

Abstract
The interaction of activated G protein-coupled receptors with G proteins is a key event in signal transduction. Here, using a fluorescence resonance energy transfer (FRET)-based assay, we measure directly and in living cells the interaction of YFP-labeled α2A-adrenergic receptors with CFP-labeled G proteins. Upon agonist stimulation, a small, concentration-dependent increase in FRET was observed. No specific basal FRET was detected in the absence of agonist. Kinetics of the onset of receptor/G protein interaction were <100 ms and depended on expression levels of Gα. Simultaneously recorded G protein-regulated inwardly rectifying K+ channel currents revealed a maximal current response already at agonist concentrations producing submaximal FRET amplitudes. By analyzing FRET signals in the presence of a Gα mutant, which dissociates more slowly from activated receptors, it was demonstrated that only a fraction of wild-type G proteins interacts with the activated receptor at any time. Our data suggest that α2A-adrenergic receptors and G proteins interact by rapid collision coupling and indicate that there is no significant precoupling between these receptors and G proteins.
Keywords: α2A receptors, FRET, GIRK, G protein, kinetics
Introduction
G protein-coupled receptors (GPCRs) play important roles in cellular communication. Upon agonist stimulation, GPCRs interact with heterotrimeric G proteins, which, in turn, activate a variety of effectors (Bourne, 1997; Hamm, 1998). This receptor/G protein interaction represents a key event in transmembrane signal transduction; however, due to a lack of suitable methods, little is known about the dynamics of this interaction.
Originally, it was proposed that receptors and G proteins couple by collision (Orly and Schramm, 1976; Tolkovsky and Levitzki, 1978). The main feature of this model is that encounters between receptors and G proteins occur under conditions of free lateral diffusion within the cell membrane, and only activated receptors interact productively with G proteins. Since a typical cell expresses ∼100 different GPCRs (Hakak et al, 2003), a substantial number of different G protein subunits and multiple effectors, and since there is a need for directing the signal from the receptor to its cognate effector rapidly and distinctly, the collision coupling model soon was questioned (reviewed by Neubig, 1994), and more sophisticated models of signal transduction were developed (Neubig, 1994; Chidiac, 1998; Hur and Kim, 2002). One model proposes that receptor and G protein may be coupled even in the absence of agonist (‘precoupled model') (Neubig et al, 1988): this model is appealing in that it provides an inherent explanation for signaling specificity. Another model postulates that receptors, their G proteins and effectors are compartmentalized in microdomains (Neubig, 1994; Neer, 1995; Huang et al, 1997), where they diffuse freely and couple upon agonist stimulation; however, only signaling compounds within one such domain should be able to interact with each other, thereby providing the necessary specificity. The concentrations of signaling components should be locally very high and thus permits subsecond kinetics observed with GPCR signaling (Gross and Lohse, 1991).
Another issue that could not be solved until now is the duration of the individual steps of GPCR-mediated signaling. Effector activation kinetics strongly depend on the expression levels of receptors, as shown by G protein-regulated inwardly rectifying K+ (GIRK) current measurements (Bünemann et al, 1997, 2001). This, together with the phenomenon of spare receptors, suggests that at least one step in the signal transduction cascade is mediated via collision coupling (Bünemann et al, 2001). However, direct kinetic measurements on a millisecond time scale in intact cells have been, until recently, available for ion channel (effector) activation only.
The discovery of genetically encoded fluorescent proteins (Tsien, 1998) and their use for fluorescence resonance energy transfer (FRET) (Förster, 1948) has opened up the possibility to observe changes in inter- or intramolecular arrangements of molecules in intact cells. FRET techniques have been used in recent years to study protein/protein interactions in single living cells (Miyawaki, 2003). We have recently developed methods to directly monitor GPCR activation (Vilardaga et al, 2003), and methods to measure activation of G proteins have also been published (Janetopoulos et al, 2001; Bünemann et al, 2003; Yi et al, 2003; Azpiazu and Gautam, 2004; Frank et al, 2005). These assays allow time-resolved measurements of single cell signaling events. Here, using a YFP-tagged α2A-adrenergic receptor (α2A-YFP) and CFP-tagged G protein subunits, we measure directly the interaction of GPCRs with G proteins in single living cells.
Results
Fluorescence-labeled receptors and G proteins colocalize at the cell membrane and show FRET upon agonist stimulation
Our aim was to study the kinetics and extent of interaction of GPCRs with G proteins in living cells. To do so, we developed a FRET-based assay, consisting of an α2A-adrenergic receptor, whose C-terminus was fused to YFP, and a CFP-tagged Gγ2 subunit (schematically depicted in Figure 1A). After transient transfection together with Gαi1β1 in HEK293T cells, α2A-YFP and CFP-γ2 colocalized at the cell membrane (Figure 1B). Upon superfusion with norepinephrine (NE) and recording of whole-cell fluorescence, an increase in YFP fluorescence and a decrease in CFP fluorescence was observed, resulting in a small but readily detectable increase in FRET (Figure 1C). This increase in FYFP/FCFP upon NE superfusion can also clearly be seen when traces of different experiments are averaged (Figure 1D; n=8). Moreover, superfusion with different agonist concentrations ranging from 0.1 to 1000 μM showed concentration-dependent changes in FRET (Figure 1E; a concentration–response curve is given below; see Figure 2B). The ascending parts of the FRET trace represent the kinetics of the interaction, the amplitude the amount of agonist-dependent interaction, and the descending parts the off-rate of the interaction. To test whether the observed changes in FRET indeed represent changes in the interaction of receptor and G protein, we transfected only one fluorescent construct and exchanged the other with its nonlabeled counterpart and observed no FRET response upon stimulation with NE (data not shown). We also confirmed that the α2A-YFP is virtually indistinguishable from its wild-type variant with respect to GIRK channel activation (Supplementary Figure 1A), as are fluorophore-tagged G protein subunits (Bünemann et al, 2003). To further validate our assay, we transfected CFP-γ2 (plus Gαi1β2) with a membrane-anchored YFP (mYFP) instead of α2A-YFP. Both constructs showed proper membrane staining (Supplementary Figure 1B), and because both α2A-YFP and Gαi1β1 CFP-γ2 and mYFP and Gαi1β1 CFP-γ2 colocalized, it can be concluded that α2A-YFP and mYFP are also similarly distributed. Further, as judged by fluorescence, α2A-YFP and mYFP had similar expression levels under our transfection conditions. However, with mYFP, upon stimulation with 100 μM NE, no change in YFP and CFP fluorescence was detectable (Supplementary Figure 1C), indicating that the observed changes in fluorescence and hence in FRET as described above were specific and not due to quenching effects caused by movement of fluorophores relative to other proteins or the cell membrane. Moreover, we examined the behavior of a constitutively active Gαi protein mutant having decreased GTPase activity, Q204L (Coleman et al, 1994; Wang et al, 2004), and detected an agonist-induced increase in FRET, which was considerably smaller than the signal seen with the wild-type Gαi1 (Supplementary Figure 1D); the presence of a small residual signal most likely reflects the amount of remaining GTPase activity of this mutant (Coleman et al, 1994). Taken together, these data clearly show that changes in FRET in our assay reflect indeed association and dissociation of the receptor and the G protein.
