

Abstract
Peripherin/rds is an integral membrane protein required for the elaboration of rod and cone photoreceptor outer segments in the vertebrate retina; it causes a surprising variety of progressive retinal degenerations in humans and dysmorphic photoreceptors in murine models if defective or absent. (Peripherin/rds is also known as photoreceptor peripherin, peripherin/rds, rds/peripherin, rds, and peripherin-2.) Peripherin/rds appears to act as a structural element in outer segment architecture. However, neither its function at the molecular level nor its role in retinal disease processes are well understood. This report initiates a systematic investigation of protein domain structure and function by examining the molecular and cellular consequences of a series of 14 insertional mutations distributed throughout the polypeptide sequence. Protein expression, disulfide bonding, sedimentation velocity, and subcellular localization of the COS-1 cell-expressed mutant variants were examined to test the hypothesis that protein folding and tetrameric subunit assembly are mediated primarily by EC2, a conserved extracellular/intradiskal domain. Protein folding and tetrameric subunit assembly were not affected by insertion of either an uncharged dipeptide (GA) or a highly charged hendecapeptide (GDYKDDDDKAA) into any one of nine sites residing outside of EC2 as assayed by nonreducing Western blot analysis, sedimentation velocity, and subcellular localization. In contrast, insertions at five positions within the EC2 domain did cause either gross protein misfolding (two sites) or a reduction in protein sedimentation coefficient (two sites) or both (one site). These results indicate that although the vast majority of extramembranous polypeptide sequence makes no measurable contribution to protein folding and tetramerization, discrete regions within the EC2 domain do contain determinants for normal subunit assembly. These findings raise the possibility that multiple classes of structural perturbation are produced by inherited defects in peripherin/rds and contribute to the observed heterogeneity of retinal disease phenotypes.
The outer segments (OSs)1 of rod and cone photoreceptor cells act as detectors of visible light for the initial stages of the visual process and are vital for normal vertebrate vision. The OS is an architecturally complex organelle. It contains hundreds of precisely stacked membranous disks that undergo a polarized renewal process; a complete turnover of OS membrane protein occurs approximately once every 10 days in primate rod cells (1). Although the renewal process and OS stability are essential for photoreceptor viability, their underlying molecular bases remain largely undefined. Evidence from several laboratories has implicated the integral membrane protein peripherin/rds in OS morphogenesis and the renewal process (2-4). It is present in all vertebrate rod and cone photoreceptors examined to date and causes a variety of progressive retinal diseases in humans when defective (5).
Despite continued interest and study, neither the normal molecular action of peripherin/rds in OS renewal nor its role in the pathophysiology of retinal disease is well understood. Several suggestions for molecular function have been proposed based largely on the molecular genetic identification of peripherin/rds as the primary trigger for the murine retinal degeneration slow (rds) phenotype and the immunochemical localization of this protein to rod and cone OS disc rim regions (6, 7). Proposals for function include stabilization of the highly curved disc rims, maintenance of flattened disc structure through adhesive interactions, morphogenesis of disc membranes/rim regions, catalysis of disc shedding, and/or scaffolding of disc stacks (2, 8-11). The recent finding that OS morphogenesis as well as photoreceptor function can be rescued in postnatal (10-day-old) rds mice reinforces the notion that this protein is a building block for the normally continuous OS renewal process (12). In addition, the reported flattening of canine pancreatic microsomes by peripherin/rds in vitro is consistent with a direct role in OS structure (13). Current studies are directed at resolving the molecular details of peripherin/rds action.
The cloning and identification of nearly a dozen highly conserved peripherin/rds orthologs (from humans to fish) has suggested that the proteins share several distinctive features, including four hydrophobic transmembrane segments, a large extracellular/intradiskal loop domain (EC2), seven conserved cysteine residues, and a covalently attached carbohydrate moiety (3, 14-22). Hydrodynamic studies performed under reducing conditions indicate that WT peripherin/rds polypeptides can self-assemble to form homotetramers and also can co-assemble with rom-1 (a homologous polypeptide) to form heterotetramers (10, 23). The noncovalently associated tetramers can join via disulfide bonds to generate larger polymers of indeterminate size; polymerization appears to be mediated by a single conserved cysteine residue in peripherin/rds and inhibited by the presence of rom-1 (24, 25). Although tetramerization and polymerization are believed integral for function, their place in the mechanism of peripherin/rds action has yet to be established. Because OS formation is observed in the complete absence of rom-1 (26), peripherin/rds-containing tetramers appear to be the essential units of function for OS morphogenesis.
Several instances of inherited retinal degeneration have been associated with the disruption of peripherin/rds folding and subunit assembly, yet other cases do not appear to impact these processes at all (24, 27). Such observations of discrete (versus global) disruptions caused by missense mutations has led us to question whether partial loss-of-function defects may account for some of the phenotypic heterogeneity characteristic of peripherin/rds-associated retinal diseases. To improve our understanding of protein domain structure and function, we have initiated an insertional mutagenesis approach similar to one taken previously for rhodopsin (28). This report provides evidence in support of the hypothesis that the EC2 domain is particularly important for proper folding and subunit assembly of peripherin/rds.
EXPERIMENTAL PROCEDURES
Design and Construction of Phase I and Phase II Mutants—A series of 14 Phase I insertion mutants, each containing a unique Nar I endonuclease restriction site (5′-GGCGCC-3′), was constructed using sub-cloned regions of the WT peripherin/rds sequence as templates, synthetic oligonucleotide primers (Table I), and methods described previously (29). The presence of designed, and absence of spurious, mutations was confirmed by complete (single-stranded) DNA sequencing using a BigDye terminator cycle sequencing cycle kit (Applied Biosystems, Inc.). Mutagenized regions were returned to a WT peripherin/rds expression vector background by subcloning, and final constructs were confirmed by restriction mapping. Each Phase I mutation introduces a dipeptide (GA) into the WT protein sequence at the indicated position (see Fig. 1).
Table I.
