

Abstract
Gli family members mediate constitutive Hedgehog signaling in the common skin cancer, basal cell carcinoma. Snail/Snai1 is rapidly induced by Gli1 in vitro, and is co-expressed with Gli1 in human hair follicles and skin tumors. In the current study we generated a dominant negative allele of Snail, SnaZFD, composed of the zinc finger domain and flanking sequence. In promoter-reporter assays, SnaZFD blocked the activity of wild type Snail on the E-cadherin promoter. Snail loss-of-function mediated by SnaZFD or by one of several short hairpin RNAs inhibited transformation of RK3E epithelial cells by Gli1. Conversely, enforced expression of Snail promoted transformation in vitro by Gli1, but not by other genes that were tested, including Notch1, ErbB2, and N-Ras. As observed for Gli1, wild type Snail repressed E-cadherin in RK3E cells and induced blebbing of the cytoplasmic membrane. Induction of a conditional Gli1 transgene in the basal keratinocytes of mouse skin led to rapid upregulation of Snail transcripts and to cell proliferation in the interfollicular epidermis. Established Gli1-induced skin lesions exhibited molecular similarities to BCC, including loss of E-cadherin. The results identify Snail as a Gli1-inducible effector of transformation in vitro, and an early Gli1-responsive gene in the skin.
Keywords: basal cell carcinoma, hedgehog, Snail, Gli1, epithelial cell, E-cadherin
Introduction
Genetic alterations that deregulate cell fate pathways play an early, critical role in the genesis of specific forms of carcinoma (Vogelstein and Kinzler, 2004). Mutations that lead to constitutive Sonic Hedgehog (Shh) signaling, such as inactivation of the Ptch1 tumor suppressor, are a consistent step in development of cutaneous basal cell carcinoma (BCC), the most common malignancy in Caucasians, and may play a role in other tumor types (Goodrich et al., 1997; Callahan and Oro, 2001; Ruiz i Altaba et al., 2002; Berman et al., 2003; Watkins et al., 2003; McMahon et al., 2003; Pasca di Magliano and Hebrok, 2003; Karhadkar et al., 2004). Although Shh pathway defects are rate-limiting in human BCC, current mouse models exhibit a significant lag between Shh pathway activation and the outgrowth of skin tumors, indicating that defects in other pathways play an important role (Aszterbaum et al., 1999; Nilsson et al., 2000; Grachtchouk et al., 2000; Oro and Higgins, 2003; Hutchin et al., 2005). Conditional inactivation of the cell fate determinant Notch1 in the skin leads to BCC-like lesions, and p53 mutation is frequent in human BCC (Ling et al., 2001; Nicolas et al., 2003). An accurate model of BCC will incorporate these known alterations and will require identification of other genetic or epigenetic changes, the temporal order of alterations, the cell types in which they occur, and the effects of specific mutations on stem cell renewal, cell proliferation, differentiation, and the apoptotic response.
Unlike many other tumor types, BCCs are not associated with an identifiable precursor lesion, and the cell type of origin remains unclear (Miller, 1995). BCC may originate in stem cell populations found in the follicle bulge and the basal layer of the interfollicular epidermis (Blanpain et al., 2004; Tumbar et al., 2004). An origin of BCC within interfollicular epidermis is suggested by induction of BCC-like lesions in the embryonic skin of mice transgenic for Shh or Smo, a seven-transmembrane protein that transduces the Shh signal and exhibits gain-of-function mutations in BCC (Oro et al., 1997; Xie et al., 1998). However, a conditional Gli2 model showed early tumors associated with the follicle bulge (Hutchin et al., 2005).
An extensive literature points to the Gli family of zinc finger transcription factors as mediators of Shh signaling in development and in tumors (Ruiz i Altaba et al., 2002; McMahon et al., 2003; Pasca di Magliano and Hebrok, 2003). Indeed, either Gli1 or Gli2 can induce BCC in transgenic mice (Nilsson et al., 2000; Grachtchouk et al., 2000; Oro and Higgins, 2003). Gli genes are thought to regulate multiple cellular processes relevant to transformation, including cell cycle progression, apoptosis, and others (Louro et al., 1999; Yoon et al., 2002; Louro et al., 2002; Duman-Scheel et al., 2002; Bigelow et al., 2004; Callahan et al., 2004). However, few downstream effectors have been extensively characterized.
As one of its multiple roles in development, Shh signals to the epithelial ventromedial somite wall to induce the connective tissue mesenchyme of the slcerotome, an example of epithelial-mesenchymal transition (EMT) (Fan and Tessier-Lavigne, 1994; Hay, 1995; Thiery, 2003). We previously utilized a conditional Gli1-estrogen receptor fusion protein (Gli1-ER) to show that Gli1 can rapidly and directly induce expression of Snail, a regulator of EMTs in embryonic development and in tumor progression (Louro et al., 2002). Snail and Gli1 transcripts were prominent in human BCCs, and in adjacent normal anagen hair follicles. Furthermore, when expressed in AT2 prostate cancer cells with low metastatic potential, Gli1 induced prominent expression of Snail and a more metastatic phenotype (Karhadkar et al., 2004). Conversely, treatment of Snail-positive, highly metastatic AT6 prostate cancer cells with the Shh pathway inhibitor cyclopamine inhibited both Snail expression and the metastatic phenotype.
In the current study, we used tetracycline (tet)-inducible strategies to correlate Gli1 and Snail expression in vitro and in vivo. In the interfollicular epidermis, Gli1 induced expression of Snail and cell proliferation within 6–12 hours of induction. By four weeks, superficial lesions with molecular and morphologic similarities to BCC were observed. A functional role for Snail in vivo was suggested by loss of E-cadherin in Gli1-induced skin lesions. Studies performed in an epithelial model in vitro supported our previous observation that Snail is directly regulated by Gli1 (Louro et al., 2002), and indicated that Snail is a limiting factor in Gli1-induced transformation that recapitulates the cell morphologic changes induced by Gli1.
Results
Rapid induction of Snail by Gli1 in vitro
Previously, we utilized an E1a-immortalized, epithelial line derived from rat kidney, termed RK3E, to identify a gene expression profile for Gli1 (Ruppert et al., 1991; Louro et al., 2002). Unlike control RK3E cells and RK3E cells transformed by c-MYC, RAS, or KLF4/GKLF, Gli1-transformed cell lines showed prominent expression of Snail. Direct regulation was suggested by rapid induction of Snail transcripts upon exposure of Gli1-ER cells to tamoxifen, even in the presence of the protein translation inhibitor cycloheximide. Although induced by Gli1, the functional significance of Snail to Gli1-induced transformation remained unclear.
To further examine Snail as a potential transcriptional target of Gli1, we isolated clones of RK3E cells in which a human Gli1 allele is induced by tet (Figure 1a). Immunoblot analysis identified several clones with a low background of Gli1 in uninduced cells and prominent expression after exposure to tet for 6 hours (left panel). Expression of transcripts corresponding to Snail and the well-established Gli1 target gene, Ptch1, were examined by semi-quantitative reverse transcription and PCR (RT-PCR, right panel). Analysis of multiple clones revealed consistent regulation of Snail and Ptch1. When RTOG10 cells were similarly analyzed over a timecourse, Snail and Ptch1 were co-regulated and induced by 1–3 hours, consistent with direct regulation of these by Gli1 (Figure 1b) (Agren et al., 2004).
Figure 1.
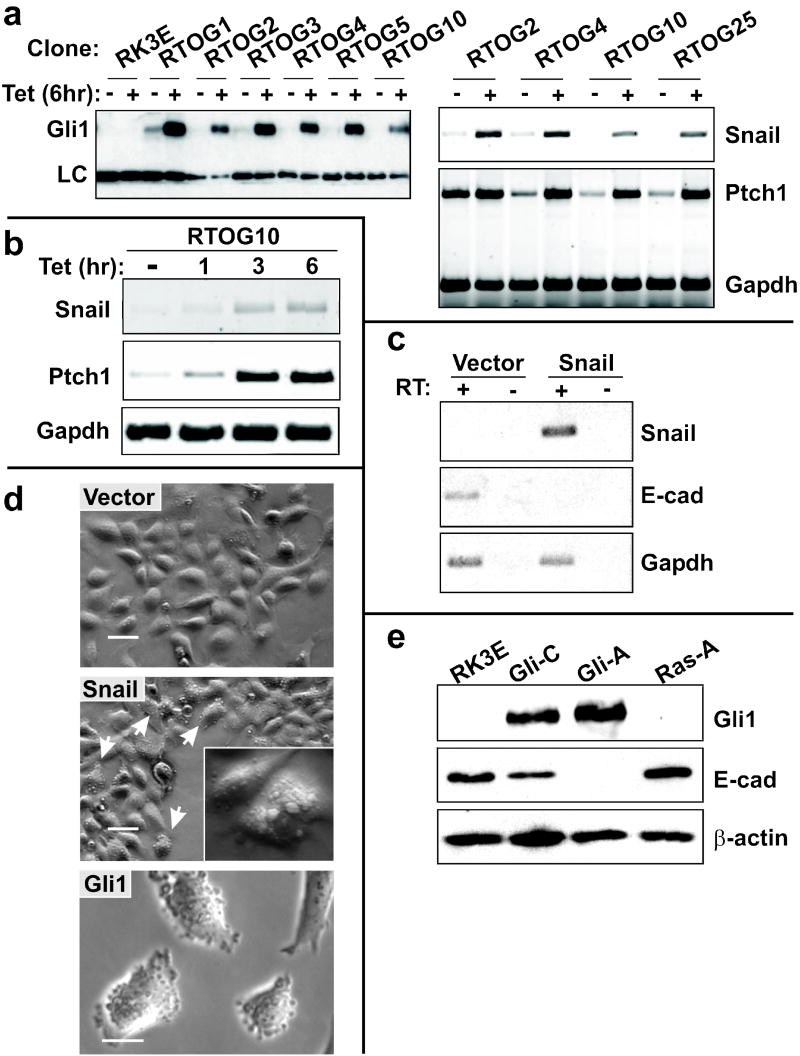
Regulation of Snail and E-cadherin by Gli1 in vitro. (a) RK3E epithelial cells were stably transduced with plasmids conferring tet-inducible Gli1 expression as described in the Materials and Methods. Cell lines were expanded from single colonies, induced with tet or vehicle control for 6 hours, and examined for Gli1 expression by immunoblot (left panel). A smaller, unidentified species detected by the antibody served as a loading control (LC). Semi-quantitative RT-PCR was used to detect the Gli1 target genes Ptch1 and Snail (right panel). (b) The time-course of Ptch1 and Snail induction was examined by RT-PCR analysis of a tet-on Gli1 cell line. Gapdh served as a control. (c) RK3E cells were stably transduced with pBpuro-Snail (Snail cells) or Vector control retrovirus (Vector cells). RT-PCR was used to detect expression of the Snail transgene, endogenous E-cadherin and Gapdh. (d) Morphology of Snail cells, Vector cells, and Gli1-transformed RK3E cells (Gli1 cells) by phase contrast microscopy. Arrows indicate examples of Snail cells with blebbing of the cytoplasmic membrane. Additional examples are shown in greater detail in the inset. Scale bars, 50μm (Vector, Snail) or 20μm (Gli1). (e) Immunoblot analysis utilized parental RK3E and transformed cell lines previously derived from oncogene-transformed foci (Louro et al., 1999). The filter was queried sequentially with the indicated antibodies. β-actin served as a loading control.
