

Abstract
Inherited defects in the RDS gene cause a multiplicity of progressive retinal diseases in humans. The gene product, peripherin/rds (P/rds), is a member of the tetraspanin protein family required for normal vertebrate photoreceptor outer segment (OS) architecture. Although its molecular function remains uncertain, P/rds has been suggested to catalyze membrane fusion events required for the OS renewal process. This study investigates the importance of two charged residues within a predicted C-terminal helical region for protein biosynthesis, localization, and interaction with model membranes. Targeted mutagenesis was utilized to neutralize charges at Glu321 and Lys324 individually and in combination to generate three mutant variants. Studies were conducted on variants expressed as 1) full-length P/rds in COS-1 cells, 2) glutathione S-transferase fusion proteins in Escherichia coli, and 3) membrane-associated green fluorescent protein fusion proteins in transgenic Xenopus laevis. None of the mutations affected biosynthesis of full-length P/rds in COS-1 cells as assessed by Western blotting, sedimentation velocity, and immunofluorescence microscopy. Although all mutations reside within a recently identified localization signal, none altered the ability of this region to direct OS targeting in transgenic X. laevis retinas. In contrast, individual or simultaneous neutralization of the charged amino acids Glu321 and Lys324 abolished the ability of the C-terminal domain to promote model membrane fusion as assayed by lipid mixing. These results demonstrate that, although overlapping, C-terminal determinants responsible for OS targeting and fusogenicity are separable and that fusogenic activity has been uncoupled from other protein properties. The observation that subunit assembly and OS targeting can both proceed normally in the absence of fusogenic activity suggests that properly assembled and targeted yet functionally altered proteins could potentially generate pathogenic effects within the vertebrate photoreceptor.
Rod and cone vertebrate photoreceptor outer segments (OSs)1 contain hundreds of neatly stacked photopigment-containing membranous disks that are essential for normal vision. A polarized renewal process replaces these membranes such that a complete turnover of OS disks in rod cells is effected approximately once every 10 days (1). Renewal is thought necessary to counter the effects of light damage and oxidation of OS lipids, and evidence from several laboratories has shown that a coordination of new disk assembly (morphogenesis) with aged disk removal (shedding) is needed to maintain normal OS structure (2, 3). Defects in disk morphogenesis or shedding that compromise the renewal process lead to disorganization of the OS layer and eventual photoreceptor cell death. Although OS renewal has been linked to environmental factors including light, temperature, and ionic milieu, the process is not currently well understood in molecular terms, particularly with regard to regulation of the membrane fusion processes required (2).
The rds (retinal degeneration slow) gene product, peripherin/rds (P/rds; also known as photoreceptor peripherin, peripherin/rds, rds/peripherin, rds, and peripherin-2), appears to be a central player in OS morphogenesis and the renewal process. First described in 1978, the rds mouse possesses a large insertion of viral DNA in chromosome 17 that results in a null allele (4-6). Heterozygous and homozygous animals display irregular and absent OSs, respectively, with otherwise normal retinal morphology at young ages (7, 8). Photoreceptor cell bodies subsequently die by apoptosis, and deterioration of the photoreceptor cell layer in homozygotes is essentially complete by 12 months of age (9, 10).
Although clearly required for OS formation, the P/rds mechanism of action at the molecular level is less obvious. Studies from several laboratories concur that it is an integral membrane protein localized to the rim regions of photoreceptor disks in normal retina (11, 12). Present in all vertebrate rod and cone photoreceptors examined to date, this protein causes a variety of progressive retinal diseases in humans when defective (13). Proposals for function include disk rim formation, disk flattening, disk stack scaffolding, disk-plasma membrane interaction, and/or catalysis of disk renewal (14-20). Compelling evidence to favor one clearly defined mode of action at the molecular level is still lacking. In some species, P/rds action can be modulated by the presence of the homologous rom-1 polypeptide; however, the molecular function of rom-1 is also unclear (21). A recent report describing the presence of a localization signal in the C terminus of P/rds (but not rom-1) suggests a rationale for why rom-1 knockout mice can elaborate photoreceptor OSs, but P/rds null mice cannot (20).
We have previously used targeted mutagenesis to investigate P/rds structure-function relationships and to show that folding and tetrameric subunit assembly are guided by discrete determinants within the extracellular/intradiskal loop 2 (EC2) domain (22). The vast majority of the predicted extramembranous polypeptide sequence, including a 66-amino acid C-terminal domain, makes no measurable contribution to protein biosynthesis. In contrast, several biophysical studies suggest that the P/rds C terminus functions to catalyze membrane-membrane fusion events (17, 23-25). Those studies indicate that the C terminus contains an amphipathic helix that functions as a fusion peptide, and a recent investigation found that truncating the protein within the helix greatly diminishes fusogenic activity (25).
Fusion peptides are short amphiphilic helical regions essential for the activity of known membrane fusion proteins (26, 27). Their amphiphilic character is thought to promote membrane destabilization via insertion into and disruption of bilayer structures; however, mechanisms of fusion peptide action are still being widely debated and studied (27). Likewise, amphiphilicity appears to be important for the proposed P/rds fusion peptide; the membrane destabilization activity of a P/rds synthetic peptide model was strongly inhibited when its amphiphilicity was reduced by decreasing hydrophobicity (24). It is noteworthy that rom-1, a peripherin/rds homolog, lacks strong amphiphilicity in this region and is unable to promote membrane fusion in vitro (28). In particular, rom-1 possesses non-polar substitutions at two positions relative to the conserved P/rds fusion peptide sequence that result in a loss of two charged residues (Glu321 and Lys324) and a decrease in predicted hydrophobic moment. The significance of charged residues in the P/rds fusion peptide has not been examined to date, although ionizable amino acids are reported to play an important role in several other fusion peptides (27, 29).
An improved understanding of P/rds C-terminal structure-function relationships is required to relate fusogenic activity measured in vitro to its proposed molecular function in the photoreceptor OS. This is an inherently difficult undertaking, as there are no facile approaches to assay the membrane fusion events involved in the vertebrate photoreceptor renewal process. This task is complicated by the recent identification of a C-terminal OS localization signal that overlaps the previously identified fusion peptide (20). This study examines the significance of two conserved charged residues, Glu321 and Lys324, to further our understanding of how the C-terminal domain contributes to P/rds function. We find that these residues are essential for P/rds fusogenic activity, but not biosynthesis, subunit assembly, or OS localization. The uncoupling of fusogenic activity from other properties suggests that properly assembled and targeted yet functionally altered proteins could generate pathogenic effects in the vertebrate photoreceptor.
EXPERIMENTAL PROCEDURES
Engineering C-terminal Helical Region (CHR) Mutants for Expression in COS-1 Cells, Escherichia coli, and Xenopus laevis—Three sets of constructs were used in this study (see Fig. 1C). The first set encodes full-length (346 predicted amino acids) bovine P/rds for expression in COS-1 cells and was used for the assay of protein biosynthesis and subunit assembly. The second set of constructs, encoding only the 66-amino acid P/rds C-terminal domain fused to glutathione S-transferase (GST), was used to express and purify proteins from E. coli and was used for assay of membrane interactions. Lack of cell-surface expression in COS-1 cells precluded the routine use of full-length mutants for membrane interaction studies. The third set of constructs, encoding the 66-amino acid P/rds C-terminal domain fused to green fluorescent protein (GFP), was utilized to drive protein expression in transgenic X. laevis for the assay of subcellular localization. Each set of constructs includes wild-type (WT) P/rds and three variants, two single missense mutations (E321L and K324A) and the combined double missense mutation (E321L,K324A).
Fig.1.
