

Abstract
This study describes a new protein digestion protocol in which a variety of detergents can be used to solubilize membrane proteins and facilitate trypsin digestion with higher efficiency. In this protocol, proteins are dissolved in solutions containing various detergents and directly incorporated into a polyacrylamide gel matrix without electrophoresis. Detergents are subsequently eliminated from the gel matrix while proteins are still immobilized in the gel matrix. After in-gel digestion of proteins, LC-MS/MS is used to analyze the extracted peptides for protein identification. The uniqueness of the protocol is that it allows usage of a variety of detergents in the starting solution without interfering with LC-MS/MS analysis. We hereby demonstrate that different detergents, including ionic SDS, non-ionic Triton X-100 and n-octyl β-d-glucopyranoside, and zwitterionic CHAPS, can be used to achieve maximum solubilization of membrane proteins with minimal interference with LC-MS/MS analysis. Enhanced digestions, i.e. improved number and intensity of detected peptides, are also demonstrated for digestion-resistant proteins such as myoglobin, ubiquitin, and bacteriorhodopsin. An additional advantage of the Tube-Gel digestion protocol is that, even without electrophoresis separation, it allows high throughput analysis of complex protein mixtures when coupled with LC-MS/MS. The protocol was used to analyze a complex membrane protein mixture prepared from prostate cancer cells. The protocol involves only a single digestion and 2.5 h of LC-MS/MS analysis and identified 178 membrane proteins. In comparison, the same membrane fraction was resolved by SDS-PAGE, and 20 gel slices were excised and individually digested and analyzed by LC-MS/MS. The more elaborate effort demanded more than 50 h of LC-MS/MS analysis and identified 268 proteins. The new Tube-Gel digestion protocol is an alternative method for high throughput analysis of membrane proteins.
Membrane proteins participate in many important biological processes including cell adhesion, signal transduction, nutrient uptake, transportation, and endocytosis (1–3). There is an increasing level of interest in comprehensive and high throughput analysis of membrane proteins (4–8). MS-based proteomics has rapidly developed in recent years as a powerful approach for the large scale identification and characterization of proteins. The commonly used bottom-up proteomic approach involves proteolytic digestion of proteins to peptides followed by mass spectrometric measurement of peptide masses and sequences (9–14). Both MS and tandem MS data are subsequently subjected to database search for identification of unknown proteins and protein post-translational modifications. The confidence of protein identification is highly dependent on the number of peptides derived from each protein, which is determined by the efficiency of the proteolytic digestion as well as the coverage and accuracy of subsequent mass spectrometric analysis. However, there are substantial technical difficulties associated with both steps when analyzing membrane proteins. The proteolytic digestion of membrane proteins usually results in low sequence coverage because of their low solubility and high hydrophobicity, which render the cleavage sites less accessible to a proteolytic agent. Detergents and organic solvents can be used to improve the solubility of membrane proteins, but they interfere with subsequent MS measurement, particularly LC-MS/MS analysis, and generate poor MS data.
Several methods have been reported for the solubilization and digestion of membrane proteins. Blonder et al. (15) used a high percentage (e.g. 60%) of methanol for the solubilization of Halobacterium purple membrane. Trypsin, a proteolytic enzyme commonly used in proteomics, retained ~20% activity in 60% methanol and was reported to be able to effectively digest bacteriorhodopsin, one of the major components of the purple membrane. Washburn et al. (16) tested the combination of 90% formic acid and cyanogen bromide, a compound that cleaves after methionine residues in proteins and remains active at acidic pH. This approach was used to analyze the membrane fraction isolated from yeast and identified 1,484 proteins, 131 of which were membrane proteins. The concerns of the above methods are the toxic reagents used as well as the compromised proteolytic enzymatic activity in organic solvents.
Detergents have been widely used for solubilizing membrane proteins. Critical disadvantages of using detergents are that, even at low levels such as 0.1%, they significantly reduce the activity of proteolytic enzymes, interfere with HPLC separation, and yield strong MS background masking peptide signals. After proteins are initially solubilized in a buffer containing high detergent concentrations, detergents are diluted to a low concentration (typically less than 0.05%) prior to trypsin digestion and LC-MS/MS analysis. The diluted low concentration of detergent introduces some but manageable background in LC-MS analysis. Han et al. (17) solubilized microsomal proteins with Tris buffer containing 0.5% SDS and performed tryptic digestion after diluting SDS to 0.05%. The tryptic peptides were subjected to cation-exchange chromatography to remove interfering SDS prior to the reverse phase HPLC MS analysis. It was unclear how efficiently the authors were able to eliminate the SDS. An acid-labile detergent was recently reported to be able to assist in solubilizing membrane proteins and then degrade spontaneously when the sample is acidified (18). The usefulness of the acid-labile detergent remains to be further evaluated.
In-gel digestion has been commonly used after proteins are resolved by SDS-PAGE (9). This protocol has been proven to be compatible with the usage of detergents in protein samples. Typically membrane proteins are solubilized with loading buffer containing 1% SDS and separated into multiple bands by SDS-PAGE with a running buffer containing 0.1% SDS. Protein bands are stained for several hours or longer, visualized, and sliced from the gel into multiple gel pieces. The gel slices are subsequently washed thoroughly by acetonitrile to remove SDS and other detergents or compounds that may interfere with subsequent procedures. Gel slices are subsequently subjected to in-gel digestion using trypsin, and the resulting tryptic peptides are extracted and subjected to LC-MS/MS analysis. In-gel digestion usually gives a clean LC-MS/MS base line as interfering substances such as SDS are effectively removed during washing steps. This approach has been effective for soluble proteins and is widely used. However, it has several limitations when applied to analyzing membrane proteins. First, it is limited to relatively low concentrations of SDS (up to 1%) because other detergents or higher concentrations of detergents distort electrophoresis separation. This limits the types and concentrations of detergents available for optimal solubilization of membrane proteins. Second, multiple protein bands are generated from SDS-PAGE and have to be analyzed individually, thus requiring significantly longer analysis time and lower throughput. Alternatively it was reported that proteins were concentrated to a band between the stacking and the separating gels by stopping electrophoresis (19, 20). This method can avoid working with multiple gel bands, but it still has the limitation of the detergents that can be used.
In this work, we developed a new digestion protocol in which membrane proteins are directly incorporated into a polyacrylamide gel without electrophoresis. This allows us to use various types of detergents at high concentrations for effective solubilization of membrane proteins. The high concentrations of detergents also effectively denature the membrane proteins and make more enzymatic cleavage sites accessible, thus increasing protein digestion efficiency and improving sequence coverage and sensitivity. Detergents are removed prior to protein digestion and will not interfere with the subsequent reverse phase or two-dimensional LC-MS/MS analysis. Another advantage of this protocol over the conventional SDS-PAGE and in-gel digestion method is that it requires significantly less LC-MS/MS analysis time, thus allowing high throughput proteomic studies. The workflow of the new protocol named Tube-Gel digestion is illustrated in Scheme 1.
Scheme 1. Workflow of the Tube-Gel digestion protocol.