When expressed in a variety of epithelial cell types, Snail inhibits expression of E-cadherin and induces features of EMT (Cano et al., 2000; Batlle et al., 2000). We generated puromycin-resistant RK3E cell populations by retroviral transduction of pBABE-puro Snail (termed Snail cells) or pBABE-puro vector (termed Vector cells). Snail cells and Vector cells exhibited similar growth rates at subconfluence, a similar frequency of apoptosis as indicated by condensed nuclei, and were similarly contact inhibited at confluence (data not shown, and see below). In contrast to Vector cells, Snail cells showed reduced expression of E-cadherin (Figure 1c), and phase-contrast microscopy revealed prominent blebbing of the cytoplasmic membrane (Figure 1d, middle panel). This phenotype was previously described in other cell types and was attributed to impaired mechanical stability of actin filaments underlying the cell membrane (Cunningham et al., 1997; Flanagan et al., 2001). Like Snail cells, Gli1 cells have reduced expression of E-cadherin (Figure 1e) and exhibit blebbing (clones Gli-A and Gli-C; e.g., Figure 1d, bottom panel).
The typical features of EMT, such as acquisition of a spindled morphology, were not apparent in Snail cells (Figure 1d). Nor was cell migration altered, as indicated by a wound healing assay in vitro (data not shown) (Cano et al., 2000). Absence of these features may be attributed to expression of E1a, which strongly promotes the epithelial phenotype (Frisch, 1994), or to lower levels of Snail protein in these cells compared with previous studies (Cano et al., 2000; Batlle et al., 2000). Transgene expression in Snail cells was not detected by immunoblot or indirect immunofluorescence using anti-Snail antibody (Santa Cruz), or else 12CA5 antibody against the C-terminal hemagglutinin [HA] epitope. Nor was protein expression observed when Snail cells were transformed with Gli1 or RAS, treated with the proteasome inhibitor MG132, or when GSK3β was inactivated with LiCl (Zhou et al., 2004; Yook et al., 2005). In these experiments, Cos-7 cells transiently expressing the same HA-tagged transgene served as a positive control, and yielded a strong signal on immunoblots (data not shown). Thus, the level of transgene-derived Snail in RK3E cells and transformed derivatives is low, even though its activity is evident as described below.
Characterization of dominant negative Snail
Mayor and colleagues generated truncated alleles of Xenopus Snail that functioned as dominant negatives (Aybar et al., 2003). To examine a role of Snail in mammalian cells, we generated fragments of mouse Snail and tested these for inhibition of the wild type protein (Figure 2a–b). Of several alleles tested in HEK293 cells, only SnaΔZFD and SnaZFD could specifically antagonize the activity of Snail on the E-cadherin promoter. Titration experiments identified SnaZFD as the more potent of the two, as E-Cadherin transcription was restored using 0.5 μg of plasmid, equal to the input of wild type Snail (Figure 2b, compare lanes 3 and 6). SnaZFD was likewise more active than SnaΔZFD when the input was twice that of wild-type (compare lanes 4 and 7), which was sufficient to saturate the effect (compare lanes 4 and 5). Consistent with inhibition of Snail by the truncated proteins, co-expression of the two was not additive or synergistic (Figure 2b, lane 8). Induction of E-cadherin promoter activity beyond levels obtained without exogenous Snail suggested the presence of an endogenous repressor (Figure 2b, compare lanes 3, 4, 5, 7, and 8 to lane 1). Indeed, Snail is expressed in HEK293 cells (Figure 2c). The activity of truncated Snail alleles on the E-cadherin promoter, their lack of activity on the Renilla control (data not shown), and their overall similarity to the Xenopus dominant negative alleles previously characterized are consistent with their function as dominant negatives.
Figure 2.
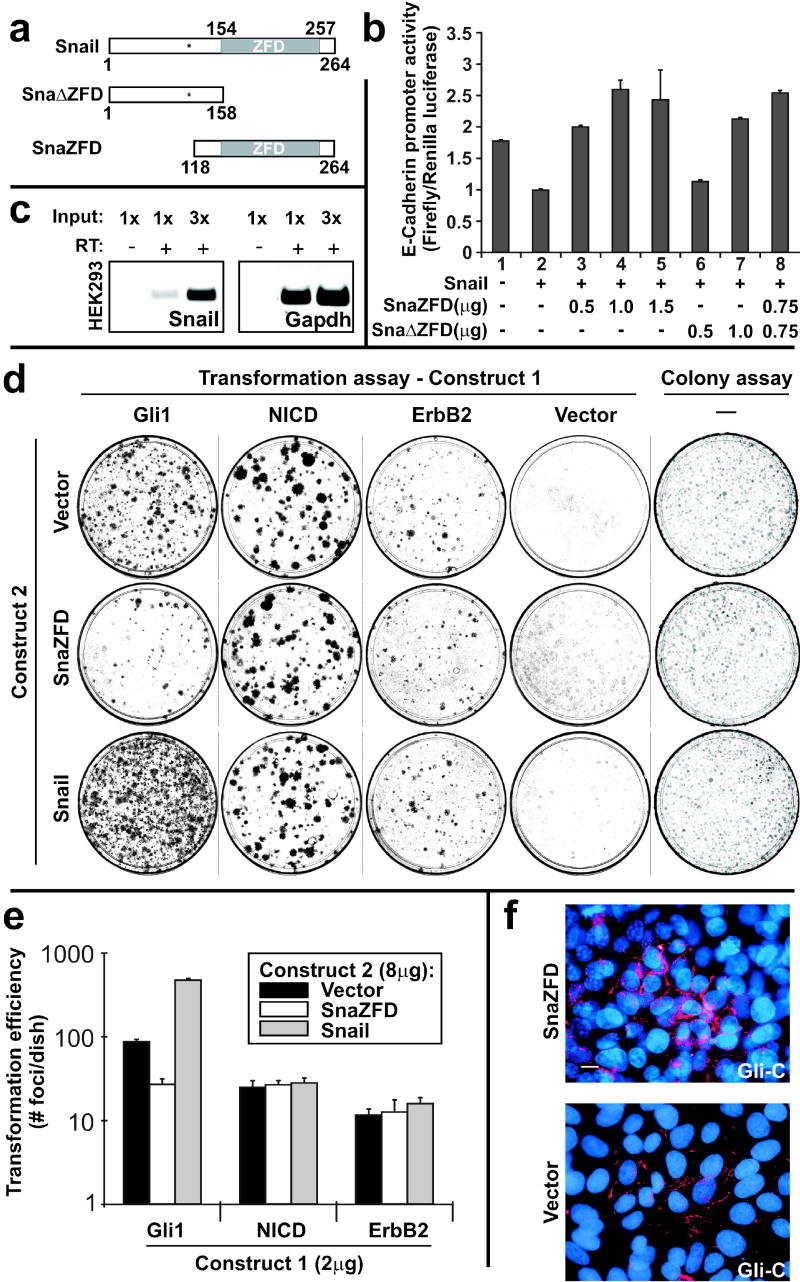
Characterization of dominant negative alleles of Snail. (a) Schematic of wild type mouse Snail, Sna ZFD and SnaZFD. The approximate position of GSK3β phosphorylation sites is indicated (asterisk) (Zhou et al., 2004; Yook et al., 2005). (b) Truncated Snail alleles were analyzed for interference with Snail-mediated repression of E-cadherin promoter activity. Plasmids were transfected into HEK293 cells, and cell extracts were analyzed using the Dual Luciferase Reporter Assay. Three experiments were performed in triplicate and standard error bars are shown. (c) Endogenous Snail in HEK293 cells was detected by RT-PCR. No product was detected without addition of RT. (d) Snail and SnaZFD were analyzed for modulation of oncogene transforming activity by lipofectamine-mediated co-transfection of the indicated plasmids (Construct 1, Construct 2) into RK3E cells. Wright-stained petri dishes contained foci of transformed cells on a monolayer of untransformed RK3E (Transformation assay, columns 1–4), or else colonies of puromycin-resistant cells that survived 7 days in selective culture media (Colony assay, column 5). (e) Transformation efficiency was plotted on a log10 scale. Results for Gli1 represent 3 independent experiments performed in triplicate, and standard error bars are shown. Results for NICD and ErbB2 represent one experiment performed in triplicate, and standard deviation bars are shown. (f) Gli-C cells were electroporated with the indicated expression plasmid. A GFP control vector indicated successful transfection of nearly all cells (not shown). E-cadherin expression (red) was visualized 48 hrs post-transfection by indirect immunofluorescence. Nuclei were stained with DAPI (blue). Scale bar, 10μ.