Synthetic oligonuclueotides used for Phase I mutagenesisSynthetic oligonucleotide primers (shown) were employed to create 14 Phase I insertion mutants using a WT peripherin/rds template; each contains a unique Nar I endonuclease restriction site (underlined). Polymerase chain reaction methods and template subclones have been described previously (24).
| Mutant | Oligonucleotide (Nar I sites are underlined) | Subclone |
|---|---|---|
| IM 1 | 5′-AATTCCACCATGGGCGCCGCGCTGCTCAAA-3′ | pHindBg |
| IM 2 | 5′-CGGGTCAAGTTGGGCGCCGCCCAAGGGCTC-3′ | pHindBg |
| IM 3 | 5′-GATGTGATGAACGGCGCCAATTCTGAGAGC-3′ | pHindBg |
| IM 4 | 5′-GCCCTGGACCCTGGCGCCGCCAAGTACGCC-3′ | pBgScKS |
| IM 5 | 5′-CTCAAGAACGGCGGCGCCATGAAATTCTAT-3′ | pBgScKS |
| IM 6 | 5′-CCAGGCCGGTGTGGCGCCTTCATGAAGAAG-3′ | pBgScKS |
| IM 7 | 5′-GGCAACAACGGCGGCGCCTTTCGGGACTGG-3′ | pBgScKS |
| IM 8 | 5′-CTGGATTTTTCCGGCGCCTCCAAAGAAGTC-3′ | pBgScKS |
| IM 9 | 5′-AATGTGGACGGGGGCGCCCGGTACCTGGTG-3′ | pBgScKS |
| IM 10 | 5′-CAGTACCAGCTCGGCGCCACCAACAACTCT-3′ | pScXho |
| IM 11 | 5′-TGGCTGCGTGGCGGCGCCTGCAGGGCCGCC-3′ | pScXho |
| IM 12 | 5′-GCGCTGGAAGGCGGCGCCATGGCCAACCCC-3′ | pScXho |
| IM 13 | 5′-GAGAGTGAGGGCGGCGCCTGGCTTCTGGAG-3′ | pScXho |
| IM 14 | 5′-CTGGGCAAGGGCGGCGCCAACCAGGTGGAA-3′ | pScXho |
Fig.1.
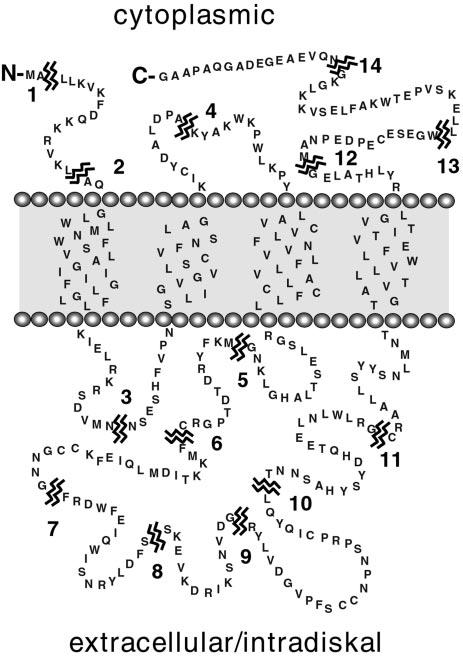
Sites of insertional mutation in peripherin/rds. The primary sequence (346 amino acids) and current folding topology model of bovine peripherin/rds are illustrated. Fourteen individual Phase I mutants (numbered) were constructed by the insertion of Nar I restriction sites at the breakpoints indicated; Phase I mutations add a Gly-Ala dipeptide to the WT sequence. Fourteen additional Phase II mutants were constructed by directional ligation of an epitope cassette coding for a nonapeptide FLAG epitope (DYKDDDDKA) into each Nar I site.
A series of 14 Phase II mutations was constructed by ligation of a FLAG epitope cassette (30) flanked by Nar I sticky ends (sense, 5′-CGACTACAAGGACGACGACGACAAGGC-3′; antisense, 5′-CGGCCTTGTCGTCGTCGTCCTTGTAGT-3′) into the Phase I mutants described. The presence, copy number, and orientation of the FLAG cassette were determined by DNA sequencing with a BigDye terminator cycle sequencing kit (Applied Biosystems, Inc.). Each of the Phase II mutants contains a nonapeptide insertion encoding a FLAG epitope (DYKDDDDKA) at the indicated position within the Phase I mutant sequences (see Fig. 1).
Generation and Characterization of Immunochemical Reagents—Anti-peripherin/rds monoclonal antibody C6 was a kind gift of Dr. John Saari and Dr. Krzysztof Palczewski of the Department of Ophthalmology at the University of Washington. Anti-rom-1 polyclonal antibody MUTT was generated by immunization of New Zealand White rabbits with an affinity-purified glutathione S-transferase fusion protein. The coding sequence for the C-terminal hydrophilic domain of bovine rom-1 was amplified from a λGT-11 bovine retinal library and then cloned in-frame into the pGEX-2T expression vector. Bacterial fusion protein expression and affinity purification were performed essentially as described (31). Each antibody was characterized with respect to its specificity in Western blot analyses using authentic proteins from rod outer segment membranes, and recombinant proteins were expressed in COS-1 cells; no cross-reactivity was observed. The anti-peripherin/rds monoclonal antibody (MAb) C6 epitope was mapped using a series of synthetic peptides kindly provided by Dr. Kathleen Boesze-Battaglia, and it was found to reside within 15 amino acids of the full-length protein C terminus. It was not expected to be affected by any of the mutations examined in this report. Peptide competition was also used to confirm MAb C6 specificity for other immunochemical procedures, including immunofluorescence localization and immunoprecipitation studies not reported here.
Assay of Expression and Disulfide Bonding in COS-1 Cells—COS-1 cells (∼1 × 106/100-mm dish) were transfected with the FuGENE 6 reagent (Roche Diagnostics Co.) and 8 μg of the indicated expression plasmid essentially as suggested by the manufacturer. Detergent extracts were prepared using 1% Triton X-100 in phosphate-buffered saline at 48 h post-transfection essentially as described (30). Methods for Western blot analysis of recombinant peripherin/rds expression and disulfide bonding have been reported previously (23).