Figure 1.

Analysis of receptor/G protein interaction by FRET. (A) FRET was measured between YFP-tagged receptors and CFP-tagged Gβγ subunits. When these constructs are excited at 436 nm, emission is shifted from 480 to 535 nm when both fluorophores are close enough to each other to permit FRET. (B) In HEK293T cells transiently expressing α2A-YFP and CFP-γ2 together with Gαi1β2 (μg transfected DNA: α2A-YFP 0.4, Gαi1 2, Gβ1 0.5, Gγ2 0.25), α2A-YFP (left) and CFP-γ2 (right) colocalize at the cell membrane (scale bar: 5 μM). (C) Upon stimulation with 100 μM NE (bar), a decrease in CFP fluorescence (FCFP) and an increase in corrected YFP fluorescence (FYFP) were observed. This resulted in an increase in FRET, assessed as the ratio of FYFP over FCFP. The FRET increase is readily reversible upon agonist washout. (D) The average increase in FRET ratio was ∼0.022 (n=8). (E) Fluorescence and FRET changes in a cell (as in panel C) in response to different NE concentrations. The FRET signal is stable over more than 400 s, and the amplitude of the FRET change depends on agonist concentration (concentrations indicated in μM; representative experiment out of eight shown). The agonist-independent increase in the YFP and CFP traces at ∼270 s is due to removal of solution from the coverslip holder; note that no increase is seen in the ratiometric FRET trace.
Figure 2.
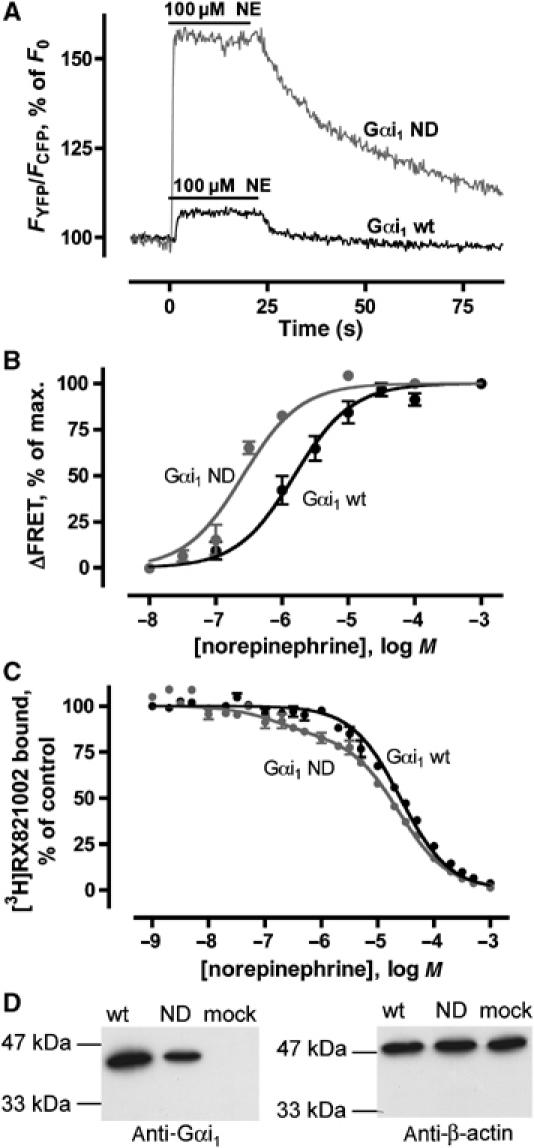
α2A-YFP interaction with wild-type Gαi1 and Gαi1ND. (A) FRET signals of individual cells in response to 100 μM NE (bar) with wild-type (black curve) and ND mutant (gray curve) Gαi1 coexpressed with α2A-YFP and Gβ1 CFP-γ2 (n=5, representative recording shown). (B) Concentration–response curves of receptor/G protein interaction for wild-type (black) and ND mutant (gray) (n=5–8) Gαi1 were determined by measuring amplitudes of FRET changes as in panel A after stimulation with different concentrations of NE. FRET responses following stimulation with 1 mM NE were set to 100%. (C) In membranes prepared from cells expressing α2A-YFP, Gβ1 CFP-γ2 and wild-type Gαi1 or Gαi1ND, high-affinity agonist binding sites were determined by competing for [3H]RX821002 binding with NE (n=3 each). Competition binding data for wild-type Gαi1 (black) were fitted best by a monophasic curve (Ki=21±1.8 μM), while a biphasic fit was significantly better (F-test) for Gαi1ND (gray; Ki,high=95±70 nM, Ki,low=17±0.8 μM, 16% high-affinity sites). (D) Western blot analysis of expression levels of HEK293T cells transfected with wild-type Gαi1 (wt) or Gαi1ND (ND); β-actin was determined in the same samples as control (n=3, representative experiment shown). Cells not transfected with Gαi1 were also analyzed (mock).