Induction of endogenous Snail is limiting for Gli1-induced transformation in vitro.
To determine if Snail has a role in transformation by Gli1, we expressed SnaZFD under control of the Moloney murine leukemia virus long terminal repeat (MMLV-LTR). Following lipid-mediated co-transfection with Gli1 expression vector, SnaZFD repressed the outgrowth of transformed foci by 69% (mean number of foci/dish: Gli1+Vector, 87; Gli1+SnaZFD, 27; Figure 2d, column 1, compare rows 1 and 2; and Figure 2e).
In multiple transformation assays, expression of Snail alone did not induce foci (Figure 2d, column 4, row 3). However, induction of Snail appeared to be a limiting factor in Gli1-induced transformation, as co-transfection of Snail with Gli1 induced focus formation by 5.5-fold (mean number of foci/dish: Gli1+Vector, 87; Gli1+Snail, 475; Figure 2d, column 1, compare rows 1 and 3; and Figure 2e).
As a complementary approach, we used sequential retroviral transduction to determine the transforming efficiency of Gli1 or N-RAS retroviral supernatants when applied to pooled populations of puromycin-resistant cells transduced with pBABE-puro (Vector cells) or pBABE-puro-Snail (Snail cells; described above, Figure 1d). In three independent experiments, each performed in duplicate, Gli1 induced an average of 5.4-fold more foci in Snail cells compared with Vector cells (mean foci/dish, 514 vs. 95), whereas the two cell lines showed a similar number of foci in response to N-RAS (92 vs. 87 foci/dish; Supplement Figure 1). Thus Snail cooperates with Gli1 but not with N-Ras.
Snail and SnaZFD may modulate Gli1 transformation in a pathway-specific fashion, or have a nonspecific effect on cell proliferation. To address this issue, we transfected these constructs into RK3E cells in combination with the intracellular domain of Notch 1 (NICD), previously reported to transform RK3E cells (Ascano et al., 2003), or an activated allele of ErbB2 that transforms these cells more efficiently than the wild type allele (unpublished data, MHK). Neither Snail nor SnaZFD altered the transforming efficiency of the controls (Figure 2d, columns 2 and 3; and Figure 2e). Thus, Snail specifically modulates transformation by Gli1.
To further address the possibility that Snail and SnaZFD may alter cell growth in a non-specific fashion, colony size and number were determined following transfection of MMLV-LTR plasmids into RK3E. Puromycin selection was applied 24 hours post-transfection, and colonies were analyzed at 7 days (Figure 2d, column 5). As indicated by colony forming efficiency, colony size, and cell density within the colonies, neither Snail nor SnaZFD exhibited an apparent growth phenotype in nontransformed RK3E. The colony assay supports the similar growth properties noted above for RK3E-Snail and RK3E-Vector cells (Figure 1c–d). In contrast to these results, transfection of Gli-C or Gli-A with SnaZFD produced fewer colonies compared with Vector (data not shown). Overall, the results shown in Figure 2d–e indicate that Snail promotes transformation in an oncogene-specific fashion, and is largely dispensable for transformation by NICD and ErbB2.
If the activity of SnaZFD in Gli1 transformation assays is due to interference with Snail, it might restore E-cadherin in Gli1 cells. We therefore examined E-cadherin by indirect immunofluorescence following transient transfection of E-cadherin-low Gli-C cells with the pcDNA3.1-SnaZFD vector (Figure 2f). For these studies, an electroporation protocol was optimized to enable transfection of >95% of cells, as indicated by a green fluorescent protein (GFP) control (not shown). Gli-C cells transduced with a vector control exhibited diffuse cytoplasmic staining and discontinuous, punctate membrane staining, but rarely exhibited continuous staining along the circumference of the cell (Figure 2f, Vector). In contrast, transfection with SnaZFD yielded cells with continuous staining along the cell periphery in nearly all fields examined (Figure 2f, SnaZFD). Immunoblot analysis revealed ~2-fold induction of E-cadherin by SnaZFD (not shown), consistent with restoration of E-cadherin expression in a subset of cells.
Snail short hairpin RNAs (shRNAs) restore E-cadherin in Gli1 cells and suppress RK3E transformation by Gli1
While inhibition of Gli1 transformation by SnaZFD is consistent with gain-of-function data indicating a role for Snail in this process, the dominant negative allele contains a conserved DNA binding domain that might compete with other E-box binding proteins (Bolos et al., 2003). To further address a specific role for Snail, we generated six Snail shRNA expression contructs (pSnash1-6) and a control (pSiCtl) by insertion of annealed oligonucleotides downstream of a U6 promoter (Table 1). As described above for SnaZFD, each was co-transfected with Gli1 into RK3E cells (Figure 3a–b, Table 1). Four constructs (pSnash2-5) dramatically decreased focus formation, one (pSnash6) was weakly inhibitory, and one (pSnash1) had no effect.
Table 1.
Structure of Snail shRNAs.
| ShRNA | Targeted regiona | Insert sequenceb (BamHI-Sense-loop-antisense-terminator-HindIII) | Reduction of Gli1 transformation (%) |
|---|---|---|---|
| sh1 | 558–576 | GATCCCGtaaggagtacctcagcctgTTCAAGAGAcaggctgaggtactccttaTTTTTTGGAAA | 4 |
| sh2 | 820–839 | GATCCCgaatgtccttgctccacaaacTTCAAGAGAgtttgtggagcaaggacattcTTTTTTGGAAA | 68 |
| sh3 | 809–827 | GATCCCGccttctcccgaatgtccttCTCAAGAGAaaggacattcgggagaaggTTTTTTGGAAA | 71 |
| sh4 | 532–552 | GATCCCggaaggccttcaactgcaaatTTCAAGAGAatttgcagttgaaggccttccTTTTTTGGAAA | 56 |
| sh5 | 536–556 | GATCCCggccttcaactgcaaatattgTTCAAGAGAcaatatttgcagttgaaggccTTTTTTGGAAA | 90 |
| sh6 | 549–567 | GATCCCatattgtaataaggagtacTTCAAGAGAgtactccttattacaatatTTTTTTGGAAA | 24 |
| Ctl | none | GATCCCGccttccggaagttgacgtttaTTCAAGAGAtaaacgtcaacttccggaaggTTTTTTGGAAA | control |
Numbering system refers to rat Snail (Genbank accession NM_053805), where the protein coding region is 81–875.
The strand shown corresponds to the final RNA Polymerase III transcript, with the +1 base in bold. Restriction sites, hairpin loop sequences, and terminator sequences are upper case.
Figure 3.

Inhibition of Gli1-mediated transformation by Snail shRNAs. (a) shRNA expression vectors were tested for modulation of Gli1 transforming activity (left) and for ability to alter expression or localization of E-cadherin in Gli-C cells (right). RK3E cells served as a positive control for E-cadherin (bottom right). The transformation assays shown were performed once in triplicate. Similar results were obtained in an independent experiment, performed in duplicate, that utilized distinct shRNA constructs. For immunostaining assays, Gli1-transformed cells were transduced by electroporation with the indicated shRNA expression vector and examined at 72 hrs post-transfection. (b) Quantitation of the transformation assays shown in panel a. Standard deviation bars are shown. (c) RT-PCR analysis of Snail transcripts in Gli1 cells following transient transfection of shRNA constructs. (d) Immunoblot analysis of E-cadherin in Gli1 cells transfected with the indicated construct. β-actin served as a control for loading.
To correlate shRNA modulation of Gli1 transformation and repression of Snail, constructs were analyzed for their ability to reduce Snail mRNA (Figure 3c) or to restore E-cadherin expression (Figure 3d) in Gli1 cells. In contrast to transient expression of pSiCtl or the non-inhibitory pSnash1, cells expressing the transformation-inhibitory constructs pSnash2 and pSnash3 exhibited increased peripheral expression of E-cadherin by immunofluorescence (Figure 3a, compare to untransformed RK3E, bottom). RT-PCR indicated that Snail transcripts were reduced by 33% or 41%, respectively, in cells expressing the Gli1 inhibitory constructs pSnash4 and pSnash5, in correlation with their relative Gli1-inhibitory activities (Figure 3c, Table 1). Immunoblot analysis showed that expression of E-cadherin, unlike β-actin, was increased in Gli1 cells by two active shRNAs but not by the inactive pSnash1 (Figure 3d). In summary, Snail shRNAs that inhibited transformation by Gli1 also inhibited Snail and induced expression of E-cadherin. Thus both gain- and loss-of-function studies indicate that Snail induction is limiting for Gli1-induced transformation.