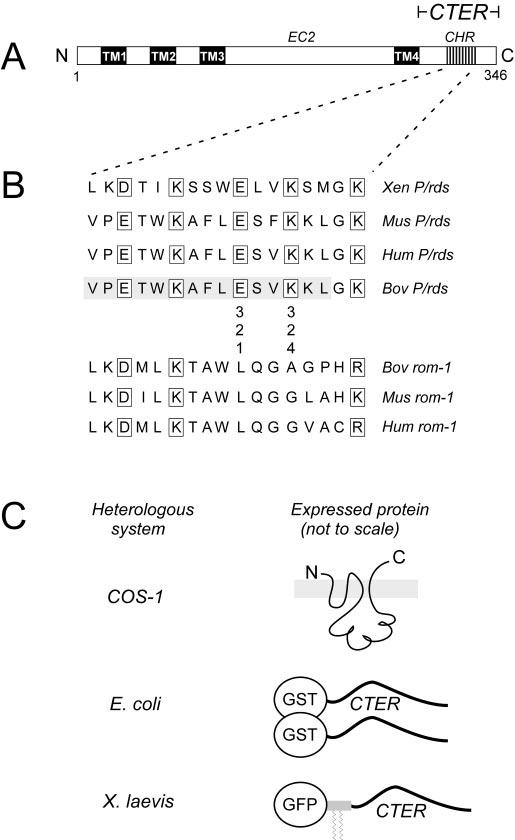
A, domain organization of P/rds. Four transmembrane domains (TM1-4) and the presence of several cysteine-containing motifs (not shown) in EC2 indicate a relationship within the tetraspanin family of proteins. CTER, C-terminal domain. B, alignment of the P/rds and rom-1 sequences for the predicted CHR at positions 312-328 within the 66-amino acid C-terminal domain. Conserved charged residues with three- to four-residue spacing are evident for P/rds. This study investigated the importance of the conserved charges at positions 321 and 324 for protein structure, localization, and fusogenic activity. Substitutions were guided by the aligned rom-1 sequences. The 15-amino acid bovine (Bov) P/rds region previously implicated as a fusion peptide is shaded. A recently reported localization signal (20) includes an additional five amino acids upstream and eight amino acids downstream of the X. laevis (Xen) sequence depicted. Mus, Mus musculus; Hum, human. C, mutant P/rds variants expressed as 1) full-length polytopic proteins in COS-1 cells for assay of protein biosynthesis, 2) C-terminal domain fusions with GST in E. coli, and 3) C-terminal domain fusions with GFP modified with a rhodopsin palmitoylation signal. This latter molecule acts as membrane-bound localization reporter in X. laevis (33).
The bacterial expression plasmids were based upon pGST-CTER, an inducible expression vector encoding GST followed by the 66-amino acid hydrophilic C terminus of WT bovine P/rds (25). Nucleotide base changes were designed to introduce point mutations into the previously identified fusion peptide region Val312-Leu326 (17, 24). In each instance, a charged residue conserved across P/rds orthologs was replaced by an uncharged residue from the homologous region of the aligned bovine rom-1 sequence (see Fig. 1B). Single and double mutants were constructed serially using synthetic oligonucleotide primers (Table I) and the QuikChange site-directed mutagenesis kit (Stratagene). Mutagenized regions were verified by DNA sequencing using a BigDye terminator cycle sequencing kit (Applied Biosystems, Inc.). Mutagenized regions (as BamHI fragments) were returned to non-mutagenized expression vector backgrounds by subcloning to generate bacterial expression plasmids pGST-E321L, pGST-K324A, and pGST-E321L,K324A. Correct assembly of the plasmids was confirmed by restriction mapping.
TABLE I.
Synthetic oligonucleotides used for targeted mutagenesis
| Variant | Oligonucleotides |
|---|---|
| pGST-E321L | 5′ -CCTTTCTGCTGAGTGTGAAGAAGCTGGGCA-3′ (forward) |
| 5′ -TGCCCAGCTTCTTCACACTCAGCAGAAAGG-3′ (reverse) | |
| pGST-K324A | 5′ -GGCCTTTCTGGAGAGTGTGGCGAAGCTGG-3′ (forward) |
| 5′ -CCAGCTTCGCCACACTCTCCAGAAAGGCC-3′ (reverse) | |
| pGST-E321L,K324A | 5′ -CTGAGTGTGGCGAAGCTGGGCAAGGGCAAC-3′ (forward) |
| 5′ -GTTGCCCTTGCCCAGCTTCGCCACACTCAG-3′ (reverse) |
The mammalian expression plasmids were based upon the bacterial expression series described above and a cDNA clone encoding the full-length WT P/rds sequence (30, 31). A 157-bp StuI/BamHI fragment containing the mutagenized CHR was excised from each variant and subcloned into the StuI/BamHI-digested subcloning vector pScXho (32). The mutagenized CHRs were then excised as SacII/XhoI fragments and ligated into SacII/XhoI-digested pcPERS, the non-mutagenized expression vector background containing the full-length protein coding region of bovine P/rds (22). Completed constructs were confirmed by restriction mapping.
The constructs designed for expression of mutant variants in transgenic X. laevis were based on the plasmid pEGFP-C1 (Clontech). The cytomegalovirus promoter was replaced with a 0.8-kb fragment of the proximal X. laevis opsin promoter (pXOP-0.8). Following the GFP cDNA, a sequence encoding the C terminus of X. laevis rhodopsin lacking the terminal five amino acids (i.e. amino acids 311-348) was inserted to create pXOP-0.8-EGFP-rhoCT. This plasmid was designed to accept the BamHI fragments encoding the 66 amino acids of bovine P/rds (described above) in-frame behind the GFP-rhodopsin sequence. The presence of a palmitoylation signal within the rhodopsin sequence creates a membrane-bound GFP reporter molecule (20). The desired constructs were selected and verified by restriction mapping. The previously described constructs GFP-rhoCT44 and GFP-rhoCT44del5 were also used as controls (33).
Assay of Expression and Disulfide Bonding in COS-1 Cells—P/rds-containing membranes were prepared at 4 °C from transiently transfected COS-1 cells as follows. Adherent cells grown on 100-mm plastic culture dishes were washed three times with Tris-buffered saline 48 h post-transfection. Cells were scraped from plates with a rubber policeman and collected into 1 ml of Tris-buffered saline containing 2.5 mm N-ethylmaleimide, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, and 5 μg/ml Pefabloc-SC (Roche Applied Science). The cells were pelleted by centrifugation at 2000 × g for 10 min at 4 °C. The cell pellets were resuspended in 1.2 ml of 10 mm Tris-HCl (pH 7.5) containing 2.5 mm N-ethylmaleimide, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, and 5 μg/ml Pefabloc-SC. The resuspended cells were passed forcefully through 26-gauge syringe needles five times, and the resulting lysates were centrifuged at 1000 × g for 10 min at room temperature. Supernatants were transferred to ultracentrifuge tubes and centrifuged at 110,000 × g for 20 min at 4 °C. The membrane pellets were resuspended by vortexing and trituration in 115-μl aliquots of Tris-buffered saline containing 2.5 mm N-ethylmaleimide, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, and 5 μg/ml Pefabloc-SC. COS-1 cell membranes prepared in this fashion were stored at -70 °C until used.
Western blot analysis of WT and mutant P/rds expression and disulfide bonding in transfected COS-1 membranes was performed using anti-P/rds monoclonal antibody (mAb) C6 and methods described previously (22). Thawed COS-1 cell membranes (3 μl/lane) were solubilized with denaturing sample buffer containing 5% β-mercaptoethanol, and samples were heated in boiling water for 5 min prior to loading onto 10% SDS-polyacrylamide gels. Analysis of disulfide bonding was performed in an analogous manner in the absence of β-mercaptoethanol.
Immunofluorescent Localization in COS-1 Cells—WT and mutant P/rds were localized in transiently transfected COS-1 cells as described previously (22). In brief, paraformaldehyde-fixed cells were permeabilized with Triton X-100 and then processed using anti-P/rds mAb C6 and a secondary anti-mouse IgG antibody covalently labeled with the Cy3 fluorophore (Amersham Biosciences). A similar procedure used a 1:200 dilution of a mouse anti-KDEL receptor monoclonal antibody (Stressgen Biotech Corp.) in conjunction with a secondary anti-mouse IgG antibody covalently labeled with the Cy2 fluorophore (Amersham Biosciences) to label cis-Golgi and intermediate Golgi membranes. Likewise, a 1:200 dilution of a rabbit anti-calnexin polyclonal antibody (Stressgen Biotech Corp.) was used in conjunction with a secondary anti-rabbit IgG antibody covalently labeled with the Cy2 fluorophore to label the endoplasmic reticulum. A Nikon Optiphot-2 microscope equipped with an epifluorescent illuminator and a SPOT RT digital imaging system (Version 3.0 software, Diagnostic Instruments Inc.) were utilized for image acquisition. Image processing was limited to adjustment of brightness and contrast.
Assay of Subunit Assembly by Velocity Sedimentation—A previously established procedure was used to measure the sedimentation velocities of detergent-solubilized WT and mutant P/rds; a Beckman Optima TLX centrifuge and TLS-55 rotor were utilized as described previously (34). In brief, Triton X-100 extracts of transiently transfected COS-1 cells were layered onto 2-ml 5-20% sucrose gradients and centrifuged overnight at 4 °C. Soluble fractions were collected by off-center puncture of tube bottoms; particulate fractions were collected by solubilization of pellets with Laemmli sample buffer. Scion Image software was utilized for image analysis of chemiluminescent Western blots; pixel summation over soluble and particulate fractions was used to calculate total P/rds reactivity (22). Peak immunoreactivities were taken for the calculation of s20,w values.
