

Abstract
Five members of a novel Ca2+-binding protein subfamily (CaBP), with 46–58% sequence similarity to calmodulin (CaM), were identified in the vertebrate retina. Important differences between these Ca2+-binding proteins and CaM include alterations within their second EF-hand loop that render these motifs inactive in Ca2+ coordination and the fact that their central α-helixes are extended by one α-helical turn. CaBP1 and CaBP2 contain a consensus sequence for N-terminal myristoylation, similar to members of the recoverin subfamily and are fatty acid acylated in vitro. The patterns of expression differ for each of the various members. Expression of CaBP5, for example, is restricted to retinal rod and cone bipolar cells. In contrast, CaBP1 has a more widespread pattern of expression. In the brain, CaBP1 is found in the cerebral cortex and hippocampus, and in the retina this protein is found in cone bipolar and amacrine cells. CaBP1 and CaBP2 are expressed as multiple, alternatively spliced variants, and in heterologous expression systems these forms show different patterns of subcellular localization. In reconstitution assays, CaBPs are able to substitute functionally for CaM. These data suggest that these novel CaBPs are an important component of Ca2+-mediated cellular signal transduction in the central nervous system where they may augment or substitute for CaM.
Among organisms as diverse as yeast and human, changes in the intracellular Ca2+ ion concentration initiate an array of signaling pathways. Ca2+ ions function as a diffusible signal that exerts its effect directly or through Ca2+-binding proteins on plasma membrane and intracellular channels, intracellular proteins involved in membrane trafficking, and a broad range of enzymes, including kinases, phosphatases, and adenylyl cyclases. Ca2+-binding proteins sense changes in [Ca2+] through either 130-amino acid (aa)1 structural elements called C2 domains, 29-aa EF-hand motifs, or through acidic regions of proteins or protein-lipid interfaces. In a growing number of eukaryotic signaling proteins, C2 and EF-hand motifs are present as either a single copy or clustered in multiple copies (1).
The largest group of Ca2+-binding proteins belongs to the calmodulin (CaM) superfamily. They are structurally related and comprise four EF-hand motifs, some of which (one or two) may be nonfunctional in Ca2+ coordination (2). Neuronal Ca2+1-binding proteins (NCBP) are a subset of the EF-hand-containing proteins, whose function is largely unknown. The sequence similarity among members of the NCBP family varies from ∼25% between CaM and visinin to ∼60% between GCAP1 and GCAP3 (3). NCBPs are acidic and similar in length. CaM and CaM-like proteins are the shortest (149–150 aa; molecular mass, 16,837 Da); other members of this family are ∼200 aa long (molecular mass, ∼23,000 Da) (2).
NCBPs also display a variety of interesting structural features. Multifunctional CaM contains a pair of N-terminal (EF-hand 1 and EF-hand 2) and C-terminal EF-hand (EF-hand 3 and EF-hand 4) motifs (4). The “dumbbell shape” of CaM undergoes a major conformational change upon Ca2+ coordination. These conformational changes result in reorientation of the two N- and C-terminal domains with respect to each other and a rearrangement of α-helices in the N-terminal domain that makes the hydrophobic target peptide binding site more accessible (5). The flexible region of the central α-helix bends and unwinds upon binding of CaM to many target proteins (6). Myristoylated recoverin, hippocalcin, frequenin, GCAPs, and other Ca2+-binding proteins comprise a separate subset of NCBPs (2). Binding of Ca2+ to recoverin induces more subtle changes in its compact structure, including the unclamping and extrusion of the myristoyl group. This so-called “myristoyl switch” may be unique to recoverin; other myristoylated NCBPs, like GCAP2, appear to have their hydrophobic group exposed to solvent permanently (7, 8). The transition is also accompanied by a 45° rotation of the N-terminal domain relative to the C-terminal domain, as well as the exposure of many hydrophobic residues (9). The main chain folds of neurocalcin and GCAP2 are similar to Ca2+-bound recoverin, except for structural differences near the N terminus (residues 2–18) and the binding of Ca2+ to EF-4 (8, 10).
Seidenbecher et al. (11) cloned the cDNA of a new neuron-specific Ca2+-binding protein, which displays ∼70% similarity with CaM within two C-terminal EF-hand motifs and high expression levels in the cerebral cortex, hippocampus, and cerebellum. This protein, named “caldendrin,” is an uncommon member of the CaM superfamily possessing a predicted molecular mass of 33 kDa. The ultra-structural localization in dendrites and the postsynaptic density were interpreted as evidence of an association with the somatodendritic cytoskeleton. Yamaguchi et al. (12) reported the cloning of a 70-aa-long form of caldendrin, termed “calbrain,” containing two putative EF-hand motifs. No reference to the caldendrin studies was provided, and thus it is difficult to evaluate whether the authors considered calbrain as a novel gene or a spliced form of caldendrin. In situ hybridization studies revealed abundant expression of mRNA containing calbrain sequence in the hippocampus, in the habenular area of the epithalamus, and in the cerebellum. Calbrain antagonized CaM in stimulation of CaM kinase II. These two studies raised an important question about the relationship between caldendrin and calbrain.
Here, we report characterization of five novel neuronal Ca2+-binding proteins: CaBP1, CaBP2, CaBP3, CaBP4, and CaBP5. These proteins display a new combination of functional EF-hand motifs and myristoylation. CaBP1, although structurally related to CaM, has a disabled EF2-hand motif and contains a consensus sequence for myristoylation. A large portion of the CaBP1 sequence is homologous to previously published caldendrin (11) and calbrain (12). We believe that caldendrin contains an extra sequence at the N terminus as a result of alternative splicing, although calbrain represents only a partial sequence of larger CaBP1. Retina-specific CaBP2 is expressed at low levels and is likely to be myristoylated. CaBP2 has a 3-aa deletion in the second EF-hand loop, possibly rendering it nonfunctional for Ca2+ binding. Insoluble human CaBP3 has an identical C-terminal segment as compared with CaBP5, contains only functional EF3- and EF4-hand motifs, and is partially encoded by the reverse and complementary DNA strand of the CaBP5 gene. mRNAs for both CaBP3 and CaBP5 were only found in the retina. This study reveals a much larger variety of genes encoding NCBPs, suggesting that these proteins may play significant roles in the physiology of neurons.
MATERIALS AND METHODS
Data Base Searches
Initially, expressed sequence tag (EST) data bases were searched with the query sequence that corresponded to the sequence of GCAP1 Ca2+-binding loops using tFasta in the GCG package (Genetics Computer Group). This search identified the AA363865 clone (M. D. Adams and colleagues, Institute for Genomic Research, Rockville, MD), which represents a partial sequence of CaBP1. The AA363865 sequence and full-length sequences of CaBPs were used in further searches.
Cloning of CaBPs
CaBPs were cloned by PCR from human, bovine, and mouse retina libraries in two overlapping fragments using primers from selected EST clones or new partially cloned CaBPs. Most CaBPs were cloned by nested PCR, employing primers that hybridized to the arm of the vector and CaBP sequences. PCR were heated for 5 min at 94 °C, followed by 35 cycles at 94 °C for 30 s, at a temperature specific to the primers for 30 s, and at 68 °C for 2.5 min followed by 7 min at 68 °C. PCR products were cloned in pCRII-TOPO vector (TOPO TA Cloning Kit, Invitrogen) and sequenced by dyedeoxyterminator sequencing (ABI-Prism, Perkin-Elmer) or purified for direct sequencing.
Construction of Directional cDNA Libraries
Total RNAs were isolated from human, bovine, or mouse retinal tissue using the UltraSpec RNA isolation system (Biotecx, Inc.). mRNAs were selected from those three RNA preparations using the mRNA separation kit (CLONTECH). Directional cDNA libraries were constructed using the Superscript™ plasmid system for cDNA synthesis and plasmid cloning (Life Technologies, Inc.) following the manufacturer's instructions. Some sequences of h-CaBPs were obtained by PCR using a λgt10 human retina cDNA library (J. Nathans, Johns Hopkins School of Medicine, Baltimore).
Characterization of CaBP Gene Structures
CaBP1—Clone 44N10 (GenBank™/EMBL accession number Z97197) was identified by searching the GenBank™/EMBL data base with CaBP1 cDNA as a query. The gene structure was solved by comparing CaBP1 cDNA with this genomic clone.
CaBP2—A BAC clone containing the CaBP2 gene was purchased from Genome Systems, Inc. Each intron was amplified separately by PCR using Takara LA Taq Polymerase (Fisher) through 35 cycles of 94 °C for 30 s, 54 °C for 30 s, and 68 °C for 20 min. Introns 1 and 2A were amplified with primers K49 (5′-CATATGGTTCAAGGGCCCATGGGAAA-3′) and K96 (5′-TTCAATCTCCTCGGGCCGC-3′); intron 2B with primers K95 (5′-GAGCTGCGGCCCGAGGAG-3′) and K63 (5′-GCTCCATCTCGGTGGGCATGTA-3′); intron 3 with K101 (5′-CGAGACCAGGACGGCTACAT-3′) and K98 (5′-CCCGGACACCGATCATGTC-3′); and introns 4 and 5 with K46 (5′-TTTGATGACTTTGTGGAGCTGATG-3′) and K16 (5′-TCAGCGAGACATCATCTTCACAAAC-3′). PCR products were sequenced directly or cloned into the pCRII-TOPO vector before sequencing by dyedeoxyterminator sequencing (ABI-Prism, Perkin-Elmer).
CaBP3 and CaBP5—A BAC clone containing the CaBP3 and CaBP5 genes was purchased from Genome Systems, Inc. A fragment covering exons 1–3 of h-CaBPs genomic DNA was amplified with primers K45 and K40 using Takara LA Taq polymerase, cloned in pCRII-Topo vector, and sequenced by dyedeoxyterminator sequencing (ABI-Prism, Perkin-Elmer). Fragments covering exons 3–4, exons 3–5, or exons 3–6 of the h-CaBP5 gene were amplified with Takara LA Taq DNA polymerase with primers K54 (5′-GGATCTGGGGAATCTCATGAGGA-3′) and K28 (5′-GGTCATCAGCTCCACAAAGTCA-3′), K54 and K29 (5′-CTCCCGGACAACCTCAGAGATC-3′), or K54 and K16 (5′-TCAGCGAGACATCATCTTCACAAAC-3′), respectively. Purified fragments were directly sequenced with primers designed in the exons. Fragments covering exons 3–4, exons 4–5, or exons 5–6 of the h-CaBP3 gene were amplified with Takara LA Taq polymerase, purified, and sequenced directly by dyedeoxyterminator sequencing.
CaBP4—The genomic clone CIT-HSP-1337H24 (GenBank™/EMBL accession number AC005849) was identified by searching the Gen-Bank™/EMBL data base with CaBPs cDNA as a query. The gene structure was solved by comparing CaBP cDNAs with this genomic clone.
Tissue Distribution of CaBPs
Northern Blot Analysis—mRNAs were prepared from bovine retinas as described above (see “Construction of cDNA Libraries”). Northern blot was prepared and hybridized with 32P-labeled h-CaBP1, h-CaBP2, h-CaBP5, or glyceraldehyde-3-phosphate dehydrogenase cDNA probe as described previously (3). A human multiple tissue Northern (MTN) containing 2 μg of poly(A)+ RNA from various human tissue (CLONTECH) was hybridized according to the manufacturer's instructions.