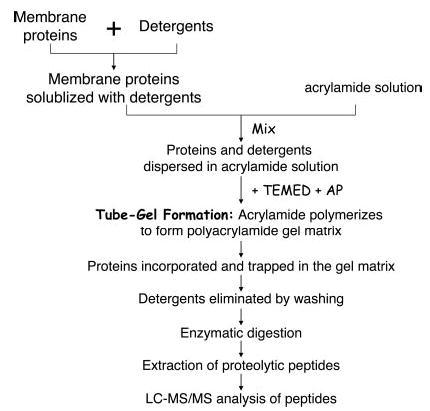
AP, ammonium persulfate.
EXPERIMENTAL PROCEDURES
Materials
Monomeric acrylamide solution (40%, 29:1) was purchased from Bio-Rad. Protein standards including BSA (99%), ubiquitin (from bovine erythrocytes, 90%), and myoglobin (from horse skeletal muscle, 95–100%) were obtained from Sigma. Bacteriorhodopsin of 75% purity from Halobacterium halobium was provided by Fluka (Milwaukee, WI). Trypsin (modified, sequencing grade) was purchased from Promega (Madison, WI). CHAPS, Triton X-100, n-octyl β-d-glucopyranoside (NOG)1 (>98%), ammonium bicarbonate (AMBIC), DTT, and protease inhibitor mixture were also from Sigma. Formic acid (>98%) was made by Merck KGaA. SDS (electrophoresis grade), heptafluorobutyric acid (HFBA), TFA, water, and ACN as well as other common reagents were obtained from Fisher.
Protein Sample Preparation
BSA, ubiquitin, and myoglobin were individually dissolved in 25 mm AMBIC (pH 8.0) at a concentration of 1 μg/μl and used as stock solutions. Bacteriorhodopsin could not be dissolved in 25 mm AMBIC but was suspended at the same concentration of 1 μg/μl so that protein quantity in this study could be accurately controlled. The protein samples used for the experiments were made by diluting the stock solution/suspension to the desired concentration with 25 mm AMBIC in the presence or absence of detergents. The protein samples were subjected to the new Tube-Gel digestion protocol as described below or other digestion methods.
Tube-Gel Protein Digestion
The above protein solutions, or the membrane fraction isolated from cell culture as described under the “Membrane Protein Isolation from PC3 Cells” section, were incorporated into a polyacrylamide gel matrix without electrophoresis. The shape of the polymerized gel matrix is a miniaturized tube, thus named “Tube-Gel,” and the protein digestion protocol described in this study is called “Tube-Gel digestion.” A 20-μl polyacrylamide gel was typically made as described below. 14 μl of the protein solution, 5 μl of acrylamide (40%, 29:1) solution, 0.7 μl of 1% ammonium persulfate, and 0.3 μl of TEMED were mixed and immediately transferred to a small glass tube with inner diameter of 1–2 mm (Fisher). The polymerization reaction was carried out for 30 min at room temperature. A gel strip, typically 6–10 mm long for a 20-μl gel mixture, formed in the glass tube, and ~1 μl of liquid remained on top of the gel strip. The 1 μl of liquid was analyzed by SDS-PAGE to assess the amount of protein that was not incorporated into the gel matrix. The gel strip was removed, cut into small pieces, and washed with 25 mm AMBIC (pH 8.0) containing 50% ACN for 15 min three times with vortexing. After being dried using a SpeedVac, the gel pieces were subjected to the standard in-gel digestion protocol as described previously (21) with minor modifications. Proteins were reduced by 10 mm DTT at 60 °C for 30 min and alkylated by 55 mm iodoacetamide (IAA) at room temperature in the dark for 30 min. Gel pieces were washed with 25 mm AMBIC, dehydrated with ACN, and dried using a SpeedVac. Proteolytic digestion was performed with 10 ng/μl trypsin dissolved in 25 mm AMBIC and was incubated at 37 °C overnight. Alternatively chymotrypsin digestion was incubated at 25 °C for 4 h. The peptides were extracted from the gel using 50 μl of 25 mm AMBIC, 100 μl of 0.02% HFBA in water, 150 μl of 0.02% HFBA in 50% ACN, and 50 μl of 100% ACN. The peptides extracted in the four steps were combined together, concentrated by SpeedVac to a desired volume (~20 μl), and subjected to LC-MS/MS analysis.
Protein Solution Digestion
Proteins were alternatively digested in solution to compare the digestion efficiency. The proteins were dissolved in 25 mm AMBIC without detergent, reduced with 5 mm DTT at 60 °C for 30 min, and alkylated with 25 mm IAA at room temperature in the dark for 30 min. Tryptic digestions were performed at a protein/trypsin ratio of 20:1 at 37 °C overnight. The protein digest was concentrated to ~20 μl.
Membrane Protein Isolation from PC3 Cells
The membrane proteins were isolated from PC3 cells, a prostate cancer cell line, by differential centrifugation. PC3 cells were generously provided by Dr. Natasha Kyprianou in the Division of Urology at the University of Kentucky and were originally obtained from ATCC. PC3 cells were cultured in RPMI 1640 medium until approaching 90% confluence. After being washed with ice-cold PBS containing 1 mm MgCl2, the cells were harvested by gently scraping the dishes and subsequently centrifuged at 400 × g for 5 min using an Eppendorf 5810R refrigerated centrifuge. Cells were incubated on ice for 15 min in hypotonic buffer (10 mm HEPES (pH 7.5), 1.5 mm MgCl2, 10 mM KCl, 1× protease inhibitor mixture, 1 mm NaF, and 1 mm Na3VO4) and lysed using a Dounce homogenizer for 30 strokes. The lysate was centrifuged in a 1.5-ml tube at 1000 × g for 5 min to remove the pellet nuclear fraction. The postnuclear supernatant was transferred to a 4-ml ultracentrifuge tube and centrifuged at 100,000 × g for 20 min using a TL-100 ultracentrifuge (Beckman) with a TLA-100.3 fixed angle rotor (22). The supernatant was discarded, and the pellet microsome fraction was resuspended in 0.1 m Na2CO3 (pH 11.5) and kept on ice for 1 h (8, 23, 24). The suspension was ultracentrifuged again at 100,000 × g for 35 min to pellet the membrane. The supernatant was discarded, and the pellet (membrane) was gently washed by pipetting with ice-cold Na2CO3 several times. The membrane was solubilized with 2% SDS in 25 mm AMBIC (pH 8.0) and subjected to Tube-Gel digestion as described above. The protein concentration was assayed using Bradford reagent at 595 nm wavelength. Approximately 500 μg of membrane proteins were consistently obtained from 10 dishes (10 cm) of PC3 cells.
Nano-LC-MS/MS and Data Analysis
Nano-flow reverse phase LC-MS/MS was performed using a capillary HPLC system (LC Packings, Amsterdam, Netherlands) coupled with a QSTAR XL quadruple time-of-flight mass spectrometer (ABI/MDS Sciex) through a nano-electrospray ionization source (Protana). Analyst QS software was used for system control and data collection. The desired volume of protein solution was injected by the autosampler and desalted on a C18 trap column (300 μm × 1 mm, LC Packings) for 6 min at a flow rate 10 μl/min. The sample was subsequently separated by a C18 reverse phase column (75 μm × 15 cm, Vydac, Columbia, MD) at a flow rate of 220 nl/min. The mobile phases consisted of water with 0.1% formic acid (A) and 90% acetonitrile with 0.1% formic acid (B), respectively. A 90-min linear gradient from 5 to 50% B was typically used. After LC separation the sample was introduced into the mass spectrometer through a 10-μm silica tip (New Objective) adapted with a nanoelectrospray source(Protana).Data were acquired in information-dependent acquisition mode. Each cycle typically consisted of a 1-s TOF MS survey from 400 to 1600 (m/z) and two 2-s MS/MS scans with mass range of 65–1600 (m/z).