We then examined whether receptors interact with PTX-inactivated G proteins. To answer this question, we transfected a PTX-sensitive Gαi1 and pretreated the cells with 50 ng/ml PTX for ⩾4 h, a treatment that completely abolished GIRK activation via α2A receptor (data not shown). Upon NE stimulation, an increase in FRET (Supplementary Figure 2) was observed. This indicates that PTX-inactivated G proteins can still associate with and dissociate from the receptor, depending on the activation state of the receptor. This new finding is in line with a recent study suggesting that βγ and α subunits of the same G protein interact sequentially with activated rhodopsin (Herrmann et al, 2004), and we speculate likewise that the βγ subunits of a complete Gαβγ heterotrimer interact first with the receptor, followed or complemented by a second interaction of the α subunit of the same heterotrimer with the receptor. While the latter interaction and hence activation of the G protein should be distorted by PTX treatment, this should not influence the first interaction; however, formal proof of the hypothesis of Herrmann et al will require further studies.
Only a small fraction of Gi proteins interacts with the receptor
A general problem with bimolecular FRET and bioluminescence resonance energy transfer (BRET) assays is that it is difficult to decide whether the observed signal amplitude is limited by distance and orientation constraints or by the fraction of partners interacting. We addressed this problem by cloning and examining a mutant of Gαi1 that is analogous to a recently published single amino-acid mutation of the yeast G protein Gpa1 (Wu et al, 2004). This mutant is still able to bind to the receptor and to hydrolyze GTP upon agonist stimulation, but shows impaired dissociation from activated receptors. By mutating the homologous amino acid of Gαi1 in the same way (N270D), we obtained Gαi1ND. Transfection of Gαi1ND instead of Gαi1 resulted in a dramatic increase in the amplitude of agonist-induced FRET (Figure 2A; for averaged traces, see Supplementary Figure 4), suggesting that the fraction of interacting partners per time is increased. Concentration–response curves of NE for wild-type Gαi1 and Gαi1ND revealed EC50 values of 1.5±0.07 and 0.26±0.06 μM for wild-type Gαi1 and Gαi1ND, respectively (Figure 2B), with Hill slopes not significantly different from 1; here, changes from basal to equilibrium FRET levels upon stimulation with different agonist concentrations are analyzed. To test whether the Gαi1ND mutation does indeed improve coupling to the receptors, we performed competition binding experiments to measure high-affinity binding sites that affect receptor/G protein complexes. While no high-affinity binding to α2A receptors was detectable for wild-type Gαi1, competition data for Gαi1ND were significantly better explained by a two-site fit (Figure 2C). Calculated Ki values for NE were 21±1.8 μM with wild-type Gαi1, and 17±0.8 μM and 95±70 nM for low- and high-affinity binding with Gαi1ND, respectively (n=3), with 16% of binding sites being of high affinity. These results confirm that Gαi1ND has a higher propensity than the wild-type protein to induce a high-affinity state of the receptor. This is further supported by the notion that the dissociation kinetics between α2A-YFP and Gαi1ND β1 CFP-γ2 is increased compared to wild-type G protein dissociation (see below). The observed effects are not due to overexpression of Gαi1ND compared to wild-type Gαi1, since Western blots showed approximately equal expression levels (Figure 2D). The HEK cells used in this study do not express detectable amounts of Gαi1 but rather other subtypes of the Gαi/o family endogenously (not shown).
Receptors and G proteins are not precoupled to a significant amount
We next asked whether receptors and G proteins are precoupled. To account for the possibility that receptors and G proteins are precoupled by some as yet unknown mechanism, we wanted to make sure that we did not overexpress functional G proteins at the plasma membrane. Therefore, we transfected only the α2A-YFP and Gβ1 CFP-γ2, thus omitting exogenous Gα subunits. In this situation, trimeric G protein content in the cell is limited by the amount of endogenous Gα subunits, and transfected Gβ1 CFP-γ2 are targeted to these Gα subunits. Some cells transfected with Gβ1 CFP-γ2 displayed proper membrane staining, indicating that endogenous Gα complemented the transfected Gβ CFP-γ subunits (Figure 3A). These cells also showed a response in FRET to agonist stimulation (Figure 3B), although the amplitude of the signal and the signal to noise ratio were reduced compared to Gα-cotransfected cells. To detect possible agonist-independent FRET as a measure of precoupling, we measured the increase in FCFP after acceptor photobleaching (see Materials and methods for rationale); an increase in FCFP of 0% would show absence of any FRET before the bleaching process. In addition to α2A-YFP, mYFP was also examined as acceptor to determine nonspecific FRET caused simply by colocalization of the fluorophores at the cell membrane. Without cotransfecting Gα and in the basal state, that is, without applying agonist, FCFP increased only slightly and independent of the acceptor (2.8±0.6% for α2A-YFP and 2.4±0.6% for mYFP; n=14 and 10, respectively) (Figure 3C). This similar increase indicates that there is no detectable precoupling of receptor and G protein at endogenous levels of G proteins, but only a small degree of nonspecific FRET. Analogous results were obtained in cells cotransfected with Gαi1 (Figure 3C; increase in FCFP: 3.2±0.3% with α2A-YFP and 3.7±0.5% with mYFP, n=17 and 33, respectively); here, the somewhat higher increase in FCFP is most likely due to a higher concentration of Gαβ CFP-γ at the cell membrane upon Gα cotransfection. Further, we measured donor fluorescence of the same Gαi1-cotransfected cell before and after photobleaching in the presence and absence of agonist. In 10 μM NE-stimulated cells, photobleaching of YFP increased the FCFP by 5.0±0.5% (Figure 3C; n=11). Compared to nonstimulated conditions (i.e. 3.2±0.3%), this increase is significant (P<0.0001, paired t-test). These measurements were also performed when Gαi1ND was cotransfected instead of Gαi1. While the increase in FCFP without agonist after acceptor photobleaching was not significantly different for Gαi1ND (4.6±0.5%, n=9) compared to wild-type Gαi1 agonist-dependent FRET was much higher for Gαi1ND (increase in FCFP 10±1.4%, n=10; P<0.001 in a one-way ANOVA followed by Tukey's multiple comparison test); this is several fold the specific increase seen with the wild-type G protein.