RNA Master Blots—Master RNA blots containing poly(A)+ RNA from 50 human tissues (CLONTECH) were hybridized with 32P-labeled hCaBP1, h-CaBP2, or h-CaBP5 cDNA probes according to the manufacturer's instructions.
Analysis by PCR—Total RNA was isolated from diverse mouse tissues using the UltraSpec RNA isolation system (Biotecx, Inc.). cDNA was prepared by reverse transcription with oligo(dT) from 3 μg of total RNA in a 20-μl reaction (Life Technologies, Inc.). PCR were carried out using 0.5 μl of cDNA and the Expand high fidelity PCR system (Roche Molecular Biochemicals) for 35 cycles with primers K65 (5′-GATGGGCAACTGCGTCAAGTCG3-′) and K7 (5′-CGGCCTCAGCGGGACATCATCC-3′) for CaBP1 (94 °C for 30 s, 60 °C for 30 s, 68 °C for 1.5 min); primers K69 (5′-CATATGGTTCAGAGACCCATGG-3′) and K72 (5′-CTCAGCGAGACATCATNCG-3′) for CaBP2 (94 °C for 20 s, 50 °C for 30 s, 68 °C for 3 min); primers K60 (5′-CATATGGGTCCTGCCTGCATCTTC-3′) and K30 (5′-CTCCCCATCTCCATTGGCA-3′) for CaBP5 (94 °C for 30 s, 68 °C for 1.5 min); and primers G3PDH-F (5′-GAAGGGCTAATGACCACAGTCCAT-3′) and G3PDH-R (5′-TAGCCATATTCGTTGTCGATCCAGG-3′) for mouse G3PDH (94 °C for 30 s, 68 °C for 1.5 min).
Chromosomal Localization of CaBP2 and CaBP3/5 by Fluorescence in Situ Hybridization
The localization of CaBP2 and CaBP3/5 was carried out by Genome Systems, Inc. using fluorescence in situ hybridization. DNA from CaBP clones were labeled with digoxigenin dUTP by nick translation. Labeled probes were combined with sheared human DNA and hybridized to normal metaphase chromosomes derived from phytohemagglutinin-stimulated peripheral blood lymphocytes in a solution containing 50% formamide, 10% dextran sulfate, and 2× SSC. Specific hybridization signals were detected by incubating the hybridized slides in fluoresceinated anti-digoxigenin antibodies followed by counter-staining with 4′,6-diamidino-2-phenylindole (DAPI). A second set of experiments was conducted in which genomic clones from the identified loci were cohybridized with CaBP clones.
Preparation of Anti-CaBP Polyclonal Antibodies
Rabbit anti-CaBP polyclonal antibodies were raised in New Zealand White rabbits by subcutaneous immunization with ∼50 μg/50 μl of the antigen solution mixed with an equal volume of Freund's adjuvant (Cocalico Biologicals, Inc., Reamstown, PA). Animals were boosted at 1–2-week intervals with 25 μg of antigen solution mixed with complete Freund's adjuvant. UW72, UW73, and UW75 were raised against bacterially expressed C-terminal region of CaBP1 (aa 25–227), UW92 against bacterially expressed CaBP2, and UW89 against bacterially expressed CaBP5.
Affinity Chromatographies
UW72, UW73, UW75, UW89, and UW92 Purification—Rabbit serum (6 ml) was diluted twice in 10 mm BTP, pH 7.5, containing 50 mm NaCl and loaded on respective CaBP-Sepharose (1 × 4 cm; 0.25 mg of protein/1 ml of the CNBr-activated Sepharose 4B). Columns were previously equilibrated in the same buffer. The antibody (∼4 mg) was eluted with 0.1 m Gly, pH 2.5.
Purification of Native Retinal CaBPs—A retinal extract containing CaBPs was prepared by homogenizing 50 bovine retinas in 50 ml of water, containing 2 mm benzamidine and 20 μg/ml leupeptin. The extract was separated from retinal particulates by centrifugation (48,000 × g for 50 min) at 4 °C. The extract was then loaded onto a UW75 polyclonal antibodies-Sepharose column (affinity purified; 2 mg of antibody per 1 ml of CNBr-activated Sepharose; 1 × 2.5 cm) equilibrated with 10 mm BTP, pH 7.5, containing 2 mm benzamidine at a flow rate of 15 ml/h. The column was then washed successively with: 1) the BTP buffer containing 200 mm NaCl and 2) the BTP buffer alone. The elution was performed with 0.1 m Gly, pH 2.5, containing 2 mm benzamidine. Fractions (1 ml) were collected, immediately neutralized with 1 m Tris/HCl, pH 8.4 (0.2 ml/fraction), and analyzed by SDS-PAGE and immunoblot with the UW72 or UW75 polyclonal antibody (CaBP1) (diluted 1:1000).
The fractions containing retinal CaBPs eluted from the affinity column were combined and concentrated to approximately 0.5 ml using a SpeedVac. CH3CN was then added to yield a final concentration of 15%, and the sample was loaded on a C4 column (W-Porex 5 C4, 4.6 × 150 mm, Phenomenex) equilibrated with 30% CH3CN in 0.1% trifluoroacetic acid. CaBPs were eluted with a linear gradient of CH3CN (30–60%) in 0.1% trifluoroacetic acid at a flow rate of 0.5 ml/min (0.5-ml fractions were collected). Final purification was obtained on a C8 column (Vydac 208TP52, 2.1 × 250 mm) equilibrated with 30% CH3CN in 5 mm BTP, pH 7.5. CaBPs were eluted with a linear gradient of CH3CN (30–60%) in 0.1% trifluoroacetic acid at a flow rate of 0.2 ml/min (0.2-ml fractions were collected). The separation of different forms of CaBPs was obtained on SDS-PAGE.
Microsequencing of Peptides Derived from CaBPs
Electrotransfer of proteins from SDS-polyacrylamide gels onto an Immobilon-PSQ transfer membrane (Millipore, Bedford, MA) was performed in 10 mm BTP, pH 8.4, containing 10% methanol at 90 V for 1 h at 4 °C. After transfer, the membrane was stained for 10 min with 0.1% Coomassie Brilliant Blue R-250 in 45% methanol, 10% acetic acid and then destained in 45% methanol, 7% acetic acid for approximately 10 min. The excised bands were in situ tryptic digested using 0.2 μg of sequencing grade trypsin (Promega) in 1% Zwittergent 3-16 (Calbiochem) in 0.1 m ammonium bicarbonate for 2 h at 37 °C (13) and fractionated by reverse phase HPLC (14) using an 0.8-μm Vydac C18 column. Selected peak fractions were analyzed by a combination of delayed extraction matrix-assisted laser-desorption/ionization reflectron time-of flight mass spectrometry (MALDI re-TOF MS; REFLEX III, Bruker-Franzen, Bremen, Germany) and automated Edman sequencing (model 477A, PE Applied Biosystems, Foster City, CA) (15, 16). Peptide sequences were compared with entries in nonredundant protein and nucleotide (including dbEST) data bases using the National Center for Biotechnology Information (NCBI) Advanced BLAST program. (17). Peptide monoisotopic masses were summed from the identified residues using ProComp version 1.2 software (obtained from Dr. P. C. Andrews, University of Michigan, Ann Arbor, MI).
Expression of CaBPs in Escherichia coli
The coding sequences for the long and short h-CaBP1 were amplified from human retina cDNA library by PCR using K57 (5′-CATATGGGCAACTGTGTCAAGTATCC-3′) and K7 (5′-CGGCCTCAGCGGGACATCATCC-3′) through 35 cycles at 94 °C for 30 s, 68 °C for 1.5 min. The coding sequence for b-CaBP1 was amplified from bovine retina cDNA library with primers K53 (5′-CATATGGGAAACTGTGCCAAGCGGCC-3′), which placed an NdeI site on the ATG and K8 (5′-TCAGTGATGGTGATGGTGATGGCGGGACATCATCCGGACAAA-3′), which adds a His6 tag at the C terminus of the protein. The PCRs were cycled 35 times at 94 °C for 30 s, 56 °C for 30 s, and 68 °C for 2 min. The coding sequence for m-CaBP5 was amplified from the mouse retina cDNA library with primers K42 (5′-CATATGCAGTTTCCAATGGGTCCTG-3′) and either K16 (5′-TCAGCGAGACATCATCTTCACAAAC-3′) or either K17 (5′-TCAGTGATGGTGATGGTGATGGCGAGACATCATCTTCACAAACTC-3′), which adds a Ths6 tag at the C terminus of the protein. The reactions were cycled 35 times through 94 °C for 30 s, 60 °C for 30 s, and 68 °C for 1.5 min. The short form of m-CaBP5 (without 1–4 aa) was amplified with primers K60 (5′-CATATGGGTCCTGCCTGCATCTTC-3′) and K16 (5′-TCAGCGAGACATCATCTTCACAAAC-3′) at 94 °C for 30 s, 60 °C for 30 s, and 68 °C for 1.5 min. The PCR product for each CaBP was cloned in the pCRII-TOPO vector and sequenced by dyedeoxyterminator sequencing (ABI-Prism, Perkin-Elmer). The coding sequences were cloned as fragments NdeI-BamHI in the pET-3b vector (Novagen). CaBPs were expressed in BL21 bacteria after induction with 0.2 mm isopropyl-β-d-thiogalactopyranoside.
Analysis of CaBP Myristoylation
CaBPs were produced in E. coli cotransfected with N-terminal myristoyltransferase (18). Bacteria were grown in the presence of 0.7 mCi of 9,10-[3H]myristic acid until A600 = 0.7. Expressions of CaBPs were then induced with 1 mm isopropyl-β-d-thiogalactopyranoside and grown at 37 °C for 4 h. Proteins were purified on Ni2+-NTA resin (CaBP2, CaBP5 and S-CaBP5) or on DEAE cellulose (S-CaBP1, L-CaBP1) and stained with Coomassie Blue. Bands with CaBPs were excised from the gel and dissolved in 30% H2O2, and mixtures were subjected for scintillation counting.
Phosphorylation of Rhodopsin and Other Substrates by Purified GRKs
bGRK2 and hGRK5 were overexpressed and purified from Sf9 cells as described (19, 20). GRK-mediated phosphorylation was assayed by incubating 0.8 pmol of GRK with either rod outer segment membranes (40 pmol of rhodopsin) or casein (5 μg) in 20 μl of 20 mm Tris/HCl, pH 8.0, 6 mm MgCl2, 0.1 mm CaCl2 (or 2 mm EGTA, where indicated) and 0.1 mm [γ-32P]ATP (1,000 cpm/pmol) in the presence of indicated concentrations of CaBPs or CaM for 4 min at 30 °C in room light. The reactions were stopped with 5 μl of SDS sample buffer and electrophoresed on 10% SDS-polyacrylamide gel. Gels were stained with Coomassie Blue, dried, and autoradiographed, and the 32P-labeled proteins were excised and counted.
Construct of Vectors Encoding CaBP-GFP Fusion Proteins
CMV promoter isolated as a fragment BglII-XhoI from the pcDNA 3.1 vector was cloned between the sites BglII-XhoI of pEGFP-1 vector, giving pEGFPF-1. The coding sequence for h-CaBP1, with part of the 5′-untranslated region and without stop codon, was amplified from human retina cDNA library with primers K85 (5′-AGCCTCCTTCATGGACCC-3′) and K94 (5′-GCGGGACATCATCCGGAC-3′). The PCR were cycled 35 times at 94 °C for 30 s, 55 °C for 30 s, and 68 °C for 2 min. Both PCR products (0.5 and 0.7 kb) were cloned in pCRII-TOPO vector and sequenced by dyedeoxyterminator sequencing (ABI-Prism, Perkin-Elmer). Both the short form (Sh-CaBP1) and the long form (Lh-CaBP1) of CaBP1 were cloned as a fragment EcoRI-EcoRI in pEGFPF-1 opened XhoI-EcoRI.
