

Abstract
Two-dimensional mean-field lattice theory is used to model immobilization and stabilization of an enzyme on a hydrophobic surface using grafted polymers. Although the enzyme affords biofunctionality, the grafted polymers stabilize the enzyme and impart biocompatibility. The protein is modeled as a compact hydrophobic-polar polymer, designed to have a specific bulk conformation reproducing the catalytic cleft of natural enzymes. Three scenarios are modeled that have medical or industrial importance: 1), It is shown that short hydrophilic grafted polymers, such as polyethylene glycol, which are often used to provide biocompatibility, can also serve to protect a surface-immobilized enzyme from adsorption and denaturation on a hydrophobic surface. 2), Screening of the enzyme from the surface and nonspecific interactions with biomaterial in bulk solution requires a grafted layer composed of short hydrophilic polymers and long triblock copolymers. 3), Hydrophilic polymers grafted on a hydrophobic surface in contact with an organic solvent form a dense hydrophilic nanoenvironment near the surface that effectively shields and stabilizes the enzyme against both surface and solvent.
INTRODUCTION
Synthetic biosurfaces, in principle, should have a biomimetic ability to perform one or more biological functions that should trigger a specific required response instead of the normal and generally undesired foreign body response induced by nonspecific adsorption of proteins (1,2). Such biospecificity is generally accomplished via attachment of natural or synthetic macromolecules to the substrate, which allows for control over the concentration of the bioactive material near the surface.
One of the first applications of introducing biological specificity to a synthetic material involved the immobilization of heparin to a surface to enhance its compatibility with blood by catalyzing thrombogenic proteins into an inactive form (3). Since then, a large number of studies involving heparin and other biopolymer-modified surfaces have emerged. More importantly, studies have shown that localization of enzymes near the surface can be used to impart biospecific response as required for a particular application (4–7), although long-term enzymatic activity is, in general, found to be compromised.
Stabilization of the enzyme is necessary since the activity and native structure of proteins is largely undermined by changes in the environment. Proteins are generally inhibited by high concentrations of substrate or products and their selectivity and activity may be affected when used in non-natural processes, on non-natural substrates, and under nonconventional conditions. In particular, confinement near an untreated hydrophobic surface leads to significant loss in activity (8). Trachtenberg et al. (9) studied direct immobilization of carbonic anhydrase (CA) to nylon surfaces. They found that the relative activity of the immobilized enzyme compared to the enzyme under bulk conditions was at best ∼25%, and on most surfaces below 10%, provided that a spacer is used to isolate the enzyme from the nylon surface. Molecular dynamics simulations (10) and in vitro experiments (11,12) have shown that although mutation of the protein may have a minimal effect on enzymatic activity in bulk solution, there is a drastic decrease when the enzyme is attached to the surface. Hydrophobic or electrostatic interactions between the surface and solvent lead to a concentration partitioning in the vicinity of the surface, which is not optimal for the enzyme, and may affect its activity (10). In addition, the catalytic activity of the immobilized enzyme is often severely affected by nonspecific biological and immunological reactions (13), partial or complete inactivation of catalytic centers (10,14,15), and accumulation byproducts (15).
Partial or complete adsorption of the protein is, in general, accompanied by conformational changes resulting in denaturation and inactivation, which may trigger thrombosis and other potentially fatal conditions (16). Thus, many efforts have concentrated on stabilizing the enzymes in the vicinity of the surface by creating a controlled nanoenvironment for the protein that effectively reduces its interactions with the destabilizing environment, leading to prolonged activity.
Several strategies have been suggested for stabilizing enzymes. Multipoint covalent attachment of the protein to the substrate has been shown to stabilize the protein against extreme changes in the environment, compared with the soluble counterpart (12). Through careful control of experimental conditions, this method was shown to substantially inhibit conformational changes of the protein promoted by heat, organic solvents, pH changes, or other sources, whereas the activity is only slightly reduced (12). Fernandez-Lafuente et al. (11,14,15) found that the combined effect of both chemical modification of the immobilized enzyme in addition to modification using dextrans attached to the surface of the protein promoted a dramatic stabilization of the enzyme, associated with a minimal loss of catalytic activity. Additionally, enzymatic activity and stability of the immobilized protein against nonnative environments can be improved, e.g., by oriented immobilization (17), introducing hydrophilic groups on the hydrophobic surface (18), or mutagenesis (13)—which, however, strongly depends on the location of modification.
Crowding the protein in restricted space, e.g., within pores of silica matrix (19–22) or reverse micelles (23,24) has also been shown to induce stabilization, presumably due to nonspecific interactions (25). Reverse micelles also present a convenient way to stabilize proteins in organic solvents. The stability of the folded native state of caged proteins has also been observed using Monte Carlo simulations of model proteins (26,27). Crowding apparently shifts the folding equilibrium to the native state by restricting the conformational space of the protein (25,27). This effect is strongly supported by experimental evidence that shows faster folding kinetics in caged environment when compared with spontaneous folding in solution (28). However, for some proteins, the role of entrapped water is believed to be critical in stabilizing the structure of the caged protein (19), and retaining its bioactivity (29,30).
