

Abstract
Cancer is a heterogeneous disease in most respects, including its cellularity, different genetic alterations, and diverse clinical behaviors. Traditional molecular analyses are reductionist, assessing only 1 or a few genes at a time, thus working with a biologic model too specific and limited to confront a process whose clinical outcome is likely to be governed by the combined influence of many genes. The potential of functional genomics is enormous, because for each experiment, thousands of relevant observations can be made simultaneously. Accordingly, DNA array, like other high-throughput technologies, might catalyze and ultimately accelerate the development of knowledge in tumor cell biology. Although in its infancy, the implementation of DNA array technology in cancer research has already provided investigators with novel data and intriguing new hypotheses on the molecular cascade leading to carcinogenesis, tumor aggressiveness, and sensitivity to antiblastic agents. Given the revolutionary implications that the use of this technology might have in the clinical management of patients with cancer, principles of DNA array-based tumor gene profiling need to be clearly understood for the data to be correctly interpreted and appreciated. In the present work, we discuss the technical features characterizing this powerful laboratory tool and review the applications so far described in the field of oncology.
DNA array-based gene profiling holds great promise for the management of patients with cancer. In this review, the principles underlying this technology and the potential applications in the field of oncology are discussed.
The heterogeneity of malignant cells and the variable host background produce multiple tumor subclasses. Many analytic methods have been used to study human tumors and to classify them into homogeneous groups that can predict clinical behavior. Currently, cancer classifications are principally based on clinical and morphologic features that only partially reflect this heterogeneity, reducing the probability of the most appropriate diagnostic and therapeutic strategy for each patient. Most current anticancer agents do not differentiate between cancerous and normal cells, resulting in sometimes disastrous toxicity and an inconstant efficiency. The development of innovative drugs that selectively target cancer cells while sparing normal tissues is very promising as suggested by successful recent examples such as the use of mAb therapy against the ERBB2 receptor in breast cancer1 or the tyrosine kinase inhibitor STI571 in chronic myelogenous leukemia2 and GIST.3
The development of several gene expression profiling methods such as comparative genomic hybridization (CGH),4 differential display,5 serial analysis of gene expression (SAGE),6 and DNA arrays,7 together with the sequencing of the human genome, has provided an opportunity to monitor and investigate the complex cascade of molecular events leading to tumor development and progression. The availability of such large amounts of information has shifted the attention of scientists toward a nonreductionist approach to biologic phenomena.8 High-throughput technologies can be used to follow changing patterns of gene expression over time. Among them, DNA arrays have become prominent because they are easier to use, do not require large-scale DNA sequencing, and allow the parallel quantification of thousands of genes from multiple samples. The ultimate effects of such large repertoire of biologic variables can be impossible to be predict only looking at changes in gene expression profiles over time. Therefore, DNA array technology should be complemented with other recently developed high-throughput assays such as tissue microarray9 and proteomics.10 Hopefully, by integrating these powerful analytic tools, investigators will be able to comprehensively describe the molecular portrait of the biologic phenomena underlying tumor etiopathogenesis and clinical behavior.
DNA array technology is rapidly spreading worldwide and has the potential to drastically change the therapeutic approach to patients affected with tumor. Given the central role played by surgeons in the current management of patients with solid cancer, it is of paramount importance for them to know the principles characterizing this laboratory tool to critically appreciate the results originating from this biotechnology. We describe its main technical features, from DNA array construction to data analysis, and review some of the most important and intriguing results already achieved in the field of oncology.
DNA ARRAY TECHNICAL FEATURES
High-throughput DNA array technology allows for the simultaneous measurement of the expression level of thousands of genes in a single experiment. Each array consists of a solid support (usually nylon or glass) in which cDNA or oligonucleotides (ie, targets) are spotted in a known pattern. Fluorescent or radioactive genetic material (ie, probes) derived from mRNA are hybridized to the complementary DNA on the array. The radioactive or fluorescence emissions from the specifically bound probe are detected using an appropriate scanner, giving a quantitative estimate of each gene expression. Ultimately, these signals represent the amounts of mRNA originally present in the cell.
The process can be described in 3 steps: 1) array construction, 2) sample preparation and array hybridization, and 3) image analysis and data analysis.
Technologic Platforms
Two main implementations of DNA arrays have been applied with success. The first uses arrays of cDNA clones robotically spotted on a solid surface in the form of polymerase chain reaction (PCR) products. Several versions exist depending on the type of support (nylon, glass) and the type of target labeling (radioactivity, colorimetry, fluorescence).11–13 This approach is flexible, allowing researchers to make arrays with their own gene sets, but it requires accurate collection and storage of cDNA clones and PCR products, which may be avoided by using commercially available arrays. Glass-based full-length cDNA arrays12 are widely used, in which the DNA probes are labeled by incorporation of fluorescently tagged nucleotides. Typically, 2 probes are hybridized on a single array (so-called test or experimental probe, and reference or control probe, respectively), each labeled with 2 different fluorophores (Fig. 1). The expression of a gene in an experimental situation is then expressed as a relative ratio with respect to the control sample (ie, untreated vs, treated cell line, disease vs. normal tissue, and so on). In case of nylon arrays, an automatic gridder prints PCR amplified cDNA to positively charged nylon membranes, and RNA probes are labeled with P33 or P32-dCTP during a reverse transcription reaction. Although cheap, nylon arrays are being overlooked by investigators in favor of fluorescence-based DNA arrays, which offer the capacity for a higher throughput of samples,14 allow for significant time saving (ie, there is no exposing of phosphorscreens for days because slides are simply scanned in minutes), and need not to use radioactive isotopes.
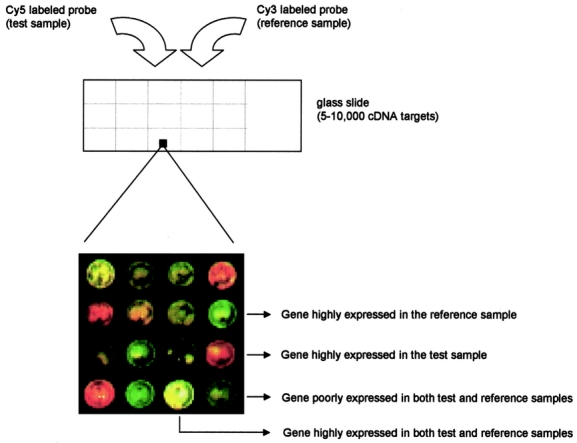
FIGURE 1. Scheme of glass-based cDNA array. DNA targets are represented by cDNA clones robotically spotted onto a solid surface. Each cDNA element on the array represents a gene. DNA probes derive from reverse-transcribed RNA extracted from biologic samples. Typically, 2 probes are hybridized on a single array: they are the control probe (usually labeled with Cy3 fluorophore) and the experimental probe (usually labeled with Cy5 fluorophore). The transcriptional levels of a given gene in an experimental situation is therefore expressed as a relative ratio with respect to the control sample. The image shown is produced by superimposing the Cy3 fluorescence image (pseudocolored green) and the Cy5 fluorescence image (pseudocolored red). Thus, red, green, and yellow colors represent respectively increased, decreased, and equal gene copy number in the experimental sample with respect to the control sample. Low fluorescence intensity is the result of low gene expression in both samples.