Mutant Expression and Subcellular Localization in Transgenic X. laevis Retina—Transgenic tadpoles were generated using a modified protocol (36) based on that of Kroll and Amaya (35). WT X. laevis adults were obtained from Xenopus I (Dexter, MI). X. laevis sperm nuclei were incubated with 0.3× high speed egg extract, 0.05 units of NotI restriction enzyme, and 100-200 ng of NotI-linearized vector DNA in a total volume of 18 μl. After 10 min, the reaction mixture was diluted to 0.3 nuclei/nl, and 10 nl was injected per egg. The resulting embryos were reared in 0.1× Marc’s modified Ringer’s solution containing 6% Ficoll and 50 μg/ml gentamycin for 48 h and then transferred to 0.1× Gerhart’s modified Ringer’s solution (37). Animals were treated with 18 μg/ml G418 for 96 h as previously described to select for transgenic animals and to reduce the extent of gene silencing (38). At 5-6 days post-fertilization, roughly corresponding to stages 40-42, tadpoles were screened for GFP expression using a Zeiss Axioskop equipped with epifluorescence optics and a GFP filter set (39). Tadpoles expressing GFP were identified by green fluorescence emitted from their eyes. All animals were raised at 18 °C on a 12/12 light cycle (8 a.m. to 8 p.m.).
Stage 48-62 transgenic tadpoles were killed between 4 and 7 p.m. (i.e. near the end of the light cycle) and fixed in 4% paraformaldehyde and 0.1 m sodium phosphate (pH 7.4). Fixed eyes were embedded in OCT tissue embedding medium (Tissue-Tek) and cryosectioned. Frozen sections (14 μm) were stained with Texas Red-conjugated wheat germ agglutinin (Molecular Probes, Inc., Eugene, OR) and Hoechst 33342 (Sigma) to label Golgi/post-Golgi membranes and nuclei, respectively, as previously described (20). Labeled sections were mounted in 50% glycerol. A Zeiss LSM 510 laser scanning confocal microscope equipped with a C-Apochromat (40× W Korr, 1.2-numerical aperture objective) and Zeiss LSM software were used to acquire images of labeled sections. At least three transgenic animals were examined by confocal microscopy for each construct.
Expression and Purification of GST-C-terminal Domain Fusion Proteins—A standardized expression and purification procedure based on methods developed for pGST-CTER was used. Thirty ml of a plasmid-transformed, freshly saturated E. coli XL1-Blue culture was used to inoculate 900 ml of Luria broth containing 100 μg/ml ampicillin. The culture was grown with agitation at 37 °C for 1.5 h, and fusion protein expression was induced for an additional 3 h (with agitation at 37 °C) by the introduction of 0.1 mm isopropyl-1-thio-β-d-galactopyranoside. Cells were collected by centrifugation and stored at -70 °C until used.
The frozen cell pellet was thawed on ice and resuspended by vortexing in 35 ml of ice-cold phosphate-buffered saline (pH 7.5) containing 1 mm dithiothreitol, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, and 1 mm phenylmethylsulfonyl fluoride. The cells were lysed using a French pressure cell, and Triton X-100 was added to a final concentration of 1%. The lysate was stirred for 20 min at 4 °C and then centrifuged at 14,000 × g for 30 min at 4 °C. The clarified supernatant was applied to a 6-ml column of packed glutathione-Sepharose 4B (Amersham Biosciences), and the column was washed with 10 bed volumes of phosphate-buffered saline. The GST-C-terminal domain fusion protein was eluted by three sequential single bed volume equilibrations of 5 min each using 50 mm Tris-HCl (pH 8.8) containing 10 mm reduced glutathione and 1 mm dithiothreitol. Combined eluates were concentrated with a stirred ultrafiltration cell; dialyzed at 4 °C against 50 mm Tris-HCl (pH 7.0), 150 mm NaCl, 1 mm EDTA, and 1 mm dithiothreitol; and stored in aliquots at -80 °C. Protein determinations were carried out with a BCA assay kit (Pierce) using bovine serum albumin to construct the standard curve.
Assay of Vesicle Fusion by Lipid Mixing—An established procedure was adapted to measure lipid mixing using fluorescence resonance energy transfer (FRET) (40). This assay is based on the dilution of a resonance energy transfer pair, N-(7-nitro-2,1,3-benzoxadiazol-4-yl)phosphatidylethanolamine (NBD-PE) and N-(lissamine rhodamine B sulfonyl)phosphatidylethanolamine (Rh-PE) (Molecular Probes, Inc.), due to vesicle hemifusion and subsequent NBD-PE dequenching. Large unilamellar vesicles (LUVs) were prepared from phosphatidylcholine/phosphatidylserine/cholesterol (4:4:1; Avanti Polar Lipids Inc., Birmingham, AL) by extrusion through a 0.1-μm filter essentially as described by Mayer et al. (41). LUVs containing 0.6 mol % Rh-PE and 0.6 mol % NBD-PE were mixed at a 1:1 molar ratio with an unlabeled (but otherwise identical) population of LUVs (10 μm phospholipid final concentration) and a purified mutant variant (8.3 μg/ml final protein concentration). The rhodamine excitation wavelength was set at 465 nm; NBD fluorescence emission was measured at 530 nm; and a 515-nm cutoff filter was used between the sample and the emission monochromator to decrease interference from light scattering. Under these conditions, vesicle hemifusion results in probe dilution and a linear increase in NBD-PE fluorescence (40). FRET was measured after 10 min (its maximal value); percent FRET was calculated as described previously (25). No above base-line changes were observed for additions of purified GST alone (8.3 μg of protein/mg of total lipid).
Assay of Membrane Destabilization by Vesicle Depolarization—Membrane bilayer destabilization was inferred from membrane depolarization studies and measured fluorometrically with the cation-sensitive dye 3,3′-dipropylthiadicarbocyanine iodide (diS-C3-(5); Molecular Probes, Inc.) as a collapse in a valinomycin-induced diffusion potential. Small unilamellar vesicles (SUVs) were prepared by sonication from phosphatidylcholine/phosphatidylserine/cholesterol at a 4:4:1 molar ratio in the presence of 100 mm K+ and then diluted into K+-free isotonic buffer (3.6 μm phospholipid final concentration). The fluorescent probe diS-C3-(5) was added to 1 nm and allowed to equilibrate at 37 °C until a stable base-line fluorescence (λex = 620 nm and λem = 670 nm) was established. The K+ ionophore valinomycin was added to 0.1 mm to create an inside negative electrochemical potential that resulted in diS-C3-(5) quenching. Purified GST fusion proteins (10 μg/ml protein final concentration) were assayed individually for their ability to induce SUV leakiness and to dissipate the diffusion potential as measured by diS-C3-(5) fluorescence recovery. A similar concentration of purified GST had no effect on fluorescence recovery. Percent fluorescence recoveries were calculated as described (42).
RESULTS
Design and Construction of CHR Mutants—We have shown previously that defects in P/rds can cause discrete and local as well as global disruptions of protein structure (22, 32, 43). Most recently, an insertion mutagenesis approach demonstrated that, although the vast majority of extramembranous polypeptide sequence makes no measurable contribution to gross protein folding or tetramerization, discrete regions within the EC2 domain do include determinants required for normal subunit assembly (22). That study aimed to identify regions making specific local contributions; therefore, mutations were inserted outside the boundaries of strongly predicted secondary structures, including an α-helix in the C terminus that promotes model membrane fusion (17).2 In this study, we examined whether targeted point mutations within the CHR affect known structural and/or functional properties of P/rds.
Seventeen highly conserved C-terminal amino acids (Val312-Lys328), including a minimal region (Val312-Leu326) previously shown to adopt a helical structure and to promote model membrane fusion (17, 24), are shown in Fig. 1B. The alternation of charged (boxed) and relatively hydrophobic residues with a three- to four-residue periodicity predicts a highly amphipathic structure, characteristic of a membrane-binding helix. In contrast, rom-1 lacks both this motif and fusogenic activity (28). Since two charged residues (Glu321 and Lys324) are missing from rom-1, we wondered whether these charges are important for P/rds fusogenic activity. We also asked whether these charges are necessary for OS localization of P/rds, given the presence of a subcellular localization signal within this same region (20).
To test these hypotheses, we constructed a set of potentially non-fusogenic P/rds variants by targeted mutagenesis of the CHR (Fig. 1B). Charged amino acid residues at two positions (Glu321 and Lys324) were substituted individually and in combination with the corresponding uncharged residues found in the aligned bovine rom-1 sequence. None of the mutant variants are predicted (using the NNPREDICT algorithm) to possess significantly altered α-helical character (44). Mutants were expressed as full-length (polytopic) proteins in COS-1 cells, as GST-C-terminal domain fusions in E. coli, and as C-terminal domain fusions with membrane-associated GFP in transgenic X. laevis photoreceptors (Fig. 1C).
