

Abstract
The recently developed subgenomic hepatitis C virus (HCV) replicons were limited by the fact that the sequence encoding the structural proteins was missing. Therefore, important information about a possible influence of these proteins on replication and pathogenesis and about the mechanism of virus formation could not be obtained. Taking advantage of three cell culture-adaptive mutations that enhance RNA replication synergistically, we generated selectable full-length HCV genomes that amplify to high levels in the human hepatoma cell line Huh-7 and can be stably propagated for more than 6 months. The structural proteins are efficiently expressed, with the viral glycoproteins E1 and E2 forming heterodimers which are stable under nondenaturing conditions. No disulfide-linked glycoprotein aggregates were observed, suggesting that the envelope proteins fold productively. Electron microscopy studies indicate that cell lines harboring these full-length HCV RNAs contain lipid droplets. The majority of the core protein was found on the surfaces of these structures, whereas the glycoproteins appear to localize to the endoplasmic reticulum and cis-Golgi compartments. In agreement with this distribution, no endoglycosidase H-resistant forms of these proteins were detectable. In a search for the production of viral particles, we noticed that these cells release substantial amounts of nuclease-resistant HCV RNA-containing structures with a buoyant density of 1.04 to 1.1 g/ml in iodixanol gradients. The same observation was made in transient-replication assays using an authentic highly adapted full-length HCV genome that lacks heterologous sequences. However, the fact that comparable amounts of such RNA-containing structures were found in the supernatant of cells carrying subgenomic replicons demonstrates a nonspecific release independent of the presence of the structural proteins. These results suggest that Huh-7 cells lack host cell factors that are important for virus particle assembly and/or release.
The hepatitis C virus (HCV) was identified as the causative agent for most posttransfusion and sporadic non-A, non-B hepatitis cases (11, 45). According to recent estimates, about 170 million individuals worldwide are infected. One striking characteristic of HCV is its strong propensity to persist in the infected host, which often leads to severe liver damage, ranging from chronic hepatitis to liver cirrhosis and even hepatocellular carcinoma (33). The possible immune evasion strategies that allow persistent viral replication in the presence of the host's immune response are not well understood, but the high variability of the virus appears to be a key determinant (38). As a consequence, HCV isolates exhibit marked sequence diversity and have been grouped according to phylogenetic analysis into six different genotypes which together form the genus Hepacivirus within the family Flaviviridae (60).
HCV particles are enveloped, have a diameter of 55 to 65 nm, and harbor an ∼9,600-nucleotide-long plus-strand RNA genome. It carries a single long open reading frame (ORF), which is flanked by highly conserved and structured nontranslated regions (NTRs), both of which have been shown to be required for RNA replication (25, 42, 70). The 5′ NTR also harbors an internal ribosome entry site (IRES) which directs the expression of a large polyprotein that is co- and posttranslationally cleaved by cellular and viral proteases into at least 10 mature viral proteins (3, 59). Analogous to other flaviviruses, the nonstructural proteins presumably form an ordered replicase complex, which associates with intracellular membranes. Even though the exact conformation of this complex is currently unknown, extensive in vitro studies have identified several enzymatic activities within the nonstructural proteins shedding light on the organization and function of the replication machinery. The NS3 protein is the key protease of HCV which in conjunction with NS4A mediates all cleavages in the NS3-to-NS5B region (4, 28, 69). Furthermore, NS3 harbors nucleoside triphosphatase and helicase activities (40, 66). While the function of NS4B in the viral replication cycle remains to be defined, NS5A was found to be a highly phosphorylated polypeptide that may be involved in the resistance to the antiviral effects elicited by alpha interferon (23, 24, 26, 27). Interestingly, a large number of adaptive mutations that increase the replication efficiency of subgenomic HCV replicons in Huh-7 cells map within the NS5A gene, in part affecting potential phosphorylation sites (9, 30, 43, 48). Although this suggests a direct role of this protein in RNA replication, the exact mechanism is currently obscure. NS5B, which is the most C-terminal cleavage product of the polyprotein, constitutes the RNA-dependent RNA polymerase (RdRp) (8, 47).
The viral structural proteins core, E1, and E2 are located in the N-terminal third of the ORF separated from the nonstructural proteins by a short hydrophobic polypeptide (p7) of unknown function. The HCV core protein has been shown to bind RNA and is believed to be responsible for genome packaging (44). Two major core species with apparent molecular masses of 23 and 21 kDa (p21 and p23) have been described, corresponding to the unprocessed precursor and the processed form that lacks at its C terminus the signal sequence of E1 (36, 46, 53, 61, 71). As p21 predominates both in transfected tissue culture cells and in virus particles from infected sera, it is believed to be the mature form (71). The glycoproteins E1 and E2 are liberated by signalase cleavages and associate to form noncovalently linked heterodimers which are retained in the endoplasmic reticulum (ER) due to the presence of specific retention signals within their respective transmembrane domains (15-18, 20, 22, 29, 58, 64; for a recent review, see reference 19). In addition to these native heterodimeric glycoprotein complexes which are stabilized by noncovalent interactions, the production of disulfide-linked E1-E2 aggregates is well documented (18, 20). Although it is generally thought that the native complexes are the prebudding form, the specific contribution of either glycoprotein form to virus assembly is currently unknown.
Due to low virus titers, isolation and characterization of HCV particles from the sera of infected individuals are difficult. Furthermore, the virus population in these sera is very heterogeneous, as viral particles associate with immunoglobulins (12, 31, 68) and β-lipoproteins (52, 57, 67), complicating direct morphological studies. Different buoyant densities of HCV derived from patient sera have been reported. HCV-immunoglobulin complexes from the sera of chronically infected patients exhibit an intermediate density ranging between 1.18 and 1.21 g/ml (12). A second population of HCV with a very low density of ∼1.08 to 1.11 g/ml in sucrose was observed and most likely represents the infectious form of the virus (10, 31, 52). Recent studies revealed the presence of significant amounts of viral particles with a buoyant density of 1.32 to 1.34 g/ml in CsCl density gradients and the physicochemical, morphological, and antigenic properties of HCV nucleocapsids in plasma samples from infected individuals (50). These structures closely resemble nonenveloped virus-like particles produced in insect cells upon infection with recombinant baculoviruses harboring cDNA for the structural region of an HCV genotype 1b isolate (7). Using this system, large amounts of both nonenveloped and enveloped virus-like particles can be prepared, which are important surrogate models that can be used to investigate virus receptor usage and entry into the host cell.
A variety of different approaches, mainly based on infection of primary human liver cells or diverse cell lines of hepatic or lymphoid origin with human patient sera have been explored in order to establish a robust in vitro infection system for HCV (5, 39). However, so far the success of these attempts has been limited due to the extremely low replication levels that do not allow detailed studies. The recent development of subgenomic HCV replicons, which allow high-level replication of HCV RNAs in cell culture, has overcome this hurdle (49). However, since the complete structural region was deleted, virus assembly and particle release could not be studied with this system. Moreover, important information about a potential influence of the structural proteins on the host cell could not be obtained.
To establish a system that allows efficient replication of the complete HCV genome in cell culture, in this study we constructed cell culture-adapted selectable full-length HCV genomes (sfl genomes). We show that these genomes stably replicate and express all viral proteins for prolonged cultivation periods. Furthermore, an authentic HCV genome that carries cell culture-adaptive mutations was generated and shown to replicate to high levels in transiently transfected cells. However, despite the release of nuclease-resistant HCV RNA into the cell culture supernatant, no evidence for virus particle assembly was found by either approach, suggesting that host cell factors important for virus production are not present in Huh-7 cells.
MATERIALS AND METHODS
Cell culture.
Monolayers of the human hepatoma cell line Huh-7 (54) were grown in Dulbecco's modified minimal essential medium (Life Technologies GmbH, Karlsruhe, Germany) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U of penicillin per ml, 100 μg of streptomycin per ml, and 10% fetal calf serum (complete DMEM). For cell lines carrying HCV replicons, various concentrations of G418 (Geneticin; Life Technologies GmbH) were added to the culture medium (100 μg/ml for cell line 20-1; 250 μg/ml for cell lines 21-5 and 21-7; 500 μg/ml for cell lines 22-1, 22-2, 22-6, and 22-16). Cells were routinely passaged twice a week at a dilution of 1:3 to 1:4, depending on confluency.
Plasmid construction.
Full-length HCV constructs were generated based on the consensus genotype 1b isolate Con1 obtained from a German patient (41) (EMBL database accession number AJ238799). To create a construct with the complete HCV ORF, we first introduced an NcoI site at the beginning of the HCV ORF into the pTMC1 vector harboring the Con1 isolate downstream of the T7 promoter (41). Due to this PCR-based mutagenesis, the second amino acid in core was changed from serine to glycine. The entire HCV ORF and the 3′ NTR were then transferred to the subgenomic replicon vector pFK-I389neo/NS3-3′/wt (48, 49) (EMBL accession number AJ242654) using NcoI and SpeI restriction sites. The resulting plasmid pFK-I389/Core-3′/wt harbors a bicistronic full-length genome with the authentic HCV 5′ NTR directing the expression of the neomycin phosphotransferase gene (neo) and an IRES element derived from the encephalomyocarditis virus (EMCV) permitting expression of the complete HCV polyprotein. To increase the replication efficiency, the SfiI fragment (positions 3615 to 8492; numbering according to the published Con1 sequence) derived from the highly adapted subgenomic replicon rep 5.1 (43) was introduced into the full-length construct. The transferred DNA carries three adaptive mutations (E1202G, T1280I, and S2197P; numbering according to the polyprotein of the published Con1 sequence) and four further mutations that do not affect replication efficiency in cell culture (43). The Con1-ET construct used for transient-replication assays is a derivative of HCV strain Con1 that was modified by PCR-based techniques to possess three cell culture-adaptive mutations (E1202G, T1280I, and K1846T; V. Lohmann et al., unpublished results). Similarly, a subgenomic replicon (replicon-ET) that carries these mutations and the sh ble gene (Zeozine resistance gene) in place of neo was created.