Expression of CaBP1-GFP Fusion Proteins in CHO Cells
Twenty micrograms of pcDNA3.1, pEGFPF-1, pSh-CaBP1-GFP, or pLh-CaBP1-GFP were transiently transfected into 50 – 80% confluent CHO cells using co-precipitates of calcium phosphate and DNA (21). Three days after transfection, cells were plated on glass coverslips, placed in culture dishes, and incubated overnight at 37 °C. Four days after transfection, fluorescent transfected cells were analyzed with a Bio-Rad MRC-600 confocal microscope.
Immunocytochemistry
Adult C57BL/6 mice were anesthetized with sodium pentobarbitol (Nembutol) delivered by intraperitoneal injection. Animals were per-fused transcardially with 20 ml of 4% formaldehyde in 86 mm NaPO4, pH 7.3. Eyes were removed and incised at the limbus to facilitate rapid fixation of the retina. After 10 min in fixative, the anterior segments and lenses were dissected from the eyes, and the eye cups were returned to the fixative. Eyes were immersed in 4% formaldehyde in 86 mm NaPO4, pH 7.3, for 4 h on ice. Residual aldehydes were removed by washing eye cups repeatedly in 137 mm NaPO4, pH 7.3 (3 × 20 min). Retinal tissue was prepared for confocal immunofluorescence labeling according to Hale and Matsumoto (22). Briefly, eye cups or peeled retinas were embedded in disposable weigh-boats containing molten low gel temperature agarose (5%) (Sigma) in phosphate-buffered saline containing 0.05% sodium azide at ∼40 °C. Agarose was solidified on ice for 1 h. Agarose blocks containing retinal tissue were trimmed and attached to metal stubs with cyanoacrylate adhesive. A Leica vibrating microtome was used to cut 100-μm sections of tissue, which were transferred to phosphate-buffered saline prior to immunolabeling. To reduce nonspecific labeling, retinal sections were incubated for 1 h in ICC buffer (phosphate-buffered saline, 0.5% bovine serum albumin, 0.5% Triton X-100, 0.01% sodium azide, pH 7.3), containing 5% normal goat serum. Sera from rabbits used for CaBP antibody production were diluted in ICC buffer as follows: UW72/CaBP1, 1:200; UW89/CaBP5, 1:400. Sections were incubated in primary antibody for 12 h at 4 °C. A monoclonal antibody to PKC (MC5 clone, Amersham Pharmacia Biotech) was diluted 1:50 and used for double-labeling studies to identify rod bipolar cells. Sections were then washed repeatedly in ICC buffer (3 × 20 min/1 × 60 min). Sections were incubated for 4 h in Cy3-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA) diluted 1:200 in ICC buffer and subsequently washed in ICC buffer 3 × 20 min and 1 × 60 min. Sections were coverslipped in 50 ml of 5% n-propyl gallate in glycerol to retard photobleaching. Sections were analyzed and images collected using a Bio-Rad 600 laser scanning confocal microscope located at the W. M. Keck Center for Advanced Studies of Neural Signaling (University of Washington School of Medicine). Negative controls for immunolabeling studies included each of the following steps: omission of primary antibody; incubation in preimmune sera at matching dilutions from rabbits used for polyclonal production; and adsorbed controls using purified CaBP1 (for UW72) or CaBP5 (UW89) (600 nm final concentration) to abolish immunolabeling.
Protein Determination, SDS-PAGE, and Immunoblotting
The protein concentration was determined by the Bradford method (23). SDS-PAGE was performed according to Laemmli (24) using 15% polyacrylamide gels. The electrotransfer of protein onto Immobilon-P (Millipore) was carried out using a Hoefer mini-gel system. For immunoblotting, membranes were blocked with 3% (w/v) gelatin in 20 mm Tris/HCl, pH 8.0, containing 150 mm NaCl and 0.05% Tween 20, and incubated for 1–2 h with primary antibody at dilutions of 1:1,000. A secondary antibody conjugated with alkaline phosphatase (Promega, Madison, WI) was used at 1:5000. Antibody binding was detected using 5-bromo-4-chloro-3-indolyl phosphate and nitro blue tetrazolium.
Models of CaBPs
Homology models of h-CaBP1, h-CaBP2, and h-CaBP5 were created on the basis of the crystal structure of h-CaM in its Ca2+-bound state (Protein Database entry: 1CLL at 1.75 Å resolution (25)), making use of the sequence alignment between these proteins (Fig. 1A). Because the N-terminal end of CaM is significantly shorter than that of the hCaBPs, for example, residues 1–19 of h-CaBP1 and residues 1–22 of h-CaBP5 were omitted from the model.
Fig.1.

Primary structure of CaBPs and phylogenetic tree. A, sequence alignment of h-CaBP1, h-CaBP2, h-CaBP3, h-CaBP4, h-CaBP5, and h-CaM. Alignment of the deduced aa sequences of h-CaBPs with CaM. The identical residues in all sequences are shown in white letters on black background. The conservative substitutions (E=D; S=T; V=M=I=L; K=R) are shown in white letters on dark gray background. The residues that are identical among CaBPs are shaded in light gray. Functional EF-hand motifs are shown in shaded boxes, and the nonfunctional EF-hand motif is in an open box. Theasterisk represents the myristoylation site, and the down arrowhead over a solid bar indicates a 4-amino acid-long extension of the central a-helix. Arrows indicate the intron/exon junctions for the h-CaBP1, 2, 4, 5, and CaM genes. Vertical bars indicate the intron/exon junctions in CaBP3 and in alternative splice forms of CaBP1 and CaBP2. L- indicates the long spliced forms of CaBPs, and the underlined aa residues are absent in the short spliced forms. The letters h and s above the sequences indicate α-helices and β-strands, respectively. B, phylogenetic tree. The tree was built with a bootstrap analysis of neighbor-joining distance using PAUPSearch in GCG (Genetics Computer Group). The sequences included, with their GenBank™/EMBL accession numbers in parentheses, are: h-CaM (A31920); h-CaM-like protein (P27482); rat caldendrin (Y17048); human recoverin (S62028); GCAP1 (L36859); GCAP2 (see Ref. 30); GCAP3 (AF110002); chicken visinin (P22728); bovine neurocalcin (JH0616); VILIP1: human visinin-like protein 1 (U14747); VILIP2: rat visinin-like protein 2 (P35332); VILIP3: human visinin-like protein 3 (P37235); NCS1: rat neuronal Ca2+ sensor 1 (P36610); CaBP4 (unpublished sequence); CaBP1, CaBP2, CaBP3, and CaBP5 are novel sequences reported in this study.
The models were generated with the HOMOLOGY module of INSIGHTII software (Molecular Simulations, Inc., San Diego, CA) using established homology modeling protocols (26). In short, protein backbone coordinates were taken from CaM for all helices, strands, EF-hands, and loops with identical lengths. The coordinates of conserved side chains were kept. Nonconserved side-chains were built from a rotamer data base.
The 4-residue insertion in the long central helix of CaBPs between the two domains of the proteins was assumed not to distort or cause significant bending of this structural element. Therefore, the residues were built in helical conformation. Experimental support for this approach comes from a mutation study with CaM in which 2 residues were deleted from the central helix. A crystal structure showed only helix shortening leading to a different separation of the two protein domains and a change in relative orientation of the domains (27). Coordinates for the loop with the 1-residue insertion were obtained by transplantation from an appropriate Protein Database entry. Finally, 2000 steps of conjugate gradient energy minimization were executed to alleviate small irregularities in the structure.
RESULTS
Molecular Cloning of Novel Subfamily of Ca2+-binding Proteins from the CaM Superfamily—A search of the EST data base with a query sequence corresponding to the Ca2+-binding loops of GCAP1 resulted in the identification of a partial sequence of a novel Ca2+-binding protein with similarity to CaM, termed CaBP1(EST AA364517 and AA363865 from a human pineal gland cDNA).
The complete sequence of h-CaBP1 was cloned by PCR from a human retina cDNA library in two overlapping fragments using primers from the EST AA364517 sequence. The full-length cDNA contains an open reading frame encoding a protein of 227 aa (Fig. 1A, Lh-CaBP1), closely related to CaM (Fig. 1B). The first ATG is set within a favorable context for translation initiation, with an A in position −3 and a G in +4 (28). PCR amplification of the full coding sequence of h-CaBP1 yielded two bands of variable intensities, one with the expected size and a shorter product with a 180-base pair deletion at the 5′-end region. The longer product encodes a protein that differs from S-CaBP1 by a 60-amino acid insert located at the N-terminal region (underlined in Fig. 1A).
b-CaBP1 and m-CaBP1 cDNA were amplified from cDNA libraries, using primers from h-CaBP1. Long and short forms of CaBP1 were cloned from bovine and mouse cDNA libraries. The amino acid sequences show a putative site for N-terminal myristoylation at Gly2 and three putative functional EF-hands, EF-hand 1, 3, and 4 motifs. Changes within the EF-hand 2 motif (Asn in position 1 instead of Asp, Gly in position 5 instead of an oxygen-containing amino acid, a flexible -Gly-Gly- sequence, and Asp in position 12 instead of Glu) may not allow Ca2+ coordination to this loop. CaBP1 shares ∼96% similarity (∼89% on the DNA level) between species and 56% similarity with CaM (∼50% on the DNA level). The predicted Mr for the long form of h-CaBP1 is 25,978 and for the short form is 19,429. CaBP1s are acidic proteins (pI ∼ 4.5) with overall aa composition similar to CaM (pI = 3.92). The 60-aa insert has no similarity to any other known protein and has an overall aa composition similar to the rest of the protein.
Clone 44N10 (GenBank™/EMBL accession number Z97197) was identified by a search of the GenBank™/EMBL data base with CaBP1 cDNA and contained the entire CaBP1 gene. The region covering the cloned CaBP1 cDNA is located between bases 170132 and 153662 of clone 44N10. The gene structure was solved by comparing CaBP1 cDNA with this genomic clone (Fig. 2A). The CaBP1 gene has an additional exon (exon 2A) in comparison with CaM. This alternatively used exon 2A is separated by two large introns and encodes the extra 60-aa fragment (Fig. 2A). The aa sequences of CaBP1 and rat caldendrin (Y17048) are highly similar within the last 151 aa and differ exactly at the exon 1/2B or exon 2A/2B junction, yielding proteins with unrelated N termini. Two additional exons encoding peptides similar to aa 91–146 and 52–90 of caldendrin were identified ∼9.5 kb upstream of the CaBP1 5′-untranslated region between bases 179,698 and 179,379 of the CaBP1 genomic clone (GenBank™/EMBL accession number Z97197; Fig. 2A). The sequence similarity between rat and human caldendrin-specific exons is only 75% (data not shown). No cDNA encoding caldendrin was detected by PCR using retina cDNA library, which suggests brain-specific expression of this splice form.
Fig.2.