Subcellular Localization in COS-1 by Immunocytochemistry—COS-1 cells (∼3 × 104/4-cm2 slide chamber) were transfected with FuGENE 6 (Roche Diagnostics Co.) and 1 μg of the indicated expression plasmid essentially as suggested by the manufacturer. Cells were fixed briefly with 4% paraformaldehyde, permeabilized with Triton X-100, and processed using MAb C6 and a secondary anti-mouse IgG covalently labeled with the Cy3 fluorophore (Amersham Pharmacia Biotech) essentially as described (23). Images were collected with a Nikon Optiphot-2 microscope equipped with an epi-fluorescent illuminator and a SPOT RT digital imaging system (Diagnostic Instruments Inc., v3.0 software).
Assay of Subunit Assembly by Velocity Sedimentation—S20, w estimates were made in a Beckman Optima TLX centrifuge using a TLA-55 rotor or in a Sorvall RC-M120EX centrifuge using an RP55-S rotor as described (29) but with the following modifications. Sucrose gradient fractions (2.1-ml) were collected by piercing tube bottoms in an offset fashion; particulate fractions were obtained by resealing punctured centrifuge tubes with laboratory film, adding 90 μl of 1× Laemmli sample buffer and vortexing vigorously. Digital analysis of chemiluminescent Western blots was performed using Scion Image software (Scion Corp.). Total peripherin/rds reactivity was calculated by pixel summation over soluble and particulate fractions.
RESULTS
Design and Construction of Phase I and Phase II Mutants— Regions of peripherin/rds consensus sequence predicted to form hydrophilic domains were evaluated for their tendency to form polypeptide secondary structure, and 14 sites within regions lacking strongly predicted structural motifs were identified (Fig. 1). A series of 14 Phase I mutants, each containing a unique Nar I endonuclease restriction site, were constructed using synthetic oligonucleotides and the polymerase chain reaction. These mutations were designed to produce minimal perturbation to protein structure as they add only two relatively small, uncharged amino acids (GA) to the WT sequence. An additional series of 14 Phase II mutants, expected to be more highly disruptive, was constructed by ligation of a single copy FLAG epitope cassette (coding for a nonapeptide, DYKD-DDDKA) into the Nar I site of each Phase I mutant.
Heterologous Expression of Insertion Mutants in COS-1 Cells—Each of the 28 mutant expression plasmids was transfected individually into COS-1 cells in culture. Protein expression was assessed by Western blot analysis using anti-peripherin/rds MAb C6. This reagent is useful as a measure of full-length protein expression because it recognizes a 15-amino acid epitope at the C terminus of peripherin/rds. Although none of the insertional mutations interrupt or lie immediately adjacent to the MAb C6 epitope, it was not known a priori whether the antibody would react with all mutants.
In fact, expression of each of the 14 Phase I mutants is detected by MAb C6 in COS-1 cells. Similar to WT peripherin/rds, the mutants typically migrate as closely spaced doublets under denaturing and reducing conditions (Fig. 2A). Moreover, peripherin/rds reactivity is also observed in extracts from COS-1 cells transfected with each of the 14 Phase II mutants (Fig. 2B). The Phase II mutants migrate somewhat more slowly than the WT protein and show a greater variation in electrophoretic mobility. The added mass and charge of the FLAG epitope insertion, heterogeneous post-translational modification, and/or residual secondary structure may be responsible for the observed differences. Identical results were obtained when Phase II mutant expression was assayed by probing Western blots with the anti-FLAG MAb M2 (not shown). Although we expected that the relatively large insertions introduced into the Phase II mutants might cause degradation and absent or reduced protein expression, reproducible differences in expression levels between the Phase I and Phase II mutants were not observed. These results demonstrate that neither minor (uncharged dipeptide) nor more major (charged hendecapeptide) insertions at any of 14 sites distributed throughout the primary sequence prevent peripherin/rds protein expression in COS-1 cells.
Fig.2.
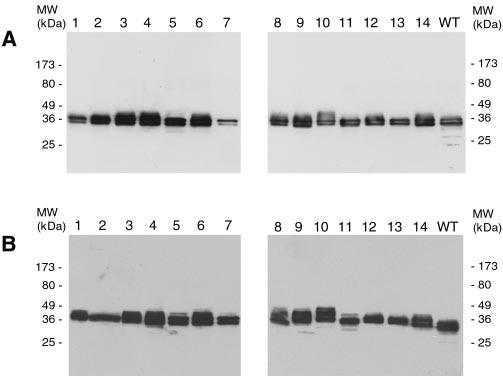
Assay of insertion mutant protein expression by Western blot analysis. Detergent extracts of transfected COS-1 cells were reduced with β-mercaptoethanol, run on a 10% SDS polyacrylamide gel and then electroblotted onto Immobilon-P membrane and probed with anti-peripherin/rds MAb C6. All 14 Phase I (A) and 14 Phase II (B) insertion mutants express peripherin/rds. Phase II mutants display reduced mobility relative to WT due to the addition of a FLAG epitope; slight variations in apparent molecular mass (MW) may be a function of heterogeneous post-translational modification and/or residual secondary structure.
WT peripherin/rds is extracted from vertebrate retina and COS-1 cells both as monomeric and disulfide-bonded dimeric forms, as assayed by nonreducing Western blot analysis. Abnormal disulfide bonding has been associated with several instances of human retinal disease. We therefore examined the mobility of each insertional mutant by Western analysis in the absence of added reducing agent. Eleven of the 14 Phase I mutants appear roughly similar to WT; they migrate primarily as both monomeric and dimeric forms (Fig. 3A). In contrast, three Phase I mutants (IM5, IM7, and IM11) show a banding pattern dissimilar from WT (Fig. 3A, lanes 5, 7, and 11). These mutants exhibit a strong tendency to form large aggregates that remain trapped in the stacking gel. Essentially identical results were obtained for the Phase II series of mutations (Fig. 3B). The combined data indicate that neither Phase I nor Phase II mutations at any of 11 insertion sites prevents normal disulfide bonding but that a mutation at any one of three sites (IM5, IM7, or IM11) disrupts normal disulfide bonding and destabilizes protein structure.