Figure 3.

Analysis of absolute FRET levels. (A) Confocal images of cells transfected with α2A-YFP (left) and Gβ1 CFP-γ2 (right; scale bar: 5 μM); here, Gαi1 was not cotransfected. (B) FRET signal mediated only by endogenous Gα subunits. In cells transfected with α2A-YFP and Gβ1 CFP-γ2, a small increase in FRET in response to agonist stimulation could be detected (representative example of 17 shown). (C) By means of measuring donor recovery after acceptor photobleaching, FRET between the α2A-YFP and CFP-γ2 was quantified. CFP fluorescence of cells expressing the indicated constructs was measured before and after acceptor photobleaching for 5 min. In the absence of agonist, the increase in FCFP was not significantly different when comparing mYFP and the α2A-YFP as acceptor, regardless of whether Gαi1 was coexpressed (n=14 and 10 for α2A-YFP and mYFP, respectively) or not (n=17 and 33 for α2A-YFP and mYFP, respectively). For cells expressing Gαi1, FCFP was also measured in the presence and absence of 100 μM NE in the same cell; here, a significant increase was observed (n=11). *P<0.0001 (paired t-test), #P<0.001 in a one-way ANOVA followed by Tukey's post test. (D) Increase in FCFP after acceptor photobleaching of cells transfected with mYFP or α2A-YFP and Gαi1-CFP β1γ2 (left; n=10 and 9) or Gαi1 Cerulean-β1γ2 (right; n=3 and 7).
To validate these results further, we examined the interaction of α2A-YFP with G proteins labeled at the α (Gαi1-CFP) or β (Cerulean-Gβ1) subunits; these constructs also showed an agonist-dependent FRET (see Supplementary Figure 3). We also determined absolute FRET levels by means of acceptor photobleaching between α2A-YFP or mYFP and Gαi1-CFP or Cerulean-Gβ1 subunits and again could not detect any significant differences (Figure 3D), confirming that indeed there is no specific interaction between receptors and G proteins in the absence of agonist.
Kinetics of receptor/G protein interaction
To determine the kinetics of receptor/G protein interaction, we stimulated transfected cells with different concentrations of NE ranging from 0.1 to 1000 μM and analyzed the corresponding increase in FRET. As demonstrated by a representative example of traces obtained from a single cell (Figure 4A), the speed of the FRET signal increased with increasing concentrations of NE. Since the traces obtained following stimulation with the lower agonist concentrations did not allow fitting of mono-exponential curves, we assessed the kinetics of the interaction using T1/2, that is, the time until the FRET ratio reached its half-maximum. We obtained values ranging from 2.4±0.36 s for 0.1 μM NE to 0.098±0.07 s for 1 mM NE, and the speed of association seems to become saturated at NE concentrations ⩾100 μM (Figure 4B; n=4–8). In order to elucidate further the temporal dynamics of signaling from the receptor to its G protein, we used a method we have already established to directly determine GPCR activation (Vilardaga et al, 2003). Measurement of receptor activation using this method also works in urea-treated membranes, which shows that receptor activation (R*) is a step different from receptor/G protein interaction (R*G) and G protein activation (Vilardaga et al, 2003). We determined the kinetics of receptor activation and receptor/G protein interaction in HEK cells using the saturating concentration of 1 mM NE. We confirmed an activation switch time constant of the receptor of 44±4.0 ms (Figure 4C; n=8) by fitting mono-exponential curves to the data. To assess whether diffusion in the cell membrane limits the kinetics of the interaction between receptor and G protein, we varied the amount of functional G proteins by cotransfecting varying amounts of Gαi1 cDNA. The time constant determined for the interaction of receptors and G proteins (at 1 mM NE) was dependent on the amount of Gαi1 cotransfected (Figure 4C; time constants: 86±10 ms (n=11), 74±2.3 ms (n=7), 58±7.0 ms (n=8), 54±3.1 ms (n=3) and 44±5.1 ms (n=10) for 0, 0.2, 0.5, 1 and 2 μg of transfected Gαi1 cDNA, respectively); this again indicates that the interaction between receptors and G proteins occurs by collision coupling. With the highest amount of Gα, the interaction of the receptor and the G protein was as fast as receptor activation itself. Figure 4D illustrates the different Gα levels by means of a Western blot against Gαi1 (left panel); β-actin was detected as loading control (right panel).
Figure 4.