Electroporation, generation of cell lines bearing sfl genomes, and transient-replication assay.
In vitro transcription was performed as described previously (49). To generate cell lines harboring sfl genomes, 4 × 106 Huh-7 cells were electroporated with 1 μg of purified in vitro transcripts. Cells were seeded into three 10-cm-diameter-dishes and after 24 h were selected with G418 at a concentration of 100, 250, or 500 μg/ml, respectively. Approximately 3 to 5 weeks postelectroporation, small colonies were visible; these colonies were isolated and expanded for further analysis. For transient-replication assays, 2.4 × 107 Huh-7 cells were electroporated with 60 μg of HCV RNA. At the time points given, cells were harvested and analyzed by Northern and Western blotting. At 96 h posttransfection, supernatants were harvested, cleared of cellular debris, and subjected to density gradient centrifugation as described below.
Northern blot analysis and quantification of HCV RNA.
The methods used for preparation of total RNA and Northern blotting have been described recently (56). HCV- and β-actin-specific bands were quantified by phosphorimaging using a BAS 2500 scanner (Fuji), and the number of replicon molecules was determined by comparison with a serial dilution of in vitro transcripts loaded in parallel onto the gel. β-Actin was used to correct for different amounts of total RNA loaded in each lane of the gel.
Amplification of sfl genome RNA, cloning of amplified DNA fragments, and sequence analysis.
The complete HCV ORF from cell lines 20-1 and 21-5 (passages 52 and 57, respectively) was amplified in two overlapping fragments by reverse transcription-PCR (RT-PCR) as described recently (48) with minor modifications. One microgram of total RNA was mixed with 50 pmol of primer A6103 (5′-GCT ATC AGC CGG TTC ATC CAC TG-3′) or A9412 (5′-CAG GAT GGC CTA TTG GCC TGG AG-3′) in a total volume of 10.5 μl. Samples were denatured for 10 min at 65°C and then subjected to reverse transcription with the Expand-RT system (Roche Biochemicals, Mannheim, Germany) as recommended by the manufacturer in a total volume of 20 μl. After 1 h at 42°C, 1/10 of the reaction mixture was used for PCR with the Expand Long Template PCR kit (Roche Biochemicals) according to the instructions of the manufacturer. Cycle conditions were as follows: an initial denaturation step for 2 min at 94°C, followed by 40 cycles, with 1 cycle consisting of 10 s at 94°C, 90 s at 54°C, and 5 min at 68°C. After 10 cycles, the extension time was increased by 10 s for each additional cycle. Finally, the reaction mixture was incubated for 10 min at 68°C. The 5′ PCR was performed with primers S59 (5′-TGT CTT CAC GCA GAA AGC GTC TAG-3′) and A4919 (5′-AGC ACA GCC CGC GTC ATA GCA CTC G-3′), and the PCR product was inserted into pFK-I389neo/NS3-3′/wt after restriction with AseI and SalI. The 3′ PCR was performed with primers S4542-HindIII (5′-CCA AGC TTG ATG AGC TCG CCG CGA AGC TGT CC-3′) and A9386-SpeI (5′-CGT ACT AGT TAG CTC CCC GTT CAT CGG TTG G-3′), and after restriction with HindIII and SpeI, the fragment was inserted into a modified pUC18 vector. Sequencing was performed with a set of 44 primers, the ABI PRISM Big Dye Terminator Cycle Sequencing Ready Reaction Kit, and ABI PRISM DNA Analyzer 3700 or 377 (PE Applied Biosystems, Warrington, United Kingdom). The Sequencher software (Gene Codes Corporation, Ann Arbor, Mich.) was used for the assembly and analysis of the sequences.
Western blotting and immunofluorescence.
Cells were washed once with phosphate-buffered saline (PBS) and detached from the plate by treatment with 0.05% trypsin-0.02% EDTA. Cells contained in a small aliquot of the suspension were counted, and the remaining cells were lysed by sonification in denaturing protein sample buffer (200 mM Tris-HCl [pH 8.8], 5 mM EDTA, 0.1% bromophenol blue, 10% sucrose, 3.3% sodium dodecyl sulfate [SDS], 2% 2-mercaptoethanol [2-ME]). Aliquots of cell lysates corresponding to 4 × 105 cells were analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (11% polyacrylamide) and transferred to a polyvinylidene difluoride membrane (PolyScreen; NEN Life Science Products, Zaventem, Belgium) using a semidry blotter (Bio-Rad, Munich, Germany) according to the manufacturer's instructions. NS5B and core protein were detected as described previously using monoclonal antibodies that were kindly provided by D. Moradpour (56). For immunofluorescence, cells were grown on glass coverslips. Three days postseeding, cells were incubated with medium containing 20 nM brefeldin A (Molecular Probes, Eugene, Oreg.) for 3 h or left untreated. Subsequently, cells were fixed with methanol-acetone, and HCV proteins were detected essentially as described previously (56). Briefly, either a core-specific mouse monoclonal antibody or a monoclonal rat antibody directed against E2, kindly provided by J. McKeating, was used at a dilution of 1:100 or 1:500, respectively. Anti-human Golgin 97 mouse monoclonal antibodies (Molecular Probes) were used at a dilution of 1:300 to stain the Golgi complex. Bound antibodies were detected with Cy3-conjugated goat anti-mouse immunoglobulin G (IgG) (Dianova, Hamburg, Germany) or fluorescein isothiocyanate (FITC)-conjugated rabbit anti-rat IgG (Sigma, Deisenhofen, Germany) at a concentration of 1:1,000 or 1:500, respectively. The cell nucleus was counterstained employing bisbenzimide (Hoechst 33342; Sigma) at a concentration of 10 μg/ml. Cells were examined with a Zeiss 510 laser scan microscope.
Metabolic radiolabeling of proteins, immunoprecipitation, and deglycosylation.
A total of 7 × 104 cells were seeded in a 3-cm-diameter culture dish in complete DMEM supplemented with Geneticin. Four days later, cells were washed, starved in methionine-free medium for 60 min, and incubated for 3 h in methionine-free DMEM supplemented with Express Labeling Mix (200 μCi/ml; NEN Life Science Products). Cells were lysed directly. For pulse-chase analysis, the cells were washed several times and incubated in complete DMEM for the time periods given in Results. Cell lysis and immunoprecipitation under nondenaturing conditions have been described previously (6). For deglycosylation studies, washed immunocomplexes were denatured for 10 min at 95°C in denaturing buffer (0.5% SDS, 1% 2-ME) and divided into two aliquots. Samples were incubated with or without 2,000 U of endoglycosidase H (New England Biolabs, Frankfurt, Germany) for 1 h at 37°C. Deglycosylation was stopped by the addition of an equal volume of protein sample buffer. Samples were heated at 95°C for 5 min, and proteins were analyzed by SDS-PAGE (10% polyacrylamide). For immunoprecipitations performed under nonreducing conditions, lysis buffer (50 mM Tris-Cl [pH 7.5], 150 mM NaCl, 1% Nonidet P-40 [NP-40], 1% sodium deoxycholate, 0.1% SDS) was supplemented with 20 mM iodoacetamide (Sigma) to prevent oxidation of thiol groups during the immunoprecipitation procedure. Prior to SDS-PAGE, samples were denatured in protein sample buffer lacking 2-ME.
EM.
The procedures used for electron microscopy (EM) and immuno-EM have been described recently (56) and were applied with the following modifications. The grids were kept overnight at 4°C in a mixture of rabbit polyclonal antiserum against E2 glycoprotein (41) and a mouse monoclonal antibody specific for the core polypeptide of HCV, kindly provided by D. Moradpour. After the grids were washed five times in TCG (150 mM NaCl, 20 mM Tris-HCl [pH 7.5], 0.5% casein, 0.2% gelatin), they were treated with a cocktail consisting of goat anti-rabbit antibodies and anti-mouse antibodies coupled to colloid gold particles with a diameter of 10 and 6 nm, respectively (Dianova). After 1 h of incubation at room temperature, the reaction was terminated by repeated rinsing in TCG. Finally, the grids were washed with distilled water, stained with 4% aqueous uranyl acetate and Reynold's lead citrate, air dried, and examined at 60 kV with a Philips CM120 EM. For controls, we used ultrathin sections of Huh-7 cells treated as described above and thin sections of cells with full-length replicons that were incubated with a cocktail consisting of irrelevant antibodies (polyclonal rabbit antiserum directed against the capsid protein of human immunodeficiency virus type 1 [HIV-1] and mouse monoclonal antibodies specific for measles virus phosphoprotein). Gold-labeled secondary antibodies were the same as described above. Additional controls were done by omitting the primary antibodies from the labeling procedure.
Ultracentrifugations and nuclease treatment of cell culture supernatants.