The LC-MS/MS data of the membrane fraction of PC3 cells were submitted to a local MASCOT server for MS/MS ion search. The peak lists from the LC-MS/MS spectra were generated by the MASCOT script embedded in the Analyst QS software using the following parameters: no smoothing, charge state determined from the MS scan, precursor ion charge states of 2+ and 3+, centroid MS/MS data, height percentage of 50%, and merge distance of 0.02 Da. The typical parameters used in the MASCOT MS/MS ion search were: Homo sapiens, maximum of two trypsin miscleavages, cysteine carbamidomethylation, methionine oxidation, a maximum of 100-ppm MS error tolerance, and a maximum of 0.15-Da MS/MS error tolerance. In light of potential ambiguous protein identifications results, rigorous identification criteria were used according to the published guideline (25). For the MS/MS ion search, proteins with two or more peptides and a score of each peptide higher than 30 were considered unambiguous identifications without manual inspection. Proteins identified with one peptide with a score higher than 45 were manually inspected and confirmed. Proteins with one peptide with a score lower than 45 were considered ambiguous and discarded. For the chymotrypsin digestion experiment, the enzyme specificity was defined in the local MASCOT server as cleaving the C termini of Pro, Phe, Tyr, Leu, or Met.
RESULTS AND DISCUSSION
Protein Incorporation into Tube-Gel
A protein solution was mixed with acrylamide monomer solution and incorporated into the polyacrylamide gel matrix (Tube-Gel) during the polymerization process without electrophoresis. It was necessary to assess the protein incorporation efficiency. A capillary glass tube (inner diameter, ~1–2 mm) was used to limit gel condensation, thus minimizing the volume of solution that was excluded from the gel matrix. 5 μg of BSA (66 kDa), myoglobin (17 kDa), and ubiquitin (8.6 kDa) were incubated in a total of 20 μl of acrylamide mixture. After gel polymerization, it was observed that ~1 μl of liquid remained on top of the gel matrix. The liquid was removed and subjected to SDS-PAGE analysis. As shown in Fig. 1, proteins excluded from the gel matrix from three independent experiments (lanes 3–5) were less than 10% of the original amount of each protein in the mixture (lane 2). The amount of protein excluded was independent of the size of the protein. In summary, more than 90% of proteins can be incorporated into the 20-μl Tube-Gel.
Fig. 1. The efficiency of protein incorporation into the Tube-Gel.
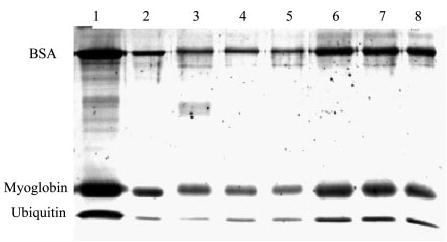
5 μg each of BSA, myoglobin, and ubiquitin were incorporated into the Tube-Gel as described under “Experimental Procedures,” and the minute portion of proteins that were excluded from the gel matrix was examined by SDS-PAGE. Lane 1 is 5 μg of each protein, and lane 2 is 0.5 μg of each protein (10% of the protein quantity in lane 1). Lanes 3–5 are proteins excluded from a 20-μl Tube-Gel in three independent experiments. Lanes 6–8 are proteins excluded from a 100-μl Tube-Gel in three independent experiments. The intensities of each band in lanes 3–5 are less than those in lane 2. The intensities of each band in lanes 6–8 are slightly greater than those in lane 2.
It is noted that the Tube-Gel preparation can be scaled up to accommodate larger quantities of proteins. Up to 100 μl of protein/acrylamide mixture was tested using a 0.6-ml Eppendorf tube. The proteins excluded from the gel matrix were slightly higher than 10% of the original amount of proteins in the acrylamide mixture (lanes 6–8). Gel density analysis estimated that the protein excluded from the gel matrix was ~15–20% of the original protein quantity; therefore more than 80% of proteins can be incorporated into a larger (100 μl) Tube-Gel.
The optimal protein quantity for the Tube-Gel digestion protocol depends on the volume of the Tube-Gel and the concentration of trypsin. For a 20-μl Tube-Gel and trypsin concentration of 10 ng/μl as typically used in this study, the total protein quantity should be ~20 μg. This will maintain the protein/trypsin ratio at ~100:1. However, if the amount of protein is more than 20 μg, this protocol can be easily scaled up by increasing the volume of the Tube-Gel matrix and/or trypsin concentration.
Compatibility of Detergents Used in the Tube-Gel Digestion Protocol with LC-MS/MS
A significant advantage of the Tube-Gel digestion protocol is that it is tolerant of a variety of detergents, salt, or other interfering substances of small molecular mass (<1000 Da). After proteins are incorporated into the Tube-Gel, the detergents and other interfering substances are eliminated by extensive washing. We tested four different detergents, SDS (ionic), NOG (non-ionic), Triton X-100 (non-ionic), and CHAPS (zwitterionic), at various concentrations (0.1–2%) in the starting BSA solutions. 1.3 μg of BSA (~20 pmol) in 25 mm AMBIC containing 2% of one of the four detergents was subjected to the Tube-Gel digestion. An aliquot of the yielded tryptic peptides containing 200 fmol of peptides was analyzed by LC-MS/MS. Fig. 2 shows the base peak HPLC chromatogram of the tryptic peptides of BSA in the presence of 2% detergent. Table I summarizes the number of peptides and sequence coverage obtained by Tube-Gel digestion of BSA in the absence and presence of detergents. It is clear from the results that the detergents were effectively removed and did not interfere with HPLC separation and MS/MS measurements. Therefore, using detergents in the initial protein sample preparation is perfectly compatible with the Tube-Gel digestion protocol. In contrast, when 10 mm NOG was present in the BSA in-solution tryptic digestion sample, the detergent dominated the mass spectrometry signals when the sample was directly infused to the mass spectrometer (data not shown). Moreover the detergent interferes with HPLC separation; thus the detergent-containing sample was not subjected to LC-MS analysis.
Fig. 2. Base peak chromatography of the tryptic peptides of BSA using Tube-Gel digestion in the presence of 2% SDS (A), n-octyl β-d-glucopyranoside (B), CHAPS (C), and Triton X-100 (D).