Conditional expression of Gli1 in the skin
Expression of Gli1 or Gli2 in mouse skin induces outgrowth of BCC-like tumors (Nilsson et al., 2000; Grachtchouk et al., 2000; Oro and Higgins, 2003; Hutchin et al., 2005). To examine early events following induction of Gli1, we employed the same tet-on strategy that we used for analysis of KLF4 (Foster et al., 2005) (Figure 4a). In this approach, an X chromosome-linked reverse tet-responsive transactivator (rtTAX) is expressed under control of a keratin 14 (K14) promoter, and administration of doxycycline (dox) results in rapid activation of tet response element (TRE)-linked transgenes.
Figure 4.
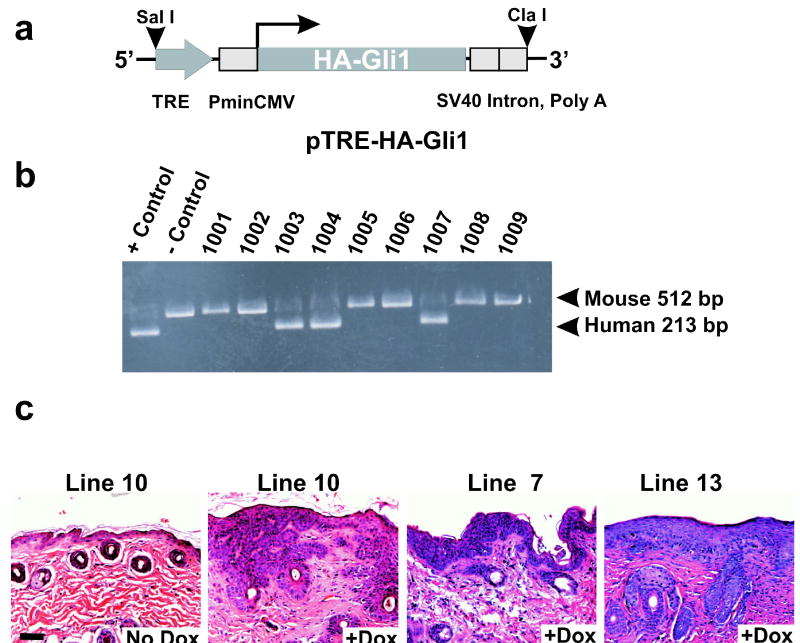
Tet-inducible Gli1 transgenic animals exhibit hyperproliferative skin lesions. (a) Schematic of the HA-tagged, human Gli1 transgene, showing the tet response element (TRE), the minimal CMV promoter (PminCMV), and the SV40 intron and polyadenylation signal. Restriction sites used for generation of the microinjection fragment are shown (SalI, ClaI). (b) PCR analysis used one pair of conserved primers, derived from exons 6 and 7, to detect both the mouse gene and the human transgene. Control DNAs were mouse genomic DNA alone or else mouse DNA admixed with a molar excess of Gli1 cDNA. (c) Histology of the skin following induction of Gli1 for 4 weeks in transgenic lines derived from 3 independent founders. A mouse of line 10 served as a control and exhibited morphologically normal skin (No dox). Scale bar, 50μ.
Founders were identified using a PCR assay in which the endogenous mouse alleles served as an internal reference (Figure 4b). Each of 5 founders was crossed to K14-rtTA animals. The progeny were induced with dox and monitored for a skin phenotype.
Gli1 rapidly induces hyperplastic lesions in the skin
Three of the five TRE-Gli1 lines showed dorsal hair loss and outgrowth of discrete skin lesions after 4 weeks of induction (Figure 4c). Grossly, lesions affected the back, neck, and ears, and appeared as clusters of flattened or slightly raised plaques, with each plaque measuring one to several mm in diameter. Lesions were yellowish in color, and microscopic analysis showed nests of basaloid cells budding from the epidermis, with extension into the dermis (Figure 4c). There was no difference in latency when 4 week inductions were initiated in mid-gestation, in 14 day-old pups, at 6 weeks, or in older adults. Thus no specific role of the hair follicle cycle was apparent (not shown). This result is in contrast to the restriction of Gli1 activity to anagen phase skin in mice with expression of Gli1 under control of a constitutive Keratin 14 promoter (Oro and Higgins, 2003). This difference between the two models may be attributed to different expression strategies and/or distinct genetic backgrounds. In the studies shown below we utilized TRE-Gli1 line #10.
Immunostaining identified similarities between Gli1-induced skin lesions, hair follicles and BCCs. Established lesions showed uniform expression of K17, a marker of follicles and BCCs, and recently implicated as a direct transcriptional target of Gli family members (Callahan et al., 2004) (Figure 5a–b). K16 marks hyperproliferative skin and is typically positive in squamous cell carcinoma (SCC), but negative in BCC. This cytokeratin was low in normal epithelial cells (Figure 5c), and likewise low in basaloid cells composing most of the lesion, but was positive in maturing keratinocytes overlying the hyperproliferative cells (Figure 5d). K1, which is normally induced upon commitment to terminal differentiation (i.e., in the parabasal cells of normal dorsal skin, Figure 5e), stained only the most superficial layer of cells within lesions, indicating a delay of differentiation, and did not stain the basaloid cells budding into dermis (Figure 5f). K14 marked the basal layer in normal skin (Figure 5g) and was uniformly positive within lesions (Figure 5h).
Figure 5.
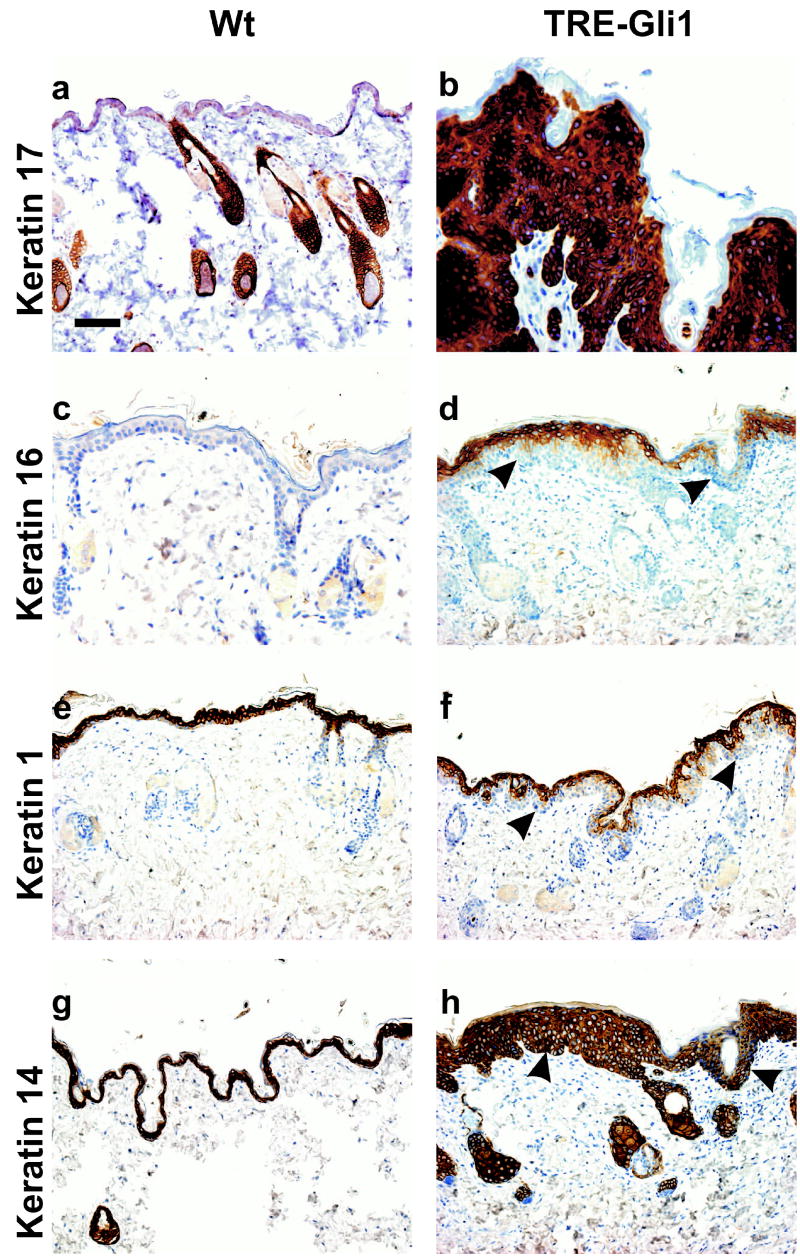
Immunostaining of Gli1-induced lesions. Dox was administered to wild type (Wt) or TRE-Gli1 mice for 4 weeks, and the indicated antibodies were applied to sections of dorsal skin. Arrowheads indicate the dermo-epidermal junction (DEJ). Scale bar, 50μ.
We utilized anti-HA antibody and indirect immunofluorescence to localize the Gli1 protein within lesions (Figure 6a). Gli1 was expressed in the interfollicular hyperproliferative epithelium, and in the outer root sheath of follicular epithelial cells. Consistent with the tight control of expression in this system (Foster et al., 2005), Gli1 was not detected without induction (Figure 6b). The strictly conditional nature of Gli1 expression in these animals is consistent with the absence of any skin phenotype in uninduced animals, and with the overall excellent health and fecundity of the transgenic lines. Primary keratinocytes obtained from bitransgenic mice exhibited prominent staining of the cytoplasm and nucleus following treatment with dox (Figure 6, c–e). At subconfluence in calcium-low culture media, these cells failed to exhibit any morphologic, proliferative, or apoptotic response to Gli1 (data not shown). The absence of any apparent phenotype of Gli1 in these cells is consistent with the limited effect of Gli1 when expressed alone in other primary rodent cells (Ruppert et al., 1991). The failure of Gli1 to affect keratinocyte morphology or growth rate at subconfluence recapitulates results obtained when Shh was expressed in primary human keratinocytes (Fan and Khavari, 1999). Our studies do not preclude more subtle effects of Gli1 such as alteration of cell cycle occupancy following a differentiation stimulus, or induction of increased cell density at confluence, both of which were observed for Shh.