Following the concept of crowding, grafted polymers are expected to contribute to the stabilization of an immobilized protein. A number of theoretical and computational studies have focused on the effect of grafted polymers on the adsorption of proteins (31–40). All these models, however, consider proteins as rigid and nondeforming, with the exception of Fang and Szleifer (33), who introduced a density functional model where the protein is modeled as a rigid sphere, which, upon adsorption, may undergo a transition to a disk configuration (32,33,39,41–44).
In this article, we introduce a semiflexible model of proteins and study their behavior and stabilization near a hydrophobic surface. We propose a relatively noninvasive approach to stabilize an anchored protein that uses grafted polymers on the surface only in the vicinity of the immobilized enzyme to stabilize it by screening, e.g., the surface-protein or solvent-protein interactions and thus provide a favorable nanoenvironment in the vicinity of the surface. Another advantage for using grafted polymers such as polyethylene oxide (PEO) is that they are known to enhance biocompatibility of the surface (9,31,45–47).
Using lattice mean-field theory, we model a proteinlike heteropolymer whose conformational space is continuous (48). The unique native conformation, which consists of a mainly hydrophobic center surrounded by a thin hydrophilic layer, is found to undergo the sharp adsorption transition characteristic of proteins, which is accompanied by denaturation to a flat conformation near a hydrophobic surface. Our model predicts that an immobilized protein, on the other hand, may be found in a partly denatured state near the surface. The designed conformation can be restored using relatively short hydrophilic polymers and low graphing densities in the vicinity of the immobilized protein. Screening of the enzyme from, e.g., solution biomatter, however, requires a bimodal grafted polymer layer, made up of short hydrophilic polymers and long block copolymers. The grafted polymers present an entropic shield for the embedded enzyme and from macromolecular adsorption in addition to providing an energetically stabilizing environment.
MODEL DEVELOPMENT
We use a two-dimensional mean-field lattice model (48) based on the theory by Scheutjens and co-workers (49,50). The protein is modeled as a copolymer made up of hydrophobic (H) and polar (P) groups that form a compact structure in the bulk. The hydrophobic monomers make up two active regions near the center of mass of the protein, thus leading to a unique conformation in the bulk, reproducing, e.g., the catalytic cleft of natural enzymes (10,17). In principle, we can model spherical proteins, elongated proteins, or other shapes in the native state. We focus on the adsorption behavior of a protein anchored to the surface using a rigid spacer. The grafted polymers are modeled as monodispersed chains with a homogeneous grafting density, σ. For complete screening of the protein from the surface and bulk material, we model bimodal grafted polymer layers composed of short hydrophilic chains and longer triblock PHP chains.
Both the grafted polymers and protein are modeled as excluded volume chains which may interact with themselves, with the solvent and with the surface. The model development closely resembles the model of Wijmans et al. (50) for grafted polymers, but has been extended to two dimensions and includes various types of interacting monomers and species. Thus, a brief outline is presented and the reader is referred to Wijmans et al. (50) for a detailed description.
The segment density distributions for the grafted polymers, solvent, and protein are obtained from the Boltzmann weighting factor Gj(x,z), where x is the coordinate parallel to the surface and z is the distance from the surface,
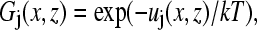 |
(1) |
where uj(x,z) includes both energetic and entropic contributions and is a function of the normalized average particle density (or volume fraction) at (x,z), 〈φ(x,z)〉, averaged over nearest-neighbor lattice sites only. The surface-protein interactions are modeled as exponential decay, whereas the entropic contribution is calculated from the number of ways that the chains can be arranged on the lattice under the given constraints. The monomer density is obtained from
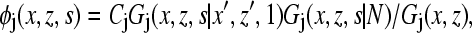 |
(2) |
where the index j refers to the different species (i.e., protein-1, polymer-2, or solvent-3). Cj is a normalization constant such that ΣjΣsφj(x,z,s) = 1. Gj(x,z,s|x′,z′,1) is the probability that segment s is at position (x,z), given that the first segment of the macromolecule (protein or polymer) is at position (x′,z′). For the grafted polymers, z′ = 0 and thus G2(x,0,1|x′,z′,1) = 1, whereas, for the protein, both x′ and z′ are fixed if it is covalently attached to the surface at xfix (which is taken to be the center of the box, i.e., 0), using a rigid spacer of length zfix. In this case G1(xfix,zfix,1|x′,z′,1) = 1. Gj(x,z,s|N) is the probability that segment s is at position (x,z), given that the last segment of the chain is anywhere on the lattice. Clearly, Gj(x,z,N|N) = 1 for both protein and grafted polymers. Additional boundary conditions are easily written down that ensure that the macromolecules are not broken. The segment densities of the grafted polymers, the protein, and the solvent are obtained self-consistently by solving Eqs. 1 and 2 with these boundary conditions. In each iteration, the positions of the active centers of the protein are relocated according to the position of center of mass. The presented results were obtained for a 99×101 size lattice.