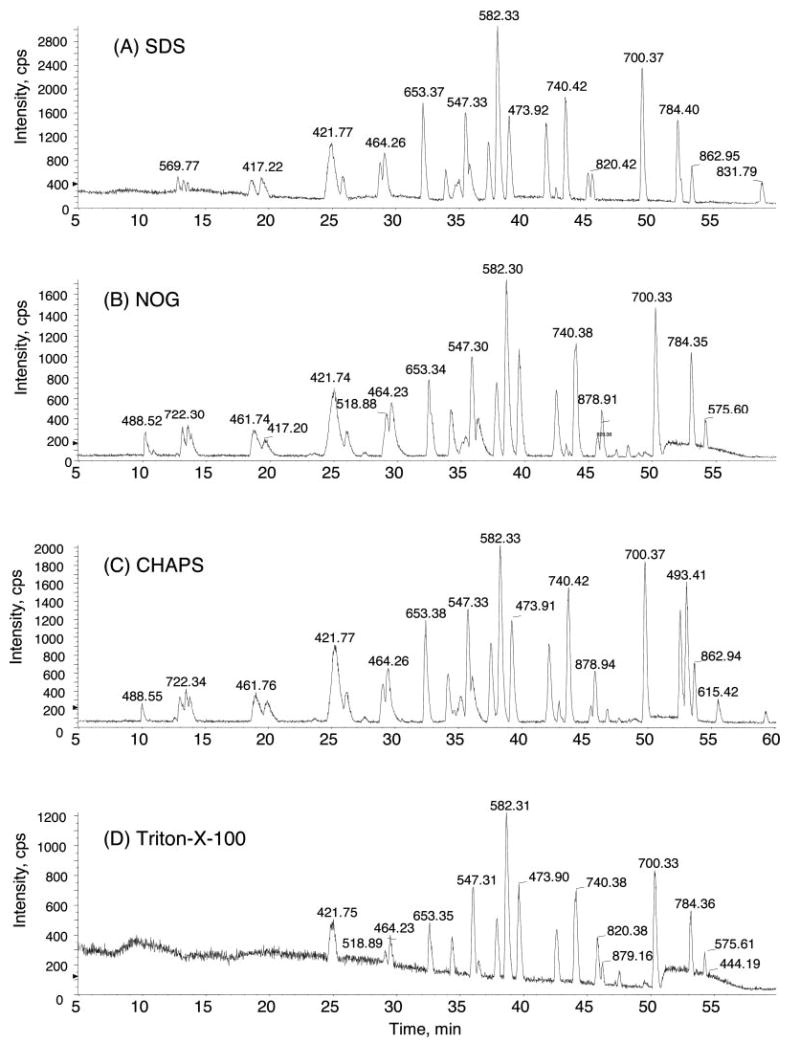
1.3 μg of BSA (69 kDa, ~20 pmol) in 25 mm AMBIC containing 2% of one of the four detergents was subjected to Tube-Gel digestion. An aliquot containing 200 fmol of the yielded tryptic peptides was subjected to the LC-MS/MS analysis. Despite 2% detergent in the starting samples, no detergent signals were observed in LC-MS/MS, and the base peaks of BSA tryptic peptides were well separated in HPLC. cps, counts per second.
Table I.
Tryptic peptides observed in LC-MS/MS analysis of Tube-Gel digestion products of BSA in the presence of 2% of different detergents
| Digestion conditions | Peptide number detected | Sequence coverage |
|---|---|---|
| % | ||
| No detergent | 36 | 66 |
| 2% SDS | 46 | 74 |
| 2% CHAPS | 42 | 69 |
| 2% Triton X-100 | 39 | 73 |
| 2% NOG | 43 | 74 |
In addition, up to 5% SDS was used to solubilize bacteriorhodopsin, a membrane protein with seven transmembrane domains. The intensities of three representative tryptic peptides (m/z values of 494.31 (2+), 726.85 (2+), and 959.92 (2+)) in the LC-MS/MS analyses of the Tube-Gel digestion samples containing different concentrations of SDS are plotted in Fig. 3. In the absence of SDS, these three peptides were undetectable. In the presence of 0.1% SDS, a significant intensity of each of the three peptides was recorded. Maximal intensities of the peptides were observed when 2% SDS was added to bacteriorhodopsin sample. Higher concentrations of SDS up to 5% in the starting protein sample did not improve Tube-Gel digestion or LC-MS/MS of bacteriorhodopsin. More importantly, 5% SDS in the starting sample was still tolerated and compatible with the Tube-Gel digestion protocol and subsequent mass spectrometric analysis. The tolerance of high concentrations of detergents can be critical to applications requiring such conditions.
Fig. 3. Mass spectrometry intensities of three representative tryptic peptides prepared by the Tube-Gel digestion of bacteriorhodopsin samples containing different concentrations of SDS.
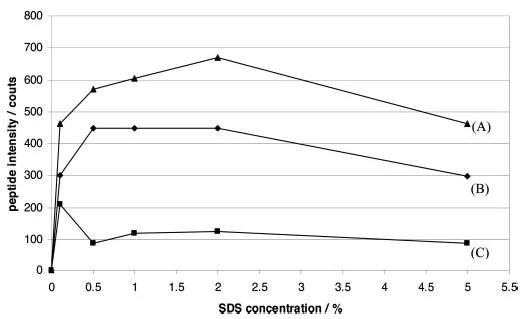
Up to 5% SDS was added in the starting bacteriorhodopsin sample, which was then subjected to Tube-Gel digestion. Equal amounts (700 fmol) of the yielded peptides were subjected to LC-MS/MS analysis in each experiment. The intensities of three representative peptides from the starting samples containing different SDS concentrations are shown in this figure. A, AESMRPEVASTFK, 2+, m/z = 726.85; B, VGFGLILLR, 2+, m/z = 494.31; C, AIFGEAEAPEPSAGDGAAATS, 2+, m/z = 959.92.
It is noted that all 35 cysteine residues in BSA were identified in the carbamidomethyl form in the above experiments using the Tube-Gel protocol, suggesting that the DTT reduction and IAA alkylation steps were highly effective in the Tube-Gel protocol. It is important to maintain the cysteine residues in a uniform form so that better sample homogeneity will be achieved and the sensitivity of the overall analysis will be improved.
Enhanced Digestion Efficiency Using Detergents in the Tube-Gel Protocol
To test whether detergents used in the Tube-Gel protocol can significantly enhance the proteolytic digestion efficiency, three standard proteins were subjected to three different digestion protocols and subsequent LC-MS/MS analysis. Myoglobin, ubiquitin, and bacteriorhodopsin were selected in this study because they have been reported to be moderately or highly resistant to tryptic digestion (18). The proteins were subjected to Tube-Gel digestion in the presence of SDS, Tube-Gel digestion in the absence of SDS, or in-solution digestion with SDS. The resulting tryptic peptides were analyzed by LC-MS/MS using identical LC and MS parameters.
The standard proteins were first prepared in 25 mm AMBIC (pH 8.0) at 1 μg/μl as stock. Each individual protein stock was divided into two parts: one was diluted with 2% SDS in 25 mm AMBIC, and the other was diluted to the same concentration with 25 mm AMBIC without SDS. It should be noted that bacteriorhodopsin was insoluble in 25 mm AMBIC, and the stock was vortexed for 1 min prior to dilution; thus the protein stock sample was homogenous, and the dilution was accurate and consistent among different aliquots. Equal quantities of proteins were subjected to trypsin digestion using the Tube-Gel protocol in the presence of SDS, the Tube-Gel protocol in the absence of SDS, or an in-solution digestion method in the absence of SDS. All reactions were performed at 37 °C overnight using approximately the same protein/trypsin ratio (50:1). The tryptic peptides were extracted in parallel, and the same quantities of peptides were subjected to the subsequent LC-MS/MS analyses using identical LC and MS/MS parameters as described under “Experimental Procedures.”
