

Abstract
Leptin gains access to the central nervous system where it influences activity of neuronal networks involved in ingestive behavior, neuroendocrine activity, and metabolism. In particular, the brain melanocortin (MC) system is important in leptin signaling and maintenance of energy balance. Although leptin or MC receptor insensitivity has been proposed to be associated with obesity, the present study compared central leptin and MC receptor stimulation on some of the above-mentioned parameters and investigated whether these treatments predict proneness to diet-induced obesity (DIO) in out-bred Wistar rats. Third-cerebroventricular administration of equi-anorexigenic doses of leptin and of the MC agonist melanotan-II caused comparable increases in plasma ACTH and corticosterone levels and c-Fos-labeling in approximately 70% of paraventricular hypothalamic (PVN) neuronal cell bodies containing CRH. This reinforces involvement of paraventricular CRH neurons in the short-term neuroendocrine and ingestive effects of leptin and melanocortins. In the DIO prediction study, anorexigenic efficacy of melanotan-II was not correlated with any parameter linked to DIO but was highly correlated with MC in situ binding (with labeled [Nle4,d-Phe7]α-MSH) as well as CRH immunoreactivity in the PVN of DIO rats. This suggests intricate relationships among MC signaling, the CRH system, and ingestive behavior unrelated to DIO. In the same animals, leptin’s anorexigenic efficacy was not correlated with PVN MC in situ binding or CRH immunoreactivity but correlated inversely to post-DIO plasma leptin, liver weight, and abdominal adiposity, the latter being correlated to insulin resistance. Thus, differences in leptin but not MC signaling might underlie DIO, visceral obesity, and insulin resistance.
Abbreviations: AgRP, Agouti-related protein; Arc, arcuate hypothalamus; AUC, area under the curve; cFLI, c-Fos-like immunoreactivity; CNS, central nervous system; DIO, diet-induced obesity; HED, high-energy diet; HPA, hypothalamo-pituitary-adrenal; i3cv, third cerebral ventricle; IVGTT, iv glucose tolerance test; Mag, magnocellular; MC, melanocortin; MTII, melanotan-II; Par, parvocellular; POMC, proopiomelanocortin; PVN, paraventricular hypothalamus; VMH, ventromedial hypothalamus
Leptin, the 167-amino-acid product of the OB gene, is synthesized and secreted mainly by adipose tissue roughly in proportion to the size of adipose tissue mass in the body (1, 2). There is compelling evidence that this hormone is involved in the long-term regulation of energy balance (3). Its actions in the central nervous system (CNS) are thought to close a feedback loop from adipose tissue to neuronal circuitry that regulates food intake, neuroendocrine outflow, and metabolism in a coordinated fashion (3). This idea is based on findings that infusion of a relatively low dose of leptin into the third cerebral ventricle (i3cv) of rats causes profound reductions in food intake (4, 5) and body fat mass even independent of food intake effects (6). The fact that rodents and humans with genetic deficiencies in the leptin system become morbidly obese supports the importance of leptin in regulation of energy balance (7, 8).
A key neuronal circuit involved in the signaling of leptin originates from neuronal cell bodies in the arcuate hypothalamus (Arc) that synthesize proopiomelanocortin (POMC). POMC is cleaved into several splice products among which α-MSH may be most relevant for regulation of energy balance. α-MSH acts agonistically on specific brain melanocortin MC3 and MC4 receptors (9). Other neurons in the Arc produce agouti-related protein (AgRP), which acts antagonistically on these receptors (9). Because leptin up-regulates expression of POMC mRNA and down-regulates AgRP mRNA in Arc neuronal cell bodies (6, 10), these opposing actions of α-MSH and AgRP on MC receptors probably exert a dual input on brain MC receptors. The idea that MC receptors act downstream from leptin signaling is consistent with the finding that co-infusion of the MC3–4 receptor antagonist with leptin prevents leptin’s anorexigenic efficacy and neuronal activation of the paraventricular hypothalamic nucleus (PVN) (4). The latter is known to be an important structure in regulation of food intake, neuroendocrine outflow, and metabolism and includes, among others, neuronal cell bodies that synthesize CRH (11).
The MC system received considerable attention over the last several years because mutations in the DNA encoding MC receptors, POMC, or POMC splicing enzyme have been associated with moderate to severe forms of human (12) and animal (9) obesity. Important evidence demonstrating that reduced MC receptor stimulation in the CNS leads to obesity is consistent with our observation that central, but not peripheral, administration of the MC receptor antagonist SHU9119 in rats causes dramatic increases in food intake and body weight, and it leads to several metabolic derangements associated with obesity (13). Although spontaneous genetic MC deficiencies render humans relatively frequently hyperphagic and obese (for example, relative to spontaneous deficiencies in leptin synthesis or reception), still the majority of screened obese subjects has no apparent mutation in the MC system. It might be hypothesized, however, that signaling of endogenous MC receptor ligands varies dramatically among subjects (e.g. as a result of differences in receptor number or downstream signaling cascades), and those with the lowest sensitivity could be most obesity prone, particularly when challenged with a high-energy diet (HED).
To further explore whether differences in MC receptor signaling might be relevant for the etiology of diet-induced obesity (DIO), we performed two experiments in rats. In the first experiment, we characterized short-term anorexigenic efficacies and activation patterns of PVN neurons (by using c-Fos and CRH double staining) and neuroendocrine outflow after i3cv administration of different doses of the synthetic MC receptor agonist melanotan-II (MTII) (Ac-Nle4-c [Asp5,d-Phe7,Lys10]α-MSH-(4–10-NH2). In the second experiment, it was investigated whether reduced sensitivity to one of the doses of MTII (with relative suppression of food intake as readout parameter) correlates to the development of DIO and associated metabolic derangements in rats challenged with a HED. MC receptor in situ binding [with 125I-labeled [Nle4,d-Phe7]α-MSH (NDP-MSH)] was performed after HED exposure to reveal whether reduced MC receptor binding in certain brain regions might be associated with DIO. These studies are important considering the fact that MC agonists are candidate drugs in the prevention or treatment of obesity. In both studies, the effects of MTII were compared with those of leptin, anticipating that we could reproduce the findings of Levin et al. (14) who previously reported that leptin insensitivity is a marker for DIO.
Materials and Methods
Animal preparation
A total of 47 male Wistar rats weighing between 360 and 390 g were individually housed in Plexiglas cages (25 × 25 × 30 cm) in a colony room with a 12-h light, 12-h dark cycle and normal ambient temperature (20 × 2 C). Pelleted chow and water were continuously available (except where noted). All animals were given a 21-gauge stainless steel cannula (Plastics One, Roanoke, VA) aimed at the third cerebral ventricle (at 1.5 mm posterior to Bregma and 7.5 mm ventral to dura) according to methods previously described (5). One week after surgery, brain cannula placements were confirmed by i3cv administration of 10 ng an-giotensin II in 1 μl sterile saline. This and all other i3cv administrations were performed with an injector that extended 1 mm beyond the tip of the brain cannula. Animals that did not drink 5 ml of water within 60 min after angiotensin II administration were excluded from the experiment (n = 1). Continuing rats were allowed to recover for at least 2 wk, during which they all returned to above presurgical body weights. In addition to the brain cannula, 36 rats also received a silicon catheter (od, 0.95 mm; id, 0.50 mm) inserted into the right jugular vein according to techniques described elsewhere (15). This catheter allowed frequent repeated blood sampling and/or infusion of fluids in undisturbed freely moving rats. Patency of the blood sampling catheters was checked weekly. Before experiments, animals were accustomed on 3 different days to brain injection and/or blood sampling procedures. All these and other procedures mentioned below are in accordance with the guidelines of the Animal Ethics Committee of the University of Groningen.
Experimental design
Assessment of MTII and leptin effects on food consumption
On 2 experimental days, food was removed 2 h before the dark phase and rats (n = 10, with i3cv cannulas only) were weighed. On the first experimental day, rats were divided into two groups matched for body weight (n = 5). One group was i3cv infused with sterile saline (3.0 μl), and the other was i3cv infused with leptin [3.0 μg human leptin (PreproTech, London, UK) in 3.0 μl saline]. After 1 wk, five rats (two and three randomly assigned rats from, respectively, the saline-infused and leptin-infused group) were i3cv infused with 0.05 μg MTII (Sigma Chemical Co., St. Louis, MO), whereas the remaining five animals were i3cv infused with 0.5 μg MTII. This order of administration was chosen because we have previously shown that i3cv MTII treatment may have some aversive side effects (16). For infusion, an animal was taken from its cage between 2 and 1 h before lights off. While sitting unrestrained on the experimenter’s lap, the obturator was removed and a 26-gauge infusion cannula connected to PE-20 infusion tubing was inserted into the guide cannula. This infusion cannula extended 1.0 mm beyond the tip of the guide cannula. Solutions were manually infused with a Hamilton syringe over 60 sec. Immediately after infusion, the injector was replaced by the obturator, and animals were returned to their home cage. Food was weighed and returned to the cage at lights off, and food intake was assessed after 3 h in the dark phase and overnight.