Supernatant (10 to 30 ml) derived from ∼1 × 107 to 3 × 107 cells was harvested 4 to 7 days postseeding, and cellular debris was removed by low-speed centrifugation at 13,000 × g for 30 min at 4°C, followed by filtration with 0.45-μm-pore-size filters (Millipore, Molsheim, France). Supernatant bearing infectious bovine viral diarrhea virus (BVDV) was harvested 24 h postinfection of R.D.420 bovine testicle cells (kindly provided by R. Donis) with virus stocks of cytopathic BVDV strain NADL (kindly provided by R. Donis). For nuclease resistance assays, samples were spun in an L8-80 M Beckman ultracentrifuge (Beckman, Palo Alto, Calif.) at 110,000 × g at 4°C for 6 h. Sediments were resuspended in PBS, and different aliquots were supplemented with various amounts of RNase A and NP-40 as indicated in Results. After 4 h of incubation at room temperature, RNA was prepared by phenol-chloroform extraction as described below. For density gradient analyses, 30 ml of cleared culture fluid was spun over a 40% (wt/vol) iodixanol (Optiprep; Invitrogen, Karlsruhe, Germany) density cushion (ρ = 1.215 g/ml), prepared in CSM (0.85% [wt/vol] NaCl, 10 mM Tricine-NaOH [pH 7.4]; ρ = 1,006 g/ml) for 6 h at 52,000 × g at 4°C. The supernatant was discarded, and the interface and density cushion (∼2.5 ml) were transferred to the bottom of a fresh tube. Samples were overlaid with a linear iodixanol gradient (28 to 0%) and spun for 16 h at 110,000 × g at 4°C. Twelve fractions (1 ml each) were taken from the top of the tube, and RNA was prepared as described below.
RNA preparation and quantitative Taqman RT-PCR.
Gradient fractions and resuspended sediments were supplemented with 0.1% SDS and extracted with an equal volume of a phenol-chloroform mixture (1:1). After 15 min of centrifugation at 18,000 × g, the nucleic acids in the aqueous phase were transferred to a new tube, precipitated with isopropanol, washed once with 70% ethanol, and resuspended in 20 μl of double-distilled water. Five microliters of the sample was used for quantitative RT-PCR analysis using an ABI PRISM 7700 Sequence Detector (Taqman; Perkin-Elmer, Wellesley, Mass.). Amplifications were conducted in duplicate with the One Step RT-PCR Kit (Qiagen, Hilden, Germany) using the following primers and 3′-phosphate-blocked, 6-carboxyfluorescine (6-FAM)- and tetrachloro-6-carboxyfluorescine (TAMRA)-labeled probes (TIB Molbiol, Berlin, Germany): BVDV Taqman probe, 5′-6FAM-ATA GCA GCT CTG AGC TTT GGT TGA GGC AT-TAMRA-3′; BVDV-S6291, 5′-CCG GGA TAA CCT ATG CAT CAT ACG-3′; BVDV-A6401, 5′-TGG CAC AAT GGT ATT CAT CTA AGA-3′; HCV Taqman probe, 5′-6FAM-TCC TGG AGG CTG CAC GAC ACT CAT-TAMRA-3′; HCV-S66, 5′-ACG CAG AAA GCG TCT AGC CAT-3′; and HCV-A165, 5′-TAC TCA CCG GTT CCG CAG A-3′. Reactions were performed in three stages under the following conditions: stage 1, 60 min at 50°C (reverse transcription reaction); stage 2, 15 min at 95°C (heat inactivation of reverse transcriptase and activation of Taq polymerase); stage 3, 40 cycles, with 1 cycle consisting of 15 s at 95°C and 1 min at 60°C (PCR amplification). The total volume of the reaction mix was 15 μl and contained the following components: 2.66 μM 6-carboxy-X-rhodamine (Rox, passive reference), 4 mM MgCl2, 0.66 mM deoxynucleoside triphosphates, 0.266 μM probe, 1 μM (each) sense and antisense primer, and 0.6 μl of enzyme mix. The amounts of HCV and BVDV RNA were calculated by comparison to serially diluted in vitro transcripts included in the RT-PCR analysis.
RESULTS
Establishment of Huh-7 cells carrying sfl genomes.
To allow analysis of the complete viral life cycle in vitro, we wished to establish cell lines that harbor autonomously replicating full-length HCV genomes. Initially, we generated a selectable HCV genome that was based on the original sequence of the Con1 isolate and that had an organization similar to that of the subgenomic replicons described previously (49). However, we failed to establish viable cell clones after transfection of this RNA and selection with G418 (data not shown). Therefore, after we had identified cell culture-adaptive mutations that increase HCV RNA replication dramatically, an sfl genome that carried an SfiI fragment derived from the highly adapted subgenomic replicon rep 5.1 was generated (Fig. 1A) (43). In addition to the heterologous sequences introduced, the resulting HCV genome differed from the original genome by six amino acid substitutions. While three of these mutations did not affect replication, the combination of the three remaining ones synergistically increased RNA replication efficiency (E1202G and T1280I in NS3 and S2197P in NS5A) (43). Upon transfection of Huh-7 cells with this sfl genome, we were able to generate a panel of G418-resistant colonies. However, it is noteworthy that the efficiency of colony formation (ECF) per microgram of transfected RNA was about 3 to 4 orders of magnitude lower than the ECF of the respective subgenomic replicon rep 5.1. Despite the low number of colonies (<100/μg of RNA), several independent cell lines could be established. As can be seen in the Northern blot shown in Fig. 1B, all cell lines examined carried an HCV RNA of correct length, demonstrating that the sfl genomes were capable to replicate efficiently in Huh-7 cells.
FIG. 1.

Generation of Huh-7 cell lines bearing sfl genomes. (A) Schematic representations of the structures of the HCV genomes and selectable HCV RNAs used in this study. The length of the construct (number of nucleotides) is given in parentheses to the right of each construct. The parental HCV genome Con1 is shown at the top. The proteins of the polyprotein encoded by the viral RNA are shown (open boxes), and cleavage sites between given proteins are indicated by vertical lines. 5′ and 3′ NTRs flanking the polyprotein are indicated (thick lines). The Con1-ET construct is a derivative that carries three cell culture-adaptive mutations (E1202G, T1280I, and K1846T; Lohmann et al., unpublished) indicated by black dots. The selectable HCV RNAs depicted below both contain an internal EMCV IRES (E-I) directing the expression of core to 5B (sfl genome) or NS3 to 5B (replicon-ET). The selectable marker (neo or zeo, respectively) is expressed via the HCV IRES. While replicon-ET carries the same combination of adaptive mutations as Con1-ET, the sfl genome harbors the SfiI fragment (shaded area) derived from the highly adapted subgenomic replicon rep 5.1 (contributing adaptive mutations E1202G, T1280I, and S2197P). Note that the HCV IRES extends into the core coding region and therefore the selectable markers (neo and zeo) are expressed as fusion proteins carrying the initial 12 amino acids of HCV core. (B) Northern blot of cell lines carrying an sfl genome. Total RNA from seven different sfl cell lines (lanes 2 to 8) was prepared, and 2 μg was analyzed by Northern blotting using 32P-labeled riboprobes complementary to a region within the NS5B gene of HCV and β-actin. For a reference, 2 μg of total RNA derived from naive Huh-7 cells (lane 1) or from subgenomic replicon cell line 9-13 (49) (lane 9) as well as serial dilutions of the original in vitro transcripts (subgenomic replicon [lanes 10 to 12] and sfl genome [lanes 13 to 15]) were analyzed in parallel.
To determine the replication levels of sfl genomes in the established cell lines more precisely, cells of each cell line were harvested at different time points between 3 and 7 days postseeding and analyzed by Northern blotting. This procedure was necessary because HCV RNA replication is tightly coupled to host cell proliferation and therefore varies significantly depending on cell growth (56). The average levels of HCV RNA in the cell lines examined ranged between ∼1.5 × 107 to 2.5 × 107 molecules per μg of total RNA (data not shown), which is about fivefold lower than the copy number of a subgenomic replicon present in cell line 9-13 (∼108 molecules per μg of total RNA) (49).
Stability of sfl genomes and genetic drift.
To ensure stable high-level replication of sfl genomes in established cell lines, the cells were continuously maintained in the presence of G418. Under these conditions, only those cells with sufficient amplification of the RNA that carries the selectable marker (neo) could propagate. Hence, selective pressure assured only the maintenance of a functional replicase complex, whereas the viral structural proteins that did not contribute to viral replication might even pose a disadvantage because of potential cytopathogenicity or the increased length of the RNA. In view of these considerations, we wanted to assess whether the sfl genome was stable over extended periods of time. Therefore, six different cell lines carrying an sfl genome were cultured under continuous selection for almost 6 months, corresponding to about 50 passages. Total RNA was isolated from cells at various time points and probed by HCV-specific Northern blotting. As shown in Fig. 2A, sfl genomes in all cell lines included in this follow-up were stable. Interestingly, in cell lines 20-1 and 22-1, shorter RNA molecules coreplicated with the full-length genomes, but at later time points these shorter RNAs disappeared, evidence that the deletions had no selective advantage (data not shown). In summary, these data show that the sfl genome is stably maintained for at least 6 months.
FIG. 2.

Stability of sfl genomes during continuous cell culture. (A) Six different cell lines were continuously cultured in the presence of G418. At the indicated time points (days [d]), cells were lysed, total RNA was prepared, and 4 μg was analyzed by Northern blotting. For comparison, given amounts of RNA containing subgenomic replicon transcribed in vitro were analyzed in parallel. To allow detection of sequence variants and of smaller HCV RNA pieces, the entire blot was hybridized under low-stringency conditions. (B) Genetic drift of the sfl genome in cell line 21-5. Cells were continuously cultured, and after 57 passages (corresponding to ∼6 months), total RNA was prepared. The coding region of the HCV polyprotein was amplified by long-distance RT-PCR in two overlapping fragments shown as bars below the ORF. For each fragment, two independent clones were sequenced. The boundary of the sequenced clones is indicated by a dashed vertical line. Amino acid substitutions are drawn as vertical lines at their respective positions within the HCV polyprotein. The hypervariable region 1 (HVR1) present at the N terminus of E2 is depicted as a shaded area. Six mutations were conserved between both sequenced clones, and they are indicated (stars).