RESULTS AND DISCUSSION
Protein adsorption
In the majority of the results presented we consider a dimeric protein designed as two copolymer strands, each consisting of 40 segments of which 25 are hydrophobic. The grafted polymer chain length varies to comply with a desired application. The conformation of the protein far from the surface is presented in Fig. 1 a as an image of majority polar (P) and hydrophobic (H) regions, whereas the contour lines present the total monomer density. The two polar ends of the dimeric protein are immobilized at xfix = 0 and a given zfix. The conformation far from the surface displays a dense polar center at the fixation point, surrounded by a mixed HP region with two dense hydrophobic regions representing the active sites of the protein. A low density, mostly hydrophilic, cloud makes up the outer region of the protein that is exposed to the solvent. The effect of the hydrophobic surface on the structure of the protein immobilized using spacers of different lengths is shown in Fig. 1, b–d. Insignificant conformational changes are observed for zfix > 20 (note that at zfix = 20 the bottommost part of the protein is only three lattice units away from the surface, but is not yet adsorbed). However, during the initial stages of adsorption, the protein becomes slightly elongated as outer H segments adsorb on the surface while the P segments remain in the bulk, leading to an increase in its cross-section area (Fig. 1 b). Yet, for the range of 14 < zfix < 20 a significant density of the H active sites is still retained. Immobilization of the enzyme even closer to the surface, however, leads to partial unfolding (51) of the protein due to local interactions of the hydrophobic centers with the surface (Fig. 1 c). For zfix < 10, the protein collapses on the surface in a flat disklike conformation (Fig. 1 d).
FIGURE 1.

Conformation and density contours (given as log φ) of an immobilized dimeric protein interacting with a surface (χ23 = ±0.5, χ2s = ±2.0, where s = surface): (a) bulk conformation, zfix = 55; (b) conformation during initial adsorption, zfix = 15; (c) conformation before complete loss of active centers, zfix = 12; and (d) adsorbed in a collapsed conformation, zfix = 0.
A large number of kinetic and thermodynamic studies have demonstrated partial conformational changes or collapse of various proteins upon adsorption on hydrophobic surfaces (52). Although direct measures of interfacial protein structure are not currently possible, a number of indirect, mainly spectroscopy, methods can be used to infer conformational changes, and in combination with kinetic studies, the effects of adsorption on bioactivity can be correlated. A particularly useful technique to observe conformational changes appears to be time-of-flight secondary ion-mass spectroscopy, which displays peaks that are characteristic to each amino-acid group and can be used to study the conformation, orientation, and degree of denaturation of an adsorbed protein (53). Using this technique, it was shown that albumin adsorbed in a denatured rearranged configuration that exposes the hydrophobic residues to the polycarbonate surface to maximize the protein-surface interactions (53), similar to the observed adsorbed configuration shown in Fig. 1. Partially denatured configuration of bovine serum albumin near the silica surface was observed using time-resolved evanescent wave-induced fluorescence spectroscopy (54). In this study, the adsorbed protein appears increasingly more coiled and retains its hydrophobic sites further away from the surface, displaying a hydrophobicity gradient in the adsorbed layer normal to the surface. These results are again in excellent qualitative agreement of the adsorbed conformations in Fig. 1. As a final example, Michael et al. (55) carried out kinetic adsorption studies that indicate fast adsorption followed by substantial rearrangement of the protein to maximize favorable surface contacts. More importantly, they found high correlation between conformational changes and lower activity of the protein (probed using enzyme-linked immunosorbent assay), especially when adsorbed on a hydrophobic surface.
The adsorbed fraction and cross-sectional area of the protein as a function of distance from the surface are plotted in Fig. 2. Solid and open symbols correspond to a protein immobilized on a clean hydrophobic surface and a surface grafted with hydrophilic polymer (discussed below), respectively. Concentrating on the former, the curves show a sharp adsorption transition accompanied by drastic conformational changes when the hydrophobic core of the protein approaches a critical distance from the surface (zfix = 15 for the particular case studied). The cross-sectional area of the protein is slightly reduced before adsorption as a result of the unfavorable interactions between the hydrophobic surface and the enveloping hydrophilic cloud of the protein. As adsorption sets in, the curve of the cross-sectional area of the protein as a function of spacer length reveals a metastable state at the transition (at ∼12 ≤ zfix ≤ 15), where the protein is stretched and partially adsorbed, but the hydrophobic centers are still retained (Fig. 1 b). For zfix ≤ 12 the protein collapses to a flat conformation on the surface. This behavior is in excellent qualitative agreement with results obtained from Monte Carlo simulations for the adsorption of proteinlike random heteropolymers (RHPs) on nonhomogeneous surfaces (56), reproduced in the inset of Fig. 2. The conformational order parameter calculated in the simulations indicates an increase in the number of conformations sampled by the protein (i.e., unfolding) when adsorption begins, corresponding to the increase in cross-sectional area observed in our study. In its adsorbed state, the protein adopts a few conformations that are dictated by surface characteristics, corresponding to the decrease in cross-sectional area as the protein adopts an increasingly flat globular conformation dictated by the interactions with the surface.
FIGURE 2.