Assessment of MTII and leptin effects on neuroendocrine and PVN neuronal activity
At 4.5 h before lights off, other rats (n = 22) were briefly taken from their cages and connected with their jugular vein catheter to blood sampling tubing (PE 50, length 50 cm) and i3cv infusion tubing such that these extended out of their cages. Upon return, their food was removed, and animals were left alone for at least 30 min. During a 2-h window ending 2 h before lights off, a blood sample (1 ml) was taken and immediately thereafter, groups matched for body weight were i3cv infused with one of four solutions (leptin, 3.0 μg, n = 5; MTII, 0.05 μg, n = 7; MTII, 0.5 μg, n = 6; or vehicle, n = 6). Second and third blood samples (each 1 ml) were taken at 45 and 90 min after i3cv infusion. Blood samples were collected for measurement of glucose, ACTH, corticosterone, catecholamines, glucagon, leptin, and insulin in vials kept on melting ice, which contained aprotinin and EDTA, homogenized, and immediately centrifuged (15 min at 4300 rpm at 4 C), and stored at −80 C until determination. After each blood sample was taken, a transfusion of a similar volume of heparinized donor blood (taken from undisturbed donor rats) was iv infused into the animals to avoid reduction of blood volume. After the last blood sample, animals were anesthetized with sodium pentobarbital (6%, 0.5 ml, iv) and transcardially perfused with isotonic PBS followed by 4% paraformaldehyde in 0.1 m phosphate buffer. Brains were removed and postfixed for approximately 24 h and then processed for c-Fos-like immunoreactivity (cFLI) and CRH-like immunoreactivity.
Assessment of sensitivity to MTII and leptin and their predictiveness for DIO and related derangements
After rats (n = 12) with i3cv cannulas and jugular vein catheters had surpassed their preoperative body weights minimally at 1 wk, the anorexigenic efficacies of MTII (0.05 μg) and leptin (3.0 μg) relative to saline were investigated. These doses of MTII and leptin were chosen based on their similar anorexigenic efficacies as shown in the previous experiments. Food was removed 2 h before the dark phase, and between 2 and 1 h before lights off, rats were weighed and i3cv infused with MTII or leptin in randomized order with 1 wk between successive experiments. Before and in between drug treatments, rats were i3cv infused with saline (3 μl). After each i3cv infusion, food was weighed and returned to the cage at lights off, and food intake was assessed overnight. The efficacy of drugs to reduce food intake in each individual animal was expressed as follows: [(food intake after drug − average food intake after saline)/average food intake after saline] × 100%. After the sensitivity tests, rats were given, in addition to their normal chow, free access to a palatable liquid HED over a period of 16 wk. This diet consisted of a 10% sucrose solution in which corn oil was emulsified. This diet was based on the work of Lucas and Sclafani (17) and Smith et al. (18) and causes DIO in rats (19). The concentration of corn oil was doubled every 2 wk starting with 4% and finally ending up at 32% corn oil over the last weeks. The diet was freshly made each day, and specifics for preparation can be found elsewhere (19). Food intake (chow and HED) and body weight of rats were assessed on the final day of each dietary step.
During the final week of HED exposure, all rats were challenged with an iv glucose tolerance test (IVGTT). Specifically, food hoppers and the HED were removed at lights on, and 4 h later rats were connected to the blood sampling tubing and again left alone for 1 h. After taking a baseline blood sample (each 150 μl), 1.5 ml of a 10% glucose solution was iv injected over a period of 15 sec (t = 0) and blood samples were taken at t = 1, 3, 5, 7, 10, and 15 min. Thereafter, rats were disconnected from their blood sampling tubing and access to food was allowed again. The samples were immediately transferred to heparinized tubes (kept on melting ice) from which 50 μl blood was directly taken for determination of blood glucose. After centrifugation (15 min at 4300 rpm at 4 C), the remaining plasma was stored at −20 C until analysis for plasma insulin concentration. Three days after the IVGTT, animals were anesthetized with CO2 between 2 and 4 h after lights on and decapitated within 30 sec, and their brains were removed from the crania and deeply frozen on dry ice and stored at −80 C. Trunk blood was collected for measurement of ACTH, corticosterone, leptin, insulin, and glucose in vials (kept on melting ice) containing aprotinin and EDTA, centrifuged (15 min at 4300 rpm at 4 C), and stored at −80 C until determination. Weights of liver, gastrointestinal tract (full and empty), and fat pads (epididymal, mesenteric, and retroperitoneal) were assessed.
Chemical determinations and immunohistochemistry
Determination of plasma concentrations of insulin, ACTH, glucagon, and leptin were performed using sensitive RIAs (Linco Research, St. Charles, MO). Plasma concentrations of glucose, cholesterol, free fatty acids, and liver triglyceride content were assessed enzymatically (Boehringer kits; Boehringer, Mannheim, Germany). Plasma corticosterone (20) and catecholamines (21) were analyzed by HPLC. Determination of liver glycogen content was assessed as previously described (13).
For c-Fos immunohistochemistry and CRH double labeling, 40-μm coronal slices were cut on a vibrotome from the forebrain and included sections through the PVN. The sections were collected as alternate sets, with one set processed for immunoreactivity and the other stained with cresyl-violet for anatomical definition. Tissues were rinsed in PBS, incubated for 20 min in 0.3% H2O2 in absolute methanol to quench endogenous peroxidase, rinsed, and incubated 1 h in 1% gelatin, 5% normal goat serum in PBS. Slices were then transferred without rinsing to the primary antibody solution consisting of 0.005 g/ml polyclonal rabbit antiserum (Santa Cruz Biotechnology, Santa Cruz, CA) (1:20,000), which recognizes residues 3–16 of the c-Fos protein. After approximately 48 h incubation on ice (4 C), slices were rinsed and processed using the ABC method (Vector Laboratories, Burlingame, CA). Slices were transferred to biotinylated goat antirabbit antibody for 1 h, rinsed, and developed with diaminobenzidine substrate intensified with nickel sulfate (6 min), which produced dark cFLI nuclei. After an overnight rinse in PBS, sections were run through a second assay using the same procedure as above, except that the primary antibody was rabbit anti-CRH (1:10,000) and sections were developed in diaminobenzidine substrate without the nickel sulfate (which produced light-brown cell bodies). Thus, double-labeled cells were seen as dark nuclei surrounded by light-brown cell bodies. Finally, slices were rinsed (10× PBS), mounted on slides, and coverslipped with Permount. Camera lucida drawings from c-Fos immunoreactive sections were prepared and scored by a blind rater who recorded the number and location of c-Fos-positive nuclei in one slice of each rat revealing the PVN most optimally at 1.7 mm caudal from Bregma according to the brain atlas of Paxinos and Watson (22). In these slices, counts were also made of CRH-positive cells and of cells that coexpressed the c-Fos and the CRH proteins.
Brains from rats that were rendered diet-induced obese were cut as 25-μm coronal slices (using a cryostat microtome), which included the area covering the PVN to the ventromedial hypothalamus (VMH) (from −1.5 to −3.5 relative to Bregma), and immediately taken up on gelatin-coated slides. Two sets were postfixed with 4% paraformaldehyde in 0.1 m phosphate buffer on glass and dried (48 h at 30 C) for cresyl violet staining and CRH immunohistochemistry. A third set was dehydrated with silica gel used for in situ binding with 125I-labeled NDP-MSH. CRH immunohistochemistry was performed according to the same techniques as above. CRH-positive neurons were found after peroxide-antiperoxide labeling of the complex (peroxidase-antiperoxidase immunoprocessing). The assay for [125I]NDP-MSH in situ binding was performed according to the methods described by Vrinten and colleagues (23). In short, sections were prewashed, incubated with [125I]NDP-MSH (106 cpm/ml) in binding buffer for 1 h, washed six times to stop binding reactions, and rapidly air dried. Also sections were incubated with [125I]NDP-MSH in the presence of noniodinated NDP-MSH to determine the specificity of tracer. All binding assays were on the same day in one experimental session. Autoradiography was performed by exposing an x-ray film directly to slices for 2 wk. Autoradiograms harboring the PVN and the VMH were digitized and quantitatively analyzed using a Leica Quantimet Image Analysis System (Quantimet Q-600HR; Leica, Wetzlar, Germany). The PVN and VMH were found and outlined using adjacent cresyl-violet-stained sections for the VMH and an adjacent CRH-stained section for the PVN. Absorbance levels within the outlined bilateral PVN and VMH were determined, and specific binding was calculated by subtraction of the mean background value, determined within the ventral-lateral thalamus of the same sections. The absorbance values were compared with those found in a linear calibration curve, and levels fitted well within the linear part of this curve. The level of CRH immunohistochemistry was assessed in four to six PVNs bilaterally of each animal. This was done by assessing the absorbance over the entire PVN region using the Leica Quantimet Image Analysis System. Background absorbance was also determined in the area in close vicinity of the PVN and was subtracted from the value obtained from the PVN region.