Table II summarizes the protein sequence coverage results obtained from the three different digestion methods. It is clearly evident that, compared with the two other protocols, the usage of detergent in the Tube-Gel digestion protocol greatly enhanced the digestion efficiency, thus resulting in more observed peptides and higher sequence coverage. The sequence coverage of myoglobin was improved from ~50% (without SDS) to 84% (with SDS). Moreover the intensities of the detected tryptic peptides using Tube-Gel with SDS were at least 4 times greater than those of corresponding peptides using protocols without SDS. The data are shown in Fig. S1 in the supplemental results.
Table II.
Comparison of protein sequence using different digestion protocols
| Protein | Tube-Gel digestion with 2% SDS | Tube-Gel digestion without SDS | In-solution digestion without SDS |
|---|---|---|---|
| Bacteriorhodopsina | 5 peptides/21% | 0 peptides/0% | 0 peptides/0% |
| Ubiquitinb | 10 peptides/86% | 0 peptide/0% | 0 peptide/0% |
| Myoglobinc | 12 peptides/84% | 6 peptides/52% | 7 peptides/50% |
Bacteriorhodopsin (28 kDa), 400 ng of which was digested and 20 ng (~700 fmol) of which was analyzed by LC-MS/MS.
Ubiquitin (8.6 kDa), 130 ng of which was digested and 2.6 ng (~300 fmol) of which was analyzed by LC-MS/MS.
Myoglobin (17 kDa), 1.3 μg of which was digested and 13 ng (~700 fmol) of which was analyzed by LC-MS/MS.
The most significant improvements were achieved for ubiquitin and bacteriorhodopsin. No peptides were detected for either protein when SDS was not used. In contrast, 86% (10 peptides) and 21% (five peptides) sequence coverage was accomplished for ubiquitin and bacteriorhodopsin, respectively, when they were subjected to Tube-Gel digestion in the presence of 2% SDS.
The LC-MS/MS results of ubiquitin tryptic digestion using the above three protocols are shown in Fig. 4. No peptides from ubiquitin were detected in LC-MS/MS analysis of 300 fmol of ubiquitin when SDS was not used (Fig. 4, B and C). When 2% SDS was used in the protein solubilization step, the subsequent Tube-Gel digestion and LC-MS/MS analysis yielded 10 peptides with intense peptide signals in TOF MS scans (Fig. 4A) and high quality MS/MS spectra in MS/MS scans.
Fig. 4. Comparison of base peak chromatography of the tryptic peptides of ubiquitin prepared by Tube-Gel digestion with 2% SDS (A), Tube-Gel digestion without SDS (B), and in-solution digestion without SDS (C).
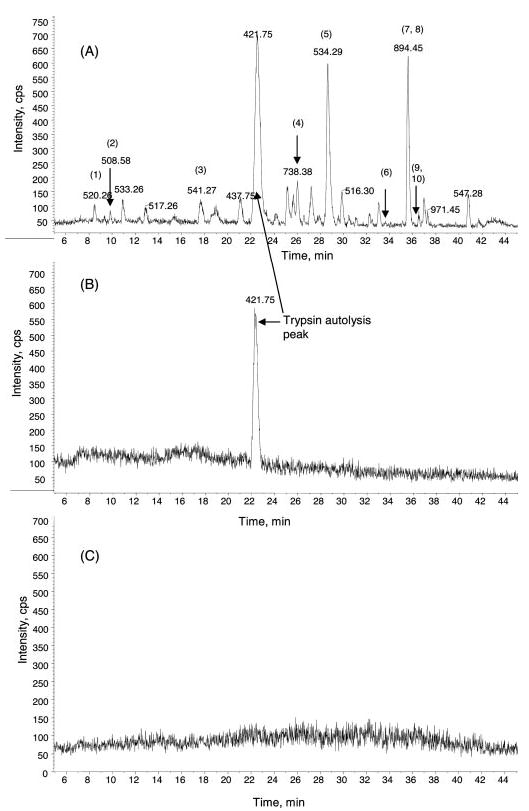
Equal amounts of ubiquitin were subjected to trypsin digestion using the three different protocols, and equal amounts of digestion products were subjected to further LC-MS/MS analysis. No tryptic peptides from ubiquitin were detected using the methods in B or C. 10 unique tryptic peptides from ubiquitin were observed using the Tube-Gel digestion in the presence of 2% SDS. They are as follows: 1, EGIPPDQQR (m/z 520.26, 2+); 2, IQD-KEGIPPDQQR (m/z 508.58, 3+); 3, TLSDYNIQK (m/z 541.27, 2+); 4, WQIFVK (m/z 410.71, 2+); 5, ESTLHLVLR (m/z 534.29, 2+); 6, TI-TLEVEPSDTIENVKAK (m/z 663.01, 3+); 7, TITLEVEPSDTIENVK (m/z 894.45, 2+); 8, TLTGKTITLEVEPSDTIENVK (m/z 763.38, 3+); 9, TLS-DYNIQKESTLHLVLR (m/z 533.28, 4+; m/z 710.70, 3+); 10, QLEDGRTLSDYNIQKESTLHLVLR (m/z 707.85, 4+; m/z 566.47, 5+). cps, counts per second.
Bacteriorhodopsin is a membrane protein with seven trans-membrane domains (26). It is insoluble in 25 mm AMBIC in the absence of SDS or other detergents. It was not surprising that no peptides were detected no matter how the bacteriorhodopsin suspension was digested. When 2% SDS was used to solubilize bacteriorhodopsin, the subsequent Tube-Gel digestion and LC-MS/MS analysis yielded five peptides with high quality MS/MS spectra (see Fig. S2 in the supplemental results). Although the sequence coverage is relatively low at 21%, it is a significant improvement compared with the other two methods in which no peptides were detected. The relatively low sequence coverage is likely because of the limited number of trypsin cleavage sites (lysine and arginine residues) present in the protein sequence, resulting in a low number of tryptic peptides that are suitable to be extracted from the gel matrix and be analyzed by LC-MS/MS. Other proteolytic enzymes such as chymotrypsin can be used to solve this problem, which will be described in the next section.
The above experiments demonstrate that the inclusion of detergents in the Tube-Gel digestion protocol significantly enhances the proteolytic digestion efficiency, thus improving the sequence coverage of proteins analyzed. The unique feature of the Tube-Gel digestion protocol is its compatibility with high concentrations of detergents; for instance 2% SDS was used in the above experiments. Detergents have two functions in this protocol: improving the solubility of hydrophobic proteins and denaturing proteins. Greater solubility will result in larger quantities of proteins incorporated into the gel matrix and subjected to digestion. More denatured proteins will yield greater accessibility to cleavage sites in the protein structure. The combination of both will significantly enhance the efficiency of proteolytic digestion using the new Tube-Gel digestion protocol.