Fig.3.
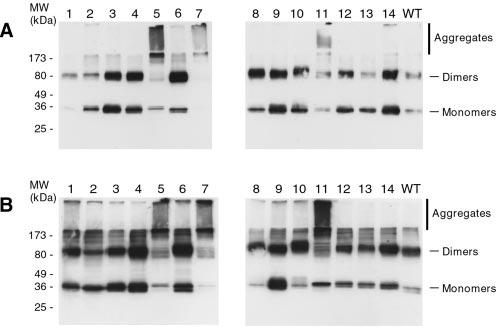
Assay of insertion mutant disulfide bonding by nonreducing Western blot analysis. Detergent extracts of transfected COS-1 cells were treated with N-ethylmaleimide to block free thiols, subjected to nonreducing 10% SDS-PAGE, and then electroblotted onto Immobilon-P membrane and probed with anti-peripherin/rds MAb C6. All 14 Phase I (A) and 14 Phase II (B) insertion mutants are observed as several species. Three mutants show a pronounced tendency relative to WT to form higher order aggregates: IM5, IM7, and IM11. Similar results are observed for the Phase I and Phase II mutations.
Assay of Insertion Mutant Subunit Assembly by Velocity Sedimentation—WT peripherin/rds is assembled as a tetrameric protein in both retinal photoreceptors and transfected COS-1 cells. Tetramerization appears to be essential for function as impaired subunit assembly has been linked to dysmorphic OSs in mice and several forms of retinal degeneration in humans (24, 27). We have previously developed a reliable method for the assay of tetramer formation in COS-1 by characterizing protein sedimentation velocity (29) and apply it here to determine whether any of the insertional mutations affect this process.
Phase I and Phase II mutants were centrifuged through sucrose density gradients, and fractionated gradients were analyzed by Western blotting using MAb C6. This approach generated sedimentation profiles that fall into four main classes; raw data for particular Phase II mutants typical of each class are presented (Fig. 4, B-E). A representative sedimentation profile for WT peripherin/rds is given for comparison (Fig. 4A). Sedimentation coefficients (S20, w values) calculated for each hydrodynamically distinct species observed are presented in Table II.
Fig.4.
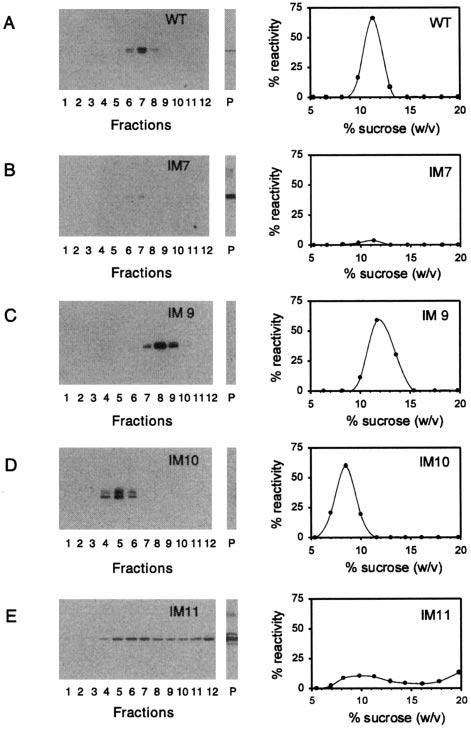
Assay of Phase II mutant subunit assembly by velocity sedimentation. Detergent extracts from transiently transfected COS-1 cell cultures were sedimented under reducing conditions in 5-20% (w/w) sucrose gradients. Fractionated gradients and particulate fractions (P) were assayed for peripherin/rds by Western blot analysis with MAb C6. Chemiluminescent blots and corresponding plots generated by image analysis are shown (A-E); four classes of sedimentation profile are evident. WT peripherin/rds (A) sediments as a single major peak characteristic of a tetrameric stoichiometry (24). Most insertional mutants, including IM9 (C), gave similar results, indicating that their subunit assembly is normal. Two mutants, including IM10 (D), displayed sedimentation profiles more characteristic of dimeric peripherin/rds (37). Insertions at two sites, including IM7 (B), caused gross aggregation as protein was only recovered from the particulate fraction (P). Insertion at one site, IM11 (E), was characterized by variable behavior. Both dimeric and aggregated species are evident; this site appears to destabilize protein structure in a significant yet incomplete fashion. Sedimentation coefficient estimates for all 28 Phase I and Phase II mutants were calculated as described previously (24) and are reported in Table II.
Table II.
Sedimentation coefficients (S20, w) of peripherin/rds mutantsSedimentation coefficients (S20, w) for peripherin/rds variants expressed in COS-1 cells were determined as described under “Experimental Procedures.” The number of independent transfection/sedimentation trials performed to obtain each value is given by n; standard deviations are given where appropriate. Mutants that were recovered from the particulate fraction are indicated (P).
| Phase I | Phase II | |
|---|---|---|
| WT | 5.4 ± 0.1 (3)a,b | 5.4 ± 0.1 (3)a |
| IM1 | 5.5 (2) | 5.7 (2) |
| IM2 | 5.5 (2) | 5.5 ± 0.3 (4) |
| IM3 | 5.5 (2) | 5.8 (2) |
| IM4 | 5.5 (2) | 5.6 (2) |
| IM5 | P (2) | P (3) |
| IM6 | 5.4 (2) | 5.6 (2) |
| IM7 | P (2) | P (2) |
| IM8 | 4.5/5.3c (4) | 3.9 ± 0.2 (3) |
| IM9 | 5.3 ± 0.3 (3) | 5.7 (2) |
| IM10 | 4.2 ± 0.3 (3) | 3.8 ± 0.2 (3) |
| IM11 | 5.6 (2)/Pd | Variable (6)/Pe |
| IM12 | 5.6 (1) | 5.5 (2) |
| IM13 | 5.4 (2) | 5.7 (2) |
| IM14 | 5.6 (2) | 5.6 (2) |
Determined in a previous study (24).
