoreductase from Helicobacter pylori. J. Bacteriol. 177:3953-3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes, N. J., C. L. Clayton, P. A. Chalk, and D. J. Kelly. 1998. Helicobacter pylori porCDAB and oorDABC genes encode distinct pyruvate:flavodoxin and 2-oxoglutarate:acceptor oxidoreductases which mediate electron transport to NADP. J. Bacteriol. 180:1119-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorgensen, M. A., J. Manos, G. L. Mendz, and S. L. Hazell. 1998. The mode of action of metronidazole in Helicobacter pylori: futile cycling or reduction? J. Antimicrob. Chemother. 41:67-75. [DOI] [PubMed] [Google Scholar]
- 31.Kather, B., K. Stingl, M. E. van der Rest, K. Altendorf, and D. Molenaar. 2000. Another unusual type of citric acid cycle enzyme in Helicobacter pylori: the malate:quinone oxidoreductase. J. Bacteriol. 182:3204-3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly, D. J. 1998. The physiology and metabolism of the human gastric pathogen Helicobacter pylori. Adv. Microb. Physiol. 40:137-189. [DOI] [PubMed] [Google Scholar]
- 33.Kelly, D. J., N. J. Hughes, and R. K. Poole. 2001. Microaerobic physiology: aerobic respiration, anaerobic respiration, and carbon dioxide metabolism, p. 113-124. In H. L. T. Mobley, G. L. Mendz, and S. L. Hazell (ed.), Helicobacter pylori: physiology and genetics. ASM Press, Washington, D.C. [PubMed]
- 34.Marais, A., G. L. Mendz, S. L. Hazell, and F. Mégraud. 1999. Metabolism and genetics of Helicobacter pylori: the genome era. Microbiol. Mol. Biol. Rev. 63:642-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcelli, S. W., H. T. Chang, T. Chapman, P. A. Chalk, R. J. Miles, and R. K. Poole. 1996. The respiratory chain of Helicobacter pylori: identification of cytochromes and the effects of oxygen on cytochrome and menaquinone levels. FEMS Microbiol. Lett. 138:59-64. [DOI] [PubMed] [Google Scholar]
- 36.Marshall, B. J. 2001. One hundred years of discovery and rediscovery of Helicobacter pylori and its association with peptic ulcer disease, p. 19-24. In H. L. Mobley, G. L. Mendz, and S. L. Hazell (ed.), Helicobacter pylori: physiology and genetics. ASM Press, Washington, D.C. [PubMed]
- 37.McGee, D. J., F. J. Radcliff, G. L. Mendz, R. L. Ferrero, and H. L. Mobley. 1999. Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity. J. Bacteriol. 181:7314-7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendz, G. L. 2001. Nucleotide metabolism, p. 147-158. In H. L. Mobley, G. L. Mendz, and S. L. Hazell (ed.), Helicobacter pylori: physiology and genetics. ASM Press, Washington, D.C. [PubMed]
- 39.Mendz, G. L., B. P. Burns, and S. L. Hazell. 1995. Characterisation of glucose transport in Helicobacter pylori. Biochim. Biophys. Acta 1244:269-276. [DOI] [PubMed] [Google Scholar]
- 40.Mendz, G. L., and S. L. Hazell. 1995. Aminoacid utilization by Helicobacter pylori. Int. J. Biochem. Cell Biol. 27:1085-1093. [DOI] [PubMed] [Google Scholar]
- 41.Mendz, G. L., and S. L. Hazell. 1991. Evidence for a pentose phosphate pathway in Helicobacter pylori. FEMS Microbiol. Lett. 84:331-336. [Google Scholar]
- 42.Mendz, G. L., and S. L. Hazell. 1993. Fumarate catabolism in Helicobacter pylori. Biochem. Mol. Biol. Int. 31:325-332. [PubMed] [Google Scholar]
- 43.Mendz, G. L., and S. L. Hazell. 1993. Glucose phosphorylation in Helicobacter pylori. Arch. Biochem. Biophys. 300:522-525. [DOI] [PubMed] [Google Scholar]
- 44.Mendz, G. L., S. L. Hazell, and B. P. Burns. 1994. The Entner-Doudoroff pathway in Helicobacter pylori. Arch. Biochem. Biophys. 312:349-356. [DOI] [PubMed] [Google Scholar]
- 45.Mendz, G. L., S. L. Hazell, and B. P. Burns. 1993. Glucose utilization and lactate production by Helicobacter pylori. J. Gen. Microbiol. 139:3023-3028. [DOI] [PubMed] [Google Scholar]
- 46.Mendz, G. L., S. L. Hazell, and S. Srinivasan. 1995. Fumarate reductase: a target for therapeutic intervention against Helicobacter pylori. Arch. Biochem. Biophys. 321:153-159. [DOI] [PubMed] [Google Scholar]
- 47.Mendz, G. L., S. L. Hazell, and L. van Gorkom. 1994. Pyruvate metabolism in Helicobacter pylori. Arch. Microbiol. 162:187-192. [DOI] [PubMed] [Google Scholar]
- 48.Mendz, G. L., B. M. Jimenez, S. L. Hazell, A. M. Gero, and W. J. O'Sullivan. 1994. De novo synthesis of pyrimidine nucleotides by Helicobacter pylori. J. Appl. Bacteriol. 77:1-8. [DOI] [PubMed] [Google Scholar]
- 49.Mendz, G. L., B. M. Jimenez, S. L. Hazell, A. M. Gero, and W. J. O'Sullivan. 1994. Salvage synthesis of purine nucleotides by Helicobacter pylori. J. Appl. Bacteriol. 77:674-681. [DOI] [PubMed] [Google Scholar]