Statistical analyses
ANOVA was used to investigate effects of i3cv saline vs. MTII (two doses) vs. leptin on overnight food intake, neuronal activation, and changes in blood parameters. Spearman’s ρ nonparametric analysis was used to assess correlations of the anorexigenic efficacy of i3vt administered MTII and leptin with hormonal and metabolic parameters linked to DIO, as well as with the intensity of 125I-labeled NDP-MSH in situ binding and paraventricular CRH immunostaining.
Results
Effects of MTII and leptin food consumption
The effects of i3cv saline, leptin, and the two dose of MTII on 3-h and overnight food intake are presented in Table 1. ANOVA revealed a significant drug effect on both 3-h (F3,22 = 10.77; P < 0.0001) as well as overnight (F3,22 = 20.47; P < 0.0001) food intake. Post hoc tests revealed that i3cv leptin and the two doses of MTII produced significant reductions in food intake compared with the saline group. Whereas food intake was comparably reduced with leptin and the 0.05-μg dose of MTII at 3 h (respectively, −57 and −65%) and overnight (respectively, −35 and −45%), the 0.5-μg dose of MTII produced a significantly more pronounced reduction in overnight food intake (respectively, −89 and −79%) relative to 0.05 μg MTII and leptin treatment.
TABLE 1.
Three-hour and overnight (12-h) food intake in rats after i3cv administration of drugs
| Treatment | 3 h | 12 h |
|---|---|---|
| Saline (n = 5) | 3.9 ± 0.5 | 15.5 ± 1.5 |
| Leptin (3.0 μg, n = 5) | 1.7 ± 0.6a | 9.9 ± 1.0a,d |
| MTII (0.05 μg, n = 5) | 1.4 ± 0.1b | 8.4 ± 0.7b,d |
| MTII (0.5 μg, n = 5) | 0.4 ± 0.3c | 3.3 ± 0.9c |
Values are means ± sem expressed in grams.
P < 0.05;
P < 0.01;
P < 0.001 relative to saline treatment.
P < 0.05 relative to MTII 0.5 μg treatment.
Assessment of MTII and leptin effects on neuroendocrine and PVN neuronal activity
The effects of i3cv saline, leptin, and the two doses of MTII on plasma levels of ACTH, corticosterone, and leptin are presented in Fig. 1. Two animals could not be used for blood sampling (one in the vehicle and one in the 0.05-μg MTII group) because of occlusion of blood sampling catheters. Repeated-measures ANOVA revealed significant effects on plasma corticosterone (time × drug: F6,38 = 5.88; P < 0.0001), ACTH (time × drug: F6,38 = 7.08; P < 0.0001), and leptin (time × drug: F6,38 = 12.39; P < 0.001). Plasma levels of corticosterone after i3cv leptin and MTII were higher relative to vehicle treatment. The 0.5-μg dose of MTII caused significantly higher levels of plasma corticosterone relative to the 0.05-μg dose of MTII. Significantly higher levels of ACTH were found at all time points after MTII and leptin relative to the vehicle. The 0.5-μg dose of MTII caused higher levels of plasma ACTH relative to the 0.05-μg dose and leptin at time point t = 45 min. Only higher plasma levels of leptin were found after i3cv leptin administration.
Fig. 1.
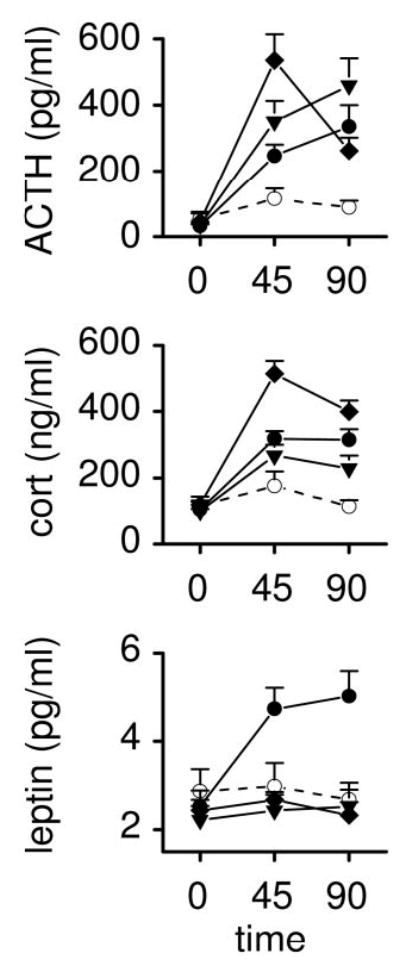
Effects of i3cv administration of saline (○, n = 5), leptin (•, 3.0 μg, n = 5), and MTII (▾, 0.05 μg, n = 6; ▪, 0.5 μg, n = 6) on plasma levels ACTH, corticosterone (cort), and leptin. Solutions were given in 3.0-μl volumes at t = 0 min.
The effects of i3cv saline, leptin, and the two doses of MTII on plasma levels of glucagon, epinephrine, and norepinephrine are presented in Fig. 2. Repeated-measures ANOVA revealed significant effects on plasma epinephrine levels (time × drug: F6,38 = 4.62; P = 0.001). Post hoc analysis revealed significantly higher levels of plasma epinephrine after i3cv MTII but not after i3cv leptin. Although an ANOVA including all groups did not attain a significant effect on plasma glucagon levels, there appeared to be an elevation of plasma glucagon with i3cv MTII (0.5 μg) (F2,20 = 3.65; P = 0.045). ANOVA did not reveal any effects on norepinephrine or on plasma insulin and glucose (the latter are not shown).
Fig. 2.
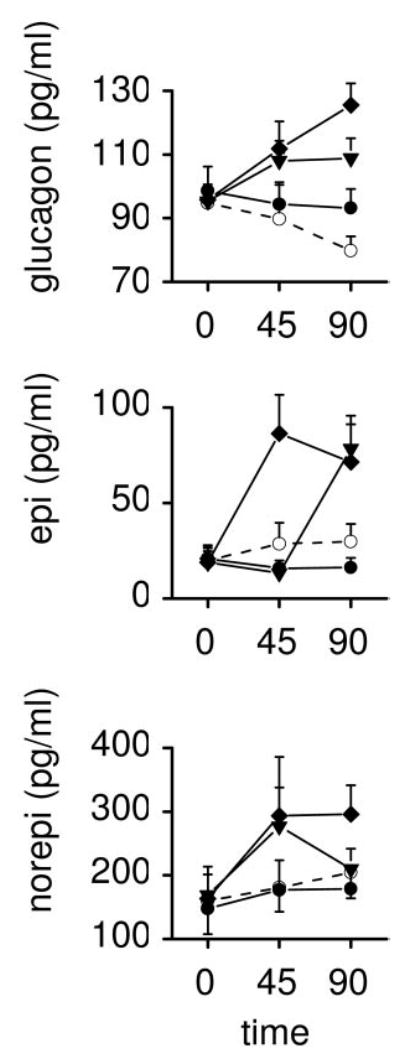
Effects of i3cv administration of saline (○, n = 5), leptin (•, 3.0 μg, n = 5), and MTII (▾, 0.05 μg, n = 6; ▪, 0.5 μg, n = 6) on plasma levels glucagon, epinephrine (epi), and norepinephrine (norepi). Solutions were given in 3.0-μl volumes at t = 0 min.
Areas under the curve (AUC) of the significantly affected plasma parameters were calculated and presented in Table 2. ANOVA revealed significant effects of treatment on the AUC of plasma ACTH (F3,21 = 6.06; P = 0.005), corticosterone (F3,21 = 14.22, p = <0.0001), leptin (F3,21 = 21.35; P < 0.0001), and epinephrine (F3,21 = 3.74; P < 0.03). Post hoc analysis revealed that both doses of MTII and leptin caused increases in the AUC of the plasma ACTH and corticosterone responses, whereas only leptin caused an increase in the AUC of leptin. Only the highest dose of MTII caused an increase in the AUC of the plasma epinephrine response. I3cv saline treatment did not result in an increased AUC of any assessed parameter.
TABLE 2.