Mean ± S.D. from n samples (number in parentheses).
In half the experiments, reactivity sedimented with a mean of 4.5, in others it sedimented with a mean of 5.3.
In each experiment, approximately half the reactivity was found in the pellet.
The proportion of reactivity found in the pellet varied (10-90%) between experiments.
Neither Phase I nor Phase II insertional mutations affected velocity sedimentation profiles at most (9 of 14) sites, including IM1, IM2, IM3, IM4, IM6, IM9, IM12, IM13, and IM14. All were essentially indistinguishable from WT (Fig. 4, A and B and Table II). These sites are distributed throughout the polypeptide primary sequence (Fig. 1), and their lack of effect demonstrates that subregions within every predicted hydrophilic domain can tolerate both small (uncharged) and large (highly charged) insertional mutations without global disruption of WT structure.
In contrast, either large or small insertions into any one of five sites, all contained within the EC2 domain, resulted in largely non-WT sedimentation behavior. Insertions into two sites, IM5 and IM7, caused increased sedimentation velocities and recovery of the aggregated mutants from the particulate fraction (Fig. 4B and Table II). These effects illustrate sites at which insertional mutations sufficiently reduce protein structural stability to generate global misfolding. More interestingly, mutants at IM8 and IM10 sediment with a velocity substantially less than that of WT (Fig. 4D and Table II). Mutations at these sites result in sedimentation coefficients that are more characteristic of peripherin/rds dimers than tetramers, and they appear to disrupt normal subunit assembly without impairing protein folding. Finally, IM11 mutants revealed more complex and variable behavior than insertions in other sites (Fig. 4E). Phase I (IM11) insertions generated both grossly misfolded and WT species, in an approximately equal ratio, whereas Phase II (IM11) insertions produced variable amounts of aggregated protein and hydrodynamic species of variable (non-WT) mobility (Fig. 4E and Table II). In sum, these results demonstrate that insertional mutations at five of seven sites within EC2 cause measurable changes in protein structure; these insertions produced either global structural disruption (IM5 and IM7), altered subunit assembly (IM8 and IM10), or both (IM11).
Subcellular Localization of Insertion Mutants by Immunofluorescence Microscopy—WT peripherin/rds expressed in COS-1 cells is retained largely within intracellular membranes; a previous study (23) reported a distinctly perinuclear subcellular distribution and suggested that the protein was localized within the Golgi apparatus. Our more recent co-localization studies utilize a Golgi-specific marker and support this interpretation.2 Because quality control mechanisms are known to prevent misfolded membrane proteins from exiting the endoplasmic reticulum and entering the Golgi apparatus (32), we speculated that mutations that alter peripherin/rds folding and subunit assembly might also affect its subcellular localization.
We used immunofluorescence microscopy to examine whether insertions in EC2 that were disruptive for protein folding or assembly also affected subcellular localization. It should be noted that fluorescence localization patterns vary widely over populations of transfected cells, and the images presented (Fig. 5) are representative of the most commonly observed distribution for each variant. Because results were similar for Phase I and Phase II mutations, raw data are presented for selected Phase II mutants only.
Fig.5.
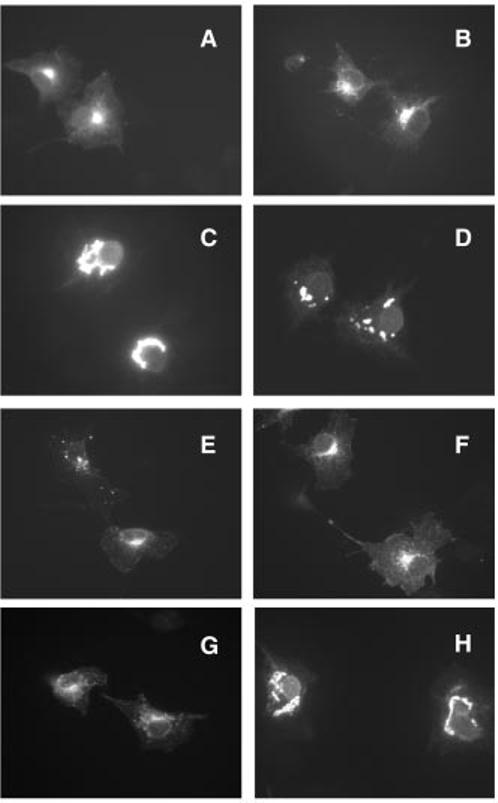
Subcellular localization of Phase II insertion mutants by indirect immunofluorescence microscopy. COS-1 cells transiently transfected with the indicated mutant expression vectors were fixed with paraformaldehyde, permeabilized with Triton X-100, and labeled with anti-peripherin/rds MAb C6. WT peripherin/rds (A) has previously been localized to internal membranes, a perinuclear localization pattern typical of distribution within Golgi membranes is apparent. Insertional mutations that do not result in grossly misfolded protein, including IM2 (B), IM8 (E), IM9 (F), and IM10 (G), display similar perinuclear distributions. Insertions that do result in grossly misfolded protein, including IM5 (C), IM7 (D), and IM11 (H), produce larger and more numerous fluorescent blobs that are distributed less focally and are characteristic of localization within the endoplasmic reticulum.