Adsorption isotherms and cross-sectional area changes of the protein as a function of distance from the surface. (Inset) Adsorbed fraction (solid line), and conformation order parameter (dashed line) as a function of surface loading for random heteropolymer (RHP) with strong specific intersegment interactions, reproduced from Srebnik et al. (56). The conformation order parameter accounts for the number of available conformations for the RHP. When it equals unity, the entire conformational space is sampled; when it is less than unity, the RHP are restricted to a small number of energetically favored conformations. For the case of strong specific interactions between the heteropolymer segments and surface, it is seen that a sharp adsorption transition is accompanied first by restricting further the conformational space available for the RHP, followed by unfolding of the RHP and refolding into a surface-matched compact conformation. This situation is akin to adsorption of a protein in its native state, followed by “unraveling” of the folded protein before adsorbing into conformation determined by the interactions with the surface.
The free energy of the protein provides information on the relative stability of the conformational state of the protein at various distances from the surface. Assuming that the native state has the lowest free energy, we define the excess Helmholtz free energy as
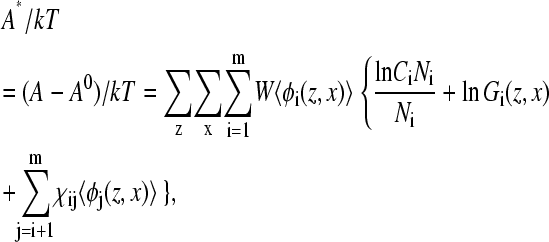 |
(3) |
where A0 is the free energy of the protein in its native conformation, W is the width of the simulation box, and χij is the Flory Chi interactions between species i and j. The third summation is over the different species. A* and the normalized average density of the active sites are plotted as a function of distance from the surface in Fig. 3. The excess free energy curve indicates that initial stages of adsorption (12 ≤ zfix ≤ 15) are accompanied by an increase in energy and a sharp decrease in the density of hydrophobic centers. This state corresponds to a stretched configuration (Fig. 1, b–c) where the hydrophobic segments extend to the surface. This state is transitional between the bulk conformation and the compact adsorbed conformation that is not observed for free proteins. Immobilization at distances closer to the surface results in loss of the hydrophobic centers and large energetic gain as the hydrophobic centers adsorb on the surface.
FIGURE 3.

Relative density of active sites (normalized by bulk density) (triangles) and excess free energy (circles) as a function of distance of the center of the protein from the surface for an immobilized protein for N = 16, σ = 0.1, and χ12 = ±0.2.
Stabilization against hydrophobic surface
The role of the spacer is twofold—attach the enzyme to the surface and render it accessible (57). In practice, relatively short spacers are used to localize the enzymes near the surface (58), which leads to significant deactivation, as has been discussed in the Introduction. Our results presented in the previous section support experimental evidence that suggests that immobilization close to the surface leads to adsorption and denaturation of the protein.
In general, the various stabilization techniques that have been reported in the literature require direct modification of the protein, which are rather demanding and may lead to substantial deactivation of the enzyme. We suggest a layer of end-grafted hydrophilic polymers in the vicinity of the immobilized enzyme at relatively low grafting densities (σ = 0.1) as a minimally invasive approach that stabilizes the protein against the hydrophobic surface. Our results suggest that hydrophilicity of the polymers is necessary, and a mere steric barrier, i.e., neutral polymers, is not sufficient (48). In addition, we find that there is a narrow range of polymer lengths and grafting densities for which stabilization occurs. Longer polymers lead to collapse of the protein on the surface, whereas shorter polymers and lower densities do not stabilize the enzyme. Our results should be supported by crowding theory, which suggests that confined environment favors a globular conformation (25).
Fig. 4 presents the contour density profile of a protein immobilized on a hydrophobic surface with a rigid spacer of length lfix = 12 in the presence of a grafted hydrophilic polymer layer. Comparison with Fig. 1 c, which presents the protein immobilized at the same distance but on a clean surface, shows that the protein structure is largely preserved within the grafted polymer layer, resembling the native structure in Fig. 1 a. In particular, the density of the hydrophobic monomers in the active sites is retained. The open symbols in Fig. 2 correspond to the adsorption and surface area isotherms of the protein in presence of the grafted polymer. Both curves indicate that the grafted polymer layer allows for immobilization up to three lattice units closer to the surface, before the protein denatures. The free energy of the stabilized protein shows the same qualitative behavior of an anchored protein, but again shifted by three lattice units (Fig. 5). Thus, for the particular parameters chosen for the analysis, the protein can be immobilized somewhat closer to the surface before it adsorbs and denatures. The stabilizing effect depends on brush length and grafting density as well as polymer-protein interactions. Our computational method is limited to short chains at relatively low grafting densities. Presumably, better stabilization can be achieved by higher grafting densities than those simulated here, which we intend to analyze using computer simulation methods.
FIGURE 4.

Conformation and density contours (given as log φ) of the protein immobilized at, zfix = 12 within a hydrophilic grafted polymer layer (N = 16, σ = 0.1, χ12 = ±0.2, χ23 = ±0.5, and χ2s = ±2.0).
FIGURE 5.