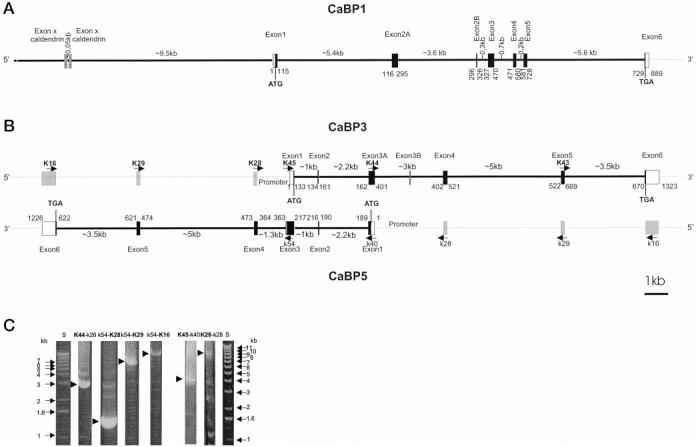
Organization of h-CaBPs genes. The coding regions are shown in black boxes, and noncoding regions of exon 1 and exon 6 are shown in white boxes. Introns are shown as thick lines. A, organization of the h-CaBP1 gene. The gray outlined box indicates the position of the closest exon specific to the caldendrin gene. The numbers below CaBP1 indicate the position of the exon/intron junctions in the CaBP1 cDNA. B, organization of h-CaBP3 and h-CaBP5 genes. Gray boxes indicate the position of exons in nonoverlapping complementary strands. The gray outlined box indicates the extra exon (exon 3B) of the CaBP3 gene that is part of exon 3 in the CaBP5 gene. Arrows indicate selected primers (K, forward primers; k, reverse primers) used to amplify the introns. Numbers below CaBP3 and above CaBP5 indicate the position of the exon/intron junctions in the CaBP3 and CaBP5 cDNAs. C, agarose-gel separation of PCR products for CaBP3 and CaBP5 gene analysis. Selected PCR products (indicated by arrows) generated with primers indicated above the gel, were cloned or purified and partially sequenced to determine the organization of the CaBP3 and CaBP5 genes.
The 5′-end of b-CaBP2 was cloned by PCR from a bovine retina cDNA library with primers from m-CaBP1 cDNA. The 3′-end of CaBP2 was then cloned as an overlapping fragment by PCR with primers from the 5′-end of b-CaBP2. h-CaBP2 and m-CaBP2 were amplified by PCR from the respective cDNA libraries with primers from b-CaBP2. The full-length cDNA contains an open reading frame encoding a protein of 221 aa (Fig. 1A), which has 72% similarity with CaBP1. CaBP2 shares 91–98% similarity between species, and it is closely related to CaM (56% similarity; Fig. 1B). A consensus sequence for myristoylation is present on a Gly adjacent to the initiation codon. PCR amplification of the full-length coding sequence of mCaBP2 yielded two products, the expected 221-aa-long product and a shorter product encoding for a protein of 160 aa. The extra 61 aa, present in the long form of m-CaBP2, localize at the same position where the extra 60 aa are present in the long form of CaBP1. A 10-aa-long fragment (LV/LGPACIFLR) of this extra peptide is 90% similar between both CaBPs. The aa sequences show three putative functional EF-hands, EF-hands 1,3, and 4, with the EF-hand 2 motif partially deleted in CaBP2. The predicted Mr are 24,834 and 18,279 for the long form and short forms of m-CaBP2, respectively. Screening of a human BAC genomic library with CaBP2 cDNA probe resulted in a BAC genomic clone containing this gene. The locations of the introns were identified by comparison with the genomic structure of CaBP1. Each intron was amplified by PCR with primers designed in the exons, and the PCR products were used to sequence the exon/intron junctions. All the exon/intron junctions were at the same position compared with the other CaBP genes (Fig. 1A). Like CaBP1, the alternatively spliced exon 2A of CaBP2 is isolated from exon 1 and exon 2B by 2 introns (not shown). The deletion of part of the EF-hand 2 motif occurred just at the position of an intron but conserved the open reading frame. The CaBP2 gene is much smaller (∼5 kb) than the CaBP1, CaBP3, and CaBP5 genes (∼16 kb) (sequences deposited in the GenBank™/EMBL Data Bank).
Using the sequence of CaBP2, we identified a genomic clone CIT-HSP-1337H24 (GenBank™/EMBL accession number AC005849), containing a novel CaBP4 (∼ 75% similarity with other CaBPs, Fig. 1, A and B). The intron/exon junctions of h-CaBP4 (Fig. 1A) were in identical positions as in other CaBPs, as determined by comparing cDNA sequences with the genomic sequence. CaBP4 is highly conserved between species (data not shown). It appears that CaBP4 gene is expressed at low levels in the retina, as well as in other tissues. CaBP4 was not characterized in this study.
Unusual Structures of CaBP3 and CaBP5 Genes That Partially Overlap on Complementary Strands of DNA—Cloning of CaBP5 was helped by EST W22993 (deposited by Dr. J. Nathans, John Hopkins School of Medicine, MD) obtained from adult human retina cDNA. This clone was found through searching EST data bases with CaBP1 as a query sequence. EST AA318398 (deposited by A. R. Kerlavage, the Institute for Genomic Research, Rockville, MD) also covers part of h-CaBP5. h-CaBP5 was cloned by PCR from a human retina cDNA library in two overlapping fragments using a primer from EST W22993 and a primer from the arm of the vector. The translation initiation ATG is difficult to assign, as there is no consensus pattern for eukaryotic translation initiation. Moreover, the second in frame Met residue shows a putative myristoylation site. The first ATG has been assigned (Fig. 1A) as the first in frame Met residue because the sequence is conserved between species from this first ATG and, furthermore, the presence of Gly close to the second ATG is not myristoylated as shown below in myristoylation analysis. This open reading frame codes for a protein of 173 aa with three putative functional EF-hands (EF1, EF3 and EF4). b-CaBP5 and m-CaBP5 have been cloned from bovine and mouse retina cDNA libraries, respectively, following the same procedure. h-CaBP5 shares 96% similarity with m-CBP5 and b-CaBP5. h-CaBP5 shares 58% similarity with h-CaM (Fig. 1B).
Using primers from EST W22993, covering part of h-CaBP5, a fifth CaBP cDNA has been cloned by PCR from human retina cDNA library. The open reading frame encodes a protein of 192 aa with only two putative functional EF-hands, EF3 and EF4 (h-CaBP3, Fig. 1A). Attempts to clone CaBP3 from bovine and mouse have not been successful so far. Two-thirds of CaBP3 cDNA has 99.5% similarity to CaBP5 in both translated and untranslated regions at the cDNA 3′-end. At the 5′-end, CaBP3 sequence is complementary and reverse to CaBP5 sequence, suggesting an overlapping organization of both genes.
A BAC genomic clone was obtained by screening of a human BAC genomic library with the full-length CaBP3 cDNA probe. The exon/intron junctions of CaBP3 gene were obtained by sequencing of PCR products or the BAC clone. The CaBP3 gene consists of 7 exons spanning > 15 kb of the genomic clone (Fig. 2B). The sequence of CaBP3, complementary and reverse to CaBP5, is encoded by exons 1–3A. To prove that both CaBP genes are on complementary strands, a PCR was carried out with a single primer (K28) from exon 4, located outside the overlapping region. An ∼8-kb PCR product (Fig. 2C, K28-k28) was obtained, which is the size expected if both genes are overlapping (Fig. 2B). The identity of the product has been confirmed by partial sequencing.
The CaBP5 gene consists of 6 exons spanning >14 kb of the genomic clone. In comparison, CaBP3 has an extra exon (exon 3B) of 10 bases which is part of exon 3 in CaBP5. Exons 4, 5 and 6 of CaBP3 have 99.5% similarity with exons 4, 5, and 6 of CaBP5. The lengths of CaBP5 introns are the same as the lengths of CaBP3 introns except for intron 3. Intron 3 of CaBP5 corresponds to introns 3A and 3B of CaBP3. Intron 3 is ∼1 kb for CaBP5 (Fig. 2B, k54-K28). Intron 3A + 3B covers ∼3 kb in CaBP3 (Fig. 2B, K44-K28). All intron/exon junctions of CaBP5 follow the gt-ag rule. One consequence of the partial overlapping organization of these CaBP genes is that the overlapping intron/exon junctions of CaBP3 do not have consensus gt-ag sequences (U2-type intron) but have complementary ct-ac sequences, which is not the rare consensus at-ac sequence.
The introns of CaBP5 (Fig. 1A, arrows above the sequences) localize at the same positions as the introns of CaM except intron 4, which is 2 aa upstream from intron 4 of CaM (Fig. 2C, arrows below the sequences). The location of CaBP5 introns is identical to the position of CaBP1 introns (Fig. 1A).
CaBP Genes Are Not Clustered on One Chromosome—In contrast to S100 proteins (chromosome 1Q12–26) (29) or GCAP1 and GCAP2 (chromosome 6p.21.1) (30), CaBPs do not cluster on one chromosome in humans. The CaBP1 gene is located on chromosome 12, based on the characterization of the 44N10 genomic clone (GenBank™/EMBL accession number Z97197); the CaBP2 gene was found on band 11q13.1 by fluorescence in situ hybridization (FISH) analysis using a BAC genomic clone as a probe (present studies, data not shown); CaBP3 and CaBP5 genes were localized to band 19q13.33 by FISH analysis using a BAC genomic clone as a probe (present studies, data not shown); and the CaBP4 gene is located on chromosome 11, based on the characterization of the genomic clone CIT-HSP-1337H24 (GenBank™/EMBL accession number AC005849).
The Expression Patterns among CaBPs Differ Significantly—Tissue distribution of CaBPs mRNA was analyzed through diverse approaches. Northern blot analyses with CaBP cDNAs as probes showed a transcript of 1.7 kb in retina and in brain for CaBP1 (Fig. 3A), weak bands of 1.5 and 1.3 only in the retina for CaBP2 (Fig. 3B), and a product of ∼1.9 kb only in retina for CaBP3/5 (Fig. 3C). A Master RNA Blot containing poly(A)+ RNA from 50 human tissues was hybridized with CaBPs as probes. No signal was detected above the background for CaBP2 and CaBP3/5. Hybridization of the Master RNA Blot with the CaBP1 probe showed a signal in whole brain, occipital lobe, and cerebral cortex (data not shown). Reverse transcriptase-PCR was performed on total RNA isolated from several mouse tissues. CaBP1 reverse transcriptase-PCR products were observed in the retina and cerebellum (Fig. 3D). CaBP2 and CaBP5 products were seen only in retina (Fig. 3, E and F). G3PDH was amplified as the control in every tissue (data not shown). The CaBP mRNA tissue distribution analyzed by different methods consistently showed specific expression of CaBP2 and CaBP5 in retina. CaBP1 mRNA was present in retina and also in brain.
Fig. 3.