Like all other RNA viruses, HCV encodes an RdRp that lacks a proofreading function. As a consequence, replication is error prone and characterized by high mutation rates. This elevated mutability is a general feature of RNA viruses and has been shown to range from 10−1 to 10−4 substitutions per genome site per year (32). While our long-term study established that deletions within the sfl genome occur only rarely, we wanted to obtain a more detailed picture of genetic drift. Hence, we prepared total RNA from two different cell lines bearing an sfl genome (20-1 and 21-5) after 52 and 57 passages, respectively, corresponding to a cultivation period of ∼6 months. HCV RNA was amplified by RT-PCR in two overlapping fragments, and several clones of each fragment were sequenced. For both the structural and nonstructural regions of the genome, only ∼40% of all nucleotide substitutions detected were silent. An example of the amino acid substitutions found in sfl genomes that were recovered from cell line 21-5 that illustrates the distribution of amino acid changes throughout the complete HCV polypeptide is shown in Fig. 2B. The coding region for the structural proteins appeared to be slightly more likely to acquire mutations than the nonstructural portion of the genome with the exception of the NS5A protein. Interestingly, six substitutions were present in both sequenced clones (K6R, V1282A, G1308S, E1709V, S2327L, and W2405R), which implies that they are dominant in the population of RNA molecules replicating in the cell line at the time the cells were analyzed (passage 57) (Fig. 2B). In summary, our sequencing analysis revealed a high genetic variability throughout the entire polyprotein with an average mutation rate of 8.5 × 10−3 substitutions per amino acid position per 6 months. The highest drift rates (12.2 × 10−3 to 17.4 × 10−3 amino acid changes) were found with the core protein, E1, and p7, implying that the structural region is more variable than the remainder of the sfl genome. This result is in keeping with the notion that only the viral replicase complex is under selective pressure, whereas the structural proteins are subject to free genetic drift.
Characterization of structural protein expression in cell lines with an sfl genome.
Having developed cell lines that harbor stably replicating HCV sfl genomes, we next investigated the expression and maturation of the structural proteins because this is a critical prerequisite for virus formation. As shown in Fig. 3A, the core protein was efficiently expressed in four different cell lines with sfl genomes as a single species with an apparent molecular mass of 21 kDa. This protein most likely corresponded to the processed form of core (positions 1 to 174) from which the signal sequence of E1 had been removed. As evidenced by the pulse-labeling experiment performed on cell line 21-5 and subsequent nondenaturing immunoprecipitations, E2 was expressed as a rather stable E2-p7-NS2 precursor, which could be detected with a chase period of 6 h (Fig. 3B). Furthermore, E2 released from the precursor and small amounts of coprecipitating E1 and NS2 were observed for at least 12 h. Deglycosylation of immunocomplexes with endoglycosidase H revealed the absence of terminally glycosylated forms of both E1 and E2 in the cell line (Fig. 3B). This observation suggests that the glycoproteins were retained in the ER and/or early cis-Golgi compartment and is consistent with earlier reports which report ER retention of both envelope proteins due to the presence of retention signals within their respective transmembrane domains (15, 16, 18, 22, 51).
FIG. 3.

Analysis of structural protein expression in cell lines carrying sfl genomes. (A) Detection of HCV core protein by Western blotting. Lysates, each corresponding to 2 × 105 cells of the given cell lines, were loaded onto a gel, separated by electrophoreses, blotted, and probed with an HCV core-specific monoclonal antibody. (B) Pulse-chase and endoglycosidase H analysis of HCV glycoprotein complexes. Cells were incubated with [35S]methionine/cysteine-containing medium for 1 h, and after the cells were washed extensively, nonradioactive medium was added for the times specified above the lanes. Glycoprotein complexes were isolated by immunoprecipitations from cell lysates under nondenaturing conditions and were either mock treated (− EndoH) or deglycosylated by endoglycosidase H digestion (+ EndoH). After SDS-PAGE, proteins were detected by autoradiography. The deglycosylated forms of the proteins are labeled with asterisks. (C) Analysis of HCV glycoprotein complexes under reducing and nonreducing conditions. Immunoprecipitations were performed in the presence of 20 mM iodoacetamide in the lysis and wash buffer. Prior to loading, half of each sample was boiled in denaturing buffer with or without 2-ME as indicated. Proteins were detected as described above.
As alluded to above, HCV glycoproteins fold by two different pathways, leading to the formation of native E1-E2 heterodimeric complexes or misfolded disulfide-linked glycoprotein aggregates (13, 16, 18, 20, 21). These aggregates can be detected by immunoprecipitations and SDS-PAGE under nonreducing conditions, because the disulfide linkages are maintained and thus, a significant proportion of these E1-E2 aggregates have a high molecular weight and barely enter the gel. However, the migration pattern of the glycoprotein complexes isolated from cells with an sfl genome was not influenced by reducing conditions, suggesting that the majority of the E proteins formed native heterodimers (Fig. 3C). Taken together, these data indicate that the viral structural proteins were efficiently expressed and that E1 and E2 predominantly formed noncovalently linked native glycoprotein complexes.
Morphology of cells with an sfl genome and subcellular localization of structural proteins.
In order to investigate potential cytopathic effects exerted by the replication of sfl genomes and the expression of all HCV proteins, we first analyzed cultures of parental Huh-7 control cells by EM. Naive Huh-7 cells displayed a loose inconspicuous cytoplasmic architecture with variable numbers of vesicles of various sizes (Fig. 4B). The degree of vesiculation was apparently dependent on the cell density and correlated with the culture time after seeding. The vesicles were heterogeneous in size and contained homogenous material that exhibited gray staining. Similarly, cell lines 20-1 and 21-5 with an sfl genome and cells with a subgenomic replicon revealed strong cytoplasmic vesiculation. Since the frequency and distribution of vesicles in the cytoplasm did not depend on the presence of replicons, we concluded that these structures represented a general feature of Huh-7 cells (56) (data not shown). However, vesicles in cells bearing an sfl genome were delimited to different degrees by an electron-dense region resembling a thickened membrane (Fig. 4A) that distinguished them from the structures observed in parental Huh-7 cells and cells bearing subgenomic replicons, which are devoid of this electron-dense rim (compare Fig. 4A and B). Additionally, some cells exhibited discrete hyperplastic modifications of ER cisternae or altered mitochondria of abnormal, irregular shape which were sometimes vacuolized or with thinned and fragmented cristae similar to those observed by others (not shown) (2). Particular morphological alterations of nuclei potentially caused by apoptosis as described by others for HepG2 cell transfected with full-length HCV RNA (37) could not be detected.
FIG. 4.

Morphology of cell lines with an sfl genome and localization of viral proteins. (A) Ultrathin section obtained from cell line 21-5 after glutaraldehyde-osmium fixation and ERL embedding. Magnification, ×28,500. The cells exhibit numerous lipid vesicles (V) of different sizes surrounded to different degrees by electron-dense rims resembling a thickened membrane. (B) Ultrathin section of parental Huh-7 cells (fixation, embedding, and magnification as for panel A). Note the absence of the electron-dense rim around the border of the vesicles. (C to E) Localization of HCV E2 (C) and core (D) proteins and the Golgi-resident protein Golgin 97 (E) in cell line 21-5 by indirect immunofluorescence. Representative sections are shown at a magnification of ×304. Bar, 50 μm. Nuclei were counterstained using bisbenzimide (Hoechst). Note the granular staining pattern of the core protein that to some extent resembles the distribution of the Golgi marker Golgin 97. (I and J) Specificity control for the anti-core and anti-E2 sera. Cells harboring a subgenomic replicon were fixed and stained as cells with the sfl genome. Magnification, ×285. (F to H) Localization of E2, core, and Golgin 97 in cell line 21-5 treated with 20 nM brefeldin A for 3 h. Detection of Golgin 97 is abrogated (H), while the distribution of E2 and core remains essentially unchanged (F and G, respectively). Magnification, ×304.
To further characterize structural protein expression in cells with an sfl genome, we studied their subcellular localization in greater detail by indirect immunofluorescence using monoclonal antibodies against E2 and core protein on sfl genome-bearing cell line 21-5 and as a specificity control on a cell line harboring a subgenomic replicon (cell line 9-13). Both E2 and core are efficiently expressed in the cytoplasm of cells with an sfl genome (Fig. 4C and D, respectively), whereas essentially no staining was observed in the control cells (Fig. 4I and J). While E2 was disseminated throughout the cytoplasm rather evenly, core displayed a strikingly granular staining pattern that to some extent resembled the localization of Golgin 97, which is a peripheral membrane protein resident on the cytoplasmic face of the Golgi apparatus (Fig. 4E).