- 50.Mendz, G. L., D. J. Meek, and S. L. Hazell. 1998. Characterization of fumarate transport in Helicobacter pylori. J. Membr. Biol. 165:65-76. [DOI] [PubMed] [Google Scholar]
- 51.Mendz, G. L., A. J. Shepley, S. L. Hazell, and M. A. Smith. 1997. Purine metabolism and the microaerophily of Helicobacter pylori. Arch. Microbiol. 168:448-456. [DOI] [PubMed] [Google Scholar]
- 52.Mitchell, H. M. 2001. Epidemiology of infection, p. 7-18. In H. L. Mobley, G. L. Mendz, and S. L. Hazell (ed.), Helicobacter pylori: physiology and genetics. ASM Press, Washington, D.C. [PubMed]
- 53.Mobley, H. L., G. L. Mendz, and S. L. Hazell (ed.). 2001. Helicobacter pylori: physiology and genetics. ASM Press, Washington, D.C. [PubMed]
- 54.Nedenskov, P. 1994. Nutritional requirements for growth of Helicobacter pylori. Appl. Environ. Microbiol. 60:3450-3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pitson, S. M., G. L. Mendz, S. Srinivasan, and S. L. Hazell. 1999. The tricarboxylic acid cycle of Helicobacter pylori. Eur. J. Biochem. 260:258-267. [DOI] [PubMed] [Google Scholar]
- 55a.Price, N. D., J. A. Papin, and B. O. Palsson. 2002. Determination of redundancy and systems properties of the metabolic network of Helicobacter pylori using genome-scale extreme pathway analysis. Genome Res. 12:760-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reynolds, D. J., and C. W. Penn. 1994. Characteristics of Helicobacter pylori growth in a defined medium and determination of its amino acid requirements. Microbiology 140:2649-2656. [DOI] [PubMed] [Google Scholar]
- 57.Schilling, C. H., J. S. Edwards, D. Letscher, and B. O. Palsson. 2000. Combining pathway analysis with flux balance analysis for the comprehensive study of metabolic systems. Biotechnol. Bioeng. 71:286-306. [PubMed] [Google Scholar]
- 58.Schilling, C. H., J. S. Edwards, and B. O. Palsson. 1999. Towards metabolic phenomics: analysis of genomic data using flux balances. Biotechnol. Progress 15:288-295. [DOI] [PubMed] [Google Scholar]
- 59.Schilling, C. H., D. Letscher, and B. O. Palsson. 2000. Theory for the systemic definition of metabolic pathways and their use in interpreting metabolic function from a pathway-oriented perspective. J. Theor. Biol. 203:229-248. [DOI] [PubMed] [Google Scholar]
- 60.Schilling, C. H., and B. O. Palsson. 2000. Assessment of the metabolic capabilities of Haemophilus influenzae Rd through a genome-scale pathway analysis. J. Theor. Biol. 203:249-283. [DOI] [PubMed] [Google Scholar]
- 61.Schilling, C. H., and B. O. Palsson. 1998. The underlying pathway structure of biochemical reaction networks. Proc. Natl. Acad. Sci. USA 95:4193-4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skouloubris, S., A. Labigne, and H. De Reuse. 1997. Identification and characterization of an aliphatic amidase in Helicobacter pylori. Mol. Microbiol. 25:989-998. [DOI] [PubMed] [Google Scholar]
- 63.Skouloubris, S., J. M. Thiberge, A. Labigne, and H. De Reuse. 1998. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect. Immun. 66:4517-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stark, R. M., J. Greenman, and M. R. Millar. 1995. Physiology and biochemistry of Helicobacter pylori. Br. J. Biomed. Sci. 52:282-290. [PubMed] [Google Scholar]
- 65.Stark, R. M., M. S. Suleiman, I. J. Hassan, J. Greenman, and M. R. Millar. 1997. Amino acid utilisation and deamination of glutamine and asparagine by Helicobacter pylori. J. Med. Microbiol. 46:793-800. [DOI] [PubMed] [Google Scholar]
- 66.Tomb, J. F., O. White, A. R. Kerlavage, R. A. Clayton, G. G. Sutton, R. D. Fleischmann, K. A. Ketchum, H. P. Klenk, S. Gill, B. A. Dougherty, K. Nelson, J. Quackenbush, L. Zhou, E. F. Kirkness, S. Peterson, B. Loftus, D. Richardson, R. Dodson, H. G. Khalak, A. Glodek, K. McKenney, L. M. Fitzegerald, N. Lee, M. D. Adams, E. K. Hickey, D. E. Berg, J. D. Gocayne, T. R. Utterback, J. D. Peterson, J. M. Kelley, M. D. Cotton, J. M. Weidman, C. Fujii, C. Bowman, L. Watthey, E. Wallin, W. S. Hayes, M. Borodovsky, P. D. Karp, H. O. Smith, C. M. Fraser, and J. C. Venter. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539-547. [DOI] [PubMed] [Google Scholar]
- 67.Varma, A., and B. O. Palsson. 1994. Metabolic flux balancing: basic concepts, scientific and practical use. Bio/Technology 12:994-998. [Google Scholar]
- 68.Varner, J., and D. Ramkrishna. 1999. Mathematical models of metabolic pathways. Curr. Opin. Biotechnol. 10:146-150. [DOI] [PubMed] [Google Scholar]