The second implementation (Fig. 2) uses arrays of oligonucleotides either directly synthesized in situ on a support15,16 or robotically spotted.17 Probe design requires knowledge of the gene sequences. Their length (oligonucleotides of 20–80 bp) allows for differential detection of members of gene families or alternative transcripts not distinguishable with full-length cDNA arrays.16,18 The main drawback remains the elevated cost. This technology uses a different system to label the probe. Message RNA is converted in biotinylated complementary RNA before being hybridized to the array. Each sample is hybridized to a different array and every array is incubated with an avidin-conjugated fluorophore. Current commercial products include sets of arrays that monitor 36,000 potential mouse genes and 60,000 potential human genes.
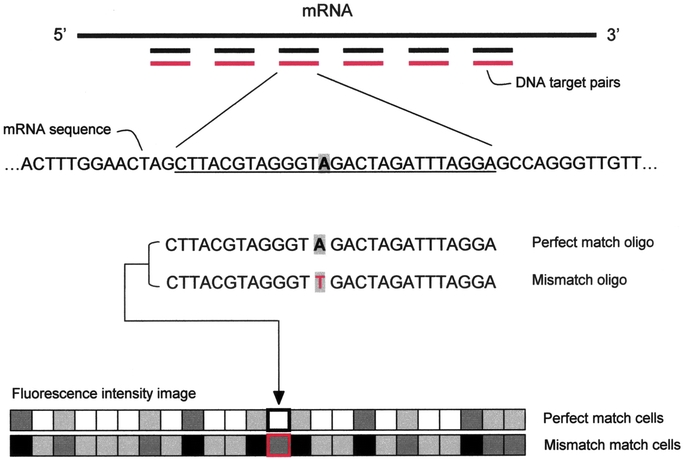
FIGURE 2. Oligonucleotide array scheme. The key point for this DNA array platform is the targeted design of probe sets. Using as little as 200 to 300 bases of gene, cDNA, or EST sequence, independent 25-mer oligonucleotides are selected to serve as unique, sequence-specific detectors. The arrays are designed in silico, and as a result, it is not necessary to prepare, verify, quantitate, and catalog a large number of cDNAs, polymerase chain reaction products, and clones, and there is no risk of a misidentified tube, clone, cDNA, or spot. Crucial for this approach is the use of target redundancy, which is not meant as the deposition of the same piece of DNA in multiple locations on an array, but rather the use of multiple oligonucleotides of different sequences designed to hybridize to different regions of the same RNA. The use of multiple independent detectors for the same molecule greatly improves signal-to-noise ratios, improves the accuracy of RNA quantitation, reduces the effects of crosshybridization, and drastically decreases the rate of false-positives. For each gene monitored, 32 oligonucleotides are synthesized, using photolithography, directly onto the chip. Oligonucleotides are arranged as 16 pairs: each pair includes a perfect match 25mer, which is an exact complement to the gene sequence, and a control oligonucleotide, which differs from the perfect match oligo at the 13th base. The reported hybridization intensity is a composite of the 16 perfect match–mismatch differences per gene. This redundancy considerably increases the statistical power of the technology and data can be analyzed using standard statistical techniques18 or by taking advantage of the changes in relative hybridization between perfect match and mismatch oligos to define genes as absent, present, increased, or decreased according to a set of heuristic rules.16 The mismatch probes act as specificity controls that allow the direct subtraction of both background and crosshybridization signals, and allow discrimination between “real” signals and those resulting from nonspecific or semispecific hybridization, which are more likely to occur with single-spot strategy DNA arrays (eg, cDNA array platform). In the presence of even low concentrations of RNA, hybridization to the perfect match/mismatch pairs produces recognizable and quantitative fluorescent patterns. The strength of these patterns directly relates to the concentration of the RNA molecules in the complex sample (even without a competitive hybridization or 2-color comparison).
STUDY DESIGN
The correlation observed between gene expression levels from duplicate spots on a single array usually exceeds 95%. This is often interpreted as a demonstration of reproducibility. However, if the same sample is split and hybridized to 2 different arrays, the correlation across hybridizations is likely to fall to the 60% to 80% range. Correlations between samples obtained from individual inbred mice may be as low as 30%. If the experiments are carried out in different laboratories, the correlations may be even lower. These decreasing correlations reflect the cumulative contributions of multiple sources of variation.19 The main sources of variability are biologic and technical variation. As for the former, it is generally appropriate to take steps to vary the conditions of the experiment, for example, by assaying multiple animals, to ensure that the effects that do achieve statistical significance are real and will be reproducible in different settings.
Identifying the independent units in an experiment is a prerequisite for a proper statistical analysis, because any hidden correlations in the data can lead to bias and inflated levels of statistical significance. In general, 2 measurements may be regarded as independent only if the experimental materials on which the measurements were obtained could have received different treatments, and if the materials were handled separately at all stages of the experiment when variation might have been introduced. For instance, consider a cell line that is divided into 8 equal samples. Four are assigned to 1 treatment and the remaining 4 receive a second treatment. The 8 aliquots are handled separately throughout the entire experimental procedure, and each is measured in triplicate. This results in 24 total observations, but there are 8 experimental units. Now consider a cell line that is divided into 2 aliquots, each one receiving a different treatment. The material is further subdivided into 4 aliquots per treatment group, each of which is processed and then measured in triplicate. Again we have 24 observations, but now there are only 2 independent experimental units.
A simple way to assess the adequacy of a design is to determine the degrees of freedom (df). This is done by counting the number of independent units and subtracting from it the number of distinct treatments (count all combinations that occur if there are multiple treatment factors). If there are no degrees of freedom left, there may be no information available to estimate the biologic variance, and the statistical tests will rely on technical variance alone. Five degrees of freedom or more are generally recommended for a statistical analysis to be considered sound19 (Fig. 3).