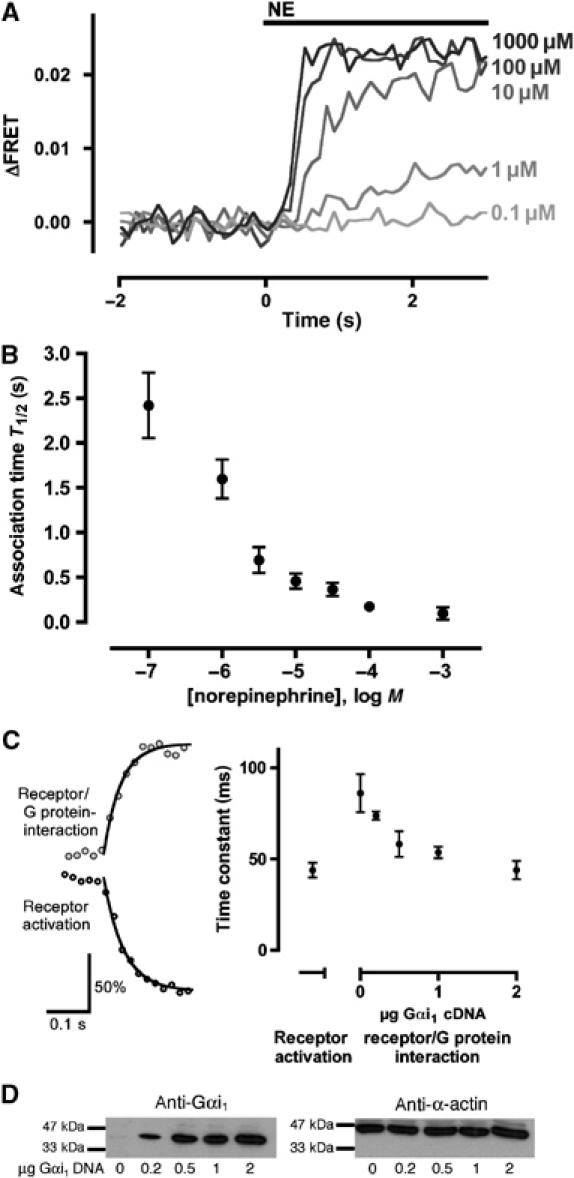
Kinetics of the α2A-adrenergic receptor/Gi interaction. (A) Cells transfected with α2A-YFP and Gαi1β1 CFP-γ2 were superfused with different NE concentrations. Activation traces of the same cell after stimulation with different concentrations of NE are shown (representative experiment out of eight). (B) Times required to achieve a half-maximal FRET response (T1/2) are plotted as a function of agonist concentration; P<0.0001 (one-way ANOVA). (C) Kinetics (left) of receptor activation (dark gray; determined with cells expressing the α2A-CFP/YFP; Vilardaga et al, 2003) and receptor/G protein interaction (light gray) after stimulation with 1 mM NE (representative experiments of 8 and 10, respectively). The maximal amplitude of both traces was set to 100%. Averaged time constants (right) were 44±4.0 ms for receptor activation; for receptor/G protein interaction, the time constants depended on the amount of Gαi1 and were 86±10, 74±2.3, 58±7.0, 54±3.1 and 44±5.1 ms for 0, 0.2, 0.5, 1 and 2 μg transfected Gαi1 cDNA, respectively; P=0.0022 for different Gα amounts (one-way ANOVA). (D) Western blot of lysates from cells transfected with α2A-YFP, Gβ1 CFP-γ2 (for amounts, see Materials and methods) and indicated amounts of Gαi1 (left). In the same samples, β-actin was determined as a control.
We also analyzed the off-rate of the FRET signal (see Figure 2A), presumably reflecting dissociation kinetics of receptor and G protein, which were assessed by fitting a mono-exponential decay curve to the FRET trace. The time constant was not dependent on the concentration of the agonist applied before (0.1 μM–1 mM NE tested, n=33) and was determined as 13±2.1 s. When Gαi1ND was used instead of Gαi1, a significantly longer deactivation time was observed (time constant 30.3±2.9 s, n=22; P<0.0001 in a two-tailed t-test versus wild-type G proteins; Figure 2A), indicating a slower dissociation of this mutant G protein from receptors.
The kinetics of receptor deactivation were determined using the α2A-FlAsH-CFP construct (Hoffmann et al, 2005) and were found to be 2.24±0.12 and 1.88±0.25 s with cotransfection of wild-type and ND mutant G proteins, respectively (n=11 each). These values are not significantly different from each other. Taken together, receptor deactivation kinetics are markedly faster than the interaction kinetics.
Effector measurements
Last, we compared the kinetics of the receptor/G protein interaction to effector kinetics and correlated their temporal relationship by measuring GIRK channel activation, which is directly mediated via βγ subunits of Gi proteins (Wickman et al, 1994). To assess the interaction of the α2A-YFP with G proteins and the effector response simultaneously, we measured agonist-dependent FRET between receptor and G protein while patch-clamping the cell in the whole-cell configuration. After stimulation with 100 μM NE, an increase in FRET and in GIRK currents was observed; both effects were readily reversible upon washout of the agonist (Figure 5A). Moreover, the deactivation kinetics of the receptor/G protein interaction are much faster than the deactivation of the GIRK channel, which follows G protein deactivation (Bünemann et al, 2003). Next, we compared receptor–G protein FRET and GIRK current responses for stimulations with 1 and 100 μM NE. Consecutive experiments in the same cell showed the same maximal GIRK response to 1 and 100 μM NE (with the response to 100 μM being much faster), while the 1 μM NE-evoked FRET signal amounted to only 25% of the signal observed with 100 μM (Figure 5B). This indicates that a submaximal receptor/G protein interaction can be sufficient to produce a maximal GIRK response.
Figure 5.

Analysis of the simultaneously recorded receptor/G protein interaction and GIRK currents. (A) In cells expressing α2A-YFP, Gαi1β1 CFP-γ2 and GIRK1+4, FRET responses (thick curves) and GIRK currents (thin curves) were measured simultaneously after stimulation with 100 μM NE (bar; complete reversal of GIRK currents not shown). (B) FRET responses (thick curves) and GIRK currents (thin curves) were recorded during two subsequent stimulations with 1 and 100 μM NE (gray and black curves, respectively) in the same cell. The amplitudes obtained with the 100 μM NE were set to 100%, and the FRET and GIRK current traces following 1 μM NE stimulation were normalized accordingly (n=6, representative recording shown).
Discussion
The interaction between agonist-occupied GPCRs and G proteins constitutes a key event that leads to signal transduction from the extracellular to the intracellular side of the cell. Using a FRET-based approach, we were able to directly measure the interaction between receptors and G proteins in living cells, where an increase in interaction of G proteins with the receptor can be visualized in real time as an increase of the FRET ratio. To do this, we established an assay consisting of a YFP-tagged α2A-adrenergic receptor and a CFP-tagged Gβγ. The tagged receptor is functional with respect to plasma membrane targeting, ligand binding and signaling (shown by unaltered GIRK channel activation), and, likewise, the labeled G protein is fully functional (Ruiz-Velasco and Ikeda, 2001; Bünemann et al, 2003). These constructs exhibited no significant specific FRET when coexpressed; however, upon stimulation with NE, a small but significant increase in FRET was detected. Since the kinetics of this signal are in line with kinetics of receptor and effector activation (Bünemann et al, 2001; Vilardaga et al, 2003), and depend on the amount of Gαi1, the measured signal indeed appears to originate from receptor–G protein complexes. This is further reinforced by the observation that a constitutively active mutant of Gαi1 (Q204L), which possesses only little remaining GTPase activity (Coleman et al, 1994), shows a markedly smaller FRET signal upon agonist stimulation. Moreover, this remaining FRET signal can also be explained through FRET between G proteins consisting of Gβγ complemented with endogenous Gα subunits and receptors. After agonist withdrawal, the kinetics of the termination of the interaction of receptor and G protein were about 10-fold slower compared to the kinetics of receptor deactivation; this reflects the dissociation of G proteins from receptors in their high-affinity state.