AUC of significantly affected hormone responses after i3cv administration of drugs
| Treatment | ACTH (ng/ml·min) | Corticosterone (μg/ml·min) | Leptin (pg/ml·min) | Epinephrine (ng/ml·min) |
|---|---|---|---|---|
| Saline (n = 5) | 3.6 ± 2.6 | 2.5 ± 3.5 | 1.0 ± 4.8 | 0.61 ± 1.06 |
| Leptin (3.0 μg, n = 5) | 16.5 ± 2.9a | 12.8 ± 2.4a, d | 155.5 ± 26.7c | −0.33 ± 0.53d |
| MTII (0.05 μg, n = 6) | 23.7 ± 4.3b | 9.7 ± 1.9a,d | 7.9 ± 15.5e | 1.08 ± 1.57d |
| MTII (0.5 μg, n = 6) | 27.0 ± 5.3c | 24.2 ± 1.8c,d | 16.5 ± 7.2e | 4.22 ± 1.57a |
Values are means ± SEM.
P < 0.05;
P < 0.01;
P < 0.001 relative to saline treatment.
P < 0.05 relative to MTII 0.5 μg treatment.
P < 0.0001 relative to leptin treatment.
Gross inspection of brain slices clearly revealed CRH-positive neurons (stained as light brown) in the PVN (see Fig. 3A), mostly in the parvocellular (PVN-Par) division (Fig. 3B), but only to a small extent in the magnocellular (PVN-Mag) division. This is in agreement with the data of Sawchenko et al. (24). cFLI could be observed as black staining of nuclei within CRH-positive as well as in CRH-negative cells. Data from cFLI in PVN-Par and PVN-Mag are presented in Table 3. ANOVA revealed significant drug effects on cFLI in PVN-Par (F3,18 = 7.56; P = 0.0017) as well as PVN-Mag (F3,18 = 7.35; P = 0.002). Post hoc tests comparing the control group with drug-treated groups revealed that leptin (P = 0.014) (Fig. 3, C and D) and the two doses of MTII [0.05 μg, P = 0001 (Fig. 3, E and F); 0.5 μg, P = 0.0025 (Fig. 3, G and H)] produced increased cFLI in the PVN-Par. With respect to the PVN-Mag, only the two doses of MTII (0.05 μg, P = 0.0035; 0.5 μg, P = 0.0078) produced reliably increased cFLI relative to that observed after saline treatment, whereas leptin effects on cFLI were not different from saline treatment. In addition, both doses of MTII (0.05 μg, P = 0.012; 0.5 μg, P = 0.019) also produced increased cFLI relative to leptin treatment. The percentages of cFLI-positive neurons that contained CRH were significantly elevated in all treatment groups relative to saline (F3,18 = 69.06; P < 0.00001). Levels of double labeling were not different among groups despite differences in c-Fos labeling.
Fig. 3.
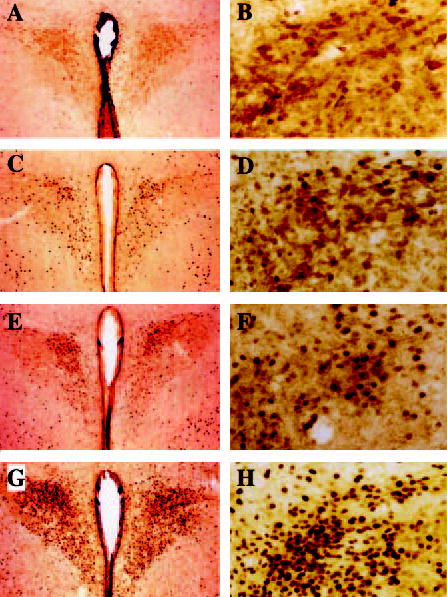
Representative photomicrographs of the parvocellular and magnocellular region of the PVN. Neuronal cell bodies were immu-nostained for CRH (indicated as light brown) and c-Fos (indicated as dark nuclear staining) of rats treated i3cv with saline (A and B), leptin (3.0 μg, C and D), and MTII (0.05 μg, E and F; 0.5 μg, G and H).
TABLE 3.
cFLI of PVN-Par and PVN-Mag neuronal cell bodies and its percentage (expressed as percent CRH/cFLI), which is doubly labeled for CRH after i3cv administration of drugs
| Treatment | PVN-Par | PVN-Mag | % CRH/cFLI |
|---|---|---|---|
| Saline (n = 5) | 22.4 ± 3.7 | 7.4 ± 0.7 | 5.4 ± 0.5 |
| Leptin (3.0 μg, n = 5) | 113.4 ± 26.0a | 15.0 ± 2.9 | 69.6 ± 5.9c |
| MTII (0.05 μg, n = 6) | 126.8 ± 22.5c | 51.0 ± 10.1c,d | 76.0 ± 3.5c,d |
| MTII (0.5 μg, n = 6) | 181.0 ± 34.5b | 58.5 ± 13.6c,d | 76.8 ± 11.8c,d |
Values are means ± sem expressed in grams.
P < 0.05;
P < 0.01;
P < 0.001 relative to saline treatment.
P < 0.05 relative to leptin treatment.
Assessment of sensitivity to MTII and leptin and their relation to DIO and related derangements
Before, between, and after sensitivity tests with MTII and leptin, 12 rats were i3cv treated at three different times with saline. On consecutive occasions, the mean overnight food intake was 17.2 ± 0.6, 16.9 ± 0.9, and 16.9 ± 1.7 g, respectively, with the individual intakes replicated within relatively narrow margins. Because these outcomes were similar, the effect of saline treatment on food intake was expressed as average of the three outcomes for each individual animal. Overnight food intake of rats after i3cv MTII (0.05 μg) and leptin (3.0 μg) was 7.5 ± 1.7 and 9.7 ± 0.9 g, respectively, and these effects were not different from each other. Although there were large individual differences in the anorexigenic efficacy of leptin (between −84 and-12% relative to saline) and MTII (between −93 and −3%), these sensitivities were not correlated (see Table 4). No correlations were found of anorexigenic efficacies of leptin and MTII with plasma leptin levels before HED exposure.
TABLE 4.
Correlations of pre- and post-DIO parameters in rats (n = 12) subjected to a HED for 16 wk
| Pre-DIO
|
Post-DIO
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Leptin sensitivity | MTII sensitivity | Plasma leptin | AUC insulin | Visceral obesity | Liver weight | PVN MC receptor binding | VMH MC receptor binding | PVN CRHimmuno-reactivity | |
| Pre-DIO | |||||||||
| Leptin sensitivity | 1.000 | ||||||||
| MTII sensitivity | 0.042, P = 0.897 | 1.000 | |||||||
| Post-DIO | |||||||||
| Plasma leptin | 0.741, P = 0.006 | −0.056, P = 0.863 | 1.000 | ||||||
| AUC insulin | 0.287, P = 0.366 | 0.147, P = 0.649 | 0.399, P = 0.199 | 1.000 | |||||
| Visceral obesity | 0.608, P = 0.036 | 0.007, P = 0.983 | 0.867, P < 0.0001 | 0.629, P = 0.028 | 1.000 | ||||
| Liver weight | 0.720, P = 0.008 | 0.294, P = 0.354 | 0.608, P = 0.036 | 0.434, P = 0.159 | 0.587, P = 0.045 | 1.000 | |||
| PVN MC receptor binding | 0.385, P = 0.217 | −0.664, P = 0.018 | 0.434, P = 0.159 | −0.049, P = 0.880 | 0.399, P = 0.199 | 0.077, P = 0.812 | 1.000 | ||
| VMH MC receptor binding | 0.084, P = 0.795 | −0.056, P = 0.863 | 0.322, P = 0.308 | 0.070, P = 0.829 | 0.210, P = 0.513 | 0.483, P = 0.112 | 0.350, P = 0.265 | 1.000 | |
| PVN CRH immunoreactivity | 0.175, P = 0.587 | 0.755, P = 0.005 | 0.147, P = 0.649 | −0.021, P = 0.948 | 0.280, P = 0.379 | −0.084, P = 0.795 | 0.685, P = 0.014 | −0.154, P = 0.633 | 1.000 |
Correlation coefficient (Spearman’s ρ) and corresponding P values are given for each pairing. Significant correlations are given in bold.
HED exposure caused an increase in energy intake with each dietary step (relative to a group of nontreated controls that are not shown in the current study) to finally a level at the 16th week of exposure, which was on average 128% higher than intake at baseline, and this was solely a result of the increased intake of the sucrose/corn oil mixture. There was, in fact, a slight decrease in the intake of chow (12% suppression relative to basal intake). This is consistent with one of our previous studies using the same paradigm, which finally led to an 82% increase in body weight relative to a group of rats that were left on chow (19).