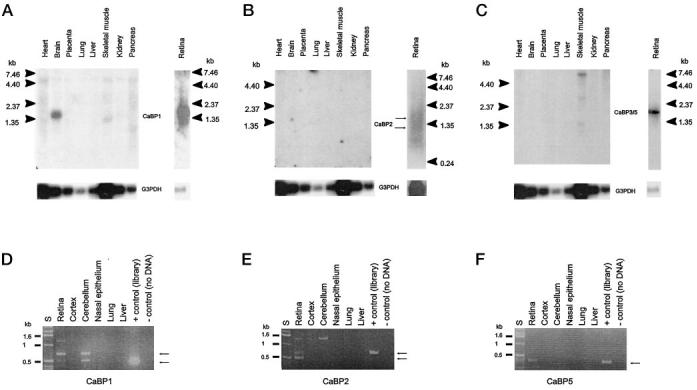
Tissue distribution of CaBP mRNAs. A–C, Northern blot analysis of CaBP mRNAs. Poly(A)+ RNA from various human tissues (CLONTECH), from bovine (A and C), and from human retinas (B) were probed with a 32P-labeled human CaBP1, CaBP2, or CaBP3 cDNA. Control hybridization with a 32P-labeled G3PDH probe is shown at the bottom. The migration of molecular size standards (in kilobases) are shown on both sides. D–F, PCR analysis of CaBPs transcripts distribution in various mouse tissues. Reverse-transcribed total RNA from multiple mouse tissues was used to amplify by PCR the CaBP1 full coding sequence of CaBP1 and CaBP2 or a partial CaBP5. A positive control was carried with G3PDH-specific primers (data not shown)
Retinal Neurons Express CaBP1 and CaBP5—Bacterially expressed CaBPs were purified and used to generate polyclonal antibodies in rabbits. One of these antibodies, UW72, was used to immunolabel CaBP1-expressing neurons in mouse retina. The antibody immunolabeled two distinct subsets of neurons in the inner nuclear layer (INL) (Fig. 4A). One group of CaBP1-positive cells has somata located in the center of the INL and the other, at the inner border of the INL. CaBP1-positive cells located at the inner border of the INL have a morphology and distribution characteristic of amacrine cells. The middle of the INL contains somata from a mixed population of neurons (rod and cone bipolar neurons) and glia (retinal Müller cells). In double-labeling studies undertaken to identify this subset of CaBP1-expressing cells, UW72 and anti-PKC (a marker for retinal rod bipolar cells) only rarely co-localized within the same INL neurons, indicating that CaBP1 is not expressed at detectable levels in the vast majority of rod bipolar cells. Similar double-labeling studies with antibodies to glial fibrillary acidic protein and cellular retinaldehyde binding protein (Müller cell markers) demonstrate that CaBP1 is not expressed in this cell type (data not shown). CaBP1-expressing cells in the center of the INL resemble cone bipolars, but definitive identification of these cells awaits future studies.
Fig. 4.

Immunolocalization of CaBP1 and CaBP5 in mouse retina. CaBP1 (UW72) (A) and CaBP5 (UW89) (B) expression is prominent in cell bodies located in the INL. CaBP1 expression is restricted to a small subset of INL neurons, while CaBP5 is expressed by a majority of INL neurons. No immunolabeling is visible in sections of mouse retina incubated in pre-immune sera from UW72 (C) or UW89 (D). Preincubation of UW72 sera with purified CaBP1 (600 nm) abolishes CaBP1 immunolabeling (E).Preincubation of UW89 sera with purified CaBP5 (600 nm) abolishes CaBP5 immunolabeling (F). Sections of mouse retina were double-labeled with antibodies to PKC and either CaBP1 (G) or CaBP5 (H). PKC immunolabels rod bipolar cell bodies and their processes (green). Very few CaBP1-labeled cells (red) are PKC-positive. In contrast, most CaBP5-positive cells (red) are double-labeled by antibodies to PKC (arrowheads, H), confirming their identity as rod bipolar cells. Cone bipolars are labeled by antibodies to CaBP5 but not PKC. Magnification bar: A–F, 50 μm; G–H, 25 μm.
The polyclonal antibody UW89, which was specific for CaBP5 (see Fig. 4B), immunolabeled rod, and cone bipolar cells in the retinas from a variety of mammalian species, including mouse, bovine, baboon, and human. UW89 immunolabeling is most intense in the perikarya of these retinal neurons, located in the outer half of the INL. Fine processes of these bipolar cells are also immunolabeled, including dendrites in the outer plexiform layer (OPL) and axons ramifying in three distinct strata in the inner plexiform layer (IPL). Bipolar cells comprise approximately 40% of the cells in the INL, and in rod dominant retinas like the mouse, the vast majority of these bipolar cells are rod bipolar cells. Double-labeling studies with UW89 and rod bipolar cell-specific antibodies (PKC) confirm that virtually all PKC-positive rod bipolars are also UW89 immunopositive. These double-labeled neurons have axons that terminate in layer 5 of the inner plexiform layer, a characteristic of rod bipolar cells. A subset of cells are UW89-positive but PKC-negative. These cells have nuclei located in the center of the INL and possess axons that ramify in two layers within the outer portion of the inner plexiform layer; both morphological features are consistent with cone bipolar cells. Double-labeling studies with horizontal cell markers (calbindin) and Müller cell markers (glial fibrillary acidic protein and cellular retinaldehyde binding protein) confirm that CaBP5 is not expressed at detectable levels in either of these cells types in adult animals. Specificity of UW89 antibody was confirmed by preadsorbing this antibody with an excess of purified CaBP5 prior to immunolabeling, which abolished the signal.
Two Splice Forms of CaBP Show Different Cellular Localization—To identify the subcellular localization of the long and short forms of CaBP1, two constructs carrying fused GFP to the coding regions of CaBP1 were made. pSh-CaBP1-GFP (short) and pLh-CaBP1-GFP (long) were transiently transfected into CHO cells and analyzed using confocal microscopy. S-CaBP1 localized at or near the plasma membrane (Fig. 5C), whereas L-CaBP1 was associated most likely with the cytoskeletal structures (Fig. 5D). These data suggest that the splice forms may have distinct localization patterns, producing further diversification among CaBPs.
Fig. 5.
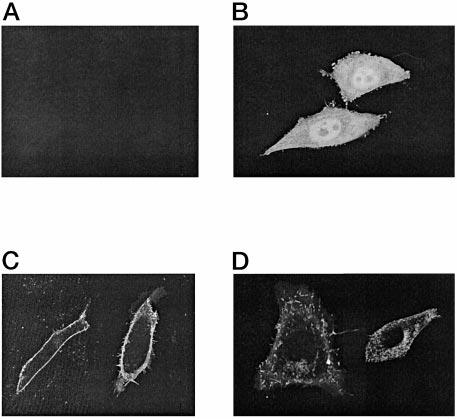
Subcellular localization of GFP fusion proteins in transfected cells. Confocal fluorescence of CHO cells transiently transfected with a vector (A), GFP (B), Sh-CaBP1-GFP fusion (C), or Lh-CaBP1-GFP fusion (D).
CaBPs Are Translated and Can Be Isolated from the Retina—To identify CaBPs in bovine retina, soluble proteins were extracted with low ionic strength buffer, separated from membranous material by ultracentrifugation and passed through a UW75-Sepharose column. UW75 was raised and affinity purified employing bacterially expressed CaBP1 (Fig. 6B). Further purification of retinal CaBP was obtained on reverse phase HPLC columns, and by SDS-PAGE (Fig. 6, C and D). Protein sequence data revealed that the higher molecular mass band (18 kDa) contained sequences identical between CaBP1 and CaBP5, whereas in the 16-kDa band, CaBP5 sequences were clearly identified. A 16-kDa form of CaBP1 was also partially purified from brain (data not shown). These results established that CaBP1 and CaBP5 are expressed in bovine retina.
Fig. 6.
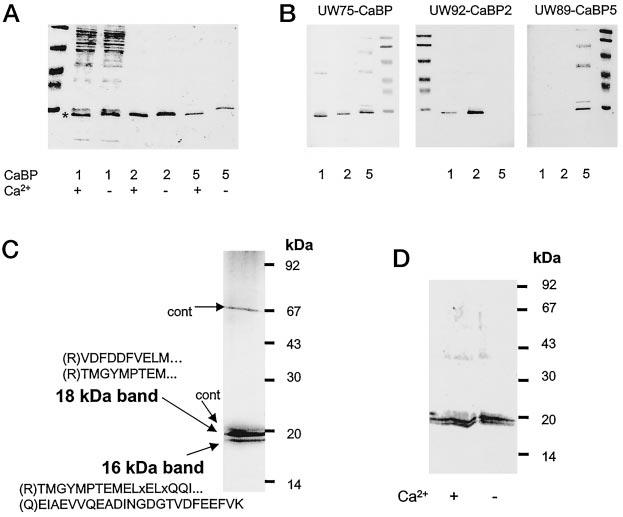
Specificities of anti-CaBP antibodies and purification of retinal CaBPs. A, Ca2+-dependent mobility change in SDS-PAGE for partially purified CaBP1 (purified on DEAE-cellulose; see “Materials and Methods”), CaBP2-His6 purified on Ni2+-chelex column, CaBP5-His6 purified on Ni2+-chelex column. B, immunoreactivity of CaBP1, CaBP2, and CaBP5 with UW75 (raised against the common domain of CaBP), UW92 (raised against CaBP2), and UW89 (raised against CaBP5). C, retinal CaBPs were purified as described under “Materials and Methods” and blotted on polyvinylidene difluoride. The protein bands were digested with trypsin, and peptides were separated on a reverse phase column. The aa sequence of peptides was obtained using a combination of Edman degradation of matrix-assisted laser desorption/ionization (MALDI; see “Materials and Methods”). In brackets are the predicted residues from the cDNA sequence and the N-terminal side of tryptic digestion: x represent an undetermined aa, dotted points show that the sequence did not reach the end of the peptide. cont, contaminants from UW72. D, Western blot of retinal CaBPs probed with UW72 in the presence or absence of 100 μm [Ca2+].
CaBP1 and CaBP2 Are Myristoylated—CaBP1 (MGNCVKYP …, where G is the site of myristoylation) and CaBP2 (MGNCAKR..) contain consensus sequences for N-terminal myristoylation (Fig. 1). The short and long forms of CaBP1 were expressed in E. coli, and cotransfected with a plasmid encoding human N-terminal myristoyltransferase. The expression of CaBPs was induced by isopropyl-β-d-thiogalactopyranoside in the presence of [3H]myristic acid. Both forms of CaBP1 were myristoylated (Fig. 7). In a similar experiment, CaBP2 was also modified; however, neither CaBP5 nor Met6-CaBP5 was myristoylated (Fig. 7). These results suggested that CaBP1 (both splice forms) and CaBP2 are myristoylated in vivo.
Fig. 7.
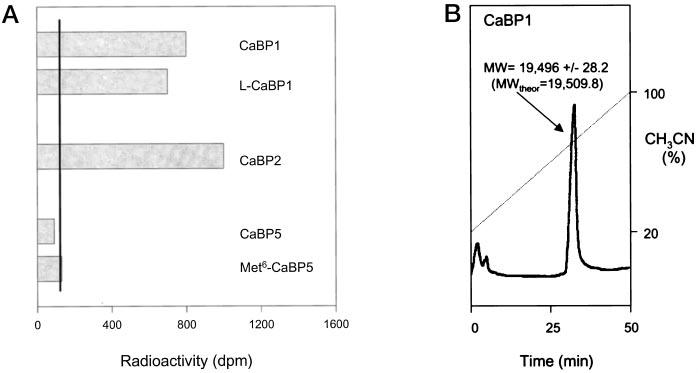
Myristoylation of CaBPs in bacteria co-expressing human N-terminal myristoyltransferase. A, the expression of CaBPs were done in 30-ml cultures in the presence of 0.7 mCi of [3H]myristic acid. The proteins were affinity purified and separated by SDS-PAGE. CaBPs were identified by Western blot, the band was cut out of the gel, and the radioactivity was measured. CaBP2 was expressed as the short form, and Met6 indicated a construct of CaBP5 with the second ATG as the initiation Met. The black line represents the basal activity obtained from similar areas on the gel but without proteins. Similar results were obtained from two independent cultures. B, HPLC purification of CaBP1. CaBP1 was expressed in E. coli, purified on the phenyl-Sepharose column (see “Materials and Methods”), and injected onto a W4 Porex C4 column (Phenomenex). Protein was eluted with 20–100% CH3CN during 50 min in the presence of 0.1% trifluoroacetic acid at a flow rate of 0.5 ml/min. The protein peak was analyzed by electrospray mass spectrometry. The predicted (theor) molecular mass was in a good agreement with the observed value, suggesting that CaBP1 was myristoylated.