To clarify whether core and/or E2 localize to the Golgi complex, cells were treated for 3 h with brefeldin A, which is known to cause the disintegration of the Golgi complex. Subsequently, cells were fixed and analyzed by indirect immunofluorescence. As can be seen in Fig. 4H, brefeldin A treatment was effective and abrogated the detection of Golgin 97. In contrast, the subcellular distributions of E2 and core were not significantly altered under these conditions (Fig. 4F and G, respectively), suggesting that E2 and core do not localize mainly to the Golgi apparatus. However, it should be mentioned that the overall intensity of the E2 staining was considerably reduced after brefeldin A treatment (data not shown), which may be caused by the toxicity of the drug or the loss of the Golgi compartment. As regards the granular distribution of core, a very similar staining pattern has been observed for core-expressing cell lines, in which an association of core with cytoplasmic lipid storage vesicles has been described (1, 34, 53). In order to determine whether the same is true in cell lines with an sfl genome and to analyze the subcellular distribution of core and E2 more precisely, we conducted immuno-EM using a rabbit antiserum directed against E2 and a mouse monoclonal antibody specific for core. Bound antibodies were detected by matched secondary antibodies complexed to colloidal gold particles with diameters of 10 nm for E2 and 6 nm for core. Since we omitted osmium tetroxide to improve the preservation of antigenicity, under these conditions improperly fixed lipids were extracted. Interestingly, by using this method, i.e., EM contrast, the vesicular structures in cells embedded for immunostaining appeared devoid of any content (Fig. 5A and C), suggesting that they did indeed contain lipids. The 6-nm-diameter gold particles specific for core localized primarily to the border of these empty vesicles (Fig. 5A), consistent with the studies performed by Barba and coworkers showing the association of core with lipid droplets in transiently transfected cells (1). In contrast, E2 was not found to associate with these vesicles but rather at stacked cisternae, suggesting that E2 localizes primarily to the ER and early Golgi compartments (Fig. 5B). It was shown previously, that HCV replication and/or translation are tightly coupled to the cell cycle (56). Therefore, in an asynchronously growing replicon cell population, HCV antigen levels can differ dramatically from cell to cell. Consequently, in our immunogold labeling experiments, not all labeled sfl genome-containing cells carried detectable levels of gold particles, which indicates the specificity of our staining (data not shown). This specificity was confirmed by the result shown in Fig. 5C in which cells of the sfl genome-bearing cell line 21-5 were incubated with isotype-matched primary antibodies directed against HIV and measles virus proteins, followed by the same gold-labeled secondary antibodies. In conclusion, these data show that cells with an sfl genome exhibit several morphological peculiarities, most notably the presence of large amounts of lipid vesicles that are delimited by an electron-dense rim. The majority of core did not colocalize with E2, since only minor staining was detected at the ER, while most of the antigen was tightly associated with cytoplasmic lipid vesicles.
FIG. 5.

Subcellular localization of HCV proteins by immuno-EM. (A and B) Localization of HCV core (6-nm-diameter gold particles) and E2 (10-nm-diameter gold particles) in cell line 21-5. (A) The majority of the core protein is found on the surfaces of lipid vesicles (V), and only minor staining is detected at the ER. No 10-nm-diameter gold specific for E2 can be seen in the area around the vesicles. Note that no osmium postfixation was used, and therefore, the lipid vesicles appear empty, due to removal of the content by alcohol dehydration and embedding medium. (B) The 10-nm-diameter gold marker specific for E2 associated with elongated, cisterna-like structures in the cytoplasm presumably belonging to the ER and early compartments of the Golgi complex. Note the absence of 6-nm-diameter gold marker specific for core. (C) Specificity control of the immuno-gold labeling. Cell line 21-5 was labeled with primary antibodies directed against HIV and measles virus proteins, which share the isotypes of the antibodies used in panels A and B. Subsequently, the same gold-labeled secondary antibodies were used. Magnification, ×92,150.
Cell lines carrying HCV replicons release RNA-containing structures with a buoyant density of 1.04 to 1.11g/ml that are resistant to nuclease treatment.
Since the sfl genome stably replicated in cells and expressed all viral proteins, we explored the possibility that the HCV particles were released. To this end, we used cell-free supernatants derived from several cell lines with an sfl genome to infect parental Huh-7 cells. For a stringent control for nonspecific transduction of G418 resistance, supernatants from a cell line carrying a subgenomic HCV replicon were applied in parallel. At 24 h postinoculation, culture fluid was removed and replaced by G418-containing medium. Using this approach, we observed transduction of both sfl genomes and subgenomic replicons at a very low frequency (data not shown), indicating a nonspecific release of HCV RNAs from selected cell lines and the nonspecific uptake of these nucleic acids by naive Huh-7 cells. On the other hand, as it is currently unknown whether Huh-7 cells are infectible, we could not exclude the possibility that we failed to observe specific infectivity due to the nonpermissiveness of our target cells. Therefore, we attempted to demonstrate virus particles in the supernatant by biochemical methods. In the first set of experiments, we searched for nuclease-resistant HCV RNA in the supernatants of several cell lines with an sfl genome. For negative controls, we used supernatants from a subgenomic replicon-bearing cell line and naive Huh-7 cells. Five days postseeding, the supernatants of these cells were harvested, and particulate material was pelleted by ultracentrifugation as described in Materials and Methods. Sediments were resuspended in PBS, and aliquots were incubated for 4 h with various amounts of RNase A in the presence or absence of detergent. RNA was prepared and analyzed by real-time RT-PCR. As shown in Fig. 6, cell lines with an sfl genome released significant amounts of HCV RNA into the supernatant (∼5 × 104 molecules per ml of medium). Interestingly, in the absence of detergent, this RNA was resistant to RNase A up to a concentration of 100 μg/ml but was completely degraded upon the addition of 0.5% NP-40 (compare Fig. 6C and A). While the detergent-labile nature of the otherwise protected HCV RNA was principally consistent with the notion that it is encased in a virus particle, we were surprised to detect HCV RNA with similar characteristics in the supernatant of cells carrying subgenomic replicons that lack the HCV structural proteins (∼105 molecules per ml of medium [Fig. 6B]). This result indicated that these cell lines were capable of releasing large amounts of nuclease-resistant HCV RNAs by as yet unknown nonspecific mechanisms.
FIG. 6.
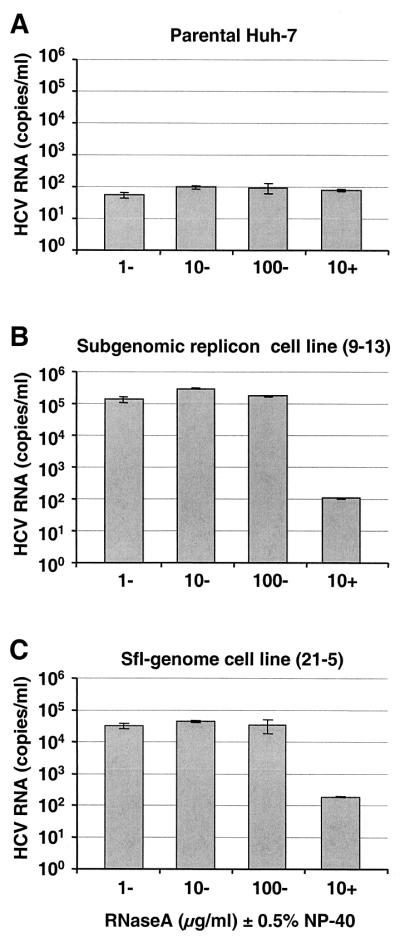
Detection of nuclease-resistant HCV-RNAs in supernatants of various cell lines. Supernatants from parental Huh-7 cells (A), cell line 9-13 harboring a subgenomic replicon (B), and cell line 21-5 with an sfl genome (C) were harvested 6 days postseeding, cleared by low-speed centrifugation, filtered through 0.45-μm-pore-size filters, and pelleted by high-speed ultracentrifugation. Pellets were resuspended in PBS supplemented with carrier RNA, and aliquots were incubated for 4 h at room temperature with RNase A at the concentrations given below the bars. Some aliquots were also supplemented with 0.5% NP-40 (indicated by + [−, no NP-40 added]). HCV RNA was measured by quantitative RT-PCR performed in duplicate. Mean values of duplicates representing the number of HCV RNA molecules per milliliter of supernatant are given, and the error bars indicate the standard errors of the means.
To distinguish between nonspecifically released RNA and true virus particles, we attempted to separate them by their buoyant densities. Supernatants from cell lines with an sfl genome or a subgenomic replicon were harvested 5 days postseeding and subjected to density gradient centrifugations, and 12 fractions of each gradient were analyzed by quantitative RT-PCR. For a reference for an infectious virus from the family of Flaviviridae, we included BVDV (strain NADL) obtained 24 h postinfection of bovine testicle cells (R.D.420 cells). In previous studies, BVDV has been shown to have a buoyant density of 1.09 to 1.15 g/ml in sucrose gradients (35), which is very similar to the low-density forms reported for infectious HCV (1.06 to 1.11 g/ml) (10, 31). In our analyses, we employed iodixanol (Optiprep) as gradient material, as it is less osmotically active than sucrose and therefore presumably allows separation under gentler conditions. Figure 7 depicts the distribution of HCV RNAs contained in the supernatants from two cell lines bearing a subgenomic replicon (5-15 and 9-13 [Fig. 7A]) (49) and two cell lines bearing an sfl genome (20-1 and 21-5 [Fig. 7B]) and of BVDV RNA (Fig. 7C) in linear iodixanol gradients (0 to 40%). High titers of BVDV (∼109 RNA molecules per ml of gradient fraction) were found in the range between 1.04 to 1.11 g/ml with a peak at a density corresponding to 1.06 g/ml. Interestingly, HCV RNA structures derived from sfl genome-bearing cell lines 20-1 and 21-5 had similar low densities with peaks of 1.04 and 1.05 g/ml, respectively. This shows that HCV RNA released into the supernatants of sfl genome-bearing cell lines is most likely incorporated into lipid-containing structures that have biophysical properties comparable to those of infectious virus particles of BVDV. However, the amount of HCV RNA was about 3 orders of magnitude lower than that observed 24 h after a productive infection with cytopathic BVDV (∼106 copies/ml for HCV versus ∼109 copies/ml for BVDV [compare Fig. 7B and C]). Furthermore, the HCV RNAs present in supernatants of cell lines with subgenomic replicons had similar low densities (1.06 and 1.07 g/ml for cell lines 5-15 and 9-13, respectively), and the RNA amounts were at least fourfold higher, reflecting their higher replication efficiency. These results suggest the nonspecific release of lipid- and HCV RNA-containing structures (vesicles) into the medium of cell lines harboring subgenomic replicons and sfl genomes.
FIG. 7.