Excess free energy as a function of distance of the center of the protein from the surface for an immobilized protein (solid triangles), and an immobilized protein stabilized by hydrophilic grafted polymers (open triangles) for N = 16, σ = 0.1, and χ12 = ±0.2.
Nonfouling biocompatible surfaces
Several studies have shown that the most important parameters in preventing protein adsorption using grafted polymers are grafting density (59) and chain length (60,61). Sofia et al. (60) argue that polymer chains of roughly the same size as the protein will prevent their adsorption, due mainly to steric hindrance, and that for a given polymer chain length, there is a corresponding grafting density that will prevent protein adsorption which is weakly dependent on protein size. According to Szleifer (61), if the surface is not attractive, then protein adsorption behavior would be the same in the same range of grafting density despite the difference in PEO chain length. Due to the attractive forces, higher grafting densities are needed for smaller chain length to achieve the chain overlap, indicating that there is a specific dependence of protein adsorption on chain length and grafting density. In addition, protein size must be considered, since small proteins could penetrate the grafted layer and adsorb, deforming the polymers.
In the case of an adsorbing hydrophobic surface, our study suggests that if the protein penetrates far enough into the grafted polymer layer, then the protein will adsorb in a flat and denatured conformation and the polymer layer will deform to accommodate the adsorbed protein. The density profiles of the protein and grafted polymer shown in Fig. 6 reveal that the polymer is considerably deformed around the denatured protein. Whereas a native protein may not be able to place itself between sufficiently close polymer anchors, a denatured one that is free to sample various conformation may adsorb surrounding a number of polymer anchors and is not constrained between the spacing of neighboring ones. There appears to be a critical penetration distance for a given grafting density and chain length, beyond which both polymer and protein will deform to accommodate the protein on the surface. Thus, perhaps an additional criterion apart for grafting density and polymer chain length should be the polymer stiffness. Although the effect of rigidity of the polymers on the behavior of a protein near the surface is beyond the scope of this article, we can anticipate that a stiff polymer would deform less and thus would prevent penetration of the protein to that critical distance (62).
FIGURE 6.

Density profiles of the adsorbed protein (circles), grafted polymer far from the adsorbed protein (triangles), and grafted polymer at the axis of center of mass of the protein, xfix (squares). The density profile of the native protein (dashed line) is given as a reference on the secondary x axis. Parameters: N = 16, σ = 0.1, and χ12 = ±0.2.
For flexible polymers the degree of penetration determines whether a protein will adsorb or not. That is, beyond a critical penetration depth, the protein will adsorb in a denatured flat conformation, whereas small penetrations will result in repulsion of the protein back to the bulk solution. In Fig. 7 the normalized density below and above the center of mass of the protein are plotted as a function of distance from the surface. As the protein approaches the grafted polymer surface, steric repulsion between the polymer layer and the protein leads to slight shift of the density of the protein away from the surface and into the transverse direction. This trend intensifies as the repulsion between the outer hydrophilic segments of the protein and the grafted polymer begins to be felt. However, at this point the protein is not adsorbed and still essentially retains its nativelike compact structure. Initial adsorption is accompanied by a sharp shift of the density toward the surface (increase in zfix−1) as well as elongation of the protein, indicated by the decreasing average density of the transverse, x, direction. The last stage of adsorption, or collapse of the protein, results in a flat, disklike conformation. That is, both protein and polymer will deform for strong enough surface-protein interactions. Therefore, we may conclude that, in general, the tethered polymers will exclude the protein from the surface. However, in practice, a small amount of protein may still penetrate the polymer layer due to, e.g., grafting defects or fluctuation cavities formed as a result of mobility of the chain ends, which could lead to serious complications in medical applications. The results presented in Fig. 7 suggest that even slight penetration to z = 12 (brush density is negligible for z > 13, results not shown) leads to significant concentration shift of (mainly hydrophobic) segments toward the surface.
FIGURE 7.

Density profiles of the protein as a function of distance from the surface in the presence of a grafted hydrophilic polymer layer (N = 16, σ = 0.1, and χ12 = ±0.2) one lattice unit toward the surface measured from the center of mass of the protein (open circles); one lattice unit away from the surface (solid circles); and average density in the transverse direction (solid line).
Screening from bulk biomaterial
We have shown that in addition to providing biocompatible medium, short hydrophilic grafted polymers can prevent the adsorption and denaturation of a protein that is immobilized on a hydrophobic surface. However, when placed in a biological medium, such surfaces, although designed to promote a specific bioreaction, do not inhibit nonspecific reactions (2). Hence, the creation of a favorable nanoenvironment near the surface is necessary to stabilize the protein from the surface and provide a hydrophilic medium exposed to the aqueous solution, as well as provide a barrier to nonspecific biomolecular reactions.