Tube-Gel Digestion of Bacteriorhodopsin with Chymotrypsin
To improve the sequence coverage of the Tube-Gel digestion and subsequent LC-MS/MS analysis of bacteriorhodopsin, chymotrypsin was used to produce more proteolytic peptides. Chymotrypsin is a proteolytic enzyme with preferential cleavage sites at the C terminus of Trp, Phe, and Tyr. Bacteriorhodopsin was solubilized in 25 mm AMBIC containing 2% SDS and subjected to Tube-Gel digestion using chymotrypsin at 37 °C for 4 h. For the same quantity of bacteriorhodopsin as used in trypsin digestion (700 fmol, 20 ng), 42 proteolytic peptides were detected in the LC-MS/MS analysis, covering 83% of the bacteriorhodopsin sequence. Fig. 5 shows the base peak chromatogram of the LC-MS/MS analysis of the chymotrypsin Tube-Gel digestion of bacteriorhodopsin and the sequence coverage of the analysis. The results demonstrate that chymotrypsin is suitable for digestion of membrane proteins because it can digest highly hydrophobic proteins more completely, yielding a greater number of proteolytic peptides. It also proves that the Tube-Gel digestion protocol is versatile and that different proteolytic enzymes can be used in the method.
Fig. 5. Chymotrypsin digestion of bacteriorhodopsin using the Tube-Gel protocol in the presence of 2% SDS.
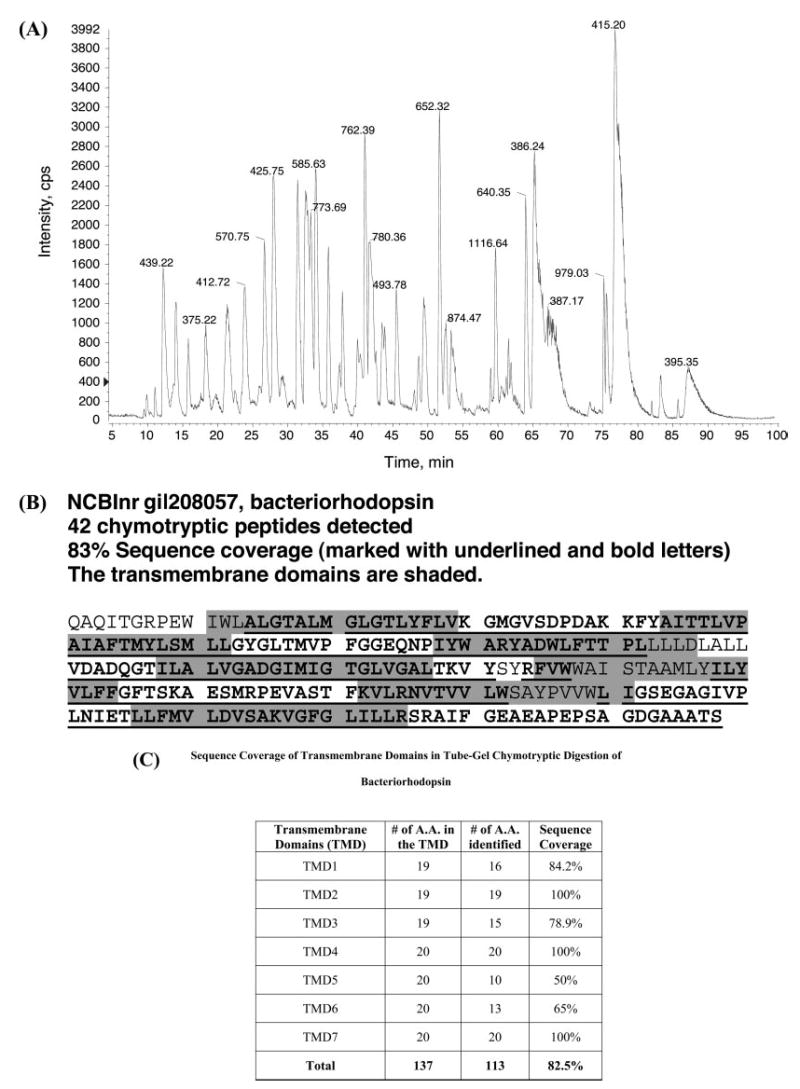
A, base peak chromatography of LC-MS/MS. B, sequence coverage of chymotrypsin peptides detected by LC-MS/MS. Digestion and analysis of 20 ng (700 fmol) of bacteriorhodopsin yielded 42 observed chymotrypsin peptides (underlined) with 83% sequence coverage. The transmembrane domains are shaded, and it is obvious that all seven transmembrane domains are represented in the identified chymotryptic peptides. C, sequence coverage of the transmembrane domains in Tube-Gel chymotryptic digestion of bacteriorhodopsin. cps, counts per second; TMD, transmembrane domain.
More interestingly, 26 of a total of 42 identified chymotryptic peptides contain amino acid residues from all seven transmembrane domains. The transmembrane domains are illustrated by shading in Fig. 5B, and the shaded sequences are obviously overlapped with the underlined sequences. The numbers of animo acid residues from each transmembrane domain that were identified in the LC-MS/MS analysis of chymotryptic peptides are shown in Fig. 5C. Overall the sequence coverage of the seven transmembrane domains is 82.5%. The results demonstrate that the Tube-Gel is an efficient method to analyze membrane proteins with significant coverage of transmembrane domains.
To examine the efficiency and sensitivity of the Tube-Gel digestion protocol, lower quantities of bacteriorhodopsin, including 400, 120, 40, 10, and 4 fmol, were digested using the Tube-Gel protocol as described above and analyzed by LC-MS/MS. When 400 fmol of bacteriorhodopsin were subjected to digestion, the same number of peptides (42 peptides, 83% sequence coverage) were observed in the subsequent LC-MS/MS analysis. 22 peptides (61% coverage) and 16 peptides (46% coverage) were observed when 120 and 40 fmol of bacteriorhodopsin were subjected to digestion, respectively. No peptides were identified at 10 fmol or lower. It should be noted that the detected peptide number and sequence coverage are also determined by the detection limit of LC-MS and the performance of database searching algorithm. With our system, QSTAR XL and MASCOT database searching algorithm, the detection limit of standard peptides such as [Glu1]-fibrinopeptide B (EGVNDNEEGFFSAR) is typically at 10 fmol. The results of the Tube-Gel chymotrypsin digestion of bacteriorhodopsin were close to the detection limit of LC-MS/MS system used in the study; thus the Tube-Gel digestion protocol is highly efficient.
Application of Tube-Gel Digestion Protocol to Analyzing Prostate Cell Membrane Proteins
The Tube-Gel digestion protocol and subsequent LC-MS/MS analysis were applied to characterizing membrane proteins isolated from PC3 cells (a prostate cancer cell line). The membrane fraction was prepared by 100,000 × g centrifugation of the postnuclear supernatant. The pellet was further treated with 0.1 m Na2CO3 (pH 11.5) for 1 h on ice. This isolation method can produce a highly enriched membrane fraction (8, 23, 24), including proteins that are usually difficult to solubilize and digest. The membrane pellet was dissolved in 25 mm AMBIC (pH 8.0) containing 2% SDS until no pellet was observed after gentle mixing. The solubilized membrane proteins were subjected to 15,000 × g centrifugation for 20 min, and no particulate objects were observed. Protein quantity was estimated using the Bradford assay. Approximately 20 μg of the membrane proteins were incorporated into a 20-μl Tube-Gel and subjected to trypsin digestion as described above. The tryptic peptides were extracted and subjected to reverse phase LC-MS/MS analysis.