During the last week of HED exposure, rats were subjected to IVGTTs. Upon iv injection of glucose, blood glucose levels rose from 6.84 ± 0.24 mmol/liter to a maximum level of 14.38 ± 0.49 mmol/liter at t = 1.5 min and then returned to basal levels at t = 15 min. Whereas the blood glucose responses to the IVGTT did not vary to a large extent between animals, the plasma insulin responses to IVGTTs, however, varied dramatically among rats. Expressed as AUC relative to baseline levels, insulin responses varied between 82.7 and 348.4 ng/ml·15 min. Postmortem assessment of parameters related to nutritional status revealed that abdominal fat mass varied between 36.0 and 71.8 g, liver weights between 17.8 and 22.5 g, and plasma levels of leptin between 5.3 and 33.1 ng/ml. Within the hypothalamus, 125I-labeled NDP-MSH binding revealed strong signaling in the PVN and ventro-medial hypothalamus (VMH), and CRH immunohistochemistry revealed strong labeling only of PVN neurons.
Correlation analysis of post-DIO parameters revealed significant interactions between abdominal fat mass and leptin concentration and the AUC of the plasma insulin response to the IVGTT. The anorexigenic efficacy of leptin, but not of MTII, before the DIO appeared to be highly inversely correlated to the post-DIO plasma leptin concentration, post-DIO abdominal fat mass, post-DIO liver weight (Fig. 4), and hepatic glycogen content (P = 0.005; r = 0.74) but not to hepatic triglyceride storage (P = 0.08; r = 0.21). The anorexigenic efficacy of MTII, but not of leptin, was correlated to post-DIO CRH labeling in the PVN and to the post-DIO 125I-labeled NDP-MSH binding specifically in the PVN but not to the VMH (Fig. 5). All correlation coefficients and levels of significance not indicated here are presented in Table 4.
Fig. 4.
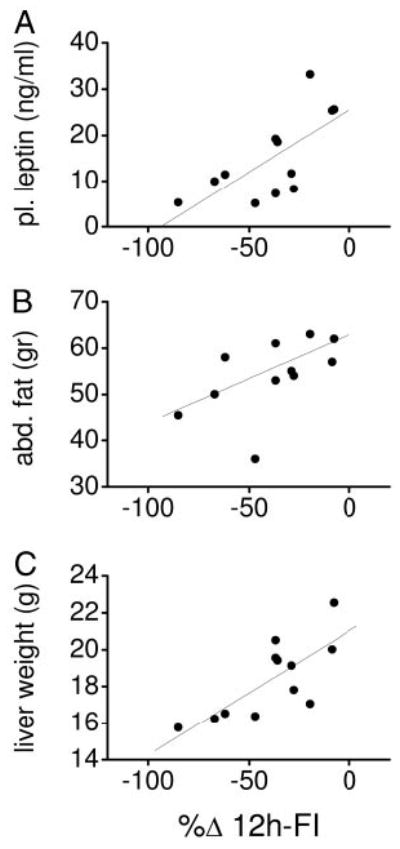
Leptin sensitivity (n = 12) expressed as reduction in 12-h food intake after i3cv leptin administration relative to saline (%Δ 12h-FI) but not MTII sensitivity (data not shown) before the HED exposure was significantly correlated to the plasma leptin concentration (A), abdominal fat mass (B), and liver weight (C) after the HED exposure.
Fig. 5.
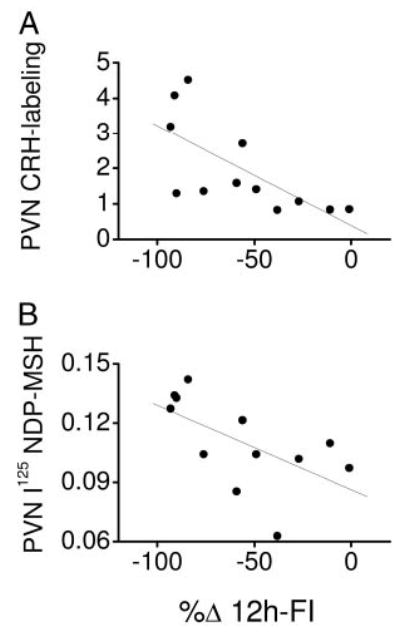
MTII sensitivity (n = 12) expressed as reduction in 12-h food intake after i3cv MTII administration relative to saline (%Δ 12h-FI) but not leptin sensitivity (data not shown) before the HED exposure was significantly inversely correlated to CRH labeling (A) and [125I]NDP-MSH binding (B) in the PVN after the HED exposure.
Discussion
A number of reports suggest that the effects of leptin are mediated, in part, via activation of PVN neurons that synthesize and secrete CRH (25–29). One hypothesis proposes that reduced sensitivity of brain MC receptors might underlie the development of DIO (30). The present study was designed to explore the relevance of interactions between MC receptor sensitivity and PVN CRH neurons in the etiology of DIO and associated metabolic derangements. The latter is an extremely important issue in public health, because the prevalence of obesity and the metabolic syndrome are increasing to epidemic proportions (31, 32), and treatment is not yet available.
In the present study, equi-anorexigenic doses of leptin (3.0 μg) and MTII (0.05 μg) caused identical increases in cFLI of PVN-Par neurons. Double-labeling immunohistochemistry revealed that approximately 70% of these cFLI-positive PVN neuronal cell bodies colabeled CRH with either treatment. A higher dose of MTII (0.5 μg) with a notably stronger anorexigenic efficacy caused stronger cFLI in the PVN. The ratio of CRH-positive and cFLI-positive neuronal cell bodies remained unaltered with the higher MTII dose, which indicates that in total more CRH neurons were activated relative to that caused by the lower dose of MTII or leptin. The presumably increased CRH output caused by the high MTII dose may also be the result of a stronger activation of CRH neurons, which would be stimulated with the lower dose of MTII as well. Unfortunately, c-Fos immunohistochemistry is not a suitable method to clarify this issue. Nevertheless, together with observations by others that the anorexigenic effects of leptin (26, 27) as well as MTII (28) can be prevented by co-infusion of the CRH receptor antagonist, α-helical-CRH (9–41), these data lend further support to the idea that CRH-producing PVN neurons, at least in the short term, are key in leptin receptor-mediated as well as MC receptor-mediated reductions in food intake (29). The effects of MTII on CRH neurons is probably direct, because MC4 receptors have been demonstrated to exist on PVN neuronal cell bodies that express CRH (28). Thus, in the case of stimulation of CRH neurons by leptin, this probably requires activation of POMC-containing neuronal cell bodies in the Arc (10, 33), which then release the endogenous MC receptor ligand α-MSH from their nerve terminals adjacent to MC4 receptors on PVN neuronal cell bodies (28). Indeed, α-MSH has previously been demonstrated to increase phospho-cAMP response element binding protein in paraventricular CRH neurons (29). The idea that leptin-mediated activation of PVN CRH neurons depends on the integrity of the MC system is in agreement with the observations of Seeley et al. (4) that i3cv co-infusion of the MC3/4 receptor antagonist SHU9119 blocked not only leptin’s anorexigenic efficacy but also its stimulatory effects on cFLI in the PVN.
Whereas the projections of PVN CRH neurons as well as their subclasses of CRH receptors that contribute to the regulation of food intake are currently being unraveled (e.g. Refs. 34 and 35), the classical contribution of PVN CRH neurons to the activity of the hypothalamo-pituitary-adrenal (HPA) axis has been already known for over two decades (36). The similar acute activation patterns of PVN CRH neurons by equi-anorexigenic doses of leptin and MTII were associated with similar increments in the plasma levels of ACTH and corticosterone. Although leptin presumably influences neuropeptide/neurotransmitter systems that regulate food intake and neuroendocrine activity independent from the MC-CRH axis (e.g. Refs. 37–39), one implication of these data may be that these parallel systems are less important within a time frame of less than a few hours. Indeed, the magnitudes by which food intake, HPA axis activity, and PVN CRH neuronal activity were affected by i3cv administered leptin and MTII were markedly similar. In the long term, however, leptin and MTII might greatly differentiate with respect to their anorexigenic efficacies, CRH and HPA axis activation, and other endocrine and metabolic parameters. In fact, leptin has been shown in usually more chronic infusion paradigms (i.e. with osmotic minipump infusion over several days to weeks) to decrease CRH mRNA expression and/or HPA axis activity (e.g. Refs. 40–42), as opposed to the effects of MC receptor agonists (de Vries, K., and G. van Dijk, unpublished observations). It might be possible that the short-term effects of leptin and MTII to stimulate paraventricular CRH neurons may not engage a physiological mechanism to cause a reduction in food intake but that it signals an emergency situation. Indeed, a variety of psychological and physical stressors increase the expression of CRH in the PVN, and leptin and MTII may simply tie into this mechanism. However, it needs to be mentioned here that involuntary overfeeding in rats causes increased activation of the MC-CRH axis (43, 44). Although the exact mechanism underlying this effect remains to be investigated, it may be argued that the dramatic increase in leptin under conditions of involuntary overfeeding signals an increase in the nutritional status that animals might have difficulties in dealing with in a homeostatic sense (45). Thus, along these lines, it might very well be possible that a sudden increase in the ambient leptin level caused by exogenous application may signal an abrupt increase in the nutritional status and would thus be relayed as an emergency situation. A potential effect of leptin to induce discomfort, nausea, or pain per se is less likely because a similar dose of leptin used in one of our previous studies was not capable of inducing a conditioned taste aversion (46).