Figure 6.
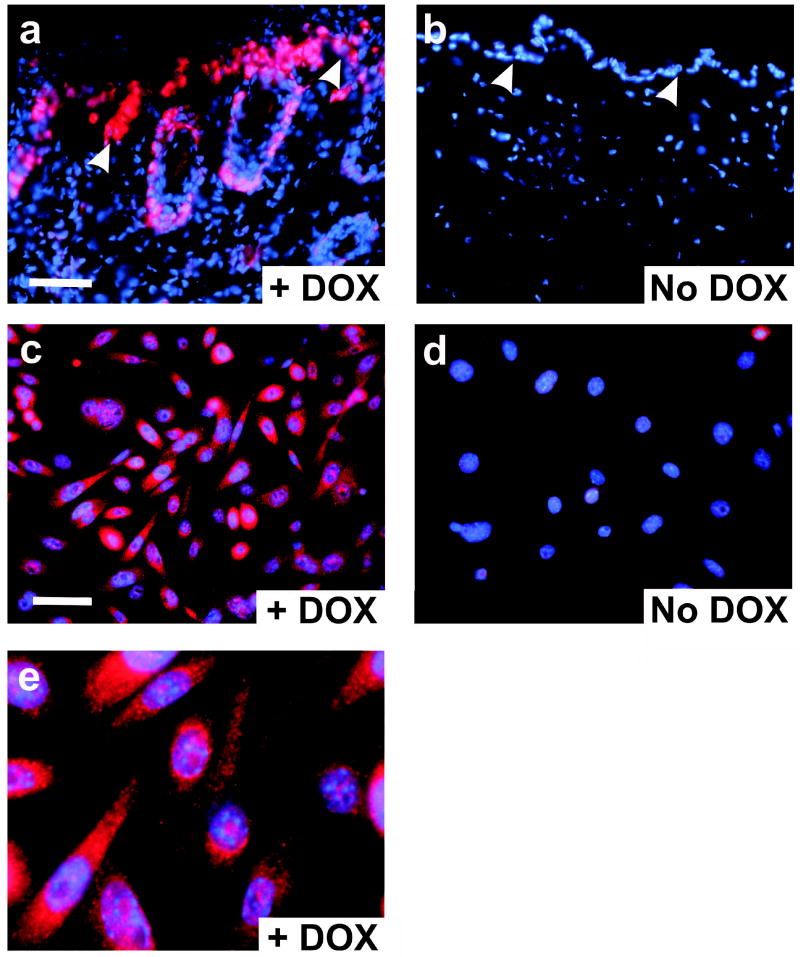
Expression of Gli1 in tissues and cells by indirect immunofluorescence. Antibody to the aminoterminal HA epitope was used to localize Gli1. All panels show merged red (antibody) and blue (nuclei) images. (a) Frozen section of a Gli1-induced skin lesion following 4 weeks of dox. Arrowheads indicate the DEJ. (b) Control skin from an animal that was not induced with dox. (c) Primary keratinocytes from a K14-rtTAX;TRE-Gli1 newborn mouse were induced with dox overnight. (d) Uninduced keratinocytes from a K14-rtTAX;TRE-Gli1 mouse. (e) Further magnification of the image shown in c. Scale bars, 50μ.
While these rapidly-induced, diffuse basaloid lesions exhibit certain molecular and morphologic similarities to BCC, they failed to form the large, focal tumors observed following longer term expression of Gli1 or Gli2 (Nilsson et al., 2000; Grachtchouk et al., 2000; Hutchin et al., 2005). We therefore sought to modify the phenotype by introducing genetic alterations observed in human tumors. We introduced deficient alleles of p53 and Ptch1, respectively, to obtain K14-rtTAX;TRE-Gli1;p53+/− and K14-rtTAX;TRE-Gli1;Ptc1+/− mice. In p53+/− animals, Gli1-induced lesions were significantly enhanced, with a 2.3-fold increased in thickness (mean 194 mm vs 83.3 mm, P<0.0001; Supplement Figure 2). These enhanced lesions had prominent neovascularization, perhaps due to the increased growth potential of mesenchymal cells in p53+/− animals, and/or to proliferative signals emanating from the neoplastic cells. In contrast to p53, the Gli1-induced skin phenotype was not altered in Ptc1+/− animals (data not shown).
Gli1 rapidly induces Snail transcripts and cell cycle progression in the interfollicular epidermis
To temporally correlate activation of Gli1 with induction of Snail in vivo, we examined expression of the human Gli1 transgene and endogenous Snail and K17, and monitored BrdU incorporation at intervals following administration of dox to bitransgenic mice (Figure 7). For these studies, littermates were induced at 40 days of age, when the hair follicle cycle is still synchronized and in the telogen phase.
Figure 7.
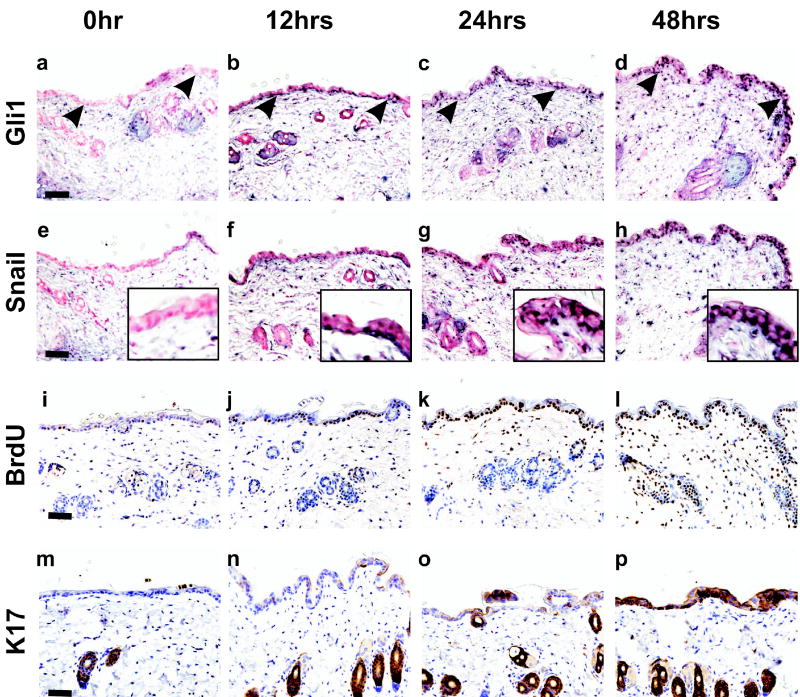
Skin alterations in the period immediately following induction of Gli1. Dox was added to the drinking water of 40 day old K14-rtTAX;TRE-Gli1 littermates for the indicated interval, and the telogen-phase skin was analyzed. Expression of Gli1 (a–d) and Snail (e–h) were monitored by in situ hybridization using anti-sense RNA probes. A Snail sense probe served as negative control and exhibited no signal in any section (not shown). BrdU incorporation (i–l) and K17 expression (m–p) were determined by immunostaining. Sections taken at 6 hrs were similar to the zero timepoint, indicating that Gli1, Snail, and BrdU were co-induced at 6–12 hrs. Arrowheads point to the DEJ. Insets (e–h) show the epidermis at higher magnification. Scale bars, 50μ.
Prior to induction there was no detectable expression of Gli1, Snail, or K17 in the interfollicular epidermis, and, of these, only K17 was present in normal follicle cells (Figure 7a, e, i, m). In the 6–12 hr interval following induction with dox, interfollicular cells concomitantly upregulated Gli1 and Snail, and incorporated more BrdU (Figure 7b, f, j). These increased until 48 hrs (Figure 7, a–l). Sections from t = 6 hr mice stained like t = 0 hr (not shown). Telogen follicle cells were slower to respond, showing increased Gli1, Snail, and BrdU signals only at 48 hrs. Compared to Snail and BrdU incorporation, K17 was slower to respond in the interfollicular cells, and became prominent only at 48 hrs. Thus, K17 may not be directly regulated by Gli1 in this setting, or its expression in interfollicular cells is restricted (Figure 7, m–p).
To determine if Snail is active during outgrowth of skin lesions, we examined E-cadherin in tissue sections from these same animals and from an animal with established lesions. Prior to induction, E-cadherin was similarly expressed in interfollicular cells and follicle cells (Figure 8a). E-cadherin largely persisted through the first 48 hrs of induction, although at this timepoint expression was lower in the interfollicular epidermis compared with adjacent follicle cells on the same slide (Figure 8b). This differential effect is consistent with the more rapid induction of Gli1, Snail, and BrdU in interfollicular epidermis compared to telogen follicles (Figure 7). More strikingly, E-cadherin was markedly reduced in established lesions, particularly in dysplastic basaloid cells infiltrating the dermis (arrowheads, Figure 8c). In the interfollicular cells of these animals, E-cadherin staining was lower where lesion thickness was greater (Figure 8c). These results recapitulate the loss of E-cadherin expression in infiltrating human BCC (Pizarro et al., 1994), and indicate that Snail may repress E-cadherin in response to Gli1 in vivo.
Figure 8.