The subcellular distribution of WT peripherin/rds is shown for comparison; the protein displays a stereotypical Golgi localization pattern, forming a small “cap” over the nuclei of expressing COS-1 cells (Fig. 5). Not surprisingly, insertional mutants that do not affect protein folding or subunit assembly (IM2 and IM9) generate fluorescence distributions similar to WT. In addition, mutants that appear to preserve normal folding but alter subunit assembly (IM8 and IM10) also show WT distributions. These results indicate that tetramerization is not required for export from the endoplasmic reticulum. In contrast, a pattern strikingly dissimilar from WT is observed for those mutants that inhibit normal folding and cause protein aggregation (IM5, IM7, and IM11). Mutations at these sites produce large blobs of peripherin/rds that tend to encircle the nuclei. The extent to which blobs were present was variable; in the most extreme cases, the cytoplasm of the expressing cells was virtually blob-filled. This type of labeling was rarely if ever observed for other insertion mutants or WT peripherin/rds. These results demonstrate that subcellular localization patterns of peripherin/rds are altered by mutations that disrupt protein folding but not subunit assembly.
DISCUSSION
The EC2 region of peripherin/rds has been the subject of considerable previous attention; it was there that the rds defect was discovered and also there that the first human disease-related defects were described (5). More recent studies of recombinant peripherin/rds in COS-1 support the notion that this region is highly significant (24, 25, 27, 29, 33, 34). The current investigation uses two series of insertional mutations distributed throughout the peripherin/rds polypeptide sequence to assess the relative importance of the EC2 region for protein structure. We find that EC2 is critical for proper protein folding and that specific determinants within this region guide tetrameric subunit assembly.
Insertional mutations at 9 of 14 sites distributed throughout the peripherin/rds polypeptide sequence (IM1, IM2, IM3, IM4, IM6, IM9, IM12, IM13, and IM14) had no discernible effect on protein folding or subunit assembly in COS-1 cells (Fig. 6, white ovals). These mutations did not prevent protein expression, impair disulfide-mediated dimerization or tetrameric subunit assembly, or prevent Golgi processing, and they include sites both within and outside of the EC2 domain. These results suggest that regions other than EC2 make relatively minor contributions to folding and subunit assembly. The observed lack of effects may also reflect the limited resolution of our experimental design. Spacing between sites of insertional mutation in hydrophilic domains ranged from 10 to 25 amino acids, and the intervening sequences may contain additional areas of consequence. Since our goal was to identify regions that make specific local contributions, insertion sites were designed where possible to lie outside the boundaries of strongly predicted secondary structures. For example, we avoided disrupting a predicted amphipathic helix in the C terminus that displays fusogenic activity in vitro (35). Our data do not exclude the possibility that this as well as other region(s) may contribute to the folding and subunit assembly of peripherin/rds.
Fig.6.
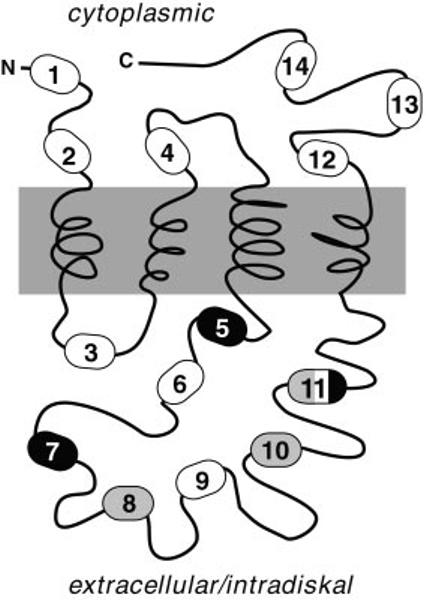
Molecular consequences of insertional mutations on peripherin/rds structure. Combined findings from investigations of disulfide bonding, subunit assembly, and subcellular localization are integrated to summarize current conclusions. The majority of peripherin/rds polypeptide sequence can tolerate either small uncharged (dipeptide) or larger, negatively charged (hendecapeptide) insertions without measurable effects on protein folding or tetrameric subunit assembly (white ovals). Mutations that do affect protein structure all reside within EC2, the second extracellular/intradiskal domain. Insertions caused either grossly misfolded protein (black ovals) or dimeric (versus tetrameric) subunit assembly (gray ovals). One site, IM11, produced each effect at variable ratios.
Insertions at IM5 and IM7 consistently caused abnormal disulfide bonding, retention within the endoplasmic reticulum, and irreversible protein aggregation. We conclude that these proteins are grossly misfolded (Fig. 6, black ovals). A previous study (24) pointed out the importance of EC2 for recombinant protein structure; rapidly sedimenting species and protein trapped in stacking gels suggested that substitution of any one of six conserved cysteines (by serine) caused protein instability and aggregation. We expect that grossly misfolded peripherin/rds would generally be retained and eventually degraded within the endoplasmic reticulum and produce retinal disease by haploinsufficiency. Alternatively, if misfolded protein preserved some ability to form covalent and/or noncovalent interactions with WT peripherin/rds, a dominant negative effect would be predicted. Although not directly examined here, we propose that a graded capacity of misfolded and WT protein to interact represents a potential mechanism for producing phenotypic heterogeneity in peripherin/rds-associated retinal disease.
Mutations at two sites, IM8 and IM10 (Fig. 6, gray ovals), inhibit normal tetrameric subunit assembly and produce proteins with significantly decreased sedimentation coefficients. Since these molecules are characterized by normal solubility and disulfide bonding, we conclude that they are essentially properly folded but contain disruptions in regions required for tetrameric assembly. They, similar to an L185P mutation associated with defective subunit assembly and digenic retinitis pigmentosa (27, 36), display sedimentation coefficients most characteristic of a dimeric stoichiometry (37). We were somewhat surprised to find these mutants released to the Golgi because quality control mechanisms typically retain incompletely assembled oligomeric membrane proteins within the endoplasmic reticulum (32). These observations raise the possibility that WT peripherin/rds is also exported from the endoplasmic reticulum in a dimeric form and that tetramerization normally occurs in the Golgi. Polytopic membrane protein oligomerization in the Golgi has been documented previously (38). The recent finding that the EC2 region of CD81, a tetraspanin superfamily member related to peripherin/rds, crystallizes in a dimeric form (39) indicates that self-assembly is a general property of the tetraspanins, and this finding strengthens the likelihood that peripherin/rds subunit assembly represents a dimerization of dimers (37). At least two fates can be envisioned for dimerized peripherin/rds. Previous studies (27, 36) have shown that L185P dimers can be recruited (rescued) into normally sedimenting tetramers by rom-1 or WT peripherin/rds; in this example, little or no disease phenotype is displayed. Alternatively, some pathogenic mutations may prevent a rescue of the dimerized protein; it might be recycled into the endoplasmic reticulum for degradation or perhaps be exported to the OS. The possibility that incompletely assembled protein (dimers) could participate in OS renewal in a deficient fashion (i.e. a partial function mutation) offers a second putative mechanism for the generation of variability in peripherin/rds-related disease.