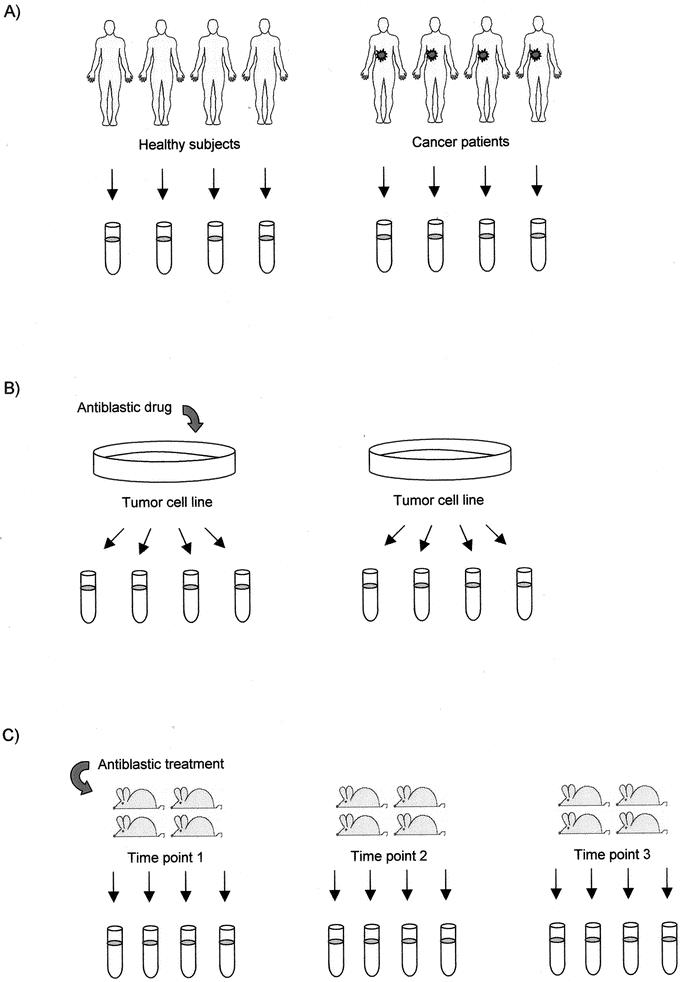
FIGURE 3. Study design for DNA array-based experiments. (A) Pairwise comparison. In the search for carcinogenesis-related genes, the gene profile of tumor biopsies from 4 patients is compared with that of normal tissue biopsies from 4 healthy subjects, providing 6 (8 independent experimental units minus 2 experimental conditions) residual degrees of freedom (df). (B) In this experiment, the gene profile of 2 samples (1 treated with an antiblastic drug) of the same tumor cell line are compared to dissect the mechanism of action of the antiblastic drug. Four aliquots are obtained from each cell line and directly compared on pairs of DNA arrays. The experiment lacks biologic replication because the aliquots are not independent (2 independent experimental units, 2 treatments, df = 0). The design could be improved by using 8 independent cell lines, 4 of them treated with the antiblastic agent (in this case, df would be 6). (C) In a time-course study on the effects of an antiblastic treatment on the gene profile of the tumor microenvironment, 3 time points are considered, 4 mice being sampled from each of them (12 independent experimental units, 3 experimental conditions, df = 9).
To increase DNA array result reproducibility, the issue of technical variability should also be addressed while designing experiments. Although this can be achieved by repeating the experiment, high-throughput DNA array experts suggest that the use of spot replicates within the same array is the best way to deal with this issue.20,21 In particular, biostatistical analysis has shown that a minimum of 3 replicates should be used to reduce the number of false-positive and false-negative results generated by studies performed without replication.22
DATA ANALYSIS
Comparison of Independent and Paired Samples
The comparison of 2 independent samples (eg, diseased vs. normal tissue) is the simplest experimental situation. Although a number of statistical tests are available to assess the significance of the observed differences, most of the groups active in this field use filtering rules based on arbitrarily assigned fold difference criteria. This strategy lies on the unverified assumption that a less than X-fold difference in gene expression is not associated with a significant biologic effect. Despite the good results yielded with this method,12,23,24 it is possible that the application of a simple fold-based rule leads to false-positive results.25 Classic statistical techniques can be adopted to test the significance of the observed differences.26,27 For example, if 2 independent samples are compared, a standard t test is appropriate. The genes in the array can be ranked according to increasing P values and an appropriate threshold can be chosen depending on the percentage of false-positives that we are prepared to tolerate. If the 2 samples to compare are somehow related to each other (ie, they both come from the same individual), then a paired t test would be needed to assess the significance of the differences. More complex experimental situations may involve the comparison of multiple samples. Appropriate statistical tests are available, but in analogy with the 2-sample comparison case, thresholding rules are often used.25
Classification of Gene Expression Data
DNA arrays deliver several thousands of measurements per experiment. The analysis, interpretation, and meaningful display and storage of such a large volume of data are particularly challenging. Although genes that display extreme expression changes between samples may require specific analysis, the true strength of high-throughput experiments in revealing the complexity of tumor/host relation derives from the mathematical identification of expression patterns (called “signatures”) within profiling data. In the context of gene expression studies, this involves finding similar gene expression patterns by comparing profiles. Dedicated software developed for this task includes the “unsupervised” and “supervised” varieties.28 Unsupervised methods (eg, cluster analysis,29 self-organizing map [SOM],30 principal component analysis [PCA]31) define classes without any a priori intervention on data, which are organized by clustering genes and/or samples simply according to similarities in their expression profiles. The resulting sample classification often correlates with a general characteristic of the sample as defined by large sets of genes and not necessarily with the particular feature of interest, generally identified by a smaller set of genes. By defining relevant classes before analysis, supervised techniques (eg, support vector machines,32 weighted votes,33 and neural networks34) bypass this issue. These algorithms incorporate external information related to samples studied to identify the optimal set of genes that best discriminate between experimental samples. Unsupervised clustering techniques for analyzing microarrays are useful for initial data exploration and have been validated under certain circumstances by their successful “rediscovery” of known classes of genes.14,29 In particular, unsupervised techniques can be effectively adopted in oncology when the aim of the study is to identify new prognostic subgroups.14 However, these methods have certain shortfalls. Because prior biologic knowledge is not incorporated, all measurements within the expression profile contribute equally to the analysis. Thus, measurements that have little or nothing to do with distinguishing the groups of interest can confound the placement of an example into the correct category. The advantage of supervised classification for gene profile analysis is its ability to incorporate biologic knowledge. For example, a supervised approach might be used to predict whether a gene's product is involved in protein synthesis by comparing its expression profile to the profiles of both genes known to be involved and genes known not to be involved in protein synthesis. Yet, if we measure gene-expression patterns using RNAs collected from various patients for which there is, for example, tumor-stage classification or survival data, we can use the DNA array data to “train” an algorithm that can then be applied to the classification of other previously unclassified samples. This approach could lead to the development of “molecular expression fingerprinting” for disease classification. In cancer diagnosis, the ability to produce a molecular expression fingerprint of each tumor might prove to be extremely important because histologically similar tumors might in fact be the result of substantially different genetic changes, which might profoundly influence the progression of the tumor and its response to treatment. Recent reports have demonstrated the ability of supervised classification to type leukemia (myeloid vs. lymphoid)33 and assign functions to genes35 based on DNA array data.
Cluster Analysis
Depending on the way in which the data are clustered, we can distinguish between hierarchical and nonhierarchical clustering. Hierarchical clustering, which is the most commonly adopted unsupervised method, allows detection of higher-order relationships between clusters of profiles. By contrast, the majority of nonhierarchical classification techniques (eg, quality cluster36 or k-means.37) work by allocating gene expression profiles to a predefined number of clusters (supervised methods).38 For example, an attempt to classify patients with 2 morphologically similar (eg, breast carcinoma) but clinically distinct (eg, favorable vs. poor outcome) diseases using microarray expression patterns can be imagined. By using k-means clustering on experiments with k = 2, the data will be partitioned into 2 groups. The challenge then faced is to determine whether or not, from the genetic viewpoint, there are really only 2 distinct groups represented in the data.
The possibility of exploring different levels of the hierarchy has led many authors to prefer hierarchical clustering to the nonhierarchical alternatives. In particular, aggregative hierarchical clustering is still the preferred choice for the analysis of patterns of gene expression.29,39–46 It produces a representation of the data with the shape of a binary tree, in which the most similar patterns are clustered in a hierarchy of nested subsets (Fig. 4).