Analysis of released HCV RNA by density gradient centrifugation. (A and B) Cell culture supernatants from two different cell lines carrying subgenomic replicons (5-15 and 9-13) (A) and two cell lines carrying sfl genomes (20-1 and 21-5) (B) were harvested 3 days postseeding and processed as described in Materials and Methods. RNA was prepared and subjected to quantitative RT-PCR. (C) For a control, supernatant from R.D.420 cells productively infected with BVDV (strain NADL) was analyzed in parallel. Copies of HCV and BVDV RNA detected per milliliter of gradient fraction (mean value of duplicates) are plotted against the density. The density of the peak fraction is given at the top of each graph.
Transient replication of a full-length HCV genome in Huh-7 cells.
The lack of virus production in cell lines carrying sfl genomes could be caused by several factors or reasons. For instance, the presence of the heterologous sequences in the 5′ region of the sfl genomes may destroy a specific cis-active packaging signal or yield an RNA genome that exceeds the packaging limit, therefore abrogating virus assembly. Alternatively, it is possible that the high genetic drift renders the structural proteins nonfunctional due to the accumulation of inactivating mutations. To circumvent these potential problems, we used full-length HCV genomes lacking heterologous sequences and conducted transient-replication assays. In these experiments, we transfected parental Huh-7 cells with in vitro-transcribed full-length RNA of the unmodified Con1 genome (41), which has recently been shown to be infectious (J. Bukh, unpublished results). Additionally, we transfected a full-length genome derivative harboring a new combination of adaptive mutations that confers maximal replication efficiency in Huh-7 cells (E1202G, T1280I, and K1846T) (Lohmann et al., unpublished results) (Fig. 1A). For a stringent negative control for virus release, a selectable subgenomic replicon with the equivalent set of mutations was used (Fig. 1A). Cells were harvested at various time points and processed for Northern and Western blotting. At 96 h posttransfection, cell-free supernatants were harvested and analyzed for the presence of HCV RNA by density gradient centrifugation and quantitative RT-PCR as described above. Both the adapted full-length HCV genome and the adapted subgenomic replicon replicated to high levels, as significant amounts of RNA and protein could be detected for at least 96 h posttransfection (Fig. 8A and B). In contrast, the wild-type Con1 genome replicated poorly and did not allow detection of proteins and RNA for more than 24 h after transfection. In each gradient analysis of supernatants harvested 96 h after transfection, residual amounts of input RNA were found in fractions of high density (>1.13 g/ml). Notable amounts of a low-density RNA species were detected only from cells carrying a subgenomic replicon; this low-density RNA species presumably corresponds to the HCV RNA-containing structures released from cell lines carrying stable replicons. In summary, we conclude that cell culture-adapted full-length HCV genomes, despite efficient transient replication in Huh-7 cells, are unable to assemble and/or secrete virus particles.
FIG. 8.

Transient replication of a subgenomic replicon and full-length HCV genomes. Huh-7 cells were transfected with the authentic HCV Con1 genome (Con1), a cell culture-adapted full-length RNA (Con1-ET), or a selectable subgenomic replicon (replicon-ET), and cells were harvested at the indicated time points. (A and B) Transient replication was monitored by Northern blotting (A), as described in the legend to Fig. 1B, and NS5B-specific Western blotting (B). (C) At 96 h posttransfection, supernatants from three 10-cm-diameter dishes were harvested, cleared of cellular debris, and subjected to density gradient centrifugation as described in Materials and Methods. HCV RNA in each fraction was measured by quantitative RT-PCR. The average value of duplicate experiments is plotted against the density of the corresponding fraction.
DISCUSSION
In the present study, we established and characterized several Huh-7 cell clones that harbor autonomously replicating selectable full-length HCV genomes. While our first attempts with the wild-type Con1 sequence failed, we were successful with an sfl genome that carries cell culture-adaptive mutations. However, although these mutations cause a strong enhancement of RNA replication and also a highly elevated ECF when introduced into a selectable subgenomic replicon (43), the ECF observed with the sfl genomes was very low. In several independent experiments, a value of <100 colonies/μg of transfected RNA was obtained, which is 3 to 4 orders of magnitude lower than that for the subgenomic replicons. Principally, this could be due to cytotoxic effects caused by the expression of the structural proteins of HCV. It is conceivable that cytotoxicity limits the number of cell colonies that can be obtained after electroporation either because mutations are required to ablate such toxicity of the structural proteins or because only a few transfected cells are resistant to these potential cytotoxic effects. Alternatively, the presence of the heterologous sequences which render the RNA molecule significantly larger than the wild-type genome (11,076 versus 9,605 nucleotides) may impede replication and thus reduce the ECF. Interestingly, despite the low ECF, the number of sfl genomes per cell was only two- to fivefold lower than that of cell lines carrying a subgenomic replicon, indicating that the length of the sfl genomes per se does not preclude efficient replication.
Recently, Chung and coworkers described the replication of HCV in CV-1 and HepG2 cells after transfection of a plasmid that carries the full-length H77 genome flanked at the 3′ end by the hepatitis delta virus ribozyme (14). This system is based on transcription of the HCV genomic RNA by T7 RNA polymerase that is provided by infection with a recombinant vaccinia virus (vTF7-3). Although this is an interesting approach, it has some limitations. For instance, infections with vTF7-3 are cytopathic and may have pleiotropic effects. Furthermore, the specific detection of the minus-strand RNA is complicated due to possible copy-back RNA synthesis by T7 RNA polymerase and the presence of large amounts of double-stranded plasmid DNA used for the transfection. In addition, since T7 RNA polymerase-based transcription is error prone, it is difficult to differentiate between genetic drift caused by error-prone RNA synthesis of NS5B RdRp and errors introduced by the low fidelity of T7 polymerase. Finally, vTF7-3 itself can be blocked by alpha interferon, making the distinction between inhibition of HCV RNA replication and the recombinant vaccinia virus problematic (14). Therefore, we think that the self-replication of full-length HCV genomes after transfection of RNA as described in the present study is superior. The availability of the cell lines described in this study for the first time permitted the analysis of HCV structural proteins expressed from autonomously replicating full-length HCV RNAs which closely mimics the in vivo situation within an infected hepatocyte. We found that the dominant core protein within cells bearing an sfl genome is p21, which presumably results from a cleavage event around amino acid residue 174. This observation is in direct accordance with previous data which clearly established that the major core species both in tissue culture cells expressing HCV core and in virus particles derived from human sera is p21 (71). Immunolocalization experiments revealed an exclusively cytoplasmic distribution of the polypeptide, with a strikingly granular pattern that was independent of brefeldin A treatment. This is in contrast to a report by Yasui et al. (71) that reports some core staining also within the nuclei of cell lines stably or transiently expressing HCV core. However, since we used the anti-core monoclonal antibody C7-50, which according to Yasui and coworkers recognizes only core protein expressed in the cytoplasm (71), we cannot rule out the possibility that a certain proportion of core also localizes to the nucleus in cells with the sfl genome.
The increased resolution provided by immuno-EM established that the majority of core protein localized to the boundary of cytoplasmic lipid storage vesicles, whereas only minor staining was found at the ER. This finding directly correlates with previous studies which show a tight association of the core protein with lipid droplets. Since this observation was made using core protein from different HCV isotypes expressed by alternative approaches in various cell lines (1, 53, 71) and as the association of HCV core with lipid droplets was also observed in specimens obtained from chronically infected chimpanzees, this appears to be a general phenomenon that also takes place in infected hepatocytes in vivo.
It was shown that folding of HCV E1 and E2 glycoproteins can proceed by two pathways: (i) a slow and inefficient productive pathway that leads to the assembly of noncovalently linked heterodimers which are believed to be the prebudding forms of E1 and E2 (18-21) and (ii) a nonproductive pathway that leads to the formation of disulfide-linked heterogeneous E1-E2 aggregates (18, 20). In either case, the complexes are retained in the ER, the presumed site of virus budding, due to retention signals present in the transmembrane domains of both E1 and E2 (15-17, 22). As shown in this report, the majority of E1-E2 complexes present in cell lines with an sfl genome folded by the productive pathway. The absence of glycoprotein aggregates in these cells might be due to a lowered propensity of the Con1-derived glycoproteins to fold nonproductively or to preferential binding of our antiserum to native E1-E2 complexes. However, it should be kept in mind that the expression level of the HCV glycoproteins from the sfl genome is significantly lower than in the vaccinia virus-based expression systems frequently employed to analyze HCV glycoproteins (data not shown). Since productive folding of E1-E2 heterodimers is a slow process that involves the ER chaperone calnexin (20, 21), it is conceivable that the high expression levels reached in these systems may overload cellular factors required for proper folding, while the lower expression in our system may not exceed the capacities of these factors in the folding pathway. Although the majority of the glycoproteins detected appeared to fold along the productive pathway, only a weak coprecipitation of E1 with E2 was found. Again, this could be an inherent feature of our Con1 HCV isolate, or it could be due to the antibodies used. However, this is unlikely, because we observed much more efficient coprecipitation of E1 with E2 after expression of the glycoproteins in Huh-7 cells with recombinant vaccinia virus, employing the same antiserum for immunoprecipitation (41). Therefore, the weak association between E1 and E2 may be ascribed to the presence of mutations that have accumulated during prolonged passage of these cell lines. This notion is further supported by the high genetic drift observed with the sfl genomes.