CaBPs Are Sensitive to Ca2+—There are several lines of evidence in addition to the consensus sequences for Ca2+-binding, that these proteins are indeed active in Ca2+ coordination. CaBPs showed small but reproducible shifts in mobility during SDS-PAGE (Fig. 6A). Similar to CaM, CaBP1 was bound to Phenyl-Sepharose in the presence of Ca2+, and eluted when Ca2+ was removed. This procedure allowed the purification of CaBP1 to apparent homogeneity in one step and with ∼50% yield (data not shown). CaBP2 was also efficiently purified on this column (the overall yield ∼20%), whereas CaBP5 interacted very strongly with phenyl-Sepharose in the presence and absence of Ca2+. Equilibrium dialysis experiments are in progress, and they indicate high affinity Ca2+-binding sites within CaBPs. Collectively, these data indicate that CaBPs are sensitive to Ca2+ and function as a Ca2+-sensitive sensor within the cells.
Activation of CaM kinase II by CaBPs—To identify potential targets for CaBPs, we have tested known effector proteins that are regulated by CaM. This approach was justified by close similarity of CaBPs with CaM. Furthermore, it was reported that calbrain inhibited the CaM protein kinase II activity (12). In our assay conditions, all CaBPs were able to stimulate CaM kinase II (Table I, Fig. 8A). These differences most likely resulted from the fact that calbrain represents only a partial clone of CaBP1. From all CaBPs, CaBP2 was the most effective in the CaM kinase II stimulation (Fig. 8A). CaBP2 also bound most avidly to a peptide derived from the CaM-binding domain of CaM kinase II (Fig. 8B), whereas CaBP1 and CaBP5 did not bind significantly.
Table I.
Activation of CaM kinase II by CaBPs The activity of CaM kinase II from rat brain was measured as a Ca2+-dependent phosphorylation of syntide-2 (BioMol Inc.) for 30 min at 30 °C (12). The reaction was carried out in 50 mm Hepes, pH 7.5, containing 10 mM MgCl2, 100 μm [γ-32P]ATP (800,000 cpm/nmol), 2 mm CaCl2, 80 μm syntide-2, 0–2 μm Ca2+-binding proteins, and 0.4 ng/μl CaM kinase II (purchased from BioMol Inc.). ND, not determined.
| Ca2+-binding protein | Maximal activity | Activation EC50 |
|---|---|---|
| nmol/min/mg | nM | |
| – | 0.91 ± 0.4 | – |
| CaM | 187.5 ± 19 | 138 ± 12 |
| CaBP1 | 14.5 ± 3 | 360 ± 120 |
| CaBP2 | 62.5 ± 12 | 241 ± 16 |
| CaBP5 | 6.25 ± 1.1 | ND |
Fig. 8.
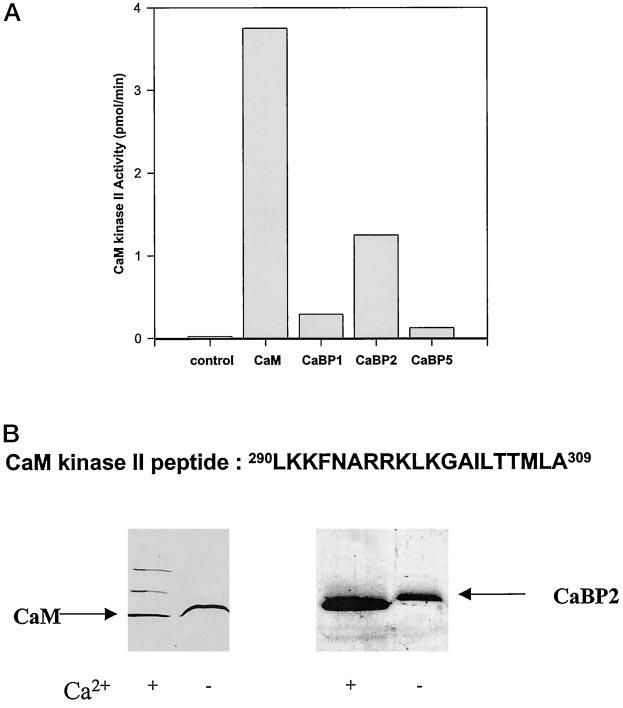
Activation of CaM kinase II by CaBPs and interaction of CaBP2 with the peptide derived from the CaM-binding domain of CaM kinase II. A, the activity of purified CaM kinase II was measured in the presence of 2 μm Ca2+-binding proteins using syntide-2 as a substrate (Table I). The short forms CaBP1 and CaBP2 were used in the assays. B, binding of CaBP2 to the peptide derived from the CaM-binding domain of CaM kinase II. The peptide 290LKKFNARRKLKGAILTTMLA309 was coupled to CNBr-activated Sepharose, and CaM and CaBP2 were added in the presence of 50 mm Ca2+. The column was washed with 10 mm BTP, pH 7.5, containing 100 mm NaCl. Bound proteins were eluted with 1 mm EGTA. CaBP1 and CaBP5 did not bind to this column.
Inhibition of GRK2 and GRK5 Activities by CaBPs—We also tested whether CaBPs can inhibit GRK activity, known to be potently modulated by CaM (reviewed in Ref. 31). At the highest concentrations tested (CaBP1 ∼ 0.8 μm, CaBP2 ∼ 6 μm, CaBP5 ∼ 20 μm) none of the CaBPs significantly inhibited the activity of GRK2 in either the absence or presence of Gβγ (data not shown). However, CaBPs inhibited GRK5-mediated phosphorylation of rhodopsin (Fig. 9). CaBP1 appears to have a much higher affinity for GRK5 (IC50 ∼0.12 μm) compared with CaBP2 (IC50 ∼5.5 μm) and CaBP5 (IC50 ∼2.5 μm) (Fig. 9). CaBPs also inhibited GRK5 phosphorylation of a soluble substrate, phosvitin, although significantly less efficiently; at 0.8 μm, CaBP1 inhibited ∼47% of phosphorylation, while at 20 μm, CaBP5 inhibited ∼34% of phosvitin phosphorylation (data not shown). Also unlike CaM, which activates GRK5 autophosphorylation (32), CaBPs had no significant effect on GRK5 auto-phosphorylation (data not shown). These experiments showed that CaBPs can substitute for CaM in some assays and showed some degree of specificity among CaBPs.
Fig. 9.
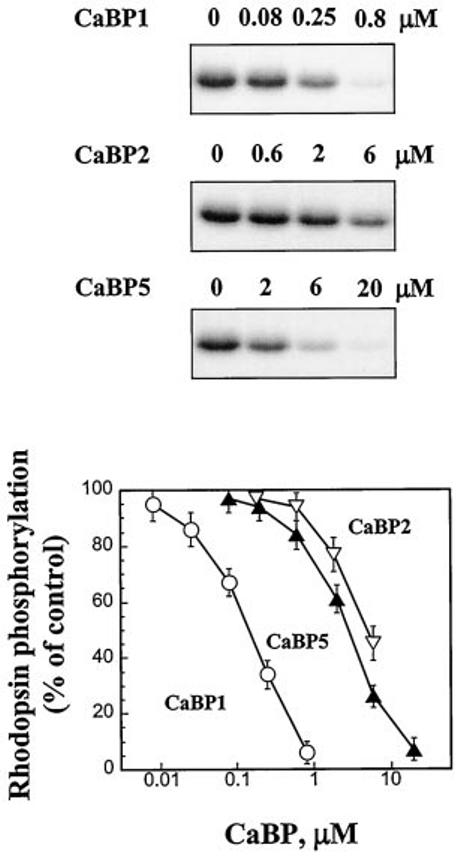
Inhibition of GRK5 activity by CaBP1. Purified GRK5 (0.8 pmol) were used to phosphorylate rod outer segment membranes (2 μm rhodopsin) in the presence of indicated concentrations of CaBPs. Proteins were separated on a 10% SDS-polyacrylamide gel and visualized by autoradiography. 32P incorporation into proteins was determined by excising and counting the radioactive bands. The activity of GRK5 in the presence of CaBPs is expressed as a percentage of the rhodopsin phosphorylation in the absence of CaBPs. The short forms CaBP1 and CaBP2 were used in the assays.
DISCUSSION
A Novel Family of CaBPs—Changes in [Ca2+] are a key factor in cellular regulation. These changes are sensed, either directly by proteins, including enzymes, or through specific Ca2+-binding proteins. To understand Ca2+-signaling on the molecular level, it is essential to identify all molecules that respond to changes in [Ca2+], including sensing proteins and their physiological targets. A large number of Ca2+-binding proteins have been identified, and by far, the largest subfamily are CaM-like proteins. Members of this subfamily are small (∼200 aa), acidic (pI ∼ 4–5) proteins that contain four EF-hand motifs, from which only selected motifs are functional. Frequently, they are specifically expressed in subsets of neurons (NCBP:GCAPs, recoverin, neurocalcin, and others).
In this study, we identified five related novel genes encoding Ca2+-binding proteins and their several alternatively spliced forms, yielding an even larger group of related proteins. In addition, a peptide sequence related to CaBP was obtained from sequencing retinal proteins purified on an anti-CaBP-specific antibody column. So far, this sequence has not been assigned to any specific gene reported in this study,5 suggesting that the subfamily of CaBPs, is perhaps even larger.
Structural Differences between CaBPs and CaM—CaBPs are most closely related to the ancestor CaM and other neuron-specific Ca2+-binding proteins (Fig. 1B), but they differ from them in three major ways. First, some of the CaBPs are myristoylated, or have a Cys residue that could be palmitoylated. CaBP1 and CaBP2 are also produced in two splice forms (or three for CaBP1, considering caldendrin), respectively. As shown in this study, each of the two CaBP1 variants display unique localization to specific regions of the cell (e.g. cytoskeleton versus the plasma membrane) (Fig. 5). In general, the N-terminal part of CaBPs is the most divergent from CaM. Second, the central α-helix is extended by one turn (4 aa) as compared with the corresponding 28-amino acid-long central helix in CaM (Fig. 10). Because this α-helix in CaM undergoes major conformational changes upon Ca2+ coordination and is involved in the interaction with the target molecules, further structural studies will be important to elucidate what structural changes this additional turn may cause to the overall conformation of these proteins. Third, the EF2-hand motif is likely to be nonfunctional because of the following changes in this loop: the presence of a Gly-Gly motif, only one negative charge within this Ca2+-binding loop, and substitution of Glu in 12 position by Asp (CaBP1); deletion of 3 aa followed by a flexible -Gly-Gly- allowing a shorter Ca2+ loop (CaBP2); Arg/Lys substitution of a critical Asp in position 1 (CaBP4 and CaBP5). Inactivation of specific EF-hand motifs of CaM may have provided an evolutionary mechanism for diversification Ca2+ signaling (Figs. 10 and 11). Similar examples of inactivation may be found in the EF1-hand motif (GCAPs), the EF1-and EF-4 hand motifs (recoverin), or the EF2-hand motif (CaBPs). This selective EF-hand inactivation may contribute to specificity in the interaction with the target molecules, and for intracellular compartmentalization. The C-terminal two-thirds of the CaBP sequence is very similar to CaM (∼60%). CaBPs also contain a high percentage of Met; for example, 7.5% in CaBP5, or 6.5% in CaBP1. These values are similar to those found in CaM (6.6%) but much higher than for recoverin (2.5%), which undergoes rather minor conformational changes (9), or for a typical globular protein (1.5%). These Met residues are well conserved among CaBPs, but only four Met residues are conserved with CaM. It is believed that Met residues are important because of a more polarizable -S-CH3 group than -CH2-CH3 in other hydrophobic aa residues. These properties of Met are also crucial for specific interactions with the target molecules through van der Waals interactions. Met residues are also more easily hydrated, allowing major structural conformational changes upon Ca2+ binding/dissociation when the hydrophobic interior is exposed in these proteins (6). It appears that CaBPs may utilize the same property of Met.