Mutations at one site within EC2, IM11, produced both protein misfolding-retention within the endoplasmic reticulum and altered subunit assembly in variable ratios (Fig. 6, black/white/gray oval). The proximity of this site to an essential cysteine and the interexperimental variability displayed suggest that a frequent but incomplete failure to form proper disulfide bonds causes substantial protein misfolding; the variable population of protein molecules that do fold appear to be heterogeneous in size. We speculate that comparable fractional misfolding in photoreceptors might constitute a third mechanism that contributes to phenotypic heterogeneity in individuals with inherited defects in peripherin/rds.
Our current observations (that disruption of normal disulfide formation and folding are caused by the mutation of EC2 but not other regions) are consistent with previous reports and support the notion that EC2 plays a predominant role in structuring this molecule. These results are generally reminiscent of findings for rhodopsin. Insertions into cytoplasmic domains were generally nondisruptive, but extracellular/intradiskal regions, including a cysteine pair involved in a disulfide bond, were found to be crucial for structure and function (28, 40, 41). EC2 has previously been proposed to be structurally significant (34). The finding that peripherin/rds and the homologous polypeptide, rom-1, assembled both with themselves and with each other led to the hypothesis that subunit interactions may be mediated by conserved structural motifs within their EC2 regions (23). The structural (versus functional) importance of this region was also demonstrated by the effects of a chimeric protein in transgenic mice; that study (34) found that the peripherin/rds EC2 region (in a rom-1 context) was necessary, yet not sufficient, for rescue of the rds phenotype. We interpret those results to reflect both the presence of structural determinants within EC2 regions of peripherin/rds and rom-1 and the existence of a region that is required for protein function, which is distinct from EC2 and is found only in peripherin/rds.
Every region of peripherin/rds has now been associated with human retinal disease; more than 40 pathogenic mutations have been reported to date (5). Thus, it is not unexpected that most of the 14 insertional mutation sites described here lie proximal (or adjacent) to one or another disease-associated defect. Consistent rules for predicting disease phenotype based on whether a defect causes gross protein misfolding or merely impairs subunit assembly are not apparent from inspection of the current data. Interestingly, pathogenic defects also lie near to several sites (i.e. IM1, IM4, IM6, IM9, IM12, and IM13) that do not affect structure as measured here. Although this may merely reflect insensitivity of our methods, it may also indicate sites of primarily functional (versus structural) significance.
The current report demonstrates that defects in peripherin/rds can cause discrete and local, rather than global, disruptions of protein structure. Moreover, our findings reveal that a variety of molecular consequences are available to non-WT peripherin/rds. These results, along with an emerging picture of specialized functional domains in peripherin/rds, lead us to speculate that inherited defects can preferentially affect one or another aspect of the overall photoreceptor OS renewal process to generate heterogeneous retinal pathophysiologies. This report initiates a systematic process for defining domain structure-function relationships to test whether positive correlations can be made between particular classes of disruption and characteristic disease phenotypes.
Acknowledgments
Acknowledgments—The MAb C6 hybridoma line was generously provided by Dr. Jack Saari and Dr. Krzysztof Palczewski; synthetic peptides were a kind gift of Dr. Kathleen Boesze-Battaglia. We thank Dr. Visvanathan Ramamurthy, Dr. Joseph Corless, Dr. Shu-Chu Chen, Dr. Jing Huang, Daniel Possin, Timothy Bruggeman, and the spirit of Linda Munar for technical assistance and discussion.
Footnotes
This work was supported by generous grants (Grants NIH R01 EY13246 to A. F. X. G. and NIH R01 EY06641 to J. B. H.) from the NEI National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- OS