Encouraged by the high replication levels, efficient expression of the structural proteins, and formation of native glycoprotein complexes, we examined whether cells carrying the sfl genome produced and released HCV particles. To this end, we directly transferred cleared cell culture supernatant to parental Huh-7 cells but failed to detect specific infectivity. The same was found using cell lysates generated after repetitive freeze-thaw cycles. However, since it is not known whether Huh-7 cells are susceptible to HCV infection, we cannot rule out the possibility that we may have missed infectious virus due to a flawed assay system. On the other hand, cell lines that have been reported to be permissive for HCV infection do not support replication of sfl genomes and are thus not suitable as target cells for infectivity assays. Therefore, as an alternative, we examined whether cells with an sfl genome released nuclease-resistant HCV RNA. Interestingly, such RNAs were indeed found in the supernatants of these cells (>104 copies per ml). Moreover, these RNAs had a very low density similar to the density reported for infectious HCV (31) and, as shown here, also to that of the pestivirus BVDV. However, in supernatants of cell lines with subgenomic HCV replicons, HCV RNA-containing structures with equivalent characteristics were found, indicating that their release is a general feature of replicon-bearing Huh-7 cells that is not mediated by the viral structural proteins. The exact nature of these RNA-containing structures is currently unknown, but their biophysical properties suggest that they correspond to membranous vesicles. In addition, the overall yield of these structures is rather low. While intracellular HCV RNA levels range from ∼200 to 500 copies per cell, the number of extracellular HCV RNA-containing vesicles amounts to ∼0.1 to 1 vesicle per cell in supernatants harvested 5 to 7 days postseeding. Although we cannot strictly rule out the possibility that sfl genome cells also produce HCV particles, this is unlikely for two reasons. First, the overall low level of released HCV RNA suggests a nonspecific process, and second, the amount of HCV RNA in the supernatant did not increase due to the presence of the structural proteins, which would be expected for productive assembly and release. In fact, both in transient-replication assays and in cell lines carrying sfl genomes or subgenomic replicons, we consistently found slightly higher levels of HCV RNA in the supernatant of the latter. Thus, the amount of these RNA species correlated with the intracellular replication level, rather than the presence or absence of the structural proteins, suggesting that RNAs were released by nonspecific mechanisms.
There are several factors or reasons that may cause the inability of the cell lines with an sfl genome to produce HCV particles. First, one could imagine that the serine-to-glycine mutation that was introduced at the second position of the core protein due to the cloning procedure may inactivate the protein and thus abrogate virus assembly. However, this is unlikely, as second-generation cell lines with an sfl genome that harbor constructs with an authentic core protein displayed similar phenotypes in our assays and thus apparently also did not yield virus particles (unpublished results). Alternatively, the presence of the heterologous sequences (EMCV IRES and neo) may disrupt a packaging signal or increase the size of the RNA beyond the packaging limit. Finally, the high genetic variability observed with sfl genomes may quickly lead to the accumulation of mutations that inactivate the assembly competence of the structural proteins. However, our transient-replication assays with full-length unmodified Con1 RNA and a variant of this RNA bearing only three adaptive mutations in the nonstructural region also did not reveal evidence for virus assembly in Huh-7 cells. Thus, since these RNA molecules did not carry heterologous sequences and because genetic variability can be ruled out due to the transient nature of these assays, we conclude that the lack of virus assembly is likely attributable to other factors. Recently, it was shown that the Con1 isolate is infectious in vivo (Bukh, unpublished), indicating that the structural proteins are assembly competent. Therefore, we think that the lack of virus production might be due to the inability of Huh-7 cells to support virus assembly and/or release perhaps due to the lack of certain host cell factor(s). Such a dependence of particle formation on cellular factors has been reported recently for other viruses. For instance, in the morphogenesis of adenovirus, cellular proteins are thought to be part of the viral packaging machinery (62). Furthermore, it was recently reported that cellular ubiquitin plays a vital role in a late step of the retroviral budding process (55, 63, 65). It is to be hoped that future research will soon unravel such cellular factors needed to support HCV assembly and release in cell culture, as this would finally allow construction of a model of the entire HCV replication cycle in cell culture. Furthermore, these findings may point to new virus-host interactions which may prove to be attractive targets for future antiviral treatment.
Acknowledgments
We are grateful to Ulrike Herian and Sandra Hoffmann for excellent technical assistance, to Klaus Wiegers and Ingrid Ellhof for help with the EM studies and the preparation of Fig. 4 and 5, and to Jens Bukh for permission to cite unpublished results. We also thank Darius Moradpour for providing the NS5B- and core-specific monoclonal antibodies, Ruben Donis for providing BVDV virus stocks and the R.D.420 cell line, and Jane McKeating for providing the E2 monoclonal antibody.
This work was supported in part by a grant from the European Community (QLK2-1999-00356).
REFERENCES
- 1.Barba, G., F. Harper, T. Harada, M. Kohara, S. Goulinet, Y. Matsuura, G. Eder, Z. Schaff, M. J. Chapman, T. Miyamura, and C. Brechot. 1997. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. USA 94:1200-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbaro, G., G. Di Lorenzo, A. Asti, M. Ribersani, G. Belloni, B. Grisorio, G. Filice, and G. Barbarini. 1999. Hepatocellular mitochondrial alterations in patients with chronic hepatitis C: ultrastructural and biochemical findings. Am. J. Gastroenterol. 94:2198-2205. [DOI] [PubMed] [Google Scholar]
- 3.Bartenschlager, R. 1999. The NS3/4A proteinase of the hepatitis C virus: unravelling structure and function of an unusual enzyme and a prime target for antiviral therapy. J. Viral Hepatitis 10:1-2. [DOI] [PubMed] [Google Scholar]
- 4.Bartenschlager, R., L. L. Ahlborn, J. Mous, and H. Jacobsen. 1993. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J. Virol. 67:3835-3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartenschlager, R., and V. Lohmann. 2000. Replication of hepatitis C virus. J. Gen. Virol. 81:1631-1648. [DOI] [PubMed] [Google Scholar]
- 6.Bartenschlager, R., V. Lohmann, T. Wilkinson, and J. O. Koch. 1995. Complex formation between the NS3 serine-type proteinase of the hepatitis C virus and NS4A and its importance for polyprotein maturation. J. Virol. 69:7519-7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baumert, T. F., S. Ito, D. T. Wong, and T. J. Liang. 1998. Hepatitis C virus structural proteins assemble into viruslike particles in insect cells. J. Virol. 72:3827-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Behrens, S. E., L. Tomei, and R. DeFrancesco. 1996. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 15:12-22. [PMC free article] [PubMed] [Google Scholar]
- 9.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972-1974. [DOI] [PubMed] [Google Scholar]
- 10.Bradley, D., K. McCaustland, K. Krawczynski, J. Spelbring, C. Humphrey, and E. H. Cook. 1991. Hepatitis C virus: buoyant density of the factor VIII-derived isolate in sucrose. J. Med. Virol. 34:206-208. [DOI] [PubMed] [Google Scholar]
- 11.Choo, Q. L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and M. Houghton. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359-362. [DOI] [PubMed] [Google Scholar]
- 12.Choo, S. H., H. S. So, J. M. Cho, and W. S. Ryu. 1995. Association of hepatitis C virus particles with immunoglobulin: a mechanism for persistent infection. J. Gen. Virol. 76:2337-2341. [DOI] [PubMed] [Google Scholar]
- 13.Choukhi, A., S. Ung, C. Wychowski, and J. Dubuisson. 1998. Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus glycoproteins. J. Virol. 72:3851-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung, R. T., W. He, A. Saquib, A. M. Contreras, R. J. Xavier, A. Chawla, T. C. Wang, and E. V. Schmidt. 2001. Hepatitis C virus replication is directly inhibited by IFN-alpha in a full-length binary expression system. Proc. Natl. Acad. Sci. USA 98:9847-9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cocquerel, L., S. Duvet, J. C. Meunier, A. Pillez, R. Cacan, C. Wychowski, and J. Dubuisson. 1999. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol. 73:2641-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cocquerel, L., J. C. Meunier, A. Pillez, C. Wychowski, and J. Dubuisson. 1998. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 72:2183-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cocquerel, L., C. Wychowski, F. Minner, F. Penin, and J. Dubuisson. 2000. Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J. Virol. 74:3623-3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deleersnyder, V., A. Pillez, C. Wychowski, K. Blight, J. Xu, Y. S. Hahn, C. M. Rice, and J. Dubuisson. 1997. Formation of native hepatitis C virus glycoprotein complexes. J. Virol. 71:697-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubuisson, J. 2000. Folding, assembly and subcellular localization of hepatitis C virus glycoproteins. Curr. Top. Microbiol. Immunol. 242:135-148. [DOI] [PubMed] [Google Scholar]
- 20.Dubuisson, J., H. H. Hsu, R. C. Cheung, H. B. Greenberg, D. G. Russell, and C. M. Rice. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147-6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubuisson, J., and C. M. Rice. 1996. Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J. Virol. 70:778-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duvet, S., L. Cocquerel, A. Pillez, R. Cacan, A. Verbert, D. Moradpour, C. Wychowski, and J. Dubuisson. 1998. Hepatitis C virus glycoprotein complex localization in the endoplasmic reticulum involves a determinant for retention and not retrieval. J. Biol. Chem. 273:32088-32093. [DOI] [PubMed] [Google Scholar]
- 23.Enomoto, N., I. Sakuma, Y. Asahina, M. Kurosaki, T. Murakami, C. Yamamoto, N. Izumi, F. Marumo, and C. Sato. 1995. Comparison of full-length sequences of interferon-sensitive and -resistant hepatitis C virus 1b. Sensitivity to interferon is conferred by amino acid substitutions in the NS5A region. J. Clin. Investig. 96:224-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enomoto, N., I. Sakuma, Y. Asahina, M. Kurosaki, T. Murakami, C. Yamamoto, Y. Ogura, N. Izumi, F. Marumo, and C. Sato. 1996. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N. Engl. J. Med. 334:77-81. [DOI] [PubMed] [Google Scholar]
- 25.Friebe, P., V. Lohmann, N. Krieger, and R. Bartenschlager. 2001. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J. Virol. 75:12047-12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gale, M. J., M. J. Korth, N. M. Tang, S. L. Tan, D. A. Hopkins, T. E. Dever, S. J. Polyak, D. R. Gretch, and M. G. Katze. 1997. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 230:217-227. [DOI] [PubMed] [Google Scholar]
- 27.Gale, M. J., S. M. Blakely, B. Kwieciszewski, S.-L. Tan, M. Dossett, N. M. Tang, M. J. Korth, S. J. Polyak, D. Gretch, and M. G. Katze. 1998. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanism of kinase regulation. Mol. Cell. Biol. 18:5208-5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grakoui, A., D. W. McCourt, C. Wychowski, S. M. Feinstone, and C. M. Rice. 1993. Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J. Virol. 67:2832-2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grakoui, A., C. Wychowski, C. Lin, S. M. Feinstone, and C. M. Rice. 1993. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67:1385-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo, J. T., V. V. Bichko, and C. Seeger. 2001. Effect of alpha interferon on the hepatitis C virus replicon. J. Virol. 75:8516-8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hijikata, M., Y. K. Shimizu, H. Kato, A. Iwamoto, J. W. Shih, H. J. Alter, R. H. Purcell, and H. Yoshikura. 1993. Equilibrium centrifugation studies of hepatitis C virus: evidence for circulating immune complexes. J. Virol. 67:1953-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holland, J., K. Spindler, F. Horodyski, E. Grabau, S. Nichol, and S. VandePol. 1982. Rapid evolution of RNA genomes. Science 215:1577-1585. [DOI] [PubMed] [Google Scholar]
- 33.Hoofnagle, J. H. 1997. Hepatitis C: the clinical spectrum of disease. Hepatology 26:15S-20S. [DOI] [PubMed]
- 34.Hope, R. G., and J. McLauchlan. 2000. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J. Gen. Virol. 81:1913-1925. [DOI] [PubMed] [Google Scholar]
- 35.Horzinek, M. C. 1981. Properties of virions, p. 13-110. In M. C. Horzinek (ed.), Non-arthropod-borne togaviruses. Academic Press, London, United Kingdom.