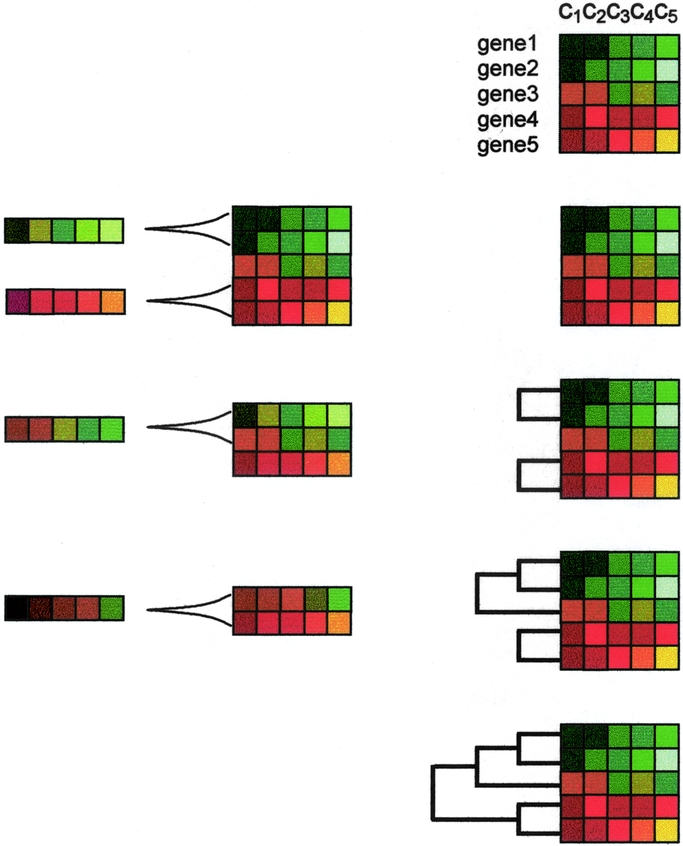
FIGURE 4. Hierarchical aggregative clustering. Using dedicated software, fluorescence ratios (see Fig. 1) are translated into color codes. Consequently, genes with unchanged expression levels are colored as black, whereas those with increasingly positive or negative expression are colored with increasingly intense red or green, respectively. Accordingly, the darker the color, the closer to unchanged expression. The figure shows an example with the color-coded expression values of 5 genes in 5 different experimental conditions (c1, c2, c3, c4, c5). In the aggregative method, the closest pair of profiles is chosen based on a given metric. Then, an average of both profiles is constructed. This defines a relationship of closeness between both profiles that remain tied by the corresponding branch of the tree. Thus, the linked profiles are substituted by the average profile, and the process continues until all the profiles are linked. The linkage relationship defines the hierarchy of the tree.
Neural Networks
Clustering methods have serious drawbacks when dealing with data containing a nonnegligible amount of noise (eg, ill-defined items with irrelevant variables and outliers). In addition, some authors have noted that aggregative hierarchical clustering suffers from a lack of robustness so that solutions may be not unique and results may be dependent on the data order.47 Unsupervised neural networks, and in particular SOM,48 provide a more robust and accurate approach to the clustering of large sets of data. They are robust with respect to noise, and they are generally independent of the shape of the data distribution. Another advantage of neural networks using SOM is the high performance displayed with large sets of data. Initially, genes are randomly allocated to the nodes but following iterative learning steps, the algorithm undergoes a training process that will result in a correct classification. During this process, the weighting of nodes change by repeated interaction with the items of the dataset in a way that captures the distribution of variability of the dataset. Thus, similar gene expression patterns map together in the network and, as far as possible, from the different patterns. At the end of the training process, the nodes of the SOM grid have clusters of related gene expression patterns assigned, and the trained nodes represent an average pattern of the cluster of data that map into it. This reduction of the data space is a very interesting property when dealing with big datasets, which is often the case with DNA array data.49
The efficacy of SOM in organizing DNA array-derived data has been demonstrated in both basic and clinical research. For instance, some investigators used SOM to compare the time-series response of 4 hematopoietic cell lines responding to an activation signal (HL-60, U937, and Jurkat were treated with PMA, whereas NB4 was treated with ATRA).47 SOM was used to classify gene expression profiles defined as the combination of the time points of the 4 experiments. SOM generated a grid of “nodes,” each containing genes with similar profiles across the data series, whereas the identification of the clusters containing genes with interesting differences/similarities between the different time courses was left to the interpretation of the researchers; they discovered that the largest group contained genes specifically induced by ATRA stimulation. These genes were not regulated in the other cell lines after stimulation with PMA, thus indicating a stimulation pathway (and a drug target) specific for that cell line. In the clinical setting, SOM has been successfully applied for the identification of acute leukemia subtypes.33
Although SOM are a useful alternative to deterministic clustering,33,47,50 they are not appropriate if the relationship between the individual profiles needs to be revealed. In analogy with the nonhierarchical methods like k-means, the number of clusters is arbitrarily fixed from the beginning and genes are allocated through a training process. Moreover, SOM produce a nonproportional classification (topology-preserving neural network).20 If irrelevant data (eg, invariant, “flat” profiles) or some particular type of profile is abundant, SOM will produce an output in which this type of data will populate most of clusters. Because of this, the most interesting profiles will map in few clusters and resolution might be low for them.
In conclusion, despite the arsenal of methods used, the optimal way of classifying such data is still open to debate, and the information potentially obtainable from these high-throughput analyses is still incompletely exploited. Likely, new computational tools will be required to define gene-interaction networks.51
APPLICATIONS TO CANCER RESEARCH
The potential applications of DNA array technology in the field of oncology are virtually unlimited, and their comprehensive description is beyond the aim of this work. The following is a brief description of some examples of how DNA array-based gene profiling data can be used to increase our knowledge on cancer biology and ultimately improve the management of patients with cancer.
Although this technology provides no information on the biologically active products of genes (ie, proteins), functional genomics studies have demonstrated a tight correlation between the function of a protein and the expression patterns of its gene,7 which represents the rational for a gene profile-based formulation of scientific hypotheses. Once a gene or (more frequently) a set of genes have been identified in a DNA array-based experiment, investigators commonly use to confirm the results with more accurate low-throughput techniques such as quantitative real-time PCR.52 To further validate gene-profiling data, the expression of proteins coded by the genes of interest is generally assessed by standard immunohistochemistry or Western blot techniques. Because translational gene expression regulation53 and posttranslational protein modifications are also of crucial importance in determining cell functions, DNA array technology should be complemented with other recently developed high-throughput assays such as tissue microarray9 and proteomics.54,55 Hopefully, by integrating these powerful analytic tools, investigators will be able to comprehensively describe the molecular portrait of the biologic phenomena underlying tumor development and progression.
Carcinogenesis and Antiblastic Drug Development
Given the multigenic complexity of carcinogenesis and the multiplicity of tumor/host interactions, high-throughput DNA array technology provides investigators with a powerful laboratory tool, which can portrait the molecular kinetics of cell transformation and tumor progression by scanning the expression of thousands of genes simultaneously.