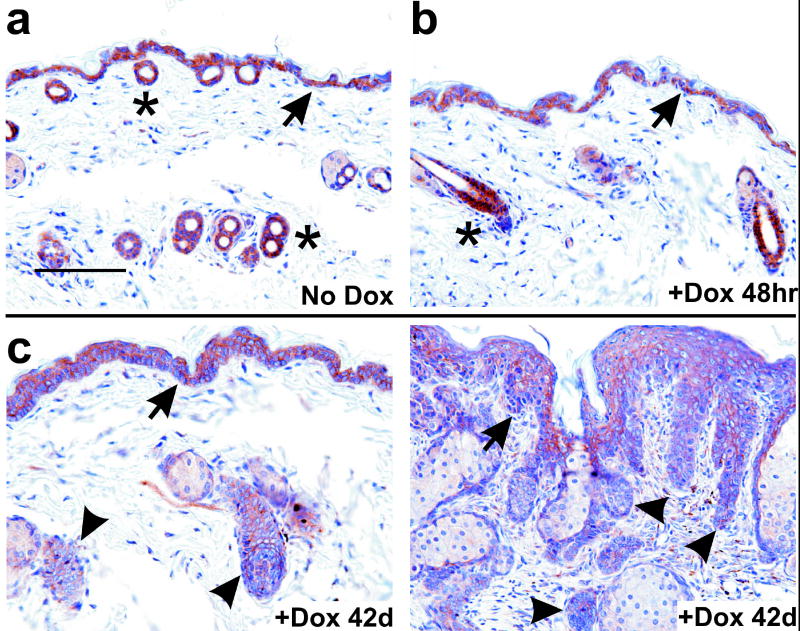
Analysis of E-cadherin expression during Gli1-induced neoplastic progression. (a–c) K14-rtTA;TRE-Gli1 mice were induced with dox for the indicated interval. Sections of skin corresponding to no treatment (No Dox); t=6, 12, or 24 hr; or t=42 days were stained in parallel with antibody to E-cadherin. Staining is indicated by a brown precipitate. (c) At 42d, adjacent areas from a single tissue section are shown, corresponding to less involved skin (left panel) or more involved skin (right panel). Arrowheads indicate epithelium with reduced staining. No signal was observed using as control a normal mouse IgG at the same concentration (not shown). Asterisks indicate hair follicles, and arrows indicate the DEJ. Scale bar, 100μ.
Discussion
Conditional expression of Gli1 in RK3E cells and in the basal keratinocytes of mouse skin demonstrated a temporal correlation of Shh pathway activity with Snail expression, and identified Snail as an early response to Gli1. Using a Snail dominant negative allele, multiple Snail shRNAs, and enforced expression of wild-type Snail, we identified Snail as a critical downstream effector of Gli1. Co-transfection of Gli1 with SnaZFD, Vector, and Snail represents a type of allelic series, with progressively increasing Snail activity. At the extremes, co-expression of SnaZFD vs. wild type Snail altered Gli1 transforming activity by 18-fold. Snail appeared specific in its ability to cooperate with Gli1, indicating that the mechanism of transformation by Gli1 is distinct from that of N-RAS, ErbB2, or Notch1. Such specificity is consistent with the induction of Snail by Gli1, but not by other genes such as KLF4 or c-MYC (Louro et al., 2002; Karhadkar et al., 2004). Our studies indicate that, in epithelial cells, the Shh signaling pathway extends to Snail to effect transformation in vitro. This signaling is a potential mechanism of E-cadherin repression in human BCC and other tumor types (Pizarro et al., 1994; Louro et al., 2002; Karhadkar et al., 2004).
EMT occurs at gastrulation and during formation of the neural tube, somites, and cardiac valves, and is essential for the establishment of complex three-dimensional tissues from epithelial precursors (Hay, 1995; Nieto, 2002; Thiery, 2003). Unlike epithelial cells, mesenchymal cells can invade and migrate through the extracellular matrix. EMT is partially or completely recapitulated invasive human carcinomas, such as BCC, and is believed to be a late event in tumor progression, when cells finally exit the epithelial compartment and become invasive and/or metastatic. EMT is regulated by signals that normally control cell fate, including the TGFβ, Notch, and Shh pathways (Hay, 1995; Thiery, 2003). During normal development and tumor progression, EMT is often linked to Snail family members such as Snail or Slug (Snai2) (Nieto, 2002).
In addition to their role in EMT, Snail family members function as regulators of proliferation or apoptosis (Cano et al., 2000; Batlle et al., 2000; Vega et al., 2004; Bachelder et al., 2005; Savagner et al., 2005; Jamora et al., 2005). Although expression in some cell types in vitro induces growth arrest, in other settings Snail induces proliferation and/or inhibits cell death. When expressed in the basal keratinocytes of transgenic mice under control of a K14 promoter, Snail activated the RAS-Mitogen-activated protein kinase pathway, repressed expression of components of the hemidesmosomes and the underlying basemement membrane, and induced proliferation (Jamora et al., 2005). GSK3β represses Snail in keratinocytes (Bachelder et al., 2005), and Slug is an effector of cell migration during wound healing in the skin (Savagner et al., 2005). Thus, the normal function of Snail family members extends beyond development to include homeostasis and healing in adult epithelium.
Both Shh and Snail are implicated in folliculogenesis. Shh-deficient mouse embryos initiate hair follicle morphogenesis, but follicles arrest at an early stage due to impaired epithelial proliferation and failure to form a dermal papilla (St Jacques et al., 1998; Chiang et al., 1999; Callahan and Oro, 2001). In addition, Shh and Gli1 are expressed in the distal portion of adult anagen follicles, probably to enable rapid downward expansion (Ghali et al., 1999; Oro and Higgins, 2003). Gli1 signaling through Snail may contribute in several respects to this process, promoting epithelial proliferation and remodeling of the basement membrane and dermis. We previously identified transcripts of Snail, Gli1, and multiple Gli1 target genes in morphologically normal anagen follicles adjacent to human BCCs (Louro et al., 2002). Similarly, Snail is transiently induced in proliferating epithelial cells of the embryonic hair bud, through the action of TGFβ2 (Jamora et al., 2005).
Our observation that Snail, like Gli1, induces membrane blebbing may help explain the minimal morphological alteration of RK3E cells upon transformation by Gli1 (Ruppert et al., 1991). In contrast to Gli1 cells, RK3E cells transformed by RAS or KLF4 show prominent membrane extensions and markedly increased spindling and refractility. Membrane blebbing has been ascribed to the inability of cells to produce more typical membrane extensions, possibly because of reduced viscosity of the peripheral actin gel (Cunningham et al., 1997; Flanagan et al., 2001; Straight et al., 2003). For melanoma cells, a tumor type that consistently expresses Snail (Poser et al., 2001), this phenotype is attributed to deficiency in the cross-linking protein Filamin A/Actin-binding protein. Our results link Snail expression to membrane blebbing, which may result from reduced expression of E-cadherin and/or from another activity of Snail (Ohkubo and Ozawa, 2004). Whether this indicates a role of Snail in regulation of Filamin A or other components of the cortical actin gel warrants further study.
Previously it was shown that constitutive Shh or Smo transgenes induced BCC-like lesions in the interfollicular epithelium of embryonic mouse skin (Oro et al., 1997; Xie et al., 1998). Our results demonstrate that Gli1 can rapidly convert interfollicular keratinocytes to proliferating, Snail-positive cells with morphological and molecular similarities to BCC. The tet-on Gli1 mouse showed clearly that adult, mammalian interfollicular epidermis, a setting relevant to human BCC tumorigenesis, is competent to rapidly respond to Shh pathway activity.
Under regulation of the same K14-rtTA transgene as used in the current study, KLF4 induced SCC-like lesions, with nuclear hyperchromicity and pleomorphism of superficially invading cells (Foster et al., 2005). The distinct phenotypes induced by Gli1 or KLF4 in the skin are consistent with expression studies and genetic data linking these genes to BCC and SCC tumorigenesis, respectively (Dahmane et al., 1997; Foster et al., 1999; Foster et al., 2000; Foster et al., 2005). Unlike Gli1-induced lesions, KLF4-induced lesions were negative for K17 and diffusely positive for K1 and K16. In contrast to the BCC phenotype induced by constitutive Gli1 transgenes (Nilsson et al., 2000; Oro and Higgins, 2003), the limited growth induced by short-term expression of Gli1 in our study suggests that other alterations are needed for progression to BCC. Such alterations may involve other Shh pathway members, such as Gli2 or Gli3, or BCC tumor suppressors such as Notch1. In addition, it will be important to examine the role of Snail in Shh-pathway mediated tumor progression in vivo, using gene deletion or conditional expression of Snail antagonists such as SnaZFD or Snail shRNAs.
Materials and Methods
Plasmid Construction
For tet-on induction in RK3E cells, an HA-tagged allele of human, wild type Gli1 was released from pBluescript (Louro et al., 2002) using XbaI and HindIII, blunted with T4 polymerase, and cloned into the EcoRV site of pcDNA4/TO (Invitrogen).
For generation of TRE-Gli1 transgenic mice, the HA-Gli1 cDNA was linker-adapted and inserted into the XbaI site of pXP2-TRE-hGFAT (a gift from J. E. Kudlow). For microinjection, a 5.7 kb fragment was released using SalI and ClaI.