Heterologous Expression and Disulfide Bonding of CHR Mutants in COS-1 Cells—Each of the full-length variants was individually transfected into COS-1 cells in culture, and protein expression was assessed by Western blot analysis using anti-P/rds mAb C6. This reagent recognizes an epitope at the distal C terminus and is therefore a good diagnostic measure of full-length protein expression (22). The mutagenized CHR lies ∼21 amino acids upstream of this epitope, and mutations do not appear to affect antibody binding. All mutants expressed robustly; the Western blot analysis shown in Fig. 2 (left panel) demonstrates that, under denaturing and reducing conditions, all mutants possessed apparent molecular masses comparable with that of WT P/rds.
Fig.2.
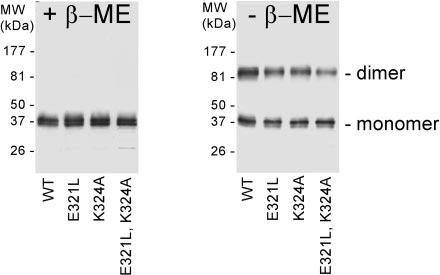
Assay of CHR mutant expression and disulfide bonding in COS-1 cells by Western blot analysis. COS-1 cells were transfected with plasmids encoding full-length mutant P/rds variants: single mutants E321L and K324A and double mutant E321L,K324A. Transfected COS-1 cell membranes were treated with N-ethylmaleimide to block free thiols and then solubilized in denaturing SDS gel loading buffer. Samples were subjected to reducing (with β-mercaptoethanol (+ β-ME)) or nonreducing (- β-ME) SDS-PAGE (10% resolving gels) and then electroblotted onto Immobilon-P membrane and probed with anti-P/rds mAb C6. All CHR mutants expressed robustly; moreover, each showed the ability to form disulfide-linked dimers, akin to WT P/rds.
Mutant protein mobility was also characterized by Western analysis in the absence of added reducing agent. It is well established that WT P/rds adopts a disulfide-bonded dimeric form both in vertebrate retina and in COS-1 cells (15, 30, 31). Several examples of human retinal disease likely involve protein misfolding as a consequence of abnormal disulfide bonding (45-48). All three C-terminal domain mutants displayed essentially WT behavior (Fig. 2, right panel); all migrated as both monomeric and (disulfide-bonded) dimeric forms. In sum, the data presented demonstrate that none of the C-terminal domain mutations measurably affect normal expression or disulfide bonding of P/rds in COS-1 cells.
Immunofluorescent Localization of CHR Mutants in COS-1 Cells—Immunofluorescence microscopy was used to examine whether neutralization of charged residues in the CHR affects subcellular localization of full-length P/rds variants expressed in transiently transfected cultured cells. Properly assembled WT P/rds displays a largely perinuclear distribution in COS-1 cells, suggesting that it remains associated with Golgi membranes; in contrast, grossly misfolded P/rds is retained within the endoplasmic reticulum (22). A recent demonstration that a targeting signal is encoded within the P/rds C terminus (20) prompted us to examine whether mutations in the CHR affect targeting in transfected COS-1 cells.
Indirect immunofluorescence was used to analyze the subcellular distribution of CHR mutants in fixed and permeabilized COS-1 cells. All CHR mutants appeared to be essentially indistinguishable from WT P/rds; we were unable to discern any effects of the mutations on protein distributions. Fluorescence distributions were somewhat heterogeneous between individual cells; images presented are representative of the most common localization patterns. Reference images of endoplasmic reticulum and Golgi immunofluorescence labeling patterns are shown for comparison, as is the distribution of grossly misfolded P/rds (Fig. 3A). The subcellular distribution of WT P/rds is also shown in Fig. 3B; the protein displayed a largely Golgi distribution, appearing as a small cap over nuclei of transfected cells. These results demonstrate that subcellular localization patterns of P/rds are not significantly altered by mutations that neutralize conserved CHR residues.
Fig.3.
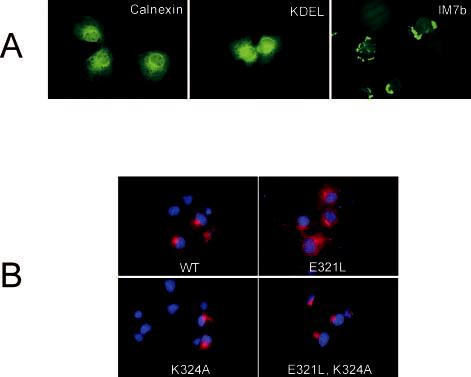
Subcellular localization of CHR mutants by indirect immunofluorescence microscopy. A, characteristic endoplasmic reticulum (anti-calnexin antibody), Golgi (anti-KDEL receptor antibody), and misfolded P/rds (anti-P/rds mAb C6, green) localization patterns are presented for reference. Misfolding of P/rds variant IM7b has been reported previously (22). B, COS-1 cells transiently transfected with WT P/rds or the indicated mutant expression vectors were fixed with paraformaldehyde, permeabilized with Triton X-100, and labeled with anti-P/rds mAb C6 (red) and 4′,6-diamidino-2-phenylindole nuclear stain (blue). All CHR mutants displayed mainly perinuclear distributions characteristic of localization within the Golgi apparatus, akin to WT P/rds.
Subunit Assembly of CHR Mutants Expressed in COS-1 Cells—Point mutations that prevent the normal formation of tetrameric P/rds cause progressive retinal disease in humans and dysmorphic OSs in transgenic mice (34, 43, 49). We recently established a primary role for EC2 in this process and showed that discrete determinants within the EC2 domain guide tetramer formation (22). Although no ill effects of C-terminal domain insertion mutations on subunit assembly were found, none of the insertions disrupted the CHR directly. Therefore, we have examined whether the neutralization of charged residues in the CHR can affect tetrameric assembly of P/rds.
Subunit stoichiometry was assessed using a velocity sedimentation technique developed previously (31). Full-length WT and mutant P/rds were expressed by transient transfection of COS-1 in culture; the cells were extracted with Triton X-100 under nondenaturing conditions; lysates were applied to sucrose gradients; and the gradients were centrifuged overnight. Fractionated gradients were assayed by Western blot analysis to determine protein mobility and to calculate sedimentation coefficients. Comparison of the mobility of a given mutant relative to that of WT P/rds was used to assess tetrameric assembly. Fig. 4 illustrates typical sedimentation profiles obtained for WT and mutant P/rds. In each case, greater than half of the P/rds reactivity remained in the soluble (versus particulate) fraction. Thus, these mutations do not cause extensive protein misfolding. In addition, the mobility of each variant is most consistent with tetrameric stoichiometry; sedimentation coefficients, calculated from immunoreactivity peaks, are presented in Table II. The results demonstrate that none of the CHR mutations significantly affected gross folding or subunit assembly of full-length P/rds in COS-1 cells.
Fig.4.
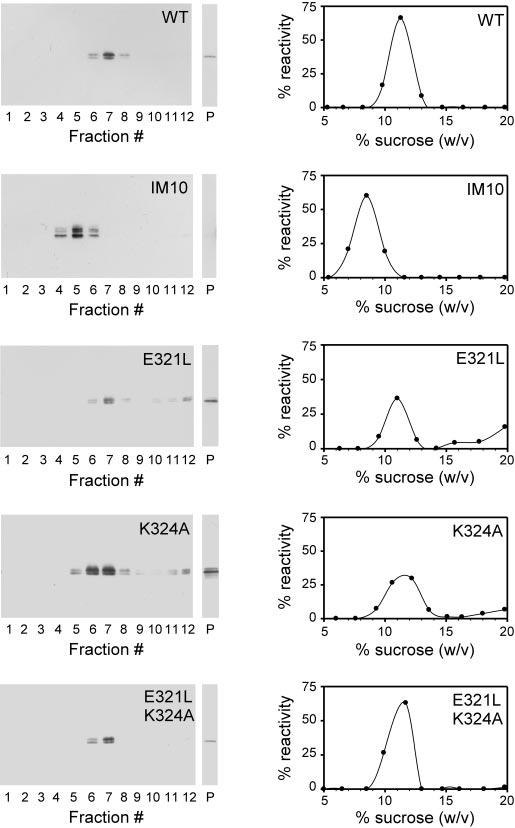
Subunit assembly assay of full-length CHR mutants by velocity sedimentation. Detergent extracts from transiently transfected COS-1 cell cultures were sedimented under reducing conditions in 5-20% (w/w) sucrose gradients. Fractionated gradients (and particulate fractions (P)) were assayed for P/rds by Western blot analysis with mAb C6. Representative chemiluminescent blots and corresponding plots generated by image analysis are shown for WT P/rds, a previously characterized dimeric insertion mutant (IM10), and the three mutant CHR variants. Immunoreactivity in sucrose gradient fractions was calculated as a percentage of the total (soluble and particulate immunoreactivity); in all cases, the majority of the P/rds reactivity remained in the soluble (versus particulate) fraction. As reported previously (22), WT P/rds sediments as a single major peak characteristic of a tetrameric stoichiometry, but IM10 displays a subunit assembly defect. All of the CHR mutants sedimented like the WT protein, indicating that each is tetrameric and that subunit assembly is unaffected by charge neutralizations in the CHR. Sedimentation coefficient estimates for all variants were calculated as described under “Experimental Procedures” and are reported in Table II.