Two major gene-profiling approaches have been so far adopted to dissect the process of carcinogenesis. The most intuitive is comparing gene profiles of each tumor histologic type with its corresponding normal tissue of origin or with cancer precursors. With this strategy, several authors have investigated esophageal,56 gastric,57 colon,58–60 ovarian,61,62 hepatocellular,63 and prostate64 carcinomas, and melanoma.65 Other investigators have addressed the question whether a common pathway of malignancy characterizes tumors arising from different tissues.41,66,67 It has been found that the most striking and most common gene expression pattern is the cell-proliferation cluster.68 This gene set contains genes involved in regulating the cell cycle and genes that encode structural protein components required for DNA replication and chromosome dynamics. The proliferation signature is correlated with cellular growth rates in vitro43 and has been identified in breast,41 lung,67,69 ovary,61 prostate,70 liver,71 and gastric carcinomas,72 as well as gliomas73 and lymphomas.74 The identification of this cluster in vivo probably represents how rapidly a given tumor is growing and, as might be expected, the high expression of these genes has been found to indicate a poor prognosis.
In most cases, the therapeutic implications of these results are yet to be defined. Pharmacogenomics should enormously benefit from tumor gene-profiling studies.75,76 The definition of tumor-associated molecular abnormalities on a genomewide scale should in fact greatly accelerate the pace of discovery of molecular targets for novel anticancer agents.77 Although several tumor-specific gene pathways have been detected with DNA array technology,78,79 this field of oncology can be still considered in its infancy, and much more work needs to be done to clinically exploit the knowledge deriving from this holistic approach to cancer biology dissection.80 Besides providing researchers with novel molecular targets to fight cancer, high-throughput DNA array technology is expected to foster in an unprecedented way the development of patient-tailored antiblastic therapy. Tumors are well known to be heterogeneously sensitive to antiblastic agents, which leads physicians to administer several lines of drug regimens in an empiric search for the “best” treatment of a given patient. Preclinical experiments have already demonstrated that, at least for a subset of antiblastic agents, the gene signature of tumor cell lines can predict malignant cell sensitivity to chemotherapeutic drugs.81,82 Similar considerations have also been made for tumor sensitivity to radiation therapy.83–85 As a corollary, the identification of molecular pathways involved in tumor chemoresistance is likely to lead to the discovery of novel drugs aimed at reversing this phenomenon.86
Prognosis
One of the major challenges in medicine is the identification of prognostic markers identifying the subgroup of patients who will recur after macroscopically radical surgery. Unfortunately, there are currently no markers that can predict accurately when or if a tumor will recur, which leaves physicians with the difficulty of choosing the best treatment options. To assure a positive outcome, therefore, individuals with early-stage cancer often receive adjuvant treatments that have their own associated morbidity and mortality. Considering the case of breast cancer, no new histoclinical factor—except the protein overexpression of ERBB2 and recently of uPA/PAI-1—has been validated as a prognostic and/or predictive factor during the past 2 decades. Although adjuvant chemotherapy improves survival in localized breast and colon cancer, a number of issues remain. In particular, patients with good prognosis need to be more accurately identified to avoid potentially toxic treatment, and patients of poor prognosis who will or will not benefit from the standard adjuvant chemotherapy currently used need to be determined.
With high-throughput DNA arrays, investigators have the possibility to molecularly portrait cancer aggressiveness and metastatic potential on a genomewide scale, thus identifying patients who really benefit from complementary therapies after surgery. Infiltrating breast carcinomas appear fairly homogenous in histologic assessments but are heterogeneous in clinical behavior. To address this issue, van't Veer et al87 profiled the primary tumors and carried out a supervised analysis in which the individuals were separated into 2 groups: those who developed metastases in less than 5 years (bad prognosis) and those who were metastasis-free for longer than 5 years (good prognosis). They identified 231 genes that correlated with this parameter. Through further statistical analysis, a discriminatory set of 70 genes was identified that showed 89% accuracy. This group of markers was found to be a statistically significant predictor in a multivariate analysis, showing that gene profiling can add value to the current list of clinical tests.
Analyzing mRNA expression of approximately 1000 candidate genes in tumor samples from 55 women who were treated with adjuvant anthracycline-based chemotherapy, Bertucci et al identified a 40-gene set whose expression distinguished 3 subclasses of tumors that, although balanced with respect to clinicopathologic features, showed significantly different 5-year survival.88 Similarly, Sorlie et al defined 5 subclasses of locally advanced breast tumors with different survival after neoadjuvant doxorubicin.66 To further explore the validity of results, investigators compared the lists of discriminator genes identified in these breast cancer prognostic studies.66,88,89 Despite several different methodologic aspects, 26 genes were found in at least 2 lists.90 It is reassuring that some of those genes have a known prognostic value (eg, ESR1, ERBB2), but most are not yet associated with prognosis and have functions making them prime candidates for novel therapeutic targets.
Two major prognostic factors of breast cancer have been investigated by comparing the molecular profiles of estrogen receptor (ER)-positive and ER-negative tumors87,88,91,92 and profiles of tumors with and without axillary lymph node metastasis.93,94 Yet, the determination of lymph node status currently relies on surgical axillary lymph dissection, which is associated with significant morbidity. Although sentinel lymph node biopsy is being evaluated to replace classic invasive dissection, false-negative results are possible. Accurate prediction of the axillary status from analysis of tumors would obviate the recourse to lymph node sampling/dissection. Among differentially expressed genes that Bertucci et al identified between tumors with and without node metastasis, some had a function in agreement with a potential role in invasion (eg, ERBB2, CDH1), whereas for others (eg, SOX4, GSTP1), the connection was not clear, calling for further investigations.93
Much work has been carried out to molecularly classify lung tumors using DNA arrays.67,69,95,96 Garber et al67 identified significant diversity in expression patterns in adenocarcinomas and defined 3 subtypes that predicted favorable, intermediate, and poor outcomes. Two of these 3 adenocarcinoma subtypes were observed by Bhattacharjee et al.69 Beer et al also identified prognostic groups within adenocarcinomas using survival as supervision.95 Investigating the gene display of primary nonsmall cell lung carcinomas, Kikuchi et al identified 40 genes whose expression could identify patients with lymph node metastasis from those without metastasis, thus confirming the ability of gene profiling to distinguish between patients who need a aggressive therapeutic approach from those who are likely to be already cured.97 Studies on renal cell carcinomas,98 prostate tumors,99,100 and hematologic malignancies101 have also identified patterns of gene expression with prognostic importance, and it is likely that other types of tumor not profiled as yet will be soon classified into subtypes of clinical importance.
Tumor Immunology
Until recently, most studies addressing the immunologic effects of vaccination in patients with cancer have looked at variations in the level of TAA-specific reactivity in circulating lymphocytes. Results from clinical trials have shown that vaccination can be quite effective in inducing tumor-specific T-cell responses that can be easily observed among circulating lymphocytes. However, the identification of such immune responses could not be consistently correlated with tumor regression.