A recent independent study using a BRET-based approach showed a similar increase in energy transfer between labeled α2A receptors and G proteins (Gales et al, 2005). This study mainly focused on β2-adrenergic receptors and observed a small agonist-independent BRET, which was attributed to the constitutive activity of the β2 receptor. In our study, we concentrated on the α2A receptor, which is known to exhibit only little if any constitutive activity (Seifert and Wenzel-Seifert, 2002). Therefore, it is not surprising that we do not observe a significant interaction between these receptors and G proteins in the absence of agonists.
We demonstrate that, in our model system, α2A receptors and G proteins are not precoupled, but rather interact upon agonist binding to the receptor. This is compatible with earlier experiments, using an activation sensor for Go (Azpiazu and Gautam, 2004). Further evidence for a collision coupling mechanism can be drawn from the observation that the interaction kinetics depend on the amount of Gα in the cell, because if receptors and G proteins were precoupled, the kinetics should be independent of G protein levels. We also obtained evidence that G proteins select for activated receptors and are not permanently associated with them in a 1:1 ratio. This can be concluded from the observed existence of spare receptors for G protein interaction: the EC50 of receptor/G protein interaction was shifted more than 10-fold to the left compared to the low-affinity Ki for agonist binding. This demonstrates that, after occupation of a small fraction of the receptors with agonist, this fraction of activated receptors is capable of interacting with a much greater number of G proteins. It is known that spare receptors exist with respect to GIRK channel activation (Bünemann et al, 2001), and it has also been shown that concentration–response curves and kinetics of G protein activation and GIRK channel activation do not differ (Bünemann et al, 2003); this indicates that there is a receptor reserve for G protein interaction and hence rules out a 1:1 coupling of receptor and G protein. A similar model is generally assumed for rhodopsin, where one activated receptor activates many G proteins (Heck and Hofmann, 2001).
Our results demonstrate that only a small percentage of G proteins is interacting with a receptor at any time during signaling. This is most evident from the data obtained with Gαi1ND, which exhibits slowed dissociation kinetics from activated receptors. The signal amplitudes obtained with this mutant are considerably higher than signal amplitudes measured with wild-type Gαi1. Since this mutant differs from the wild-type protein only at a single amino acid located in the GTP binding pocket, it is unlikely that the mutated G protein binds to the receptor sterically differently compared to wild-type Gαi1. However, we demonstrate that the mutant markedly enhances the amount of activated receptors coupled to Gαi1ND, and it has been speculated that this mutant forms a stable complex with agonist-bound receptors and Gβγ, but cannot be activated (Wu et al, 2004). Therefore, we conclude that, with wild-type Gαi1, only a fraction of the G proteins interacts with receptors at a given time during agonist stimulation; in other words, G proteins are not associated with a receptor during most of the G protein cycle. Nevertheless, this is sufficient to produce a full effector response, as detected by simultaneous GIRK current recordings.
Our results suggest collision coupling as the mechanism of receptor/G protein interaction. Therefore, we wondered whether this step may be time limiting and compared the kinetics of the activation of the α2A-adrenergic receptor (Vilardaga et al, 2003) with the kinetics of the interaction of receptor and G protein. In agreement with the collision coupling model, the time constants for the interaction of receptor and G protein decreased with increasing Gα expression. Under conditions of high levels of coexpressed Gα proteins, a substantial receptor reserve and maximal receptor activation, receptor/G protein interaction can even become as fast as receptor activation itself (both time constants ∼44 ms). Theoretically, a collision coupling model featuring a receptor reserve even allows for faster interaction kinetics compared to receptor activation, and it is tempting to speculate that similar conditions may prevail in systems where there is a need for rapid signal transduction, for example, on postsynaptic membranes in the CNS.
Taken together, our direct, time-resolved measurements of receptor/G protein interaction by FRET in living cells revealed a fast and transient agonist-induced association without detectable preformed complexes, suggesting that α2A-adrenergic receptors and Gi proteins interact in a strictly agonist-dependent manner and are driven by collision coupling.
Materials and methods
Molecular biology
The cDNA of the murine α2A-adrenergic receptor was fused C-terminally to the cDNA encoding the enhanced YFP F46L (Nagai et al, 2002) using standard PCR and cloned into pcDNA3 (Invitrogen) after removal of the stop codon using TCTAGA as a linker. Rat Gαi1, mutated at position 351 (C to I, pertussis toxin insensitive, used if not indicated otherwise), at position 270 (N to D; Wu et al, 2004) or at both positions, and human Gγ2 fused N-terminally to CFP (CFP-γ2; kindly provided by S Ikeda, Guthrie Research Institute, Sayre, PA) (Ruiz-Velasco and Ikeda, 2001) were also subcloned into pcDNA3, and human Gβ1 was cloned into pCMV. Gαi1 Q204L was purchased from cDNA Resource Center (Rolla, MO, USA). Gαi1-CFP was constructed analogous to Gαi1-YFP, and this construct and Cerulean-Gβ1 have been described earlier (Frank et al, 2005). A membrane-targeted EYFP (mYFP) was generated by fusing the EYFP-cDNA N-terminally to cDNA encoding a lipid modification site (MGCINSKRKD; kindly provided by V Nikolaev).