TABLE II.
Consequences of CHR mutations on subunit assembly and sedimentation coefficientsVelocity dsedimentation coefficients (s20,w) for full-length mutant variants are given as means ± S.D. The number of independent transfection/ sedimentation trials for each is given in parentheses. WT P/rds from detergent extracts of transfected COS-1 cells sediments as a characteristic tetramer under reducing conditions in 5–20% (w/w) sucrose gradients (31). None of the mutations assayed measurably affected P/rds subunit assembly.
| P/rds | s20,w | Stoichiometry |
|---|---|---|
| WT | 5.4 ± 0.2 (8) | Tetrameric |
| E321L | 5.3 ± 0.5 (4) | Tetrameric |
| K324A | 5.5 ± 0.2 (3) | Tetrameric |
| E321L,K324A | 5.4 ± 0.1 (3) | Tetrameric |
Targeting and Localization in Transgenic X. laevis Rod Photoreceptors—We utilized a recently developed confocal fluorescence assay to investigate whether the mutations we introduced into the CHR affect the ability of the C terminus of bovine P/rds to direct photoreceptor OS targeting and localization (20). This approach uses an X. laevis opsin promoter to drive expression of a membrane-associated GFP reporter fused to C-terminal domain variants of bovine P/rds in transgenic X. laevis rod photoreceptors (33, 38). When the WT bovine P/rds C terminus is fused to the GFP reporter, nearly all fluorescence retargets to the OS, the normal location of full-length endogenous P/rds. Importantly, the fluorescent reporter does not form complexes with full-length P/rds (20). The combined results indicate the presence of a localization signal in the P/rds C terminus that does not require other protein domains for targeting. Analysis of mutant targeting requires the use of a P/rds fragment since transgenic expression of full-length P/rds mutants would most likely result in co-assembly with the high background of endogenous P/rds and masking of potential targeting defects (22, 31, 38, 50).
Fig. 5 (A-C) shows typical localization patterns observed for the WT bovine sequence and the single mutant E321L and the double mutant E321L,K324A. As described previously (20) and as shown in Fig. 5A, the WT bovine P/rds C terminus directed the vast majority of membrane-associated GFP to the rod photoreceptor OS; only very minor amounts of GFP fluorescence are detectable in other regions (20). This distribution is similar to that generated by the WT X. laevis rhodopsin C terminus (Fig. 5D), the first OS localization signal identified (33). The subcellular distributions of the mutant P/rds variants were essentially indistinguishable from that of WT P/rds; no effect of these mutations on the ability of the C-terminal domain to direct OS localization was observed (Fig. 5, A-C). In contrast and for the sake of comparison, Fig. 5E shows the appearance of membrane-associated GFP lacking an OS localization signal. As reported previously (33), disruption of the X. laevis rhodopsin localization signal (by truncation) results in broader distributions that include both lateral plasma membranes and internal membranes of the inner segment and a clear presence in outer plexiform layer membranes. The K324A single mutant was not assayed directly; but given the lack of effect seen for the double mutant (Fig. 5C), it is likely that K324A would also have no disruptive effect.
Fig.5.

Localization of GFP-P/rds C-terminal domain fusion protein variants expressed in transgenic X. laevis rod photoreceptors. Confocal microscopy was performed on cryosections from stage 48-62 transgenic tadpole eyes expressing membrane-associated GFP fusions with WT P/rds (A), E321L (B), and E321L,K324A (C). GFP fusion proteins (green) were found almost exclusively in the outer segments (OS), with minimal localization observed elsewhere (arrowheads). Thus, neutralization of CHR charged residues at positions 321 and 324 does not disrupt the normal localization of P/rds to the OS. Shown in D is a section from a control eye showing OS localization driven by GFP-rhoCT44, which contains a rhodopsin OS localization signal (33). Shown in E is a section from a control eye showing an example of delocalization displayed by GFP-rhoCT44del5, which lacks an OS localization signal (33). In addition to OS localization, abundant GFP fluorescence was observed in the lateral plasma membranes and internal membranes of inner segments (IS) as well as outer plexiform layer (OPL) membranes (arrowheads). Shown in F is a section from a control eye in which no GFP was expressed. Red indicates Texas Red-conjugated wheat germ agglutinin, and blue indicates Hoechst 33342. Bar = 20 μm.
In Vitro Fusogenicity of CHR Mutants Expressed as GST Fusion Proteins in E. coli—We adapted a FRET approach to investigate whether charged residues in the CHR are important for P/rds fusogenic activity in vitro. Previous studies have shown that the P/rds C terminus, and more specifically, the CHR, can promote membrane vesicle fusion and that controlled trypsinolysis (or genetic truncation) results in loss of fusogenic activity (17, 24). Importantly, the homologous bovine rom-1 polypeptide sequence lacks several charged residues in this region (Fig. 1B) and does not show fusogenic activity in vitro (25). We hypothesized that neutralizing these charged residues with neutral residues present in rom-1 would inhibit P/rds ability to promote model membrane fusion. Fusogenicity was assayed using purified bacterially expressed proteins, as a lack of cell-surface expression of P/rds in COS-1 adversely affected signal-to-noise ratios in assays using COS-1 membranes.3
Each of the variants was expressed as a GST fusion protein and subsequently purified by affinity chromatography. The appearance of each relative to the GST-tagged WT P/rds C terminus (GST-WT) and GST alone is shown in Fig. 6 by Coomassie Blue staining. Each variant (including WT P/rds) ran with an apparent molecular mass of ∼28 kDa, near that of the combined predicted masses of ∼22 and ∼6 kDa of GST and the P/rds C terminus, respectively. Each preparation also contained several species of greater mobility, most likely truncation products. In each case, at least 70% of the total protein was judged to be full-length.
Fig.6.
Expression and purification of GST-P/rds C-terminal domain fusion proteins. The C-terminal domain for each variant was expressed as a GST fusion protein in E. coli by induction with isopropyl-1-thio-β-d-galactopyranoside. Purification was performed using a glutathione-agarose affinity matrix, and eluted proteins (predicted masses of ∼28 kDa) are shown separated by 15% SDS-PAGE and stained with Coomassie Blue. Each was routinely purified to >70% purity; truncated products, including GST, constitute the majority of the remaining impurities.
LUVs containing Rh-PE and NBD-PE were mixed at a 1:1 molar ratio with unlabeled LUVs (100 nm phospholipid final concentration). Fig. 7A shows how NBD-PE fluorescence intensity changed upon addition of the purified GST-C-terminal domain proteins. Consistent with a previous report (25), GST-WT promoted LUV fusion that resulted in lipid mixing, subsequent probe dequenching, and fluorescence increase. This approach was used to compare the fusogenic activity of the purified CHR mutants with that of WT P/rds. We found that fusogenic activity required both conserved charged residues in the CHR. Nearly complete inhibition resulted when single charges were neutralized at Glu321, Lys324, or both simultaneously. The net charge change (relative to the WT sequence) for these three variants is +1, -1, and 0, respectively, yet they impacted activity in an almost equivalent fashion. Thus, a simple relationship between fusogenicity, as measured by lipid mixing, and net charge change is not supported by these data. Instead, we found that any alteration of charge at these two positions largely abrogated the ability to promote model membrane fusion.
Fig.7.
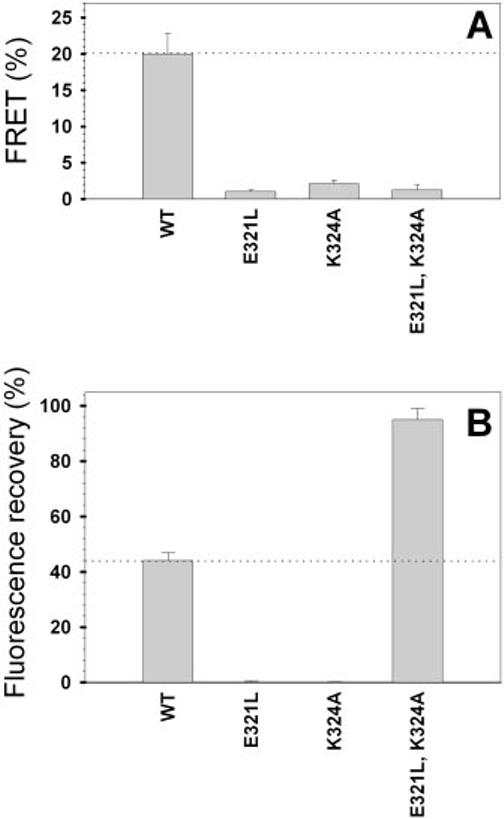
Membrane interactions of P/rds CHR mutants. A, assay of fusogenicity (lipid mixing) as measured by FRET. Affinity-purified GST fusion proteins (8.3 μg/ml final concentration) were added individually to LUVs containing the lipophilic fluorophores Rh-PE and NBD-PE in the presence of additional unlabeled vesicles (10 μm phospholipid final concentration). Neutralization of Glu321, Lys324, or both significantly impaired the ability of the P/rds C terminus to mediate vesicle fusion. Error bars represent S.D. B, membrane destabilization by GST-P/rds C-terminal domain fusion proteins. The potentiometric dye diS-C3-(5) was utilized to measure the ability of the CHR variants to induce membrane leakiness in SUVs (42). Membrane destabilization was inferred from the ability of the variants (10 μg/ml protein final concentration) to collapse the membrane potential and to allow diS-C3-(5) fluorescence recovery (25). Neutralization of either Glu321 or Lys324 abrogated the ability of the P/rds C terminus to depolarize SUVs; in contrast, simultaneous neutralization of Glu321 and Lys324 significantly enhanced SUV depolarization.