A representative LC-MS/MS chromatogram and MS and MS/MS spectra are shown in Fig. 6. In this 2.5-h LC-MS/MS experiment, 2757 MS/MS spectra were acquired. In total 178 proteins were identified using the LC-MS/MS data and MASCOT MS/MS ion search. Of these, 96 proteins (54%) have at least one transmembrane domain as assessed by TMHMM server (prediction of transmembrane helices in proteins) (27). Of the 96 proteins containing at least one transmembrane domain, 83 (86%) proteins were identified with more than three unique peptides with MASOCT peptide ion scores greater than 20. In particular, a sequence coverage of 54% was achieved for solute carrier family 25, member 5 (NCBInr accession number gi|33525218), a protein with two transmembrane domains. The results demonstrate that the Tube-Gel protocol is well suited for digesting complex membrane proteins and that the combination of this protocol with LC-MS/MS can produce a high throughput analysis of a complex mixture of membrane proteins.
Fig. 6. Representative LC-MS/MS of the Tube-Gel digestion products of the membrane fraction isolated from prostate cancer cells.
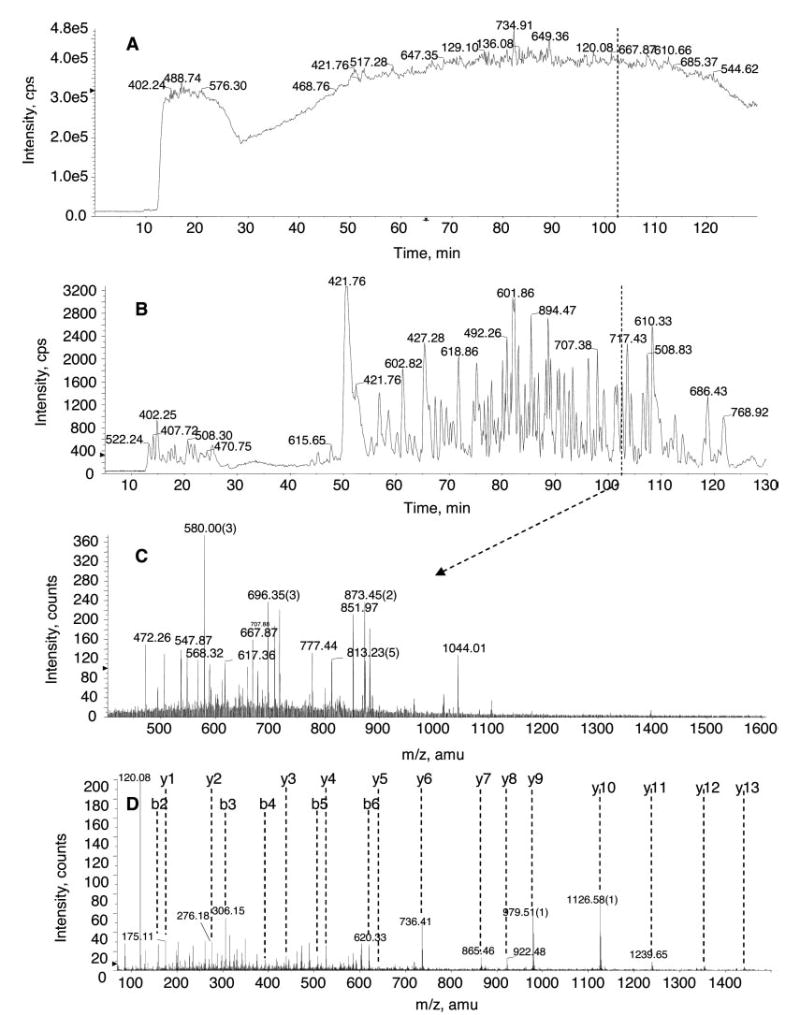
A, total ion count chromatography. B, base peak chromatography. C, a representative TOF MS survey scan at 102.3 min. D, a representative MS/MS spectrum of selected ion 873.45 (2+). The sequence of this particular peptide is SAFSNLFGGEPLSYTR, and the protein was identified as transferrin receptor (NCBInr accession number gi|4507457). Approximately 20 μg of membrane proteins prepared from prostate cancer cells were subjected to the Tube-Gel digestion protocol as described under “Experimental Procedures.” Half of the Tube-Gel digestion products (~10 μg) was analyzed by LC-MS/MS. The HPLC gradient was linear from 5 to 40% B (0.1% formic acid in 90% ACN) over 100 min and further increased to 80% at 120 min. The information-dependent acquisition experiments were composed of a 1-s TOF MS survey and three 2-s MS/MS experiments. cps, counts per second.
In comparison, the same quantity (20 μg) of membrane proteins was subjected to SDS-PAGE separation using an 8.3 × 7.3-cm polyacrylamide gel, and 20 gel slices of 1–2-mm width were cut and subjected to in-gel digestion by trypsin. Tryptic peptides from the 20 gel slices were extracted, and each of them was subjected to LC-MS/MS analysis using the same instrument settings. The experiment was accomplished in 50 h using an autosampler in the automated mode. The data from 20 LC-MS/MS experiments were pooled and subjected to MASCOT MS/MS ion search. From the much more extensive effort, 268 proteins were identified. Fig. 7 summarizes the number of proteins identified using these two approaches: Tube-Gel digestion followed by one LC-MS/MS analysis and SDS-PAGE followed by 20 LC-MS/MS analyses. All proteins identified in both methods are listed in Supplemental Table S1. 115 proteins were commonly identified in both approaches. The sequence coverages of the 115 commonly identified proteins are also compared in Supplemental Table S1. The Tube-Gel protocol produced slightly better sequence coverage even though it is a much simpler method requiring much less analysis time. It is true that the SDS-PAGE and 20 LC-MS/MS analyses identified ~50% more proteins. However, the Tube-Gel digestion followed by a single LC-MS/MS run took only 1/20 of the LC-MS/MS analysis time as the other approach. Therefore, the Tube-Gel digestion protocol provides a significantly better high throughput approach for proteomic characterization of membrane proteins.
Fig. 7. Summary of the proteins identified by two different approaches.
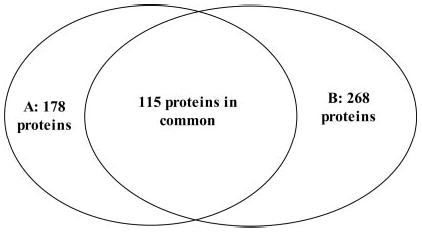
A, Tube-Gel digestion followed by only one Tube-Gel/LC-MS/MS analysis. B, SDS-PAGE followed by 20 LC-MS/MS analyses. 178 and 268 proteins were identified in each method, respectively, 115 of which are proteins that were commonly identified in both approaches.
It is noted that the resulting proteolytic peptides can be subjected to different types of LC separation and mass spectrometric analysis. Reverse phase LC-MS/MS was mainly used in this study, but other separation and mass spectrometric strategies can be readily used as necessary. For instance, MALDI mass spectrometry can be used for analyzing relatively simple samples containing membrane proteins. Alternatively if the sample is highly complex, more sophisticated separation such as two-dimensional HPLC can be used and coupled with tandem MS to achieve comprehensive characterization of complex samples.