We find that the adsorption behavior of immobilized proteins depends on the grafting density and polymer chain length in a different manner from that of proteins in solution. Whereas long polymer chains and high grafting densities prevent protein adsorption from solution, our theory predicts that grafted layers of thickness greater than the size of the immobilized protein drive the protein to a denatured adsorbed state. Therefore, to design a surface where the enzyme is fully screened both from the surface and from solution, we propose the use of grafted triblock copolymers where a short hydrophobic segment is included between two long hydrophilic ends, thus stabilizing both hydrophilic and hydrophobic segments in the outer shell of the protein (63), whereas the outer hydrophilic segments of the triblock copolymer provide for biocompatibility of the surface. In addition, short hydrophilic grafted polymers are necessary to prevent the collapse of the protein on the surface.
In Fig. 8 the density profiles of the grafted triblock copolymer, short hydrophilic polymer, and protein immobilized at zfix = 12 are presented. In Fig. 8 a, it can be seen that in absence of the protein, the short polymer is in the typical mushroom regime. The long triblock polymer, however, extends far beyond the short polymer layer due to the unfavorable hydrophobic-polar interactions. Thus, a cavity is formed that is encompassed by hydrophilic segments. It is mostly in this (deformed) cavity that the protein is positioned, as is seen in Fig. 8 b. Although the short polymer layer slightly condenses, the hydrophobic layer of the triblock polymer deforms to accommodate the protein and overlaps with the hydrophilic layer to provide a stabilizing environment for the protein. However, as is seen in Fig. 9, there is a rather narrow range of lengths of the hydrophobic block for which adsorption is minimized and the protein structure is optimized (depicted by the percent segments adsorbed on the surface). However, changes in the cross-sectional area, density at the center of mass and active sites of the protein, reveal a somewhat denser structure than native state. The optimal length of the hydrophobic block is achieved when the entropic penalty of retaining the rigid compact structure balances the stabilizing interactions between the hydrophilic and hydrophobic segments of the polymer and outer protein layer, which coincides approximately with the diameter of the protein.
FIGURE 8.
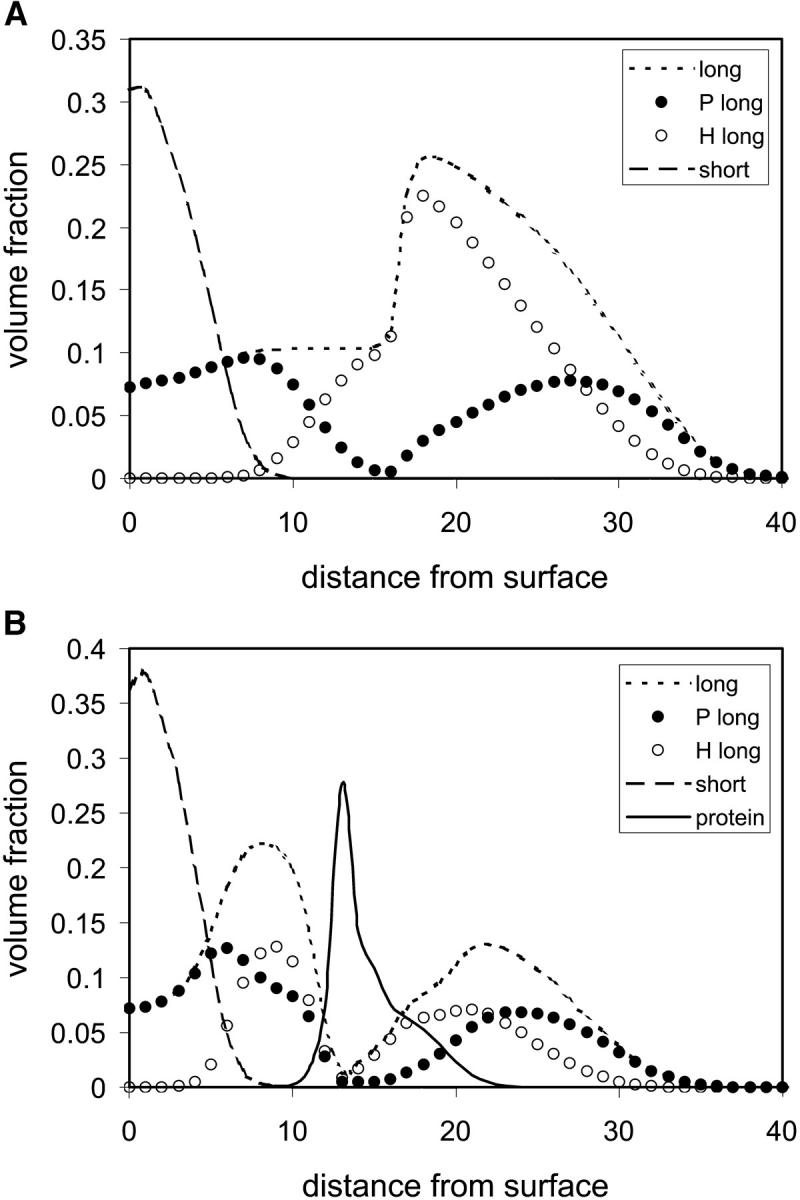
Density profiles of the grafted polymers and proteins as a function of distance from the surface. (a) Long PHP triblock copolymer grafted polymer layer (NP = 35, NH = 20, σ = 0.05, and χ12 = ±0.5) and short hydrophilic grafted polymer layer (N = 16, σ = 0.1, and χ12 = ±0.2) far from the immobilized protein. (b) Profiles of the grafted polymers and protein at the axis of immobilization (xfix).