Membrane Destabilization of CHR Mutants Expressed as GST Fusion Proteins in E. coli—Although the mechanism(s) by which the CHR promotes model membrane fusion is not understood, current evidence suggests that it involves binding and destabilization of phospholipid membranes (24, 25). Given that each of the CHR mutants we examined inhibited fusogenic activity, a vesicle depolarization assay was adopted to investigate whether any of these mutations acts by preventing membrane destabilization.
Purified GST-C-terminal domain fusion proteins were assayed for their ability to induce SUV leakiness as measured by membrane potential collapse. Fig. 7B shows the fluorescence intensity increase of the potentiometric dye diS-C3-(5) due to SUV membrane destabilization upon addition of GST-WT. We compared the destabilization activity of the purified CHR mutants relative to that of WT P/rds. As shown in Fig. 7B, we found that destabilization, like fusogenic activity, was severely inhibited by neutralization of single charges at both positions 321 and 324. Surprisingly and in contrast to fusogenic activity, destabilization activity was significantly enhanced when these two charges were neutralized simultaneously (E321L,K324A). Overall, these data indicate that, like in vitro fusogenic activity, membrane destabilization is not simply related to charge density and, furthermore, that multiple mechanisms underlie the observed loss of fusogenicity.
DISCUSSION
Early structure-function studies of P/rds focused upon the EC2 domain; this region includes the majority of the hydrophilic protein sequence (∼150 amino acids) and is the site of both the rds mouse defect and the originally described human disease-related mutations (13, 16, 34, 49, 51-57). In contrast, the smaller 66-amino acid C terminus has to date been the subject of more limited attention (17, 20, 24, 25). Recent investigations, including this study, suggest that its importance may previously have been overlooked. We used sequence analysis and engineered missense mutations to investigate the role of conserved charged residues in the amphiphilic CHR and found these sites are essential for fusogenic activity, but not biosynthesis, subunit assembly, or OS localization. The fact that these properties can be uncoupled suggests that inherited mutations in the C terminus might impair OS structure and photoreceptor viability by specifically disabling membrane fusion.
Several lines of evidence had led us to speculate previously that the C-terminal domain plays a greater role in P/rds function than in protein structure (3). First, a transgenic mouse model demonstrates that one or more domains in addition to EC2 are required for generating and maintaining OS architecture (54). Second, P/rds subunit assembly and export from the endoplasmic reticulum are both dependent upon determinants residing within the EC2 domain; insertion mutations in the C terminus do not measurably affect these processes (22). Third, reports of inherited genetic defects in humans and an engineered mutant mouse model both show that retinal physiology can be adversely affected by an abnormal C terminus (13, 59). Finally, several studies have documented the ability of the C terminus to promote model membrane fusion (17, 24, 25, 60).
This study was based in part on the presence of an amphipathic helix in the P/rds C terminus; this feature was initially suggested by sequence analysis and later confirmed by a Fourier transform IR study of a synthetic peptide (17, 24). This region, Val312-Leu326 in the original investigation, has been proposed to act as a fusion peptide, a sequence that, when triggered, can insert into and promote fusion of apposed biological membranes. Fusion peptides required for membrane fusion in viral entry are particularly well studied (26, 61). Numerous mutagenesis studies have demonstrated that alteration of fusion peptide sequence can strongly and specifically affect the fusogenic activity of the larger fusion protein and indeed the entire organism (as in a virus) (62-64). Similarly, a previous study of P/rds fusogenicity suggests that its activity is strongly impacted by changes in the fusion peptide sequence (24). Most recently, a recombinant version of the P/rds C terminus was shown to induce model membrane destabilization and fusion (25). Since the P/rds homolog rom-1 does not display this activity, we wondered whether differences in the CHR might contribute to the observed disparity in fusogenic activity (28). Therefore, we replaced conserved charged residues in P/rds with neutral residues from the aligned region of rom-1 (Fig. 1B). We reasoned that using a P/rds homolog to guide our choice of neutralizing substitutions would provide the greatest likelihood of maintaining normal protein structure. Secondary structure predictions indicated that helical structure would be preserved for each of the CHR mutants.
In fact, none of the CHR mutants produced measurable effects on any aspect of protein biosynthesis in COS-1 cells, including global folding, disulfide bonding, subcellular localization, and tetrameric subunit assembly. These findings are consistent with previous mutagenesis studies showing that gross protein structural properties, including folding, disulfide formation, subunit assembly, and endoplasmic reticulum export, are dependent upon determinants within the EC2 domain, but not the C terminus (22, 32, 43). Likewise, none of the CHR mutations examined here measurably affected OS targeting and localization in transgenic X. laevis photoreceptors. Given that the two charged residues targeted in this study reside within a previously identified OS localization signal, it is note-worthy that substitutions at the conserved Glu321 and Lys324 residues do not disrupt targeting. The high degree of conservation at these positions likely reflects their participation in other molecular activities. In addition, the demonstration that these charged residues are not required for OS localization indicates that their absence in the C terminus of the P/rds homolog rom-1 is not responsible for the lack of targeting activity observed for that molecule (20). Given current knowledge, we consider that multiple roles for the C terminus are likely and have proposed P/rds multifunctionality previously (22). A more detailed investigation will be required to determine whether expression of these mutant variants in transgenic X. laevis photoreceptors affects OS ultrastructure and GFP localization at the ultrastructural level.
In sharp contrast, all of the mutations reported here had dramatic consequences on P/rds interaction with model membranes. Mechanisms of fusion peptide action are likely manifold and are currently being debated; however, it is generally agreed that they function as oligomers and must bind and destabilize membrane surfaces (26). All three mutations described here strongly affected both membrane fusion, measured by lipid mixing, and membrane destabilization, measured potentiometrically. Neutralization of either Glu321 or Lys324 largely prevented model membrane fusion (Fig. 8). Since individual mutations at these positions also severely inhibited destabilization, it is likely that these mutations impede membrane adhesion and/or bilayer penetration. Additional studies are required to evaluate potential contributions from each of these mechanisms. Interestingly, simultaneous neutralization of charged residues in the double mutant also inhibited fusion, but significantly enhanced destabilization. Since fusogenic activity requires peptide oligomerization, but destabilization does not, these results suggest an inhibition of double mutant self-association at the membrane surface (66, 67). Although limited evidence suggests that the P/rds fusion peptide may oligomerize in aqueous solution, its self-association state while bound to membranes is not known (24). The increased destabilization produced by concurrent loss of Glu321 and Lys324 suggests that the double mutant peptide may penetrate more deeply into the bilayer, akin to effects seen for neutralizing mutations within the hemagglutinin fusion peptide (29, 62, 65). It is not apparent why the single charge neutralization mutants do not also increase destabilization, and reconciling these differences will require atomic level structural detail of the peptides in their membrane-bound states. Overall, these findings demonstrate an uncoupling of fusogenic activity from other protein properties and suggest the utility of using non-fusogenic P/rds to genetically complement rds mice to test the hypothesis that fusogenic activity is important for photoreceptor OS renewal.
Fig.8.
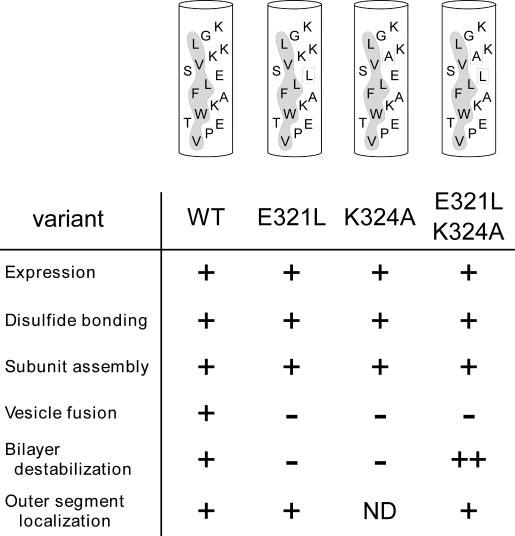
Effects of neutralizing conserved CHR residues. Helical models of WT and mutant P/rds sequences illustrate hydrophobic faces (shaded) and residues targeted in this study (boxed). The consequences of the substitutions are indicated below each variant. Data are summarized as follows: ++, enhanced; +, WT P/rds; -, inhibited; ND, not determined.