Cell culture
For GIRK current measurements, HEK293 cells stably expressing GIRK1+4 were used (Hommers et al, 2003); all other experiments were carried out in HEK293T cells. Cells were transiently transfected with (μg DNA/3 cm dish) 0.4 α2A-YFP, 2 Gαi1 (if not indicated otherwise), 0.5 Gβ1 and 0.2 CFP-Gγ2 using Effectene according to the manufacturer's instructions (Qiagen) or calcium phosphate 48 h (72 h if indicated) prior to the experiments. For receptor activation, HEK293 cells stably expressing the α2A-CFP/YFP (kindly provided by J-P Vilardaga; Vilardaga et al, 2003) and transfected with wild-type G protein subunits (μg DNA, see above) were used.
Western blot analysis
Western blots analyses were carried out according to standard procedures. Briefly, total protein was isolated from HEK293T cells 48 h after transfection, run on an SDS–PAGE and blotted onto 0.45 μM PVDF membranes (Millipore, Bedford, MA, USA). Anti-Gαi-1(I-20) (rabbit; Santa Cruz Biotechnology, Heidelberg, Germany) and anti-β-actin (mouse; Sigma-Aldrich, Taufkirchen, Germany) antibodies were used for the detection of Gαi1 and β-actin (loading control), respectively, and peroxidase-conjugated anti-mouse and anti-rabbit antibodies (DiaNova, Hamburg, Germany) were used as secondary antibodies. The bands were visualized with SuperSignal West Pico chemiluminescent solution (Pierce, Rockford, IL, USA).
Radioligand binding experiments
Saturation and competition binding experiments were performed as described (Erdbrügger et al, 1995) using (in mM) Tris 50, EDTA 0.5 and MgCl2 5, at pH 7.5 for 1 h at room temperature, and [3H]RX821002 (specific activity 60 Ci/mmol) was used as a radioligand. Mono- and biphasic curves were fitted to competition data, and biphasic curves were accepted only if they fitted the data better as judged by an F-test. Phenylephrine (10 μM) was used to determine nonspecific binding, and individual data points were determined in duplicate.
Imaging, fluorescence measurements and electrophysiology
Fluorescence microscopy was performed as described (Vilardaga et al, 2003). Briefly, cells were plated on coverslips on an Axiovert 200 inverted microscope (Zeiss, Jena, Germany) with an oil immersion × 63 objective, a dual-emission photometric system and a polychrome IV (both TILL Photonics, Planegg, Germany). Illumination time was ⩽40 ms at a frequency of 4–20 Hz. Excitation wavelength was set to 436±10 nm (beam splitter DCLP 460 nm), and emission of single whole cells was recorded at 535±15 and 480±20 nm (beam splitter DCLP 505 nm). Here, special care was taken to ensure that fluorescence levels and distribution were comparable in examined cells. FRET ratios were measured as ratios of YFP over CFP emission; YFP emission was corrected for direct excitation (YFP emission at 436 nm excitation/YFP emission at 490 nm excitation was 0.06) and spillover of CFP into the 535 nm channel (spillover of YFP into the 480 nm channel was negligible). Ratiometric FRET traces were used to analyze dynamic FRET changes; absolute FRET levels between CFP and YFP were determined by measuring donor dequenching after acceptor photobleaching for 5 min by illumination at 500 nm. This protocol bleached CFP by 3.7%, and the data are corrected for this effect.
Confocal images were taken using a Leica TCS SP2 system as described before (Krasel et al, 2005).
Cells were continuously superfused with external buffer (mM: NaCl 137, KCl 5.4, CaCl2 2, MgCl2 1, HEPES 10 at pH 7.3). For patch-clamp experiments, a different buffer (mM: NaCl 122.4, KCl 20, CaCl2 2, MgCl2 1, HEPES 10 at pH 7.3) was used; the internal buffer consisted of (mM) K+-aspartate 100, KCl 40, MgCl2 2.5, EGTA 5, HEPES 10 and ATP 5 at a pH of 7.4 supplemented with 100 μM GTP. Patch-clamp recordings in the whole-cell configuration were made as described (Bünemann et al, 2001), using an Axopatch 200B (Axon Instruments, Foster City, CA). Solutions were applied using a rapid superfusion device (Vilardaga et al, 2003) that allows for solution exchange times of 5–10 ms.
Data processing
Patch-clamp data and fluorescence intensities were acquired using CLAMPEX (Axon Instruments, Foster City, CA). Values are given as mean±s.e.m. of n experiments. Statistical analysis and curve fitting was performed using Prism (San Diego, CA, USA) or Origin (Northampton, MA, USA).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure Legends
Acknowledgments
This work was supported by a Leibniz award (Deutsche Forschungsgemeinschaft) and the Fonds der deutschen chemischen Industrie (to MJL). PH was supported by the IZKF, University Würzburg. We thank Dr G Breitwieser for helpful suggestions.