DNA array technology allowed us to find that melanoma metastases undergoing complete regression in response to peptide/IL-2-based vaccination were characterized by a higher expression of some immune-related genes as compared with those progressing.65,102 Among these genes, we focused on interleukin-10 (IL-10). Its gene overexpression was verified by quantitative real-time PCR. Then, its protein levels were assessed in metastatic melanoma biopsies by laser scanning cytometry,103,104 which confirmed the genetic findings. Although IL-10 is generally considered an immunosuppressive molecule103,105,106, several preclinical models have shown that IL-10 can also mediate tumor regression by stimulating NK cells activity.107–112 Using cDNA glass array we observed that, in vitro, IL-10 induced the expression of cytotoxicity related genes (eg, TIA-1) by human natural killer (NK) cells but not cytotoxic T lymphocytes.113 These observations led us to hypothesize that, in the presence of high intratumoral (and not systemic) levels of IL-10, the adaptive immune response (mounted by the systemic vaccination) might be favored by NK cell activation. In fact, these innate immunity cell mediators might be stimulated by IL-10 to lyse cancer cells, thus increasing TAA availability and “danger signals” delivery required by antigen-presenting cells (eg, dendritic cells) to be activated and effectively prime cytotoxic T lymphocytes against TAA peptides administered with the vaccine.114 If this hypothesis were proved to be correct, future anticancer immunotherapy strategies should address the challenging task of stimulating both the innate at the tumor site and adaptive immunity systemically. To this aim, we are currently carrying out a study in which isolated limb perfusion115,116 is used for the locoregional delivery of IL-10 during peptide-based antitumor vaccination for the treatment of murine sarcoma. This model should allow us to verify whether or not high levels of intratumoral IL-10 (as obtained in the animal model with the surgical procedure mentioned here) enhance the in vivo activity of systemic vaccination regimens.
Footnotes
Reprints: Simone Mocellin, Clinica Chirurgica Generale II, Dipartimento di Scienze Oncologiche e Chirurgiche, Università di Padova, Via Giustiniani, 2, 35128 Padova, Italy. E-mail: mocellins@hotmail.com.
REFERENCES
- 1.Baselga J. Clinical trials of Herceptin® (trastuzumab). Eur J Cancer. 2001;37(suppl 1):18–24. [PubMed] [Google Scholar]
- 2.O'Dwyer ME, Druker BJ. STI571: an inhibitor of the BCR-ABL tyrosine kinase for the treatment of chronic myelogenous leukaemia. Lancet Oncol. 2000;1:207–211. [DOI] [PubMed] [Google Scholar]
- 3.Rossi CR, Mocellin S, Mencarelli R, et al. Gastrointestinal stromal tumors: from surgical to molecular approach? Int J Cancer. 2003;107:171–176. [DOI] [PubMed] [Google Scholar]
- 4.Pinkel D, Segraves R, Sudar D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. [DOI] [PubMed] [Google Scholar]
- 5.Broude NE. Differential display in the time of microarrays. Expert Rev Mol Diagn. 2002;2:209–216. [DOI] [PubMed] [Google Scholar]
- 6.Velculescu VE, Zhang L, Vogelstein B, et al. Serial analysis of gene expression. Science. 1995;270:484–487. [DOI] [PubMed] [Google Scholar]
- 7.Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nat Genet. 1999;21(suppl):33–37. [DOI] [PubMed] [Google Scholar]
- 8.Goldenfeld N, Kadanoff LP. Simple lessons from complexity. Science. 1999;284:87–89. [DOI] [PubMed] [Google Scholar]
- 9.Kallioniemi OP, Wagner U, Kononen J, et al. Tissue microarray technology for high-throughput molecular profiling of cancer. Hum Mol Genet. 2001;10:657–662. [DOI] [PubMed] [Google Scholar]
- 10.Lawrie LC, Fothergill JE, Murray GI. Spot the differences: proteomics in cancer research. Lancet Oncol. 2001;2:270–277. [DOI] [PubMed] [Google Scholar]
- 11.Bertucci F, Bernard K, Loriod B, et al. Sensitivity issues in DNA array-based expression measurements and performance of nylon microarrays for small samples. Hum Mol Genet. 1999;8:1715–1722. [DOI] [PubMed] [Google Scholar]
- 12.Schena M, Shalon D, Davis RW, et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. [DOI] [PubMed] [Google Scholar]
- 13.Chen JJ, Wu R, Yang PC, et al. Profiling expression patterns and isolating differentially expressed genes by cDNA microarray system with colorimetry detection. Genomics. 1998;51:313–324. [DOI] [PubMed] [Google Scholar]
- 14.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. [DOI] [PubMed] [Google Scholar]
- 15.Hughes TR, Mao M, Jones AR, et al. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol. 2001;19:342–347. [DOI] [PubMed] [Google Scholar]
- 16.Lockhart DJ, Dong H, Byrne MC, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. [DOI] [PubMed] [Google Scholar]
- 17.Kane MD, Jatkoe TA, Stumpf CR, et al. Assessment of the sensitivity and specificity of oligonucleotide (50mer) microarrays. Nucleic Acids Res. 2000;28:4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Churchill GA. Fundamentals of experimental design for cDNA microarrays. Nat Genet. 2002;32(suppl):490–495. [DOI] [PubMed] [Google Scholar]
- 20.Dopazo J, Zanders E, Dragoni I, et al. Methods and approaches in the analysis of gene expression data. J Immunol Methods. 2001;250:93–112. [DOI] [PubMed] [Google Scholar]
- 21.Hess KR, Zhang W, Baggerly KA, et al. Microarrays: handling the deluge of data and extracting reliable information. Trends Biotechnol. 2001;19:463–468. [DOI] [PubMed] [Google Scholar]
- 22.Lee ML, Kuo FC, Whitmore GA, et al. Importance of replication in microarray gene expression studies: statistical methods and evidence from repetitive cDNA hybridizations. Proc Natl Acad Sci U S A. 2000;97:9834–9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heller RA, Schena M, Chai A, et al. Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc Natl Acad Sci U S A. 1997;94:2150–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teague TK, Hildeman D, Kedl RM, et al. Activation changes the spectrum but not the diversity of genes expressed by T cells. Proc Natl Acad Sci U S A. 1999;96:12691–12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Claverie JM. Computational methods for the identification of differential and coordinated gene expression. Hum Mol Genet. 1999;8:1821–1832. [DOI] [PubMed] [Google Scholar]
- 26.Glynne R, Akkaraju S, Healy JI, et al. How self-tolerance and the immunosuppressive drug FK506 prevent B-cell mitogenesis. Nature. 2000;403:672–676. [DOI] [PubMed] [Google Scholar]
- 27.Rogge L, Bianchi E, Biffi M, et al. Transcript imaging of the development of human T helper cells using oligonucleotide arrays. Nat Genet. 2000;25:96–101. [DOI] [PubMed] [Google Scholar]
- 28.Brazma A, Vilo J. Gene expression data analysis. FEBS Lett. 2000;480:17–24. [DOI] [PubMed] [Google Scholar]
- 29.Eisen MB, Spellman PT, Brown PO, et al. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nikkila J, Toronen P, Kaski S, et al. Analysis and visualization of gene expression data using self- organizing maps. Neural Netw. 2002;15:953–966. [DOI] [PubMed] [Google Scholar]
- 31.Crescenzi M, Giuliani A. The main biological determinants of tumor line taxonomy elucidated by a principal component analysis of microarray data. FEBS Lett. 2001;507:114–118. [DOI] [PubMed] [Google Scholar]