More than a dozen mutations in the P/rds C terminus have been associated with instances of human retinal disease. Defects include missense, nonsense, splice, insertion, and deletion mutations, and clinical phenotypes include autosomal dominant retinitis pigmentosa, cone dystrophy, pattern dystrophy, adult vitelliform macular dystrophy, and butterfly-shaped macular dystrophy (68-71). A mouse model for autosomal dominant retinitis pigmentosa that possesses a Δ307 frameshift mutation leads to complete loss of the CHR and compromises photoreceptor viability (59). Since a truncation of the CHR has previously been shown to inhibit fusogenic activity (25), it is possible that fusogenic loss of function could contribute to the Δ307 frameshift phenotype. This mutation generates a dominant-negative phenotype, suggesting that the resulting gene product can inhibit WT protein function. Co-assembly (in heterozygotes) of non-fusogenic P/rds with WT P/rds is likewise predicted to inhibit WT protein function since C-terminal domain mutations do not impair subunit assembly (22). Incorporation of even a single mutant polypeptide into an otherwise WT tetramer could affect activity since fusion proteins are known to function as oligomers (26). This possibility remains likely even if the Δ307 frameshift inactivates the OS localization signal since mutant polypeptide assembly into WT protein-containing tetramers is predicted to effectively mask localization signal defects. Furthermore, quality control mechanisms that prevent P/rds exit from the biosynthetic pathway fail to distinguish mutant polypeptides that are assembled into normal tetramers (58). Thus, a loss of fusogenic activity is predicted to result in a dominant-negative phenotype for individuals inheriting the Δ307 frameshift mutation. In the absence of additional information, we do not exclude the possibility that the Δ307 frameshift mutation may cause impaired targeting to the photoreceptor OS, disruption of OS protein-protein interactions, and/or deleterious new interactions generated by the novel protein sequence resulting from the frameshift.
We have addressed the importance of conserved charged residues located within the P/rds CHR and found they are critical for interactions with model membranes, but not protein biosynthesis or OS localization. The findings lead us to conclude that determinants for OS localization and fusogenic activity are overlapping yet distinct. This conclusion reinforces our notion that a multiplicity of molecular pathologies may contribute to the phenotypic heterogeneity characteristic of P/rds-associated diseases and suggests that a dominant-negative phenotype associated with a Δ307 frameshift mutation may result from an uncoupling of fusogenic activity. Determination of whether OS renewal can be restored in rds mice by transgenic complementation with non-fusogenic yet otherwise normal P/rds will be facilitated by the findings described here and will provide additional insights into P/rds action within the vertebrate photoreceptor.
Acknowledgments
Acknowledgments—We thank Drs. John Saari and Krzysztof Palczewski for the generous gift of the mAb C6 hybridoma line and Dr. Judith White for helpful discussions and critical review.
Footnotes
This work was supported in part by NEI Vision Research Infrastructure Grant EY014803 and NIH Grant EY13246 (to A. F. X. G.) and NEI Grant EY10420 (to K. B.-B.) from the National Institutes of Health; by an E. Matilda Ziegler award (to K. B.-B.); and by the Canadian Institutes of Health Research, the Karl Kirchgessner Foundation, and the Foundation Fighting Blindness of Canada (to O. L. M.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- OS
- outer segment
- P/rds
- peripherin/rds
- EC2
- extracellular/intradiskal loop 2
- CHR
- C-terminal helical region
- GST
- glutathione S-transferase
- GFP
- green fluorescent protein
- WT
- wild-type
- mAb
- monoclonal antibody
- FRET
- fluorescence resonance energy transfer
- NBD-PE
- N-(7-nitro-2,1,3-benzoxadiazol-4-yl)phosphatidylethanolamine
- Rh-PE
- N-(lissamine rhodamine B sulfonyl)phosphatidylethanolamine
- LUVs
- large unilamellar liposomes
- diS-C3-(5)
- 3,3′-dipropylthiadicarbocyanine iodide
- SUVs
- small unilamellar vesicles
Fusion is defined operationally as lipid mixing in this study. No distinction is drawn or intended between hemifusion and bona fide fusion.
K. Boesze-Battaglia, L. M. Ritter, and A. F. X. Goldberg, unpublished data.
REFERENCES
- Young RW. Investig. Ophthalmol. Vis. Sci. 1976;15:700–725. [PubMed] [Google Scholar]
- Nguyen-Legros J, Hicks D. Int. Rev. Cytol. 2000;196:245–313. doi: 10.1016/s0074-7696(00)96006-6. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Goldberg AF. Int. Rev. Cytol. 2002;217:183–225. doi: 10.1016/s0074-7696(02)17015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nie R, Ivanyi D, Demant P. Tissue Antigens. 1978;12:106–108. doi: 10.1111/j.1399-0039.1978.tb01305.x. [DOI] [PubMed] [Google Scholar]
- Demant P, Ivanyi D, Van Nie R. Tissue Antigens. 1979;13:53–55. doi: 10.1111/j.1399-0039.1979.tb01136.x. [DOI] [PubMed] [Google Scholar]
- Travis GH, Brennan MB, Danielson PE, Kozak CA, Sutcliffe JG. Nature. 1989;338:70–73. doi: 10.1038/338070a0. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Jansen HG. Neurosci. Lett. 1981;21:23–26. doi: 10.1016/0304-3940(81)90051-3. [DOI] [PubMed] [Google Scholar]
- Hawkins RK, Jansen HG, Sanyal S. Exp. Eye Res. 1985;41:701–720. doi: 10.1016/0014-4835(85)90179-4. [DOI] [PubMed] [Google Scholar]
- Sanyal S, De Ruiter A, Hawkins RK. J. Comp. Neurol. 1980;194:193–207. doi: 10.1002/cne.901940110. [DOI] [PubMed] [Google Scholar]
- Chang GQ, Hao Y, Wong F. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- Arikawa K, Molday LL, Molday RS, Williams DS. J. Cell Biol. 1992;116:659–667. doi: 10.1083/jcb.116.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierski W, Moghrabi WN, Allen AC, Jablonski-Stiemke MM, Azarian SM, Bok D, Travis GH. J. Cell Sci. 1996;109:2551–2560. doi: 10.1242/jcs.109.10.2551. [DOI] [PubMed] [Google Scholar]
- Kohl S, Giddings I, Besch D, Apfelstedt-Sylla E, Zrenner E, Wissinger B. Acta Anat. 1998;162:75–84. doi: 10.1159/000046471. [DOI] [PubMed] [Google Scholar]
- Travis GH, Sutcliffe JG, Bok D. Neuron. 1991;6:61–70. doi: 10.1016/0896-6273(91)90122-g. [DOI] [PubMed] [Google Scholar]
- Molday RS, Hicks D, Molday L. Investig. Ophthalmol. Vis. Sci. 1987;28:50–61. [PubMed] [Google Scholar]
- Wrigley JD, Ahmed T, Nevett CL, Findlay JB. J. Biol. Chem. 2000;275:13191–13194. doi: 10.1074/jbc.c900853199. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Lamba OP, Napoli AA, Jr., Sinha S, Guo Y. Biochemistry. 1998;37:9477–9487. doi: 10.1021/bi980173p. [DOI] [PubMed] [Google Scholar]
- Goldberg AFX, Molday RS. Biochemistry. 1996;35:6144–6149. doi: 10.1021/bi960259n. [DOI] [PubMed] [Google Scholar]
- Poetsch A, Molday LL, Molday RS. J. Biol. Chem. 2001;276:48009–48016. doi: 10.1074/jbc.M108941200. [DOI] [PubMed] [Google Scholar]
- Tam BM, Moritz OL, Papermaster DS. Mol. Biol. Cell. 2004;15:2027–2037. doi: 10.1091/mbc.E03-09-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke G, Goldberg AFX, Vidgen D, Collins L, Ploder L, Schwarz L, Molday LL, Rossant J, Szel A, Molday RS, Birch DG, McInnes RR. Nat. Genet. 2000;25:67–73. doi: 10.1038/75621. [DOI] [PubMed] [Google Scholar]