- 36.Hüssy, P., H. Langen, J. Mous, and H. Jacobsen. 1996. Hepatitis C virus core protein: carboxy-terminal boundaries of two processed species suggest cleavage by a signal peptide peptidase. Virology 224:93-104. [DOI] [PubMed] [Google Scholar]
- 37.Kalkeri, G., N. Khalap, R. F. Garry, C. D. Fermin, and S. Dash. 2001. Hepatitis C virus protein expression induces apoptosis in HepG2 cells. Virology 282:26-37. [DOI] [PubMed] [Google Scholar]
- 38.Kato, N., Y. Ootsuyama, H. Sekiya, S. Ohkoshi, T. Nakazawa, M. Hijikata, and K. Shimotohno. 1994. Genetic drift in hypervariable region 1 of the viral genome in persistent hepatitis C virus infection. J. Virol. 68:4776-4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato, N., and K. Shimotohno. 2000. Systems to culture hepatitis C virus. Curr. Top. Microbiol. Immunol. 242:261-278. [DOI] [PubMed] [Google Scholar]
- 40.Kim, D. W., Y. Gwack, J. H. Han, and J. Choe. 1995. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem. Biophys. Res. Commun. 215:160-166. [DOI] [PubMed] [Google Scholar]
- 41.Koch, J. O., and R. Bartenschlager. 1999. Modulation of hepatitis C virus NS5A hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J. Virol. 73:7138-7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolykhalov, A. A., K. Mihalik, S. M. Feinstone, and C. M. Rice. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74:2046-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krieger, N., V. Lohmann, and R. Bartenschlager. 2001. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75:4614-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kunkel, M., M. Lorinczi, R. Rijnbrand, S. M. Lemon, and S. J. Watowich. 2001. Self-assembly of nucleocapsid-like particles from recombinant hepatitis C virus core protein. J. Virol. 75:2119-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuo, G., Q. L. Choo, H. J. Alter, G. L. Gitnick, A. G. Redeker, R. H. Purcell, T. Miyamura, J. L. Dienstag, M. J. Alter, C. E. Stevens, G. E. Tegtmeier, F. Bonino, M. Colombo, W.-S. Lee, C. Kuo, K. Berger, R. J. Shuster, L. R. Overby, D. W. Bradley, and M. Houghton. 1989. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 244:362-364. [DOI] [PubMed] [Google Scholar]
- 46.Liu, Q. Y., C. Tackney, R. A. Bhat, A. M. Prince, and P. Zhang. 1997. Regulated processing of hepatitis C virus core protein is linked to subcellular localization. J. Virol. 71:657-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lohmann, V., F. Körner, U. Herian, and R. Bartenschlager. 1997. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 71:8416-8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lohmann, V., F. Körner, A. Dobierzewska, and R. Bartenschlager. 2001. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J. Virol. 75:1437-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lohmann, V., F. Körner, J. O. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-113. [DOI] [PubMed] [Google Scholar]
- 50.Maillard, P., K. Krawczynski, J. Nitkiewicz, C. Bronnert, M. Sidorkiewicz, P. Gounon, J. Dubuisson, G. Faure, R. Crainic, and A. Budkowska. 2001. Nonenveloped nucleocapsids of hepatitis C virus in the serum of infected patients. J. Virol. 75:8240-8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martire, G., A. Viola, L. Iodice, L. V. Lotti, R. Gradini, and S. Bonatti. 2001. Hepatitis C virus structural proteins reside in the endoplasmic reticulum as well as in the intermediate compartment/cis-Golgi complex region of stably transfected cells. Virology 280:176-182. [DOI] [PubMed] [Google Scholar]
- 52.Miyamoto, H., H. Okamoto, K. Sato, T. Tanaka, and S. Mishiro. 1992. Extraordinarily low density of hepatitis C virus estimated by sucrose density gradient centrifugation and the polymerase chain reaction. J. Gen. Virol. 73:715-718. [DOI] [PubMed] [Google Scholar]
- 53.Moradpour, D., C. Englert, T. Wakita, and J. R. Wands. 1996. Characterization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology 222:51-63. [DOI] [PubMed] [Google Scholar]
- 54.Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858-3863. [PubMed] [Google Scholar]
- 55.Patnaik, A., V. Chau, and J. W. Wills. 2000. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 97:13069-13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pietschmann, T., V. Lohmann, G. Rutter, K. Kurpanek, and R. Bartenschlager. 2001. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 75:1252-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prince, A. M., T. HuimaByron, T. S. Parker, and D. M. Levine. 1996. Visualization of hepatitis C virions and putative defective interfering particles isolated from low-density lipoproteins. J. Viral Hepatitis 3:11-17. [DOI] [PubMed] [Google Scholar]
- 58.Ralston, R., K. Thudium, K. Berger, C. Kuo, B. Gervase, J. Hall, M. Selby, G. Kuo, M. Houghton, and Q. L. Choo. 1993. Characterization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia viruses. J. Virol. 67:6753-6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reed, K. E., and C. M. Rice. 2000. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top. Microbiol. Immunol. 242:55-84. [DOI] [PubMed] [Google Scholar]
- 60.Robertson, B., G. Myers, C. Howard, T. Brettin, J. Bukh, B. Gaschen, T. Gojobori, G. Maertens, M. Mizokami, O. Nainan, S. Netesov, K. Nishioka, I. T. Shin, P. Simmonds, D. Smith, L. Stuyver, A. Weiner, et al. 1998. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. Arch. Virol. 143:2493-2503. [DOI] [PubMed] [Google Scholar]
- 61.Santolini, E., G. Migliaccio, and M. N. La. 1994. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 68:3631-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmid, S. I., and P. Hearing. 1998. Cellular components interact with adenovirus type 5 minimal DNA packaging domains. J. Virol. 72:6339-6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schubert, U., D. E. Ott, E. N. Chertova, R. Welker, U. Tessmer, M. F. Princiotta, J. R. Bennink, H. G. Krausslich, and J. W. Yewdell. 2000. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 97:13057-13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Selby, M. J., E. Glazer, F. Masiarz, and M. Houghton. 1994. Complex processing and protein:protein interactions in the E2:NS2 region of HCV. Virology 204:114-122. [DOI] [PubMed] [Google Scholar]
- 65.Strack, B., A. Calistri, M. A. Accola, G. Palu, and H. G. Gottlinger. 2000. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 97:13063-13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suzich, J. A., J. K. Tamura, H. F. Palmer, P. Warrener, A. Grakoui, C. M. Rice, S. M. Feinstone, and M. S. Collett. 1993. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J. Virol. 67:6152-6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomssen, R., S. Bonk, C. Propfe, K. H. Heermann, H. G. Kochel, and A. Uy. 1992. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. 181:293-300. [DOI] [PubMed] [Google Scholar]
- 68.Thomssen, R., S. Bonk, and A. Thiele. 1993. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. 182:329-334. [DOI] [PubMed] [Google Scholar]
- 69.Tomei, L., C. Failla, E. Santolini, R. DeFrancesco, and N. LaMonica. 1993. NS3 is a serine protease required for processing of hepatitis C virus polyprotein. J. Virol. 67:4017-4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yanagi, M., M. StClaire, S. U. Emerson, R. H. Purcell, and J. Bukh. 1999. In vivo analysis of the 3′ untranslated region of the hepatitis C virus after in vitro mutagenesis of an infectious cDNA clone. Proc. Natl. Acad. Sci. USA 96:2291-2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yasui, K., T. Wakita, K. K. Tsukiyama, S. I. Funahashi, M. Ichikawa, T. Kajita, D. Moradpour, J. R. Wands, and M. Kohara. 1998. The native form and maturation process of hepatitis C virus core protein. J. Virol. 72:6048-6055. [DOI] [PMC free article] [PubMed] [Google Scholar]