- 32.Lin K, Kuang Y, Joseph JS, et al. Conserved codon composition of ribosomal protein coding genes in Escherichia coli, Mycobacterium tuberculosis and Saccharomyces cerevisiae: lessons from supervised machine learning in functional genomics. Nucleic Acids Res. 2002;30:2599–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. [DOI] [PubMed] [Google Scholar]
- 34.Khan J, Wei JS, Ringner M, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown MP, Grundy WN, Lin D, et al. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A. 2000;97:262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heyer LJ, Kruglyak S, Yooseph S. Exploring expression data: identification and analysis of coexpressed genes. Genome Res. 1999;9:1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sherlock G. Analysis of large-scale gene expression data. Curr Opin Immunol. 2000;12:201–205. [DOI] [PubMed] [Google Scholar]
- 38.Tavazoie S, Hughes JD, Campbell MJ, et al. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. [DOI] [PubMed] [Google Scholar]
- 39.Wen X, Fuhrman S, Michaels GS, et al. Large-scale temporal gene expression mapping of central nervous system development. Proc Natl Acad Sci U S A. 1998;95:334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer VR, Eisen MB, Ross DT, et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87. [DOI] [PubMed] [Google Scholar]
- 41.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. [DOI] [PubMed] [Google Scholar]
- 42.Roberts CJ, Nelson B, Marton MJ, et al. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science. 2000;287:873–880. [DOI] [PubMed] [Google Scholar]
- 43.Ross DT, Scherf U, Eisen MB, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–235. [DOI] [PubMed] [Google Scholar]
- 44.Scherf U, Ross DT, Waltham M, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet. 2000;24:236–244. [DOI] [PubMed] [Google Scholar]
- 45.Voehringer DW, Hirschberg DL, Xiao J, et al. Gene microarray identification of redox and mitochondrial elements that control resistance or sensitivity to apoptosis. Proc Natl Acad Sci U S A. 2000;97:2680–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu S, DeRisi J, Eisen M, et al. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. [DOI] [PubMed] [Google Scholar]
- 47.Tamayo P, Slonim D, Mesirov J, et al. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci U S A. 1999;96:2907–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reibnegger G, Wachter H. Self-organizing neural networks–an alternative way of cluster analysis in clinical chemistry. Clin Chim Acta. 1996;248:91–98. [DOI] [PubMed] [Google Scholar]
- 49.Herwig R, Poustka AJ, Muller C, et al. Large-scale clustering of cDNA-fingerprinting data. Genome Res. 1999;9:1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson SB, Kent SC, Horton HF, et al. Multiple differences in gene expression in regulatory Valpha 24Jalpha Q T cells from identical twins discordant for type I diabetes. Proc Natl Acad Sci U S A. 2000;97:7411–7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. [DOI] [PubMed] [Google Scholar]
- 52.Mocellin S, Rossi C, Pilati P, et al. Quantitative real time PCR: a powerful ally in cancer research. Trends in Molecular Medicine. 2003;9:189–195. [DOI] [PubMed] [Google Scholar]
- 53.Piecyk M, Wax S, Beck AR, et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. Embo J. 2000;19:4154–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le Naour F. Contribution of proteomics to tumor immunology. Proteomics. 2001;1:1295–1302. [DOI] [PubMed] [Google Scholar]
- 55.Lawrie L, Fothergill J, Murray G. Spot the differences: proteomics in cancer research. Lancet Oncol. 2001;2:270–277. [DOI] [PubMed] [Google Scholar]
- 56.Xu Y, Selaru FM, Yin J, et al. Artificial neural networks and gene filtering distinguish between global gene expression profiles of Barrett's esophagus and esophageal cancer. Cancer Res. 2002;62:3493–3497. [PubMed] [Google Scholar]
- 57.Meireles SI, Carvalho AF, Hirata R, et al. Differentially expressed genes in gastric tumors identified by cDNA array. Cancer Lett. 2003;190:199–211. [DOI] [PubMed] [Google Scholar]
- 58.Hernandez A, Smith F, Wang Q, et al. Assessment of differential gene expression patterns in human colon cancers. Ann Surg. 2000;232:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Notterman DA, Alon U, Sierk AJ, et al. Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 2001;61:3124–3130. [PubMed] [Google Scholar]
- 60.Zou TT, Selaru FM, Xu Y, et al. Application of cDNA microarrays to generate a molecular taxonomy capable of distinguishing between colon cancer and normal colon. Oncogene. 2002;21:4855–4862. [DOI] [PubMed] [Google Scholar]
- 61.Welsh JB, Zarrinkar PP, Sapinoso LM, et al. Analysis of gene expression profiles in normal and neoplastic ovarian tissue samples identifies candidate molecular markers of epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2001;98:1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martoglio AM, Tom BD, Starkey M, et al. Changes in tumorigenesis- and angiogenesis-related gene transcript abundance profiles in ovarian cancer detected by tailored high density cDNA arrays. Mol Med. 2000;6:750–765. [PMC free article] [PubMed] [Google Scholar]
- 63.Graveel CR, Jatkoe T, Madore SJ, et al. Expression profiling and identification of novel genes in hepatocellular carcinomas. Oncogene. 2001;20:2704–2712. [DOI] [PubMed] [Google Scholar]
- 64.Welsh JB, Sapinoso LM, Su AI, et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–5978. [PubMed] [Google Scholar]
- 65.Wang E, Miller LD, Ohnmacht GA, et al. Prospective molecular profiling of melanoma metastases suggests classifiers of immune responsiveness. Cancer Res. 2002;62:3581–3586. [PMC free article] [PubMed] [Google Scholar]
- 66.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garber ME, Troyanskaya OG, Schluens K, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98:13784–13789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perou CM, Jeffrey SS, van de Rijn M, et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A. 1999;96:9212–9217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhattacharjee A, Richards WG, Staunton J, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.LaTulippe E, Satagopan J, Smith A, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–4506. [PubMed] [Google Scholar]
- 71.Chen X, Cheung ST, So S, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hippo Y, Taniguchi H, Tsutsumi S, et al. Global gene expression analysis of gastric cancer by oligonucleotide microarrays. Cancer Res. 2002;62:233–240. [PubMed] [Google Scholar]
- 73.Rickman DS, Bobek MP, Misek DE, et al. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001;61:6885–6891. [PubMed] [Google Scholar]
- 74.Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–1947. [DOI] [PubMed] [Google Scholar]
- 75.Clarke PA, te Poele R, Wooster R, et al. Gene expression microarray analysis in cancer biology, pharmacology, and drug development: progress and potential. Biochem Pharmacol. 2001;62:1311–1336. [DOI] [PubMed] [Google Scholar]
- 76.Orr MS, Scherf U. Large-scale gene expression analysis in molecular target discovery. Leukemia. 2002;16:473–477. [DOI] [PubMed] [Google Scholar]
- 77.Rothenberg ML, Carbone DP, Johnson DH. Improving the evaluation of new cancer treatments: challenges and opportunities. Nat Rev Cancer. 2003;3:303–309. [DOI] [PubMed] [Google Scholar]