- Goldberg AFX, Fales LM, Hurley JB, Khattree N.J. Biol. Chem 200127642700–42706A.F. X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Kong F, Lamba OP, Stefano FP, Williams DS. Biochemistry. 1997;36:6835–6846. doi: 10.1021/bi9627370. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Stefano FP, Fenner M, Napoli AA., Jr. Biochim. Biophys. Acta. 2000;1463:343–354. doi: 10.1016/s0005-2736(99)00226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Goldberg AF, Dispoto J, Katragadda M, Cesarone G, Albert AD. Exp. Eye Res. 2003;77:505–514. doi: 10.1016/s0014-4835(03)00151-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JM. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- Epand RM. Biochim. Biophys. Acta. 2003;1614:116–121. doi: 10.1016/s0005-2736(03)00169-x. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Gretzula C. Investig. Ophthalmol. Vis. Sci. 2003;44:4259. [Google Scholar]
- Korte T, Epand RF, Epand RM, Blumenthal R. Virology. 2001;289:353–361. doi: 10.1006/viro.2001.1108. [DOI] [PubMed] [Google Scholar]
- Connell GJ, Molday RS. Biochemistry. 1990;29:4691–4698. doi: 10.1021/bi00471a025. [DOI] [PubMed] [Google Scholar]
- Goldberg AFX, Moritz OL, Molday RS. Biochemistry. 1995;34:14213–14219. doi: 10.1021/bi00043a028. [DOI] [PubMed] [Google Scholar]
- Goldberg AFX, Loewen CJ, Molday RS. Biochemistry. 1998;37:680–685. doi: 10.1021/bi972036i. [DOI] [PubMed] [Google Scholar]
- Tam BM, Moritz OL, Hurd LB, Papermaster DS. J. Cell Biol. 2000;151:1369–1380. doi: 10.1083/jcb.151.7.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AFX, Molday RS. Methods Enzymol. 2000;316:671–687. doi: 10.1016/s0076-6879(00)16756-4. [DOI] [PubMed] [Google Scholar]
- Kroll KL, Amaya E. Development. 1996;122:3173–3183. doi: 10.1242/dev.122.10.3173. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Tam BM, Knox BE, Papermaster DS. Investig. Ophthalmol. Vis. Sci. 1999;40:3276–3280. [PubMed] [Google Scholar]
- Wu M, Gerhart J. Methods Cell Biol. 1991;36:3–18. doi: 10.1016/s0091-679x(08)60269-1. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Peck A, Tam BM. Gene (Amst.) 2002;298:173–182. doi: 10.1016/s0378-1119(02)00923-x. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal Table of Xenopus Laevis (Daudin): A Systematical and Chronological Survey of the Development from the Fertilized Egg Till the End of Metamorphosis. Garland Publishing Inc.; New York: 1994. [Google Scholar]
- Struck DK, Hoekstra D, Pagano RE. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- Mayer LD, Hope MJ, Cullis PR. Biochim. Biophys. Acta. 1986;858:161–168. doi: 10.1016/0005-2736(86)90302-0. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K. Methods Enzymol. 2000;316:65–86. doi: 10.1016/s0076-6879(00)16717-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AFX, Molday RS. Proc. Natl. Acad. Sci. U. S. A. 1996;93:13726–13730. doi: 10.1073/pnas.93.24.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller DG, Cohen FE, Langridge R. J. Mol. Biol. 1990;214:171–182. doi: 10.1016/0022-2836(90)90154-E. [DOI] [PubMed] [Google Scholar]
- Trujillo MJ, Martinez-Gimeno M, Gimenez A, Lorda I, Bueno J, Garcia-Sandoval B, Ramos C, Carballo M, Ayuso C. Hum. Mutat. 2001;17:80. doi: 10.1002/1098-1004(2001)17:1<80::AID-HUMU27>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Zhang K, Garibaldi DC, Li Y, Green WR, Zack DJ. Arch. Ophthalmol. 2002;120:485–490. doi: 10.1001/archopht.120.4.485. [DOI] [PubMed] [Google Scholar]
- Saga M, Mashima Y, Akeo K, Oguchi Y, Kudoh J, Shimizu N. Hum. Genet. 1993;92:519–521. doi: 10.1007/BF00216463. [DOI] [PubMed] [Google Scholar]
- Souied EH, Rozet JM, Gerber S, Dufier JL, Soubrane G, Coscas G, Munnich A, Kaplan J. Eur. J. Ophthalmol. 1998;8:98–101. doi: 10.1177/112067219800800208. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Moritz OL, Molday RS. J. Biol. Chem. 2001;276:22388–22396. doi: 10.1074/jbc.M011710200. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Molday RS. J. Biol. Chem. 2000;275:5370–5378. doi: 10.1074/jbc.275.8.5370. [DOI] [PubMed] [Google Scholar]
- Kedzierski W, Lloyd M, Birch DG, Bok D, Travis GH. Investig. Ophthalmol. Vis. Sci. 1997;38:498–509. [PubMed] [Google Scholar]
- Dryja TP, Hahn LB, Kajiwara K, Berson EL. Investig. Ophthalmol. Vis. Sci. 1997;38:1972–1982. [PubMed] [Google Scholar]
- Kedzierski W, Bok D, Travis GH. J. Neurochem. 1999;72:430–438. doi: 10.1046/j.1471-4159.1999.0720430.x. [DOI] [PubMed] [Google Scholar]
- Kedzierski W, Weng J, Travis GH. J. Biol. Chem. 1999;274:29181–29187. doi: 10.1074/jbc.274.41.29181. [DOI] [PubMed] [Google Scholar]
- Kedzierski W, Nusinowitz S, Birch D, Clarke G, McInnes RR, Bok D, Travis GH. Proc. Natl. Acad. Sci. U. S. A. 2001;98:7718–7723. doi: 10.1073/pnas.141124198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar GJ, Kenna P, Jordan SA, Kumar-Singh R, Humphries MM, Sharp EM, Sheils DM, Humphries P. Nature. 1991;354:478–480. doi: 10.1038/354478a0. [DOI] [PubMed] [Google Scholar]
- Kajiwara K, Hahn LB, Mukai S, Travis GH, Berson EL, Dryja TP. Nature. 1991;354:480–483. doi: 10.1038/354480a0. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Moritz OL, Tam BM, Papermaster DS, Molday RS. Mol. Biol. Cell. 2003;14:3400–3413. doi: 10.1091/mbc.E03-02-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally N, Kenna PF, Rancourt D, Ahmed T, Stitt A, Colledge WH, Lloyd DG, Palfi A, O’Neill B, Humphries MM, Humphries P, Farrar GJ. Hum. Mol. Genet. 2002;11:1005–1016. doi: 10.1093/hmg/11.9.1005. [DOI] [PubMed] [Google Scholar]
- Boesze-Battaglia K, Stefano FP. Exp. Eye Res. 2002;75:227–231. doi: 10.1006/exer.2002.2004. [DOI] [PubMed] [Google Scholar]
- Tamm LK, Crane J, Kiessling V. Curr. Opin. Struct. Biol. 2003;13:453–466. doi: 10.1016/s0959-440x(03)00107-6. [DOI] [PubMed] [Google Scholar]
- Steinhauer DA, Wharton SA, Skehel JJ, Wiley DC. J. Virol. 1995;69:6643–6651. doi: 10.1128/jvi.69.11.6643-6651.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao H, Armstrong RT, Melikyan GB, Cohen FS, White JM. Mol. Biol. Cell. 1999;10:2759–2769. doi: 10.1091/mbc.10.8.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething MJ, Doms RW, York D, White J. J. Cell Biol. 1986;102:11–23. doi: 10.1083/jcb.102.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Bushweller JH, Cafiso DS, Tamm LK. Nat. Struct. Biol. 2001;8:715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC. Cell. 1998;95:871–874. doi: 10.1016/s0092-8674(00)81710-9. [DOI] [PubMed] [Google Scholar]
- Lau WL, Ege DS, Lear JD, Hammer DA, DeGrado WF. Biophys. J. 2004;86:272–284. doi: 10.1016/S0006-3495(04)74103-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl S, Christ-Adler M, Apfelstedt-Sylla E, Kellner U, Eckstein A, Zrenner E, Wissinger B. J. Med. Genet. 1997;34:620–626. doi: 10.1136/jmg.34.8.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruning G, Millan JM, Meins M, Beneyto M, Caballero M, Apfelstedt-Sylla E, Bosch R, Zrenner E, Prieto F, Gal A. Hum. Mutat. 1994;3:321–323. doi: 10.1002/humu.1380030326. [DOI] [PubMed] [Google Scholar]
- Felbor U, Schilling H, Weber BH. Hum. Mutat. 1997;10:301–309. doi: 10.1002/(SICI)1098-1004(1997)10:4<301::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Grover S, Fishman GA, Stone EM. Ophthalmology. 2002;109:1110–1117. doi: 10.1016/s0161-6420(02)01029-1. [DOI] [PubMed] [Google Scholar]