References
- Azpiazu I, Gautam N (2004) A FRET based sensor indicates that receptor access to a G protein is unrestricted in a living mammalian cell. J Biol Chem 279: 27709–27718 [DOI] [PubMed] [Google Scholar]
- Bourne HR (1997) How receptors talk to trimeric G proteins. Curr Opin Cell Biol 9: 134–142 [DOI] [PubMed] [Google Scholar]
- Bünemann M, Brandts B, Pott L (1997) In vivo downregulation of M2 receptors revealed by measurement of muscarinic K+ current in cultured guinea-pig atrial myocytes. J Physiol 501: 549–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bünemann M, Bücheler MM, Philipp M, Lohse MJ, Hein L (2001) Activation and deactivation kinetics of α2A- and α2C-adrenergic receptor-activated G protein-activated inwardly rectifying K+ channel currents. J Biol Chem 276: 47512–47517 [DOI] [PubMed] [Google Scholar]
- Bünemann M, Frank M, Lohse MJ (2003) Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc Natl Acad Sci USA 100: 16077–16082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidiac P (1998) Rethinking receptor–G protein–effector interactions. Biochem Pharmacol 55: 549–556 [DOI] [PubMed] [Google Scholar]
- Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR (1994) Structures of active conformations of Giα1 and the mechanism of GTP hydrolysis. Science 265: 1405–1412 [DOI] [PubMed] [Google Scholar]
- Erdbrügger W, Raulf M, Otto T, Michel MC (1995) Does [3H]2-methoxy-idazoxan (RX 821002) detect more α2-adrenoceptor agonist high-affinity sites than [3H]rauwolscine? A comparison of nine tissues and cell lines. J Pharmacol Exp Ther 273: 1287–1294 [PubMed] [Google Scholar]
- Förster T (1948) Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann Phys (Leipzig) 2: 55–75 [Google Scholar]
- Frank M, Thümer L, Lohse MJ, Bünemann M (2005) G protein activation without subunit dissociation depends on a Gαi-specific region. J Biol Chem 280: 24584–24590 [DOI] [PubMed] [Google Scholar]
- Gales C, Rebois RV, Hogue M, Trieu P, Breit A, Hebert TE, Bouvier M (2005) Real-time monitoring of receptor and G-protein interactions in living cells. Nat Methods 2: 177–184 [DOI] [PubMed] [Google Scholar]
- Gross W, Lohse MJ (1991) Mechanism of activation of A2 adenosine receptors. II. A restricted collision-coupling model of receptor–effector interaction. Mol Pharmacol 39: 524–530 [PubMed] [Google Scholar]
- Hakak Y, Shrestha D, Goegel MC, Behan DP, Chalmers DT (2003) Global analysis of G-protein-coupled receptor signaling in human tissues. FEBS Lett 550: 11–17 [DOI] [PubMed] [Google Scholar]
- Hamm HE (1998) The many faces of G protein signaling. J Biol Chem 273: 669–672 [DOI] [PubMed] [Google Scholar]
- Heck M, Hofmann KP (2001) Maximal rate and nucleotide dependence of rhodopsin-catalyzed transducin activation: initial rate analysis based on a double displacement mechanism. J Biol Chem 276: 10000–10009 [DOI] [PubMed] [Google Scholar]
- Herrmann R, Heck M, Henklein P, Kleuss C, Hofmann KP, Ernst OP (2004) Sequence of interactions in receptor–G protein coupling. J Biol Chem 279: 24283–24290 [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Gaietta G, Bünemann M, Adams SR, Oberdorff-Maass S, Behr B, Vilardaga JP, Tsien RY, Ellisman MH, Lohse MJ (2005) A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nat Methods 2: 171–176 [DOI] [PubMed] [Google Scholar]
- Hommers LG, Lohse MJ, Bünemann M (2003) Regulation of the inward rectifying properties of G-protein-activated inwardly rectifying K+ (GIRK) channels by Gβγ subunits. J Biol Chem 278: 1037–1043 [DOI] [PubMed] [Google Scholar]
- Huang C, Hepler JR, Chen LT, Gilman AG, Anderson RG, Mumby SM (1997) Organization of G proteins and adenylyl cyclase at the plasma membrane. Mol Biol Cell 8: 2365–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Kim KT (2002) G protein-coupled receptor signalling and cross-talk: achieving rapidity and specificity. Cell Signal 14: 397–405 [DOI] [PubMed] [Google Scholar]
- Janetopoulos C, Jin T, Devreotes P (2001) Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science 291: 2408–2411 [DOI] [PubMed] [Google Scholar]
- Krasel C, Bünemann M, Lorenz K, Lohse MJ (2005) β-Arrestin binding to the β2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J Biol Chem 280: 9528–9535 [DOI] [PubMed] [Google Scholar]
- Miyawaki A (2003) Visualization of the spatial and temporal dynamics of intracellular signaling. Dev Cell 4: 295–305 [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20: 87–90 [DOI] [PubMed] [Google Scholar]
- Neer EJ (1995) Heterotrimeric G proteins: organizers of transmembrane signals. Cell 80: 249–257 [DOI] [PubMed] [Google Scholar]
- Neubig RR (1994) Membrane organization in G-protein mechanisms. FASEB J 8: 939–946 [DOI] [PubMed] [Google Scholar]
- Neubig RR, Gantzos RD, Thomsen WJ (1988) Mechanism of agonist and antagonist binding to α2 adrenergic receptors: evidence for a precoupled receptor–guanine nucleotide protein complex. Biochemistry 27: 2374–2384 [DOI] [PubMed] [Google Scholar]
- Orly J, Schramm M (1976) Coupling of catecholamine receptor from one cell with adenylate cyclase from another cell by cell fusion. Proc Natl Acad Sci USA 73: 4410–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Velasco V, Ikeda SR (2001) Functional expression and FRET analysis of green fluorescent proteins fused to G-protein subunits in rat sympathetic neurons. J Physiol 537: 679–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K (2002) Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol 366: 381–416 [DOI] [PubMed] [Google Scholar]
- Tolkovsky AM, Levitzki A (1978) Mode of coupling between the β-adrenergic receptor and adenylate cyclase in turkey erythrocytes. Biochemistry 17: 3795. [DOI] [PubMed] [Google Scholar]
- Tsien RY (1998) The green fluorescent protein. Annu Rev Biochem 67: 509–544 [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Bünemann M, Krasel C, Castro M, Lohse MJ (2003) Measurement of the millisecond activation switch of G protein-coupled receptors in living cells. Nat Biotechnol 21: 807–812 [DOI] [PubMed] [Google Scholar]
- Wang Y, Tawa G, Smith D, Krishnamurthy G, Young KH (2004) Mutation of cysteine 214 in Gi1 α subunit abolishes its endogenous GTPase activity. Biochem J 379: 673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickman KD, Iniguez-Lluhl JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, Gilman AG, Clapham DE (1994) Recombinant G-protein βγ-subunits activate the muscarinic-gated atrial potassium channel. Nature 368: 255–257 [DOI] [PubMed] [Google Scholar]
- Wu YL, Hooks S, Harden TK, Dohlman HG (2004) Dominant-negative inhibition of pheromone receptor signaling by a single point mutation in the G protein alpha subunit. J Biol Chem 279: 35287–35297 [DOI] [PubMed] [Google Scholar]
- Yi TM, Kitano H, Simon MI (2003) A quantitative characterization of the yeast heterotrimeric G protein cycle. Proc Natl Acad Sci USA 100: 10764–10769 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure Legends