Fig. 10.

Model of CaBPs. Ribbon diagram of CaBPs. Ribbons are shown in gray, EF-hand motifs are in yellow, Ca2+ are shown in red (figure generated using GRASP, and aa numbers are for the short form of CaBP1 (see “Materials and Methods”)). Three elements that are different from CaM are marked with arrows; these include the N-terminal myristolylation (CaBP1 and CaBP2), inactivation of EFII-hand motif (all CaBPs), and extra α-helical turn (all CaBPs).
Fig. 11.
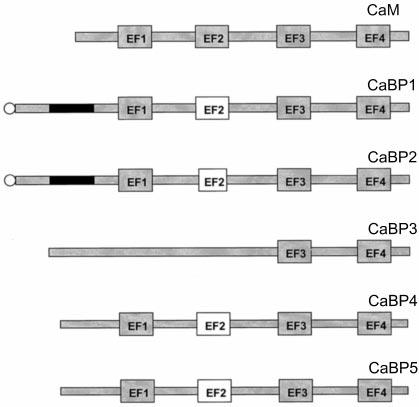
Diagram of CaBPs characterized in this study. CaBP1 and CaBP2 are expressed in two spliced forms with an ∼60-amino acid-long insert on the front of the first EF1-hand motif. The inserts are shown as black boxes. This insert sequence has 35% similarity between these two proteins. CaBP1 and CaBP2 contain a consensus sequence for myristorylation. In bacteria, they are substrates for human N-myristoyltransferase (indicated by a circle). CaM has all four EF-hand motifs functional in Ca2+ coordination, whereas all CaBPs have nonfunctional EF2-hand motif (open box), either because of deletion of 3 aa (CaBP2) or disabling mutations (CaBP1 and CaBP5). CaBP3 has dissimilar sequences with CaM and other CaBPs through the N-terminal region, EF-hand motifs 1 and 2. All CaBPs are dissimilar in sequence with CaM and other CaBPs through the N-terminal region, EF-hand motifs 1 and 2. The sequences of the C-terminal halves of all h-CaBPs are highly conserved (80%).
Genomic Structures—In contrast to S100 proteins (29), or GCAP1 and GCAP2 (30), CaBP genes do not cluster on one chromosome, although all CaBP genes are evolutionarily closely related. CaBP3 and CaBP5 genes localize close to the locus of one of the genes coding for CaM, CALM3 (33). All CaBP genes share the same genomic structure, which is also almost identical to the CaM gene structure except for one intron. The introns not only localize at the same position in the aa sequence but also at the same position relative to the reading frame in the DNA sequence. Only the fourth intron of CaM localizes 3 aa downstream from CaBPs intron 4, similarly to other genes, sharing the same genomic structure with CaM.
CaBP3 and CaBP5 genomic organization was intensively studied because we observed that CaBP3 and CaBP5 not only have identical cDNA 3′-ends but also have cDNA 5′-ends complementary to each other. We have shown that the 5′-end of CaBP3 gene overlaps the 5′-end of CaBP5 gene on the complementary strand. The exons and introns of CaBP3 are strictly complementary to the exons and introns of overlapping CaBP5. Several reports describe examples of bidirectionally transcribed genes that partially overlap at their 5′- or 3′-ends, mostly in noncoding regions or promoter regions (reviewed in Ref. 34). The BCMA gene has an organization most closely resembling CaBP3 and CaBP5 genes, with the exon/intron splicing sites shared by the sense and antisense transcripts in the coding sequence (35). In prokaryotes, natural antisense RNAs are part of a general mechanism of control of gene expression (reviewed in Ref. 36). This might also be a potential role in eukaryotes (reviewed in Refs. 34 and 37). Although many antisense RNAs have been described in eukaryotes, only a few are spliced and present an open reading frame (ORF). The h-CaBP3 gene is spliced, polyadenylated, and has an ORF of 192 aa. Moreover, the CaBP3 C-terminal half is identical to the C-terminal half of CaBP5 because of the duplication/inversion of the gene. Because no CaBP3 transcripts were detected in bovine and mouse retinas, we analyzed the complementary strand of the bovine and mouse CaBP5 for the presence of a putative ORF. No ORF similar to that in the h-CaBP3 is possible in mouse and bovine retinas because of the presence of a stop codon on the complementary strand.
Because the overlapping exon/intron junctions of CaBP3 are at the same position as those of the CaBP5 gene, the 5′-donor and 3′-acceptor sites are CT and AC, complementary to GT and AG. It is puzzling how these introns are spliced, because these splice sites cannot be recognized by the U2- or more rare U12-type spliceosome (38, 39). This gene might use a different splicing machinery, as suggested by other authors (35, 40). CaBP3 has both types of splice junctions in its gene, CT and AC in the CaBP5 overlapping part, but GT and AG in the nonoverlapping part. Because of this particular gene organization, the transcription of CaBP5 would interfere with the transcription of complementary CaBP3 through the contact of both transcription complexes. We do not know yet whether CaBP3 is translated in the retina. CaBP3 might: 1) control the level of expression of CaBP5; 2) not be expressed simultaneously with CaBP5; or 3) originate from different alleles. Monoclonal antibodies against CaBP3 will be necessary to demonstrate the expression of CaBP3 in the retina.
Expression and Localization—CaBPs have a unique expression pattern. CaBP1 is highly expressed in the brain and retina, as shown in Fig. 3A; a more detailed expression pattern is shown in Fig. 3D. mRNAs of caldendrin and calbrain, which are identical, in large part or entirely, with CaBP1, have also been found to localize to the cortex, cerebellum, and hippocampus (11, 12). In contrast, CaBP3/5 is retina-specific. CaBP2 appears to be a minor retinal component. CaBP1 and CaBP5 were also isolated and microsequenced from the retina, proving that both genes are translated.
Immunocytochemical localization of CaBPs imposes a major challenge because of high sequence similarity among these proteins. The localization data from the retina indicate that CaBP1 and CaBP5 are expressed in the inner retina. CaBP1 is present in amacrine and some bipolar cells, whereas CaBP5 is present in rod and cone bipolar cells. The development of antibodies specific for alternative splice forms will aid in the future characterization of these proteins. Biochemical experiments suggest that CaBP5 and the CaBP1 short form are at least partially soluble and can be extracted under low ionic strength buffer. Expression of the long form of CaBP1-GFP in CHO cells suggests that this protein may be associated with the cytoskeleton. The short form of CaBP1 clearly localizes to the plasma membrane. Seidenbecher et al. (11) found caldendrin to be present in cytoskeletal fractions with a further enrichment in the synapse-associated cytomatrix, suggesting an association with the somatodendritic cytoskeleton. We also observed a similar localization pattern in the retinal bipolar cells. Altogether, these data suggest that CaBPs occur within the cell as soluble proteins; significant pools of these proteins (or splice variants), however, are associated with the plasma membrane or cytoskeletal. The presence of two CaBP1 pools could be an important mechanism for targeting the effector proteins to different cellular compartments, for example, proteins that change their localization from the plasma membrane to intracellular stores (e.g. receptors) during cell stimulation.
Functional Aspects—Significant functional and structural information have accumulated on CaM, and this data may have substantial relevance for understanding CaBPs. Ca2+ binding to CaM appears to be stepwise, first to the EF3-hand and EF4-hand motifs and then to the EF1-hand and EF2-hand motifs (41). When site II was disabled by a specific mutation, there was a decrease only in the apparent affinity for the partner site I and little or no effect on sites III and IV (42). It is believed that both N-terminal binding sites can bind Ca2+ almost independently to produce different components of the Ca2+-induced conformational change (43). Once Ca2+ is coordinated in all sites, there is likely to be an interaction of the C-and N-terminal domains. Interestingly, a disabling mutation in the EF2-hand motif not only had an effect on the EF1-binding site but also accelerated the release of Ca2+ from EF3- and EF4-hand sites (44). This mutant also has decreased affinities for smooth and skeletal muscle myosin light chain kinases, adenylyl cyclase, and plasma membrane Ca2+-ATPase (45). Thus, some differences between CaM and CaBPs could be attributed to lack of functional EF2-hand and to specific sequence changes among these proteins. Finally, it may be important that CaBPs have an extended 32-aa flexible tether (central α-helical segment) versus 28 aa in CaM (Fig. 10). For CaM, this linker serves a crucial role, allowing the N- and C-terminal domains to swing around and completely enclose the target peptide from the effector molecule. It is possible that the interacting peptide fragment from the effector molecule could be larger than the one used by CaM. Because of its extraordinary plasticity, CaM binds its targets extremely tightly with a Kd up to 10−11 M; however, such tight binding may not always be advantageous when fast dissociation from the target molecule is needed. These extra 4 aa may weaken the interaction with the target enzyme/protein, providing kinetic advantage over the strength of the interaction with the target molecule.
Recently, a large body of evidence was published on the regulation of Ca2+ channels and cyclic nucleotide-gated channels by CaM (46-50). Because of these similarities to CaM, CaBPs mimic some of the interactions of CaM with the effector enzymes. Although we are cautious in claiming the physiological function of these proteins, it should be seriously considered that where in vitro CaM modulate properties of the target protein, in vivo this function may be carried out by CaBPs. As we demonstrate in this study, for example, GRK5 is potently inhibited by CaBPs (Fig. 9), and CaM kinase is activated by CaBP2 (Fig. 8). These experiments also showed clear specificity among CaBPs. Further studies on the co-localization of CaBPs and target proteins will be needed to determine whether such interactions are happening in vivo.
In summary, we describe the identification and initial characterization of a novel neuron-specific subfamily of Ca2+-binding proteins with a high similarity to CaM. Five related genes and several splice variants comprise the newest subfamily of Ca2+-binding proteins from the CaM superfamily. Their membrane localization and properties that are similar to those of CaM, but modified, suggest that some of the CaM regulatory processes observed in vitro could be carried out instead by CaBPs in vivo. The highly conserved primary and structural properties of CaBPs and CaM suggest the necessity for preserving the contact sites with the effector molecule. This exciting hypothesis requires further biochemical and structural testing to assess the importance of this subfamily in neuronal functioning.
Acknowledgments
We thank Dr. D. Brickey and Dr. T. Soderling for the initial tests of CaM kinase II.