Besides similarities in activation pattern of PVN neurons and neuroendocrine and behavioral correlates after i3cv lep-tin and MTII, we observed that i3cv leptin, but not i3cv MTII (even dosed at the 0.5-μg concentration), led to an elevation of peripheral plasma leptin levels. Although we do not know the mechanism underlying this phenomenon (see also Ref. 47), we have neither observed elevated plasma leptin levels after i3cv leptin administration in Zucker rats (van Dijk, G., unpublished observations) nor found traces of the i3cv infused human leptin in plasma in previous studies (6, 47). For these reasons, we believe that this effect is caused via a receptor-mediated action in the CNS but does not require increased activation of brain MC receptors. Another striking difference between the effects of the two treatments was that i3cv MTII but not i3cv leptin administration caused a rise in the circulating level of epinephrine and glucagon, and this was particularly evident when MTII was dosed at a higher concentration. It might be speculated that these effects are mediated via a subset of MTII-sensitive PVN neurons, which project to adrenal medullary sympathetic preganglionic neurons (48) and potentially to the preganglionics of the pancreas as well.
In a distinct group of rats, the anorexigenic efficacies of i3cv MTII and leptin were assessed relative to saline treatment, and thereafter these rats were subjected to a palatable HED known to induce DIO (19). The i3cv doses of leptin and MTII caused overall reductions in food intake that were comparable to those in the dose-response study mentioned above. There were, however, large individual differences in the responses between rats; i.e. some rats responded to i3cv leptin administration with an almost 100% reduction relative to control treatment, whereas others were markedly insensitive. This degree of variability was also observed after i3cv MTII treatment, but the individual anorexigenic efficacy of MTII in rats was not related to that of leptin. After feeding the HED for several months, a number of parameters related to obesity were highly correlated to the initial anorexigenic efficacy of leptin. Specifically, rats that were relatively insensitive to leptin’s anorexigenic actions displayed the largest HED-induced weight gain in the form of abdominal fat deposition and liver weight and had the highest plasma levels of leptin. Thus, although DIO (49) and hyperleptinemia (50) have been shown to induce leptin resistance, these data suggest that leptin insensitivity could in fact underlie hyperleptinemia and DIO, and this confirms and extends previous findings of Levin et al. (14).
The abdominal fat mass of rats after HED exposure was significantly correlated to the AUC of the insulin response during the IVGTT. An increase in the insulin response to an IVGTT may be considered as an indicator of reduced insulin sensitivity (51), and this finding is in agreement with many observations that increased abdominal obesity is associated with insulin resistance (52). Because abdominal fat deposition was inversely correlated to leptin’s anorexigenic efficacy and because increased leptin signaling (53, 54) as well as MC signaling (55) in the CNS are known to increase whole-body insulin sensitivity, one might have expected a relationship between leptin and/or MTII’s anorexigenic efficacy and insulin sensitivity. However, neither the anorexigenic efficacy of leptin nor that of MTII was directly correlated to the AUC of insulin during the IVGTT. Thus, although the IVGTTs were performed in rats food deprived no longer than 4 h, the data in the present study suggest that the primary effect of reduced central leptin sensitivity is diet-induced (visceral) obesity, and peripheral insulin resistance is presumably a secondary feature of this. In this respect, the strong inverse correlation between leptin’s anorexigenic efficacy and the post-HED liver weight is of interest, given the previous finding by Cohen et al. (56) that mice with targeted deletion of central leptin receptors have enlarged livers and are obese, and central leptin deficiency has been proposed to increase hepatic lipid storage via increased activity of the hepatic enzyme, stearoyl-CoA dehydrogenase (57). In the present study, however, we observed only a tendency of an inverse correlation between the post-HED hepatic triglyceride content with leptin’s anorexigenic efficacy. Opposite to our expectation was the finding that the post-HED hepatic glycogen content was highly inversely correlated with leptin’s anorexigenic efficacy. Because complete blockade of leptin signaling causes increased hepatic glycogenolysis leading to lower hepatic glycogen stores (58), and central leptin administration has the opposite effect (59), the insensitivity to leptin in the present study apparently does not pertain to all aspects of the metabolic syndrome. It might be suggested that the reduced central leptin sensitivity of rats in the present study is compensated by increased leptin actions in peripheral tissues.
A potentially important finding in the present study was that neither the pre-HED anorexigenic efficacy of MTII nor the post-HED 125I-labeled NDP-MSH binding in the CNS was related to any parameter linked to DIO. Although the terminal binding study does not necessarily explain any factor in the preobese state, the data may suggest that variation in MC receptor signaling (assessed by food intake suppression as well as by MC receptor binding) does not underlie the reduced leptin sensitivity of rats in the present study. Thus, although almost 4 months elapsed between i3cv MTII sensitivity testing and sampling of rat brains for 125I-labeled NDP-MSH in situ binding study, a period over which animals developed markedly different severities of DIO, it may be speculated that the differences in the activity of an anorexigenic mechanism involving MC receptor actions in the PVN is retained irrespective of differences in DIO development. This may be consistent with the maintained anorexigenic and thermogenic sensitivity to MTII in DIO rats in other studies (60). There were, however, strong correlations of MTII’s anorexigenic efficacy with the post-HED 125I-labeled NDP-MSH binding specifically in the PVN, but not in other regions studied, such as the VMH. The latter region would be of particular interest because MC receptor binding in the VMH is reduced by increasing dietary fat content and/or hyperleptinemia (61). MTII’s anorexigenic efficacy and the post-HED 125I-labeled NDP-MSH binding in the PVN correlated strongly with CRH immunoreactivity in the PVN. Thus, provided that the differences in PVN CRH immunoreactivity truly reflect differences in paraventricular CRH peptide content or release (62), there might be a tight interaction between activity of the MC system and the CRH system unrelated to the development of DIO.
Taken together, the data in the present study show that i3cv doses of leptin and MTII equated to produce similar reductions in food intake cause a similar degree of activation (assessed by cFLI and HPA axis activity) of PVN CRH neurons. Furthermore, reduced sensitivity to leptin might underlie DIO, but this does not rely on reduced sensitivity to or binding of MC receptor ligands. Thus, although leptin therapy has been disproved in this and other studies as an effective treatment strategy to counter DIO (14, 19, 63), MC receptor agonists may be more useful (50, 64, 65, 66). The success of the latter strategy may depend on interactions of MC receptor agonists with PVN CRH neurons.
Acknowledgments
We thank J. Bruggink, J. Keizer, and V. Bloks for excellent technical contributions.
Footnotes
G.v.D. was supported by grants from the Dutch Scientific Organization (NWO 903-39-157), the Royal Netherlands Academia of Arts and Sciences, and by a Career Development Grant of the Dutch Diabetes Foundation. T.T. was supported by National Institutes of Health Grants AA013573 and AA015148.