- 78.Staudt LM. Gene expression profiling of lymphoid malignancies. Annu Rev Med. 2002;53:303–318. [DOI] [PubMed] [Google Scholar]
- 79.MacDonald TJ, Brown KM, LaFleur B, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet. 2001;29:143–152. [DOI] [PubMed] [Google Scholar]
- 80.Workman P. New drug targets for genomic cancer therapy: successes, limitations, opportunities and future challenges. Curr Cancer Drug Targets. 2001;1:33–47. [DOI] [PubMed] [Google Scholar]
- 81.Staunton JE, Slonim DK, Coller HA, et al. Chemosensitivity prediction by transcriptional profiling. Proc Natl Acad Sci U S A. 2001;98:10787–10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zembutsu H, Ohnishi Y, Tsunoda T, et al. Genome-wide cDNA microarray screening to correlate gene expression profiles with sensitivity of 85 human cancer xenografts to anticancer drugs. Cancer Res. 2002;62:518–527. [PubMed] [Google Scholar]
- 83.Khodarev NN, Park JO, Yu J, et al. Dose-dependent and independent temporal patterns of gene responses to ionizing radiation in normal and tumor cells and tumor xenografts. Proc Natl Acad Sci U S A. 2001;98:12665–12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Achary MP, Jaggernauth W, Gross E, et al. Cell lines from the same cervical carcinoma but with different radiosensitivities exhibit different cDNA microarray patterns of gene expression. Cytogenet Cell Genet. 2000;91:39–43. [DOI] [PubMed] [Google Scholar]
- 85.Kitahara O, Katagiri T, Tsunoda T, et al. Classification of sensitivity or resistance of cervical cancers to ionizing radiation according to expression profiles of 62 genes selected by cDNA microarray analysis. Neoplasia. 2002;4:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weldon CB, Scandurro AB, Rolfe KW, et al. Identification of mitogen-activated protein kinase kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery. 2002;132:293–301. [DOI] [PubMed] [Google Scholar]
- 87.van ’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. [DOI] [PubMed] [Google Scholar]
- 88.Bertucci F, Nasser V, Granjeaud S, et al. Gene expression profiles of poor-prognosis primary breast cancer correlate with survival. Hum Mol Genet. 2002;11:863–872. [DOI] [PubMed] [Google Scholar]
- 89.Ahr A, Karn T, Solbach C, et al. Identification of high risk breast-cancer patients by gene expression profiling. Lancet. 2002;359:131–132. [DOI] [PubMed] [Google Scholar]
- 90.Bertucci F, Eisinger F, Houlgatte R, et al. Gene-expression profiling and identification of patients at high risk of breast cancer. Lancet. 2002;360:173–174; author reply 174. [DOI] [PubMed]
- 91.Gruvberger S, Ringner M, Chen Y, et al. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001;61:5979–5984. [PubMed] [Google Scholar]
- 92.Martin KJ, Kritzman BM, Price LM, et al. Linking gene expression patterns to therapeutic groups in breast cancer. Cancer Res. 2000;60:2232–2238. [PubMed] [Google Scholar]
- 93.Bertucci F, Houlgatte R, Benziane A, et al. Gene expression profiling of primary breast carcinomas using arrays of candidate genes. Hum Mol Genet. 2000;9:2981–2991. [DOI] [PubMed] [Google Scholar]
- 94.West M, Blanchette C, Dressman H, et al. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc Natl Acad Sci U S A. 2001;98:11462–11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. [DOI] [PubMed] [Google Scholar]
- 96.Wigle DA, Jurisica I, Radulovich N, et al. Molecular profiling of non-small cell lung cancer and correlation with disease-free survival. Cancer Res. 2002;62:3005–3008. [PubMed] [Google Scholar]
- 97.Kikuchi T, Daigo Y, Katagiri T, et al. Expression profiles of non-small cell lung cancers on cDNA microarrays: identification of genes for prediction of lymph-node metastasis and sensitivity to anti-cancer drugs. Oncogene. 2003;22:2192–2205. [DOI] [PubMed] [Google Scholar]
- 98.Takahashi M, Rhodes DR, Furge KA, et al. Gene expression profiling of clear cell renal cell carcinoma: gene identification and prognostic classification. Proc Natl Acad Sci U S A. 2001;98:9754–9759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singh D, Febbo PG, Ross K, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–209. [DOI] [PubMed] [Google Scholar]
- 100.Dhanasekaran SM, Barrette TR, Ghosh D, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–826. [DOI] [PubMed] [Google Scholar]
- 101.Shipp MA, Ross KN, Tamayo P, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8:68–74. [DOI] [PubMed] [Google Scholar]
- 102.Mocellin S, Ohnmacht GA, Wang E, et al. Kinetics of cytokine expression in melanoma metastases classifies immune responsiveness. Int J Cancer. 2001;93:236–242. [DOI] [PubMed] [Google Scholar]
- 103.Mocellin S, Wang E, Marincola FM. Cytokines and immune response in the tumor microenvironment. J Immunother. 2001;24:392–407. [PubMed] [Google Scholar]
- 104.Mocellin S, Fetsch P, Abati A, et al. Laser scanning cytometry evaluation of MART-1, gp100, and HLA-A2 expression in melanoma metastases. J Immunother. 2001;24:447–458. [DOI] [PubMed] [Google Scholar]
- 105.Akdis CA, Blaser K. Mechanisms of interleukin-10-mediated immune suppression. Immunology. 2001;103:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moore KW, de Waal Malefyt R, Coffman RL, et al. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. [DOI] [PubMed] [Google Scholar]
- 107.Berman RM, Suzuki T, Tahara H, et al. Systemic administration of cellular IL-10 induces an effective, specific, and long-lived immune response against established tumors in mice. J Immunol. 1996;157:231–238. [PubMed] [Google Scholar]
- 108.Salazar-Onfray F, Petersson M, Franksson L, et al. IL-10 converts mouse lymphoma cells to a CTL-resistant, NK-sensitive phenotype with low but peptide-inducible MHC class I expression. J Immunol. 1995;154:6291–6298. [PubMed] [Google Scholar]
- 109.Giovarelli M, Musiani P, Modesti A, et al. Local release of IL-10 by transfected mouse mammary adenocarcinoma cells does not suppress but enhances antitumor reaction and elicits a strong cytotoxic lymphocyte and antibody-dependent immune memory. J Immunol. 1995;155:3112–3123. [PubMed] [Google Scholar]
- 110.Kundu N, Beaty TL, Jackson MJ, et al. Antimetastatic and antitumor activities of interleukin 10 in a murine model of breast cancer. J Natl Cancer Inst. 1996;88:536–541. [DOI] [PubMed] [Google Scholar]
- 111.Huang S, Xie K, Bucana CD, et al. Interleukin 10 suppresses tumor growth and metastasis of human melanoma cells: potential inhibition of angiogenesis. Clin Cancer Res. 1996;2:1969–1979. [PubMed] [Google Scholar]
- 112.Petersson M, Charo J, Salazar-Onfray F, et al. Constitutive IL-10 production accounts for the high NK sensitivity, low MHC class I expression, and poor transporter associated with antigen processing (TAP)-1/2 function in the prototype NK target YAC-1. J Immunol. 1998;161:2099–2105. [PubMed] [Google Scholar]
- 113.Mocellin S, Panelli MC, Wang E, et al. The dual role of IL-10. Trends Immunol. 2003;24:36–43. [DOI] [PubMed] [Google Scholar]
- 114.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. [DOI] [PubMed] [Google Scholar]
- 115.Rossi CR, Foletto M, Pilati P, et al. Isolated limb perfusion in locally advanced cutaneous melanoma. Semin Oncol. 2002;29:400–409. [DOI] [PubMed] [Google Scholar]
- 116.Rossi CR, Mocellin S, Pilati P, et al. TNFalpha-based isolated perfusion for limb-threatening soft tissue sarcomas: state of the art and future trends. J Immunother. 2003;26:291–300. [DOI] [PubMed] [Google Scholar]