FIGURE 9.

Percent adsorbed protein segments as a function of fraction of hydrophobic segments making up the central block of the long triblock PHP grafted polymers (N = 90, σ = 0.05, and χ12 = ±0.5).
Screening from organic solvent
Since hydrophobic residues are more soluble in organic solvents than in water, hydrophobic interactions are weakened by organic solvents. The net effect of an organic solvent on protein structure, however, usually depends on the magnitude of its effect on various polar and hydrophobic interactions. At low concentration, some organic solvents can stabilize several enzymes against denaturation. At high concentrations, however, most organic solvents cause denaturation of proteins (64). However, controlled conditions that preserve a hydrated shell around the enzyme can be used to place it in an organic medium and retain high bioactivity (29).
Our model correctly predicts that, in general, nonpolar solvents lead to swelling of the protein due to dissipation of the hydrophobic active centers. In this case, the hydrophobic segments of the protein move toward the solvent-protein interface and the protein surface is made up of mainly hydrophobic residues. For weakly nonpolar solvents that lead to partial denaturation of the active centers, we find that a layer of hydrophilic grafted polymers, which form a highly dense hydrophilic environment near the surface (see Fig. 10) that extends up to the hydrophobic active centers, stabilize the hydrophobic interactions and the protein retains its nativelike conformation (results not shown). For strongly nonpolar solvents that lead to complete denaturation of the protein, we expect that bimodal grafted polymers will again be necessary to achieve stabilization in the vicinity of the surface. It is expected, however, that in this case the nonpolar block in the triblock grafted polymer will play a more decisive role in establishing the stability of the immobilized protein than for a polar solvent. However, our model is limited, so it cannot predict the collapse of the grafted polymer layer observed for poor solvents (65,66).
FIGURE 10.

Density profile of grafted polymers in a good (dashed curve) and poor (solid curve) solvent, as a function of distance from the surface normalized by polymer chain length.
CONCLUSION
Hydrophilic grafted polymers, such as PEO, are frequently used to enhance the biocompatibility of synthetic surfaces (used, e.g., for artificial organs or drug delivery) by presenting a barrier to adsorption of bulk biomaterial. To impart biofunctionality, though, binding of enzymes that serve a particular purpose have been proposed. However, the bound enzyme may quickly denature and lose its activity due to unfavorable interactions with the surface and/or material in the blood stream.
We suggest that grafted polymers can be used to provide biocompatibility as well as stabilize the immobilized enzyme from denaturing events. However, biocompatibility of the surface and the preservation of biofunctionality present conflicting effects on the immobilized protein, which have to be optimized for a particular application. We show that, depending on the application, various patterns of grafted polymers can be used to impart biocompatibility as well as biofunctionality to a surface by stabilizing the embedded enzyme from denaturing effects of the surface and/or solute.
A consequence of our model of a protein that may adopt a globular denatured state is that we predict a critical distance beyond which penetration of a protein into the grafted polymer layer will result in adsorption. This phenomenon cannot be predicted with models that treat the protein as a rigid object. Thus, for applications where even small amounts of adsorbed protein may be detrimental, immobilized protease can be used to break down proteins near the surface. In our model, structural stability is taken as an indicator of bioactivity, which may not always be the case. For example, anhydrous conditions, which may lead to increased rigidity of a protein, usually hinder its bioactivity—presumably due to interfering with the dynamics of the bound water (30). However, for aqueous proteins we always find that a hydrophilic polymer layer is necessary for stabilization, which presumably will contain sufficient water to establish proper hydration of the enzyme. Further improvement to the model would be an extension of the specific interaction potential to charged residues to examine the effect of salt concentration of the protein structure and activity. Advances in mean-field theories of polyelectrolytes (67) and polyelectrolyte brushes (68) reveal important scaling behavior in both weak and strong polyelectrolyte regimes.
Acknowledgments
The research was funded in part by the Matilda Barnett Revocable Trust, and the New York Metropolitan Research Fund. The authors acknowledge support from the Koebner-Klein Foundation.