Footnotes
This research was supported by National Institutes of Health Grants EY08061 (to K. P.) and EY06935-01 (to R. N. F.), an award from Research to Prevent Blindness, Inc. to the Department of Ophthalmology at the University of Washington, Grant-in aid Award GA99001 from Fight For Sight-Prevent Blindness America Research (to F. H.), and by the E. K. Bishop Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBank™/EBI Data Bank with accession number(s) short form of human CaBP1, AF169148; long form of human CaBP1, AF169149; short form of bovine CaBP1, AF169150; long form of bovine CaBP1, AF169151; short form of mouse CaBP1, AF169153; long form of mouse CaBP1, AF1691152; human CaBP2, AF169154; bovine CaBP2, AF169155; short form of mouse CaBP2, AF169156; long form of mouse CaBP2, AF169157; human CaBP3, AF169158; human CaBP5, AF169159; bovine CaBP5, AF169160; mouse CaBP5, AF169161; human CaBP2 genomic sequence, AF170811; exons 1, 2-3-4, 5, and 6 of human CaBP5 genomic sequence, AF170812, AF170813, AF170814, and AF170815, respectively; and exons 1-2, 3-4, 5, and 6 of human CaBP3 genomic sequence, AF170816, AF170817, AF170818, and AF170815, respectively.
The abbreviations used are: aa, amino acid(s); b-, bovine; BAC, bacterial artificial chromosome; BTP, 1,3-bis[tris(hydroxymethyl)methylamino]propane; CaM, calmodulin; CaM kinase II, Ca2+/CaM-dependent protein kinase II; CaBP, Ca2+-binding protein; CHO cells, Chinese hamster ovary cells; EST, expressed sequence tag; G3PDH, glyceraldehyde-3-phosphate dehydrogenase; GCAP, guanylyl cyclase-activating protein; GFP, green fluorescent protein; GRK, G protein-coupled receptor kinase; h-, human; HPLC, high pressure liquid chromatography; INL, inner nuclear layer; kb, kilobase pair(s); L-, long form; m-, mouse; NCBP, neuronal Ca2+-binding proteins; ORF, open reading frame; PAGE, polyacrylamide gel electrophoresis; PCR, polymerase chain reaction; PKC, protein kinase C; S-, short form.
F. Haeseleer, I. Sokal, C. L. M. J. Verlinde, H. Erdjument-Bromage, P. Tempst, A. N. Pronin, J. L. Benovic, R. N. Fariss, and K. Palczewski, unpublished observation.
Note Added in Proof—Recently, Menger et al. (Menger, N., Seidenbecher, C. I., Gundelfinger, E. D., and Kreutz, M. R. (1999) Cell Tissue Res. 298, 21–32) reported the immunolocalization of caldendrin in the retina.
REFERENCES
- 1.Nalefski EA, Falke JJ. Protein Sci. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polans A, Baehr W, Palczewski K. Trends Neurosci. 1996;19:547–554. doi: 10.1016/s0166-2236(96)10059-x. [DOI] [PubMed] [Google Scholar]
- 3.Haeseleer F, Sokal I, Li N, Pettenati M, Rao N, Bronson D, Wechter R, Baehr W, Palczewski K. J. Biol. Chem. 1999;274:6526–6535. doi: 10.1074/jbc.274.10.6526. [DOI] [PubMed] [Google Scholar]
- 4.Babu YS, Bugg CE, Cook WJ. J. Mol. Biol. 1988;204:191–199. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- 5.Wriggers W, Mehler E, Pitici F, Weinstein H, Schulten K. Biophys. J. 1998;74:1622–1639. doi: 10.1016/S0006-3495(98)77876-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikura M. Trends Biochem. Sci. 1996;21:14–17. [PubMed] [Google Scholar]
- 7.Hughes RE, Brzovic PS, Dizhoor AM, Klevit RE, Hurley JB. Protein Sci. 1998;7:2675–2680. doi: 10.1002/pro.5560071222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ames JB, Dizhoor AM, Ikura M, Palczewski K, Stryer L. J. Biol. Chem. 1999;274:19329–19337. doi: 10.1074/jbc.274.27.19329. [DOI] [PubMed] [Google Scholar]
- 9.Ames JB, Ishima R, Tanaka T, Gordon JI, Stryer L, Ikura M. Nature. 1997;11(389):198–202. doi: 10.1038/38310. [DOI] [PubMed] [Google Scholar]
- 10.Vijay-Kumar S, Kumar VD. Nat. Struct. Biol. 1999;6:80–88. doi: 10.1038/4956. [DOI] [PubMed] [Google Scholar]
- 11.Seidenbecher CI, Langnaese K, Sanmarti-Vila L, Boeckers TM, Smalla KH, Sabel BA, Garner CC, Gundelfinger ED, Kreutz MR. J. Biol. Chem. 1998;273:21324–21331. doi: 10.1074/jbc.273.33.21324. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi K, Yamaguchi F, Miyamoto O, Sugimoto K, Konishi R, Hatase O, Tokuda M. J. Biol. Chem. 1999;274:3610–3616. doi: 10.1074/jbc.274.6.3610. [DOI] [PubMed] [Google Scholar]
- 13.Lui M, Tempst P, Erdjument-Bromage H. Anal. Biochem. 1996;241:156–166. doi: 10.1006/abio.1996.0393. [DOI] [PubMed] [Google Scholar]
- 14.Elicone C, Lui M, Geromanos S, Erdjument-Bromage H, Tempst P. J. Chromatogr. 1994;676:121–137. doi: 10.1016/0021-9673(94)00089-1. [DOI] [PubMed] [Google Scholar]
- 15.Tempst P, Geromanos S, Elicone C, Erdjument-Bromage H. Methods (Orlando) 1994;6:248–261. doi: 10.1016/0021-9673(94)00089-1. [DOI] [PubMed] [Google Scholar]
- 16.Erdjument-Bromage H, Lui M, Lacomis L, Grewal A, Annan RS, McNulty DE, Carr SA, Tempst P. J. Chromatogr. 1998;826:167–181. doi: 10.1016/s0021-9673(98)00705-5. [DOI] [PubMed] [Google Scholar]
- 17.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudnicka-Nawrot M, Surgucheva I, Hulmes JD, Haeseleer F, Sokal I, Crabb JW, Baehr W, Palczewski K. Biochemistry. 1998;37:248–257. doi: 10.1021/bi972306x. [DOI] [PubMed] [Google Scholar]
- 19.Kim CM, Dion SB, Onorato JJ, Benovic JL. Receptor. 1993;3:39–55. [PubMed] [Google Scholar]
- 20.Kunapuli P, Onorato JJ, Hosey MM, Benovic JL. J. Biol. Chem. 1994;269:1099–1105. [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. pp. 16.30–16.36. [Google Scholar]
- 22.Hale I,L, Matsumoto B. Methods Cell Biol. 1993;38:289–324. doi: 10.1016/s0091-679x(08)61008-0. [DOI] [PubMed] [Google Scholar]
- 23.Bradford MM. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 24.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Chattopadhyaya R, Meador WE, Means AR, Quiocho FA. J. Mol. Biol. 1992;228:1177–1192. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- 26.Ring CS, Cohen FE. FASEB J. 1993;7:783–790. doi: 10.1096/fasebj.7.9.8330685. [DOI] [PubMed] [Google Scholar]
- 27.Tabernero L, Taylor DA, Chandross RJ, Van Berkum MF, Means AR, Quiocho FA, Sack JS. Structure. 1997;5:613–622. doi: 10.1016/s0969-2126(97)00217-7. [DOI] [PubMed] [Google Scholar]
- 28.Kozak M. Mamm. Genome. 1996;7:563–574. doi: 10.1007/s003359900171. [DOI] [PubMed] [Google Scholar]
- 29.Wicki R, Marenholz I, Mischke D, Schafer BW, Heizmann CW. Cell Calcium. 1996;20:459–464. doi: 10.1016/s0143-4160(96)90087-1. [DOI] [PubMed] [Google Scholar]
- 30.Surguchov A, Bronson JD, Banerjee P, Knowles JA, Ruiz C, Subbaraya I, Palczewski K, Baehr W. Genomics. 1997;39:312–322. doi: 10.1006/geno.1996.4513. [DOI] [PubMed] [Google Scholar]
- 31.Palczewski K. Eur. J. Biochem. 1997;248:261–269. doi: 10.1111/j.1432-1033.1997.00261.x. [DOI] [PubMed] [Google Scholar]
- 32.Pronin AN, Satpaev DK, Slepak VZ, Benovic JL. J. Biol. Chem. 1997;272:18273–18280. doi: 10.1074/jbc.272.29.18273. [DOI] [PubMed] [Google Scholar]
- 33.Berchtold MW, Egli R, Rhyner JA, Hameister H, Strehler EE. Genomics. 1993;16:461–465. doi: 10.1006/geno.1993.1211. [DOI] [PubMed] [Google Scholar]
- 34.Vanhee-Brossollet C, Vaquero C. Gene. 1998;211:1–9. doi: 10.1016/s0378-1119(98)00093-6. [DOI] [PubMed] [Google Scholar]
- 35.Laaby Y, Gras MP, Brouet JC, Berger R, Larsen CJ, Tsapis A. Nucleic Acids Res. 1994;22:1147–1154. doi: 10.1093/nar/22.7.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner EG, Simons RW. Annu. Rev. Microbiol. 1994;48:713–742. doi: 10.1146/annurev.mi.48.100194.003433. [DOI] [PubMed] [Google Scholar]
- 37.Saumon I, Reppert SM. Neuron. 1996;17:889–900. doi: 10.1016/s0896-6273(00)80220-2. [DOI] [PubMed] [Google Scholar]
- 38.Sharp PA, Burge CB. Cell. 1997;91:875–879. doi: 10.1016/s0092-8674(00)80479-1. [DOI] [PubMed] [Google Scholar]
- 39.Burge CB, Padgett RA, Sharp PA. Mol. Cell. 1998;2:773–785. doi: 10.1016/s1097-2765(00)80292-0. [DOI] [PubMed] [Google Scholar]
- 40.Senapathy P, Shapiro MB, Harris NL. Methods Enzymol. 1990;183:252–278. doi: 10.1016/0076-6879(90)83018-5. [DOI] [PubMed] [Google Scholar]
- 41.Forsin S, Vogel HJ, Drakenberg T. In: Calcium and Cell Function. Cheung E, editor. Vol. 6. Academic Press; San Diego, CA: 1986. pp. 113–157. [Google Scholar]
- 42.Starovasnik MA, Su DR, Beckingham K, Klevit RE. Protein Sci. 1992;1:245–253. doi: 10.1002/pro.5560010206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beckingham K. J. Biol. Chem. 1991;266:6027–6030. [PubMed] [Google Scholar]
- 44.Martin SR, Maune JF, Beckingham K, Bayley PM. Eur. J. Biochem. 1992;205:1107–1114. doi: 10.1111/j.1432-1033.1992.tb16879.x. [DOI] [PubMed] [Google Scholar]
- 45.Gao ZH, Krebs L, VanBerkum MF, Tong WJ, Maune JF, Means AR, Stull JT, Beckingham K. J. Biol. Chem. 1993;268:20096–20104. [PubMed] [Google Scholar]
- 46.Hsu Y-T, Molday RS. Nature. 1993;361:76–79. doi: 10.1038/361076a0. [DOI] [PubMed] [Google Scholar]
- 47.Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 48.Levitan IB. Neuron. 1999;22:645–648. doi: 10.1016/s0896-6273(00)80722-9. [DOI] [PubMed] [Google Scholar]
- 49.Peterson BZ, DeMaria CD, Yue DT. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 50.Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]