References
- 1.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 2.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Open-tanova I, Ohanessian JP, Kolaczynski JW, Bauer TL, Moore JH, Caro JF. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. Biochem Biophys Res Commun. 1996;220:735–739. [Google Scholar]
- 3.van Dijk G. The role of leptin in the regulation of energy balance and adiposity. J Neuroendocrinol. 2001;13:913–921. doi: 10.1046/j.1365-2826.2001.00707.x. [DOI] [PubMed] [Google Scholar]
- 4.Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, Baskin DG, Schwartz MW. Melanocortin receptors in leptin effects. Nature. 1997;390:349. doi: 10.1038/37016. [DOI] [PubMed] [Google Scholar]
- 5.van Dijk G, Thiele TE, Donahey JC, Campfield LA, Smith FJ, Burn P, Bernstein IL, Woods SC, Seeley RJ. Central infusions of leptin and GLP-1-(7–36) amide differentially stimulate c-FLI in the rat brain. Am J Physiol. 1996;271:R1096–R1100. doi: 10.1152/ajpregu.1996.271.4.R1096. [DOI] [PubMed] [Google Scholar]
- 6.van Dijk G, Seeley RJ, Thiele TE, Friedman MI, Ji H, Wilkinson CW, Burn P, Campfield LA, Tenenbaum R, Baskin DG, Woods SC, Schwartz MW. Metabolic, gastrointestinal, and CNS neuropeptide effects of brain leptin administration in the rat. Am J Physiol. 1999;276:R1425–R1433. doi: 10.1152/ajpregu.1999.276.5.R1425. [DOI] [PubMed] [Google Scholar]
- 7.Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–437. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- 8.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Earley AR, Barnett AH, Prins JB, O’Rahilly S. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 9.Hillebrand JJG, De Wied D, Adan RAH. Neuropeptides, food intake and body weight regulation: a hypothalamic focus. Peptides. 2002;23:2283–2306. doi: 10.1016/s0196-9781(02)00269-3. [DOI] [PubMed] [Google Scholar]
- 10.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 11.Swanson LW, Sawchenko PE. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinology. 1980;31:410–417. doi: 10.1159/000123111. [DOI] [PubMed] [Google Scholar]
- 12.Tsigos C, Kyrou I, Raptis SA. Monogenic forms of obesity and diabetes mellitus. J Pediatr Endocrinol Metab. 2002;15:241–253. doi: 10.1515/JPEM.2002.15.3.241. [DOI] [PubMed] [Google Scholar]
- 13.Adage T, Scheurink AJ, de Boer SF, de Vries K, Konsman JP, Kuipers F, Adan RA, Baskin DG, Schwartz MW, van Dijk G. Hypothalamic, metabolic, and behavioral responses to pharmacological inhibition of CNS melanocortin signaling in rats. J Neurosci. 2001;21:3639–3645. doi: 10.1523/JNEUROSCI.21-10-03639.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levin BE, Dunn-Meynell AA. Reduced central leptin sensitivity in rats with diet-induced obesity. Am J Physiol. 2002;283:R941–R948. doi: 10.1152/ajpregu.00245.2002. [DOI] [PubMed] [Google Scholar]
- 15.Steffens AB. A method for frequent sampling of blood and continuous infusions of fluids in the rat without disturbing the animals. Physiol Behav. 1996;4:833–836. [Google Scholar]
- 16.Thiele TE, van Dijk G, Yagaloff KA, Fisher SL, Schwartz M, Burn P, Seeley RJ. Central infusion of melanocortin agonist MTII in rats: assessment of c-Fos expression and taste aversion. Am J Physiol. 1998;274:R248–R254. doi: 10.1152/ajpregu.1998.274.1.R248. [DOI] [PubMed] [Google Scholar]
- 17.Lucas F, Sclafani A. Hyperphagia in rats produced by a mixture of fat and sugar. Physiol Behav. 1990;47:51–55. doi: 10.1016/0031-9384(90)90041-2. [DOI] [PubMed] [Google Scholar]
- 18.Smith JC, Fisher E, Maleszewski V, The ingestion of fat when blended with a glucose and saccharine mixture. XIIth International Conference on the Physiology of Food and Fluid Intake and the Annual Meeting of the Society for the Study of Ingestive Behavior, Pécs, Hungary, 1998 (Abstract)
- 19.Ruffin MP, Adage T, Kuipers F, Strubbe JH, Scheurink AJW, van Dijk G. Feeding and temperature responses to leptin infusion are differential predictors of obesity in rats. Am J Physiol. 2004;286:R756–R763. doi: 10.1152/ajpregu.00508.2002. [DOI] [PubMed] [Google Scholar]
- 20.Dawson R, Kontur P, Monjanr A. High-performance liquid chromatography (HPLC) separation and quantitation of endogenous glucocorticoids after solid-phase extraction from plasma. Horm Res. 1984;20:89–94. doi: 10.1159/000179979. [DOI] [PubMed] [Google Scholar]
- 21.Scheurink AJW, Steffens AB, Gaykema RPA. Hypothalamic adrenoceptors mediate sympathoadrenal activity in exercising rats. Am J Physiol. 1990;259:R470–R477. doi: 10.1152/ajpregu.1990.259.3.R470. [DOI] [PubMed] [Google Scholar]
- 22.Paxinos G, Watson C 1986 The rat brain in stereotaxic coordinates. 2nd ed. New York: Academic Press
- 23.Vrinten DH, Gispen WH, Groen GJ, Adan RA. Antagonism of the melanocortin system reduces cold and mechanical allodynia in mononeuropathic rats. J Neurosci. 2000;20:8131–8137. doi: 10.1523/JNEUROSCI.20-21-08131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawchenko PE, Imaki T, Potter E, Kovacs K, Imaki J, Vale W. The functional neuroanatomy of corticotropin-releasing factor. Ciba Found Symp. 1993;172:5–21. doi: 10.1002/9780470514368.ch2. [DOI] [PubMed] [Google Scholar]
- 25.Gardner JD, Rothwell NJ, Luheshi GN. Leptin affects food intake via CRF-receptor-mediated pathways. Nat Neurosci. 1998;1:103. doi: 10.1038/353. [DOI] [PubMed] [Google Scholar]
- 26.Uehara Y, Shimizu H, Ohtani K, Sato N, Mori M. Hypothalamic corticotropin-releasing hormone is a mediator of the anorexigenic effect of leptin. Diabetes. 1998;47:890–893. doi: 10.2337/diabetes.47.6.890. [DOI] [PubMed] [Google Scholar]
- 27.Masaki T, Yoshimichi G, Chiba S, Yasuda T, Noguchi H, Kakuma T, Sakata T, Yoshimatsu H. Corticotropin-releasing hormone-mediated pathway of leptin to regulate feeding, adiposity, and uncoupling protein expression in mice. Endocrinology. 2003;144:3547–3554. doi: 10.1210/en.2003-0301. [DOI] [PubMed] [Google Scholar]
- 28.Lu XY, Barsh GS, Akil H, Watson SJ. Interaction between α-melanocyte stimulating hormone and corticotropin-releasing hormone in the regulation of feeding and hypothalamo-pituitary-adrenal responses. J Neurosci. 2003;23:7863–7872. doi: 10.1523/JNEUROSCI.23-21-07863.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarkar S, Legradi G, Lechan RM. Intracerebroventricular administration of α-melanocyte stimulating hormone increases phosphorylation of CREB in TRH and CRH-producing neurons of the hypothalamic paraventricular nucleus. Brain Res. 2002;945:50–59. doi: 10.1016/s0006-8993(02)02619-7. [DOI] [PubMed] [Google Scholar]
- 30.Chandler PC, Viana JB, Oswald KD, Wauford PK, Boggiano MM. Feeding response to melanocortin agonist predicts preference for and obesity from a high-fat diet. Physiol Behav. 2005;85:221–230. doi: 10.1016/j.physbeh.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 31.WHO Obesity: preventing and managing the global epidemic. World Health Organization Tech Rep Ser 894. Geneva, 2000 [PubMed]
- 32.Wyatt HR. The prevalence of obesity. Prim Care. 2003;30:267–279. doi: 10.1016/s0095-4543(03)00006-x. [DOI] [PubMed] [Google Scholar]
- 33.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath T, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 34.Cone RD. The corticotropin-releasing hormone system and feeding behavior: a complex web begins to unravel. Endocrinology. 2000;141:2713–2714. doi: 10.1210/endo.141.8.7700. [DOI] [PubMed] [Google Scholar]
- 35.Bradbury MJ, McBurnie MI, Denton DA, Lee KF, Vale WW. Modulation of urocortin-induced hypophagia and weight loss by corticotropin-releasing factor receptor 1 deficiency in mice. Endocrinology. 2000;141:2715–2724. doi: 10.1210/endo.141.8.7606. [DOI] [PubMed] [Google Scholar]
- 36.Makara GB, Stark E, Karteszi M, Palkovits M, Rappay G. Effects of paraventricular lesions on stimulated ACTH release and CRF in stalk-median eminence of the rat. Am J Physiol. 1981;240:E441–E446. doi: 10.1152/ajpendo.1981.240.4.E441. [DOI] [PubMed] [Google Scholar]