SnaZFD, encoding bases 366-841 of Genbank accession M95604, was amplified from first strand cDNA of mouse NIH3T3 cells. The forward primer (5’cgcggatcccgctctggccaacatggcctcctccctggag 3’) contained a BamHI site (underlined) followed by translation initiator sequences (italics) and a region upstream of the ZFD. The reverse primer (5’ ccggaattcaagatgccagcgaggat 3’) contained an EcoRI site (underlined) and hybridized to the Snail 3’ UTR. SnaΔZFD, encoding the region up to base 488 of M95604, was released from pcDNA3-mmSnail-HA (Batlle et al., 2000) using BamHI and SspI. For use in transient transfection assays, the fragments were inserted into the pcDNA3.1+ (Invitrogen) and verified by sequencing. For stable expression, inserts were transferred to the MMLV-LTR vector pBABE-puro (Morgenstern and Land, 1990).
Employing recombinant PCR, alanine 648 (GCC) in the wild type ErbB2 cDNA (NM_004448) was altered to encode cysteine (TGC) (di Fiore et al., 1987; Kraus et al., 1987). The Xho I-adapted ErbB2 coding sequence was cloned into the Sal I site of pBABE-puro to yield pBpuro-ErbB2A648C. The mouse Notch1 cDNA (NICD), a gift of C. Jane McGlade (McGill and McGlade, 2003), was inserted into pBABE-puro.
Duplexes encoding Snail shRNAs (Table 1) were cloned into pSilencer™ 2.1-U6 neo (Ambion), transfected in XL1-Blue cells (Stratagene), and verified by sequencing. pSiCtl encodes a hairpin transcript without identity or perfect complementarity to Snail or other cellular transcripts in Genbank (release 147.0), and was constructed by reversal of sequence in pSnaSh4.
Cell transfection and retroviral transduction
pcDNA4/TO-HA-Gli1 and pcDNA6/TR (Invitrogen) were linearized with ScaI and SapI, respectively, purified from an agarose gel (Qiagen), and transfected at a 1:6 mass ratio into RK3E cells by electroporation (parameters available upon request). Colonies were selected in blasticidin (1.0 μg/ml) and zeocin (Invitrogen, 10.0 μg/ml). At 2 weeks post-transfection, individual clones were transferred to 24-well plates and expanded for further analysis.
Gli1 and H-Ras expression plasmids used for in vitro transformation assays were described previously (Ruppert et al., 1991). Focus assays were performed in 10 cm dishes, without selection, following Lipofectamine-mediated plasmid transfection (Invitrogen) (Ruppert et al., 1991). For analysis of Snail, SnaZFD and shRNAs, 2.0 μg of Gli1, NICD, or ErbB2 vector were co-transfected with 8.0 μg of the indicated plasmid. Retroviral transduction of RK3E and selection in puromycin were previously described (Foster et al., 1999; Louro et al., 2002). Colony assays following plasmid transfection utilized selection in puromycin. Transformation by retroviral transduction used the MMLV-LTR vector pLJD-HA-Gli1 (Louro et al., 2002), pCTV4-N-Ras (a gift of Robert Kay), and pLJD (a control). Phase contrast microscopic images were captured using an Axiovert 25 inverted microscope equipped with an Axiocam digital camera (Zeiss).
Transient transfection and luciferase reporter assays
Twenty-four hrs before transfection, HEK293 cells were plated at 2x105 cells per well in 6-well plates. Transfection mixtures included 0.5μg of E-cadherin promoter-luciferase reporter plasmid (Batlle et al., 2000), 0.1 μg of Renilla luciferase reporter, 0.5μg of pcDNA3.0-mmSnail-HA (Batlle et al., 2000) and the indicated amount of pcDNA3.1-SnaZFD, pcDNA3.1-SnaΔZFD, or pcDNA3.1 (Invitrogen). pcDNA3.1 was used to standardize the total amount of DNA. TransIT® -LT1 Reagent (6μl, Mirus) was used according to the manufacturer’s instructions. Extracts were prepared in Passive Lysis Buffer and luciferase activities were determined 48 hrs post-transfection.
Generation and analysis of transgenic mice
C57BL/6 (B6) X SJL (J) F2 fertilized ova were treated by microinjection in the Transgenic Animal/Embryonic Stem Cell Core Facility at the University of Alabama at Birmingham. Transgenic founders were identified by PCR. PCR primers for genotyping were 5’cctgttggggatgctggatgg3’ and 5’ggcctcagctccctggagca3’. TRE-Gli1 founders were crossed to B6;J F2 animals, and these lines were subsequently crossed to mice transgenic for X chromosome-linked alleles of K14-rtTA. Following fixation and embedding in paraffin, hematoxylin- and eosin-staining was performed by the Tissue Procurement Core Facility of the University of Alabama at Birmingham. The described gross or microscopic results were observed in five or more animals, with complete penetrance.
To generate TRE-Gli1 mice with the genotypes indicated for K14-rtTAX and p53, the three crosses used (male X female) were: p53+/− X rtTAX+/+, TRE-Gli1+/−;rtTAX+/Y X rtTAX+/+, and p53+/−; rtTAX+/Y X TRE-Gli1+/−;rtTAX+/+. Oligonucleotides, cycling parameters, and electrophoretic parameters for genotyping are available upon request. Dox (Sigma) was administered at 2.0 mg/ml in 5% sucrose water in amber bottles, and was changed three times per week.
Immunostaining
Keratins were detected in paraffin sections as described (Foster et al., 2005). K17 polyclonal antibody, a gift from Pierre Coulombe, was used at 1:2000 (McGowan and Coulombe, 1998). Immunodetection was performed using the brown chromogen diaminobenzidine (BioGenex).
E-cadherin was detected in Gli1 cells at 72 hrs post-transfection, or in RK3E cells, using primary antibody at 0.5 μg/ml (BD Biosciences #610181) and a red fluorescent secondary antibody as described (Pandya et al., 2004). For analysis of transfected cells, 20 random fields were examined at 400-fold magnification, and images corresponding to representative fields were recorded using epifluorescence. HA-Gli1 was similarly detected in primary keratinocytes using 12CA5 (Roche). For detection of HA-Gli1 in vivo, mouse skin cryosections were fixed for 10 minutes at room temperature in 4% paraformaldehyde in phosphate buffered saline (PBS), rinsed in PBS, and blocked in 20% goat serum in binding buffer (Pandya et al., 2004). The rat anti-HA antibody 3F10 (Roche) was used at 0.5 μg/ml in 2% goat serum, and bound antibody was detected using a red fluorescent secondary antibody (Molecular Probes #A11017). Nuclei were stained in DAPI and digital images were captured as described (Pandya et al., 2004).
mRNA expression studies
For RT-PCR analysis of cultured cells, total RNA was extracted using RNeasy (Qiagen). RT reactions contained 3.0 μg of total RNA per sample, oligo-dT(12–18) at 50 μg/ml, and SuperScriptTM II RT (Invitrogen). First-strand product was diluted to 200 μl, and 1–3 μl were used as PCR template. Primers are shown in Supplement Table 1. PCR reactions were terminated at intervals between 18 and 30 cycles, and products were detected by scanning of ethidium-bromide stained gels using a Typhoon 8600 (GE Biosystems). For selected samples, variation of input cDNA was used to ensure the semi-quantitative nature of the reactions.
mRNA in situ hybridization analysis of paraffin-embedded sections of mouse skin was performed using digoxigenin (dig)-labeled transcripts as described (Bardelli et al., 2003). Templates for in vitro transcription were isolated from total mRNA of mouse NIH3T3 cells by RT-PCR, and PCR primers incorporated a T7 RNA polymerase binding site. Forward and reverse primers for Gli1 were as described (Louro et al., 2002). For antisense Snail, primers were: 5’ cgtagagctgacctcgctgtccgat 3’ and 5’ ggatcctaatacgactcactatagggagacttgtggagcaaggacatgcgg 3’. For the Snail sense control probe, primers were: 5’ggatcctaatacgactcactatagggagagccggaagcccaactatagcga3’ and 5’ttcagagcgcccaggctgaggtact3’. Hybridized transcripts were detected using enzyme-antibody conjugates and the alkaline phosphatase substrate BCIP/NBT Blue (Sigma #B3804). Counterstaining was performed using Nuclear Fast Red (Sigma #N3020).
Immunoblot analysis
Cells were lysed in Laemlli buffer and extracts were quantitated as described (Sheffield et al., 1987). An immunoaffinity purified, rabbit antibody raised against residues 410–427 of human Gli1 was used at 1.7 μg/ml (Geneka/Active Motif). E-cadherin antibody (BD Biosciences #610181) was used at 0.1 μg/ml, and β-Actin antibody (Sigma #A5316) was used at 0.26 μg/ml. Bound antibodies were detected using the ECL method (Amersham Biosciences).
In vivo BrdU incorporation
BrdU reagent (Zymed #40286619, 1.0 ml/100 gr body weight) was injected into the peritoneum 2 hrs before sacrificing. BrdU was detected by immunostaining of paraffin sections as directed by Zymed (#93-3943).
Supplementary Material
Acknowledgments
We thank Antonio Garcia de Herreros, Robert Kay, C. Jane McGlade and Jeffrey E. Kudlow for sharing plasmid constructs, Bert Vogelstein and colleagues for sharing RNA in situ hybridization methods, Andrzej A. Dlugosz for advice on keratinocyte isolation, and Lawrence A. Donehower for advice on p53 genotyping. This research was supported by grants CA65686, CA094030, CA89019, P30CA13148, and P50CA097247 from the U.S. National Cancer Institute and by a gift to the Comprehensive Cancer Center from the Avon Foundation.