Conclusion
A novel Tube-Gel digestion protocol was developed in this study to allow the usage of various detergents at different concentrations to assist the characterization of membrane proteins. Four detergents (SDS, NOG, CHAPS, and Triton X-100) were tested at concentrations up to 5% with no interference being observed in the LC-MS/MS analysis. In addition to solubilizing membrane proteins, the inclusion of detergents in the protocol significantly enhances the proteolytic digestion efficiency, thus the sequence coverage of the analysis. Different proteolytic enzymes can be used in the Tube-Gel digestion protocol. The resulting proteolytic peptides can be subjected to different separation and mass spectrometric methods, allowing flexibility for further analysis. Reverse phase LC-MS/MS was used in this study, and two-dimensional LC-MS/MS analysis certainly can be used if necessary. Moreover, when analyzing complex mixtures, this novel protocol offers significantly higher throughput compared with conventional SDS-PAGE separation and multiple LC-MS/MS analyses. This Tube-Gel digestion provides a novel and better approach for high throughput proteomic studies of membrane proteins.
Supplementary Material
Acknowledgments
We thank Liu Li, Fujian Zhang, and Dr. Kei Fukada for technical assistance in culturing prostate cancer cells and for discussions. We thank Alexis Vien and Renee Kilty for reading and editing the manuscript.
Footnotes
This study was supported in part by the University of Kentucky College of Medicine (start-up funds to H. Z.), NIEHS, National Institutes of Health Grant 1R21ES12025 (to H. Z.), and National Center for Research Resources, National Institutes of Health Grant 1P20RR020171–010005 (to H. Z.). Purchase of the QSTAR XL mass spectrometer was made possible by a grant from the National Science Foundation Experimental Program to Stimulate Competitive Research and matching funds from the University of Kentucky.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
The abbreviations used are: NOG, n-octyl β-d-glucopyranoside; AMBIC, ammonium bicarbonate; HFBA, heptafluorobutyric acid; TE-MED, N,N,N′,N′-tetramethylethylenediamine; IAA, iodoacetamide.
References
- 1.Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 2.Meredith JE, Jr, Schwartz MA. Integrins, adhesion and apoptosis. Trends Cell Biol. 1997;7:146–150. doi: 10.1016/S0962-8924(97)01002-7. [DOI] [PubMed] [Google Scholar]
- 3.Martin KH, Slack JK, Boerner SA, Martin CC, Parsons JT. Integrin connections map: to infinity and beyond. Science. 2002;296:1652–1653. doi: 10.1126/science.296.5573.1652. [DOI] [PubMed] [Google Scholar]
- 4.Whitelegge JP, Gomez SM, Faull KF. Proteomics of membrane proteins. Adv Protein Chem. 2003;65:271–307. doi: 10.1016/s0065-3233(03)01023-4. [DOI] [PubMed] [Google Scholar]
- 5.Weinglass AB, Whitelegge JP, Kaback HR. Integrating mass spectrometry into membrane protein drug discovery. Curr Opin Drug Discov Dev. 2004;7:589–599. [PubMed] [Google Scholar]
- 6.Whitelegge JP. HPLC and mass spectrometry of intrinsic membrane proteins. Methods Mol Biol. 2004;251:323–340. doi: 10.1385/1-59259-742-4:323. [DOI] [PubMed] [Google Scholar]
- 7.Tu X, Huang A, Bae D, Slaughter N, Whitelegge J, Crother T, Bickel PE, Nel A. Proteome analysis of lipid rafts in Jurkat cells characterizes a raft subset that is involved in NF-κB activation. J Proteome Res. 2004;3:445–454. doi: 10.1021/pr0340779. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen PA, Olsen JV, Podtelejnikov AV, Andersen JR, Mann M, Wisniewski JR. Proteomic mapping of brain plasma membrane proteins. Mol Cell Proteomics. 2005;4:402–408. doi: 10.1074/mcp.T500002-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P, Boucherie H, Mann M. Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci U S A. 1996;93:14440–14445. doi: 10.1073/pnas.93.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 11.Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405:837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- 12.Yates JR., III Mass spectrometry. From genomics to proteomics. Trends Genet. 2000;16:5–8. doi: 10.1016/s0168-9525(99)01879-x. [DOI] [PubMed] [Google Scholar]
- 13.Williams KE, Carver TA, Miranda JJ, Kautiainen A, Vogel JS, Dingley K, Baldwin MA, Turteltaub KW, Burlingame AL. Attomole detection of in vivo protein targets of benzene in mice: evidence for a highly reactive metabolite. Mol Cell Proteomics. 2002;1:885–895. doi: 10.1074/mcp.m200067-mcp200. [DOI] [PubMed] [Google Scholar]
- 14.Wysocki VH, Resing KA, Zhang QF, Cheng GL. Mass spectrometry of peptides and proteins. Methods. 2005;35:211–222. doi: 10.1016/j.ymeth.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Blonder J, Conrads TP, Yu LR, Terunuma A, Janini GM, Issaq HJ, Vogel JC, Veenstra TD. A detergent- and cyanogen bromide-free method for integral membrane proteomics: application to Halobacterium purple membranes and the human epidermal membrane proteome. Proteomics. 2004;4:31–45. doi: 10.1002/pmic.200300543. [DOI] [PubMed] [Google Scholar]
- 16.Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 17.Han DK, Eng J, Zhou HL, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suder P, Bierczynska A, Konig S, Silberring J. Acid-labile surfactant assists in-solution digestion of proteins resistant to enzymatic attack. Rapid Commun Mass Spectrom. 2004;18:822–824. doi: 10.1002/rcm.1411. [DOI] [PubMed] [Google Scholar]
- 19.Ferro M, Salvi D, Brugiere S, Miras S, Kowalski S, Louwagie M, Garin J, Joyard J, Rolland N. Proteomics of the chloroplast envelope membranes from Arabidopsis thaliana. Mol Cell Proteomics. 2003;2:325–345. doi: 10.1074/mcp.M300030-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Marmagne A, Rouet MA, Ferro M, Rolland N, Alcon C, Joyard J, Garin J, Barbier-Brygoo H, Ephritikhine G. Identification of new intrinsic proteins in Arabidopsis plasma membrane proteome. Mol Cell Proteomics. 2004;3:675–691. doi: 10.1074/mcp.M400001-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Fukada K, Zhang F, Vien A, Cashman NR, Zhu H. Mitochondrial proteomic analysis of a cell line model of familial amyotrophic lateral sclerosis. Mol Cell Proteomics. 2004;3:1211–1223. doi: 10.1074/mcp.M400094-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Zhang W, Kho Y. Proteomic analysis of integral plasma membrane proteins. Anal Chem. 2004;76:1817–1823. doi: 10.1021/ac0354037. [DOI] [PubMed] [Google Scholar]
- 23.Wu CC, MacCoss MJ, Howell KE, Yates JR., III A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21:532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- 24.Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carr S, Aebersold R, Baldwin M, Burlingame A, Clauser K, Nesvizhskii A. The need for guidelines in publication of peptide and protein identification data. Working Group On Publication Guidelines For Peptide And Protein Identification Data. Mol Cell Proteomics. 2004;3:531–533. doi: 10.1074/mcp.T400006-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Subramaniam S, Henderson R. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 2000;406:653–657. doi: 10.1038/35020614. [DOI] [PubMed] [Google Scholar]
- 27.Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17:646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.