- 37.Boston BA, Blaydon KM, Varnerin J, Cone RD. Independent and additive effects of central POMC and leptin pathways on murine obesity. Science. 1997;278:1641–1644. doi: 10.1126/science.278.5343.1641. [DOI] [PubMed] [Google Scholar]
- 38.Sarkar S, Lechan RM. Central administration of neuropeptide Y reduces α-melanocyte-stimulating hormone-induced cyclic adenosine 5′-monophos-phate response element binding protein (CREB) phosphorylation in pro-thyrotropin-releasing hormone neurons and increases CREB phosphorylation in corticotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 2003;144:281–291. doi: 10.1210/en.2002-220675. [DOI] [PubMed] [Google Scholar]
- 39.Wanting Xu A, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005;115:951–958. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heiman ML, Ahima RS, Craft LS, Schoner B, Stephens TW, Flier JS. Leptin inhibition of the hypothalamic-pituitary-adrenal axis in response to stress. Endocrinology. 1997;138:3859–3863. doi: 10.1210/endo.138.9.5366. [DOI] [PubMed] [Google Scholar]
- 41.Arvanti K, Huang Q, Richard D. Effects of leptin and corticosterone on the expression of corticosterone-releasing hormone, agouti related protein, and proopiomelanocortin in the brain of ob/ob mouse. Neuroendocrinology. 2001;73:227–236. doi: 10.1159/000054639. [DOI] [PubMed] [Google Scholar]
- 42.Nowak KW, Pierchala-Koziec K, Tortorella C, Nussdorfer GG, Malenowicz LK. Effects of prolonged leptin infusion on rat pituitary-adrenocortical function. Int J Mol Med. 2002;9:61–64. [PubMed] [Google Scholar]
- 43.Seeley RJ, Matson CA, Chavez M, Woods SC, Dallman MF, Schwartz MW. Behavioral, endocrine, and hypothalamic responses to involuntary over-feeding. Am J Physiol. 1996;271:R819–R823. doi: 10.1152/ajpregu.1996.271.3.R819. [DOI] [PubMed] [Google Scholar]
- 44.Hagan MM, Rushing PA, Schwartz MW, Yagaloff KA, Burn P, Woods SC, Seeley RJ. Role of the CNS melanocortin system in the response to overfeeding. J Neurosci. 1999;19:2362–2367. doi: 10.1523/JNEUROSCI.19-06-02362.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woods SC. The eating paradox: how we tolerate food. Psychol Rev. 1991;98:488–505. doi: 10.1037/0033-295x.98.4.488. [DOI] [PubMed] [Google Scholar]
- 46.Thiele TE, van Dijk G, Campfield LA, Smith FJ, Burn P, Woods SC, Bernstein IL, Seeley RJ. Central infusion of GLP-1, but not of leptin, produces conditioned taste aversions in rats. Am J Physiol. 1997;272:R726–R730. doi: 10.1152/ajpregu.1997.272.2.R726. [DOI] [PubMed] [Google Scholar]
- 47.van Dijk G, Donahey JCK, Thiele TE, Scheurink AJW, Steffens AB, Wilkinson CW, Tenenbaum R, Campfield LA, Burn P, Seeley RJ, Woods SC. Central leptin stimulates corticosterone secretion at the onset of the dark phase. Diabetes. 1997;46:1911–1914. doi: 10.2337/diabetes.46.11.1911. [DOI] [PubMed] [Google Scholar]
- 48.Motawei K, Pyner S, Ranson RN, Kamel M, Coote JH. Terminals of paraventricular spinal neurones are closely associated with adrenal medullary sympathetic preganglionic neurones: immunocytochemical evidence for vasopressin as a possible neurotransmitter in this pathway. Exp Brain Res. 1999;126:68–76. doi: 10.1007/s002210050717. [DOI] [PubMed] [Google Scholar]
- 49.Widdowson PS, Upton R, Buckingham R, Arch J, Williams G. Inhibition of food response to intracerebroventricular injection of leptin is attenuated in rats with diet-induced obesity. Diabetes. 1997;46:1782–1785. doi: 10.2337/diab.46.11.1782. [DOI] [PubMed] [Google Scholar]
- 50.Scarpace PJ, Matheny M, Zolotukhin S, Tümer N, Zhang Y. Leptin-induced leptin resistant rats exhibit enhanced responses to the melanocortin agonist MTII. Neuropharmacology. 2003;45:211–219. doi: 10.1016/s0028-3908(03)00158-8. [DOI] [PubMed] [Google Scholar]
- 51.Benthem L, Keizer K, Wiegman CH, de Boer SF, Strubbe JH, Steffens AB, Kuipers F, Scheurink AJ. Excess portal venous long-chain fatty acids induce syndrome X via HPA axis and sympathetic activation. Am J Physiol Endocrinol Metab. 2000;279:E1286–E1293. doi: 10.1152/ajpendo.2000.279.6.E1286. [DOI] [PubMed] [Google Scholar]
- 52.Bjorntorp P. Body fat distribution, insulin resistance, and metabolic diseases. Nutrition. 1997;13:795–803. doi: 10.1016/s0899-9007(97)00191-3. [DOI] [PubMed] [Google Scholar]
- 53.Cusin I, Zakrzewska KE, Boss O, Muzzin P, Giacobino JP, Ricquier D, Jeanrenaud B, Rohner-Jeanrenaud F. Chronic central leptin infusion enhances insulin-stimulated glucose metabolism and favors the expression of uncoupling proteins. Diabetes. 1998;47:1014–1019. doi: 10.2337/diabetes.47.7.1014. [DOI] [PubMed] [Google Scholar]
- 54.Shi ZQ, Nelson A, Whitcomb L, Wang J, Cohen AM. Intracerebroventricular administration of leptin markedly enhances insulin sensitivity and systemic glucose utilization in conscious rats. Metabolism. 1998;47:1274–1280. doi: 10.1016/s0026-0495(98)90336-5. [DOI] [PubMed] [Google Scholar]
- 55.Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108:1079–1085. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108:1113–11121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen P, Miyazaki M, Socci ND, Hagge-Greenberg A, Liedtke W, Soukas AA, Sharma R, Hudgins LC, Ntambi JM, Friedman JM. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- 58.Aiston S, Peak M, Agius L. Impaired glycogen synthesis in hepatocytes from Zucker fatty rats: the role of increased phosphorylase activity. Diabetologia. 2000;43:589–597. doi: 10.1007/s001250051348. [DOI] [PubMed] [Google Scholar]
- 59.Liu L, Karkanias GB, Morales JC, Hawkins M, Barzilai N, Wang J, Rossetti L. Intracerebroventricular leptin regulates hepatic but not peripheral glucose fluxes. J Biol Chem. 1998;273:31160–31167. doi: 10.1074/jbc.273.47.31160. [DOI] [PubMed] [Google Scholar]
- 60.Li G, Zhang Y, Wilsey JT, Scarpace PJ. Unabated anorexic and enhanced thermogenic responses to melanotan II in diet-induced obese rats despite reduced melanocortin 3 and 4 receptor expression. J Endocrinol. 2004;182:123–132. doi: 10.1677/joe.0.1820123. [DOI] [PubMed] [Google Scholar]
- 61.Harrold JA, Williams G, Widdowson PS. Early leptin response to a palatable diet predicts dietary obesity in rats: key role of melanocortin-4 receptors in the ventromedial hypothalamic nucleus. J Neurochem. 2000;74:1224–1228. doi: 10.1046/j.1471-4159.2000.741224.x. [DOI] [PubMed] [Google Scholar]
- 62.Plotsky PM, Sawchenko PE. Hypophysial-portal plasma levels, median eminence content, and immunohistochemical staining of corticotropin-releasing factor, arginine vasopressin, and oxytocin after pharmacological adrenalectomy. Endocrinology. 1987;120:1361–1369. doi: 10.1210/endo-120-4-1361. [DOI] [PubMed] [Google Scholar]
- 63.Wilsey J, Zolotukhin S, Prima V, Scarpace PJ. Central leptin gene therapy fails to overcome leptin resistance associated with diet-induced obesity. Am J Physiol. 2003;285:R1011–R1020. doi: 10.1152/ajpregu.00193.2003. [DOI] [PubMed] [Google Scholar]
- 64.Li G, Mobbs CV, Scarpace PJ. Central pro-opiomelanocortin gene delivery results in hypophagia, reduced visceral adiposity, and improved insulin sensitivity in genetically obese Zucker rats. Diabetes. 2003;52:1951–1957. doi: 10.2337/diabetes.52.8.1951. [DOI] [PubMed] [Google Scholar]
- 65.Pierroz DD, Ziotopoulon M, Ungsunan L, Moschos S, Flier JS, Mantzoros CS. Effects of acute and chronic administration of the melanocortin agonist MTII in mice with diet-induced obesity. Diabetes. 2002;51:1337–1345. doi: 10.2337/diabetes.51.5.1337. [DOI] [PubMed] [Google Scholar]
- 66.Bluher S, Ziotopoulou M, Bullen JW, Jr, Moschos SJ, Ungsunan L, Kokkotou E, Maratos-Flier E, Mantzoros CS. Responsiveness to peripherally administered melanocortins in lean and obese mice. Diabetes. 2004;53:82–90. doi: 10.2337/diabetes.53.1.82. [DOI] [PubMed] [Google Scholar]