References
- Agren M, Kogerman P, Kleman MI, Wessling M, Toftgard R. Gene. 2004;330:101–14. doi: 10.1016/j.gene.2004.01.010. 101–114. [DOI] [PubMed] [Google Scholar]
- Ascano JM, Beverly LJ, Capobianco AJ. J Biol Chem. 2003;278:8771–8779. doi: 10.1074/jbc.M211427200. [DOI] [PubMed] [Google Scholar]
- Aszterbaum M, Epstein J, Oro A, Douglas V, LeBoit PE, Scott MP, Epstein EH., Jr Nat Med. 1999;5:1285–1291. doi: 10.1038/15242. [DOI] [PubMed] [Google Scholar]
- Aybar MJ, Nieto MA, Mayor R. Development. 2003;130:483–494. doi: 10.1242/dev.00238. [DOI] [PubMed] [Google Scholar]
- Bachelder RE, Yoon SO, Franci C, Garcia de Herreros A, Mercurio AM. J Cell Biol. 2005;168:29–33. doi: 10.1083/jcb.200409067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardelli A, Saha S, Sager JA, Romans KE, Xin B, Markowitz SD, Lengauer C, Velculescu VE, Kinzler KW, Vogelstein B. Clin Cancer Res. 2003;9:5607–5615. [PubMed] [Google Scholar]
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia de Herreros A. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Maitra A, Montes DO, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. Nature. 2003;425:846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- Bigelow RL, Chari NS, Unden AB, Spurgers KB, Lee S, Roop DR, Toftgard R, Mcdonnell TJ. J Biol Chem. 2004;279:1197–1205. doi: 10.1074/jbc.M310589200. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Cell. 2004;118:635–648. doi: 10.1016/j.cell.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. J Cell Sci. 2003;116:499–511. doi: 10.1242/jcs.00224. [DOI] [PubMed] [Google Scholar]
- Callahan CA, Ofstad T, Horng L, Wang JK, Zhen HH, Coulombe PA, Oro AE. Genes Dev. 2004;18:2724–2729. doi: 10.1101/gad.1221804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan CA, Oro AE. Curr Opin Genet Dev. 2001;11:541–546. doi: 10.1016/s0959-437x(00)00230-6. [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Chiang C, Swan RZ, Grachtchouk M, Bolinger M, Litingtung Y, Robertson EK, Cooper MK, Gaffield W, Westphal H, Beachy PA, Dlugosz AA. Dev Biol. 1999;205:1–9. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- Cunningham CC, Leclerc N, Flanagan LA, Lu M, Janmey PA, Kosik KS. J Cell Biol. 1997;136:845–857. doi: 10.1083/jcb.136.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmane N, Lee J, Robins P, Heller P, Ruiz i Altaba A. Nature. 1997;389:876–881. doi: 10.1038/39918. [DOI] [PubMed] [Google Scholar]
- di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, Aaronson SA. Science. 1987;237:178–182. doi: 10.1126/science.2885917. [DOI] [PubMed] [Google Scholar]
- Duman-Scheel M, Weng L, Xin S, Du W. Nature. 2002;417:299–304. doi: 10.1038/417299a. [DOI] [PubMed] [Google Scholar]
- Fan CM, Tessier-Lavigne M. Cell. 1994;79:1175–1186. doi: 10.1016/0092-8674(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Fan H, Khavari PA. J Cell Biol. 1999;147:71–76. doi: 10.1083/jcb.147.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan LA, Chou J, Falet H, Neujahr R, Hartwig JH, Stossel TP. J Cell Biol. 2001;155:511–517. doi: 10.1083/jcb.200105148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KW, Frost AR, McKie-Bell P, Lin CY, Engler JA, Grizzle WE, Ruppert JM. Cancer Res. 2000;60:6488–6495. [PubMed] [Google Scholar]
- Foster KW, Liu Z, Nail CD, Li X, Fitzgerald TJ, Bailey SK, Frost AR, Louro ID, Townes TM, Paterson AJ, Kudlow JE, Lobo-Ruppert SM, Ruppert JM. Oncogene. 2005;24:1491–1500. doi: 10.1038/sj.onc.1208307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KW, Ren S, Louro ID, Lobo-Ruppert SM, McKie-Bell P, Grizzle W, Hayes MR, Broker TR, Chow LT, Ruppert JM. Cell Growth Differ. 1999;10:423–434. [PubMed] [Google Scholar]
- Frisch SM. Journal of Cell Biology. 1994;127:1085–1096. doi: 10.1083/jcb.127.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghali L, Wong ST, Green J, Tidman N, Quinn AG. J Invest Dermatol. 1999;113:595–599. doi: 10.1046/j.1523-1747.1999.00729.x. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Grachtchouk M, Mo R, Yu S, Zhang X, Sasaki H, Hui CC, Dlugosz AA. Nat Genet. 2000;24:216–217. doi: 10.1038/73417. [DOI] [PubMed] [Google Scholar]
- Hay ED. Acta Anat. 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- Hutchin ME, Kariapper MS, Grachtchouk M, Wang A, Wei L, Cummings D, Liu J, Michael LE, Glick A, Dlugosz AA. Genes Dev. 2005;19:214–223. doi: 10.1101/gad.1258705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamora C, Lee P, Kocieniewski P, Azhar M, Hosokawa R, Chai Y, Fuchs E. PLoS Biol. 2005;3:e11. doi: 10.1371/journal.pbio.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- Kraus MH, Popescu NC, Amsbaugh SC, King CR. EMBO J. 1987;6:605–610. doi: 10.1002/j.1460-2075.1987.tb04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling G, Ahmadian A, Persson A, Unden AB, Afink G, Williams C, Uhlen M, Toftgard R, Lundeberg J, Ponten F. Oncogene. 2001;20:7770–7778. doi: 10.1038/sj.onc.1204946. [DOI] [PubMed] [Google Scholar]
- Louro ID, Bailey EC, Li X, South LS, McKie-Bell PR, Yoder BK, Huang CC, Johnson MR, Hill AE, Johnson RL, Ruppert JM. Cancer Res. 2002;62:5867–5873. [PubMed] [Google Scholar]
- Louro ID, McKie-Bell P, Gosnell H, Brindley BC, Bucy RP, Ruppert JM. Cell Growth Differ. 1999;10:503–516. [PubMed] [Google Scholar]
- McGill MA, McGlade CJ. J Biol Chem. 2003;278:23196–23203. doi: 10.1074/jbc.M302827200. [DOI] [PubMed] [Google Scholar]
- McGowan KM, Coulombe PA. J Cell Biol. 1998;143:469–486. doi: 10.1083/jcb.143.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP, Ingham PW, Tabin CJ. Curr Top Dev Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. 1–114. [DOI] [PubMed] [Google Scholar]
- Miller SJ. Clin Dermatol. 1995;13:527–536. doi: 10.1016/0738-081x(95)00062-k. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F. Nat Genet. 2003;33:416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- Nieto MA. Nat Rev Mol Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- Nilsson M, Unden AB, Krause D, Malmqwist U, Raza K, Zaphiropoulos PG, Toftgard R. Proc Natl Acad Sci USA. 2000;97:3438–3443. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo T, Ozawa M. J Cell Sci. 2004;117:1675–1685. doi: 10.1242/jcs.01004. [DOI] [PubMed] [Google Scholar]
- Oro AE, Higgins K. Dev Biol. 2003;255:238–248. doi: 10.1016/s0012-1606(02)00042-8. [DOI] [PubMed] [Google Scholar]
- Oro AE, Higgins KM, Hu ZL, Bonifas JM, Epstein EH, Scott MP. Science. 1997;276:817–821. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- Pandya AY, Talley LI, Frost AR, Fitzgerald TJ, Trivedi V, Chakravarthy M, Chhieng DC, Grizzle WE, Engler JA, Krontiras H, Bland KI, Lobuglio AF, Lobo-Ruppert SM, Ruppert JM. Clin Cancer Res. 2004;10:2709–2719. doi: 10.1158/1078-0432.ccr-03-0484. [DOI] [PubMed] [Google Scholar]
- Pasca di Magliano M, Hebrok M. Nat Rev Cancer. 2003;3:903–911. doi: 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- Pizarro A, Benito N, Navarro P, Palacios J, Cano A, Quintanilla M, Contreras F, Gamallo C. Br J Cancer. 1994;69:157–162. doi: 10.1038/bjc.1994.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser I, Dominguez D, de Herreros AG, Varnai A, Buettner R, Bosserhoff AK. J Biol Chem. 2001;276:24661–24666. doi: 10.1074/jbc.M011224200. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A, Sanchez P, Dahmane N. Nat Rev Cancer. 2002;2:361–372. doi: 10.1038/nrc796. [DOI] [PubMed] [Google Scholar]
- Ruppert JM, Vogelstein B, Kinzler KW. Mol Cell Biol. 1991;11:1724–1728. doi: 10.1128/mcb.11.3.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagner P, Kusewitt DF, Carver EA, Magnino F, Choi C, Gridley T, Hudson LG. J Cell Physiol. 2005;202:858–866. doi: 10.1002/jcp.20188. [DOI] [PubMed] [Google Scholar]
- Sheffield JB, Graff D, Li HP. Anal Biochem. 1987;166:49–54. doi: 10.1016/0003-2697(87)90544-6. [DOI] [PubMed] [Google Scholar]
- St Jacques B, Dassule HR, Karavanova I, Botchkarev VA, Li J, Danielian PS, McMahon JA, Lewis PM, Paus R, McMahon AP. Curr Biol. 1998;8:1058–1068. doi: 10.1016/s0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Nature. 2003;422:313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- Xie JW, Murone M, Luoh SM, Ryan A, Gu QM, Zhang CH, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH, de Sauvage FJ. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. J Biol Chem. 2005;280:11740–11748. doi: 10.1074/jbc.M413878200. [DOI] [PubMed] [Google Scholar]
- Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P. J Biol Chem. 2002;277:5548–5555. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.