- outer segment
- EC2
- extracellular/intradiskal loop 2
- MAb
- monoclonal antibody
- WT
- wild-type
- rds
- retinal degeneration slow
- IM
- insertional mutant
N. Khattree and A. Goldberg, unpublished observations.
REFERENCES
- Young RW. Invest. Ophthalmol. Vis. Sci. 1976;15:700–725. [PubMed] [Google Scholar]
- Molday RS, Hicks D, Molday L. Invest. Ophthalmol. Vis. Sci. 1987;28:50–61. [PubMed] [Google Scholar]
- Travis GH, Brennan MB, Danielson PE, Kozak CA, Sutcliffe JG. Nature. 1989;338:70–73. doi: 10.1038/338070a0. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Jansen HG. Neurosci. Lett. 1981;21:23–26. doi: 10.1016/0304-3940(81)90051-3. [DOI] [PubMed] [Google Scholar]
- Kohl S, Giddings I, Besch D, Apfelstedt-Sylla E, Zrenner E, Wissinger B. Acta Anat. (Basel) 1998;162:75–84. doi: 10.1159/000046471. [DOI] [PubMed] [Google Scholar]
- Arikawa K, Molday LL, Molday RS, Williams DS. J. Cell Biol. 1992;116:659–667. doi: 10.1083/jcb.116.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis GH, Groshan KR, Lloyd M, Bok D. Neuron. 1992;9:113–119. doi: 10.1016/0896-6273(92)90226-4. [DOI] [PubMed] [Google Scholar]
- Travis GH, Sutcliffe JG, Bok D. Neuron. 1991;6:61–70. doi: 10.1016/0896-6273(91)90122-g. [DOI] [PubMed] [Google Scholar]
- Molday RS. Invest. Ophthalmol. Vis. Sci. 1998;39:2491–2513. [PubMed] [Google Scholar]
- Goldberg AF, Molday RS. Biochemistry. 1996;35:6144–6149. doi: 10.1021/bi960259n. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Lamba OP, Napoli AA, Jr., Sinha S, Guo Y. Biochemistry. 1998;37:9477–9487. doi: 10.1021/bi980173p. [DOI] [PubMed] [Google Scholar]
- Ali RR, Sarra GM, Stephens C, Alwis MD, Bainbridge JW, Munro PM, Fauser S, Reichel MB, Kinnon C, Hunt DM, Bhattacharya SS, Thrasher AJ. Nat. Genet. 2000;25:306–310. doi: 10.1038/77068. [DOI] [PubMed] [Google Scholar]
- Wrigley JD, Ahmed T, Nevett CL, Findlay JB. J. Biol. Chem. 2000;275:13191–13194. doi: 10.1074/jbc.c900853199. [DOI] [PubMed] [Google Scholar]
- Travis GH, Christerson L, Danielson PE, Klisak I, Sparkes RS, Hahn LB, Dryja TP, Sutcliffe JG. Genomics. 1991;10:733–739. doi: 10.1016/0888-7543(91)90457-p. [DOI] [PubMed] [Google Scholar]
- Moghrabi WN, Kedzierski W, Travis GH. Exp. Eye Res. 1995;61:641–643. doi: 10.1016/s0014-4835(05)80059-4. [DOI] [PubMed] [Google Scholar]
- Kedzierski W, Moghrabi WN, Allen AC, Jablonski-Stiemke MM, Azarian SM, Bok D, Travis GH. J. Cell Sci. 1996;109:2551–2560. doi: 10.1242/jcs.109.10.2551. [DOI] [PubMed] [Google Scholar]
- Weng J, Belecky-Adams T, Adler R, Travis GH. Invest. Ophthalmol. Vis. Sci. 1998;39:440–443. [PubMed] [Google Scholar]
- Connell GJ, Molday RS. Biochemistry. 1990;29:4691–4698. doi: 10.1021/bi00471a025. [DOI] [PubMed] [Google Scholar]
- Bascom RA, Manara S, Collins L, Molday RS, Kalnins VI, McInnes RR. Neuron. 1992;8:1171–1184. doi: 10.1016/0896-6273(92)90137-3. [DOI] [PubMed] [Google Scholar]
- Gorin MB, Snyder S, To A, Narfstrom K, Curtis R. Mamm. Genome. 1993;4:544–548. doi: 10.1007/BF00364792. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Molday RS. Invest. Ophthalmol. Vis. Sci. 1996;37:352–362. [PubMed] [Google Scholar]
- Gould DJ, Petersen-Jones SM, Lin CT, Sargan DR. Anim. Genet. 1997;28:391–396. doi: 10.1111/j.1365-2052.1997.00185.x. [DOI] [PubMed] [Google Scholar]
- Goldberg AF, Moritz OL, Molday RS. Biochemistry. 1995;34:14213–14219. doi: 10.1021/bi00043a028. [DOI] [PubMed] [Google Scholar]
- Goldberg AF, Loewen CJ, Molday RS. Biochemistry. 1998;37:680–685. doi: 10.1021/bi972036i. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Molday RS. J. Biol. Chem. 2000;275:5370–5378. doi: 10.1074/jbc.275.8.5370. [DOI] [PubMed] [Google Scholar]
- Clarke G, Goldberg AF, Vidgen D, Collins L, Ploder L, Schwarz L, Molday LL, Rossant J, Szel A, Molday RS, Birch DG, McInnes RR. Nat. Genet. 2000;25:67–73. doi: 10.1038/75621. [DOI] [PubMed] [Google Scholar]
- Goldberg AF, Molday RS. Proc. Natl. Acad. Sci. U. S. A. 1996;93:13726–13730. doi: 10.1073/pnas.93.24.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borjigin J, Nathans J. J. Biol. Chem. 1994;269:14715–14722. [PubMed] [Google Scholar]
- Goldberg AF, Molday RS. Methods Enzymol. 2000;316:671–687. doi: 10.1016/s0076-6879(00)16756-4. [DOI] [PubMed] [Google Scholar]
- Chubet RG, Brizzard BL. BioTechniques. 1996;20:136–141. doi: 10.2144/96201pf01. [DOI] [PubMed] [Google Scholar]
- Smith DB, Johnson KS. Gene (Amst.) 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Molinari M, Helenius A. Science. 1999;286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- Kedzierski W, Lloyd M, Birch DG, Bok D, Travis GH. Invest. Ophthalmol. Vis. Sci. 1997;38:498–509. [PubMed] [Google Scholar]
- Kedzierski W, Weng J, Travis GH. J. Biol. Chem. 1999;274:29181–29187. doi: 10.1074/jbc.274.41.29181. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Stefano FP, Fenner M, Napoli AA., Jr. Biochim. Biophys. Acta. 2000;1463:343–354. doi: 10.1016/s0005-2736(99)00226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara K, Berson EL, Dryja TP. Science. 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Moritz OL, Molday RS. J. Biol. Chem. 2001;276:22388–22396. doi: 10.1074/jbc.M011710200. [DOI] [PubMed] [Google Scholar]
- Musil LS, Goodenough DA. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- Kitadokoro K, Bordo D, Galli G, Petracca R, Falugi F, Abrignani S, Grandi G, Bolognesi M. EMBO J. 2001;20:12–18. doi: 10.1093/emboj/20.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnik SS, Khorana HG. J. Biol. Chem. 1990;265:17520–17524. [PubMed] [Google Scholar]
- Doi T, Molday RS, Khorana HG. Proc. Natl. Acad. Sci. U. S. A. 1990;87:4991–4995. doi: 10.1073/pnas.87.13.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]