References
- 1.Ratner, B. D. 1993. New ideas in biomaterials science—a path to engineered biomaterials. J. Biomed. Mater. Res. 27:837–850. [DOI] [PubMed] [Google Scholar]
- 2.Ratner, B. D. 1996. The engineering of biomaterials exhibiting recognition and specificity. J. Mol. Recogn. 9:617–625. [DOI] [PubMed] [Google Scholar]
- 3.Leininger, I., C. W. Cooper, R. D. Falb, and G. A. Grode. 1966. Nonthrombogenic plastic surfaces. Science. 152:1625–1626. [DOI] [PubMed] [Google Scholar]
- 4.Ambrus, C. M., J. L. Ambrus, C. Horvath, H. Pedersen, S. Sharma, C. Kant, E. Mirand, R. Guthrie, and T. Paul. 1978. Phenylalanine depletion for management of phenylketonuria—use of enzyme reactors with immobilized enzymes. Science. 201:837–839. [DOI] [PubMed] [Google Scholar]
- 5.Lavin, A., C. Sung, A. M. Klibanov, and R. Langer. 1985. Enzymatic removal of bilirubin from blood—a potential treatment for neonatal jaundice. Science. 230:543–545. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe, S., Y. Shimizu, T. Teramatsu, T. Murachi, and T. Hino. 1988. Application of immobilized enzymes for biomaterials used in surgery. Methods Enzymol. 137:545–551. [DOI] [PubMed] [Google Scholar]
- 7.Ryu, G., D. Han, Y. Kim, and B. Min. 1992. Albumin immobilized polyurethane and its blood compatibility. ASAIO J. 38:M644–M648. [DOI] [PubMed] [Google Scholar]
- 8.Kiaei, D., A. Safranj, J. P. Chen, A. B. Johnston, F. Zavala, A. Deelder, J. B. Castelino, V. Markovic, and A. S. Hoffman. 1992. Immobilization of proteins on glow discharge treated polymers. Radiat. Phys. Chem. 39:463. [Google Scholar]
- 9.Trachtenberg, M. C., C. K. Tu, R. A. Landers, R. C. Willson, M. L. McGregor, P. J. Laipis, J. F. Kennedy, M. Paterson, D. N. Silverman, D. Thomas, et al. 1999. Carbon dioxide transport by proteic and facilitated transport membranes. Life Support Biosph. Sci. 6:293–302. [PubMed] [Google Scholar]
- 10.El-Sherif, H., P. L. Martelli, R. Casadio, M. Portaccio, U. Bencivenga, and D. G. Mita. 2001. Urease immobilisation on chemically grafted nylon membranes. 1. Isothermal characterisation. J. Mol. Catal., B Enzym. 14:15–29. [Google Scholar]
- 11.Fernandez-Lafuente, R., V. Rodriguez, C. Mateo, G. Penzol, O. Hernandez-Justiz, G. Irazoqui, A. Villarino, K. Ovsejevi, F. Batista, and J. M. Guisan. 1999. Stabilization of multimeric enzymes via immobilization and post-immobilization techniques. J. Mol. Catal., B Enzym. 7:181–189. [Google Scholar]
- 12.Mateo, C., O. Abian, R. Fernandez-Lafuente, and J. M. Guisan. 2000. Increase in conformational stability of enzymes immobilized on epoxy-activated supports by favoring additional multipoint covalent attachment. Enzyme Microb. Technol. 26:509–515. [DOI] [PubMed] [Google Scholar]
- 13.Ulbrich-Hofmann, R., U. Arnold, and J. Mansfeld. 1999. The concept of the unfolding region for approaching the mechanisms of enzyme stabilization. J. Mol. Catal., B Enzym. 7:125–131. [Google Scholar]
- 14.Fernandez-Lafuente, R., V. Rodriguez, C. Mateo, G. Fernandez-Lorente, P. Arminsen, P. Sabuquillo, and J. M. Guisan. 1999. Stabilization of enzymes (D-amino acid oxidase) against hydrogen peroxide via immobilization and post-immobilization techniques. J. Mol. Catal., B Enzym. 7:173–179. [Google Scholar]
- 15.Fernandez-Lafuente, R., C. M. Rosell, L. Caanan-Haden, L. Rodes, and J. M. Guisan. 1999. Facile synthesis of artificial enzyme nano-environments via solid-phase chemistry of immobilized derivatives: dramatic stabilization of penicillin acylase versus organic solvents. Enzyme Microb. Technol. 24:96–103. [Google Scholar]
- 16.Kauzmann, W. 1959. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 14:1–63. [DOI] [PubMed] [Google Scholar]
- 17.Turkova, J. 1999. Oriented immobilization of biologically active proteins as a tool for revealing protein interactions and function. J. Chromatogr. B. 722:11–31. [DOI] [PubMed] [Google Scholar]
- 18.Wang, C. C., and G. H. Hsiue. 1993. Immobilization of glucose-oxidase on polyethylene film using a plasma-induced graft-copolymerization process. J. Biomater. Sci. Polym. Ed. 4:357–367. [DOI] [PubMed] [Google Scholar]
- 19.Eggers, D. K., and J. S. Valentine. 2001. Molecular confinement influences protein structure and enhances thermal protein stability. Protein Sci. 10:250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinjo, A. R., and S. Takada. 2002. Effects of macromolecular crowding on protein folding and aggregation studied by density functional theory: statics. Phys. Rev. E. 66(3):art. no.-031911. [DOI] [PubMed]
- 21.Wei, Y., J. G. Xu, Q. W. Feng, M. D. Lin, H. Dong, W. J. Zhang, and C. Wang. 2001. A novel method for enzyme immobilization: direct encapsulation of acid phosphatase in nanoporous silica host materials. J. Nanosci. Nanotechnol. 1:83–93. [DOI] [PubMed] [Google Scholar]
- 22.Zheng, L., and J. D. Brennan. 1998. Measurement of intrinsic fluorescence to probe the conformational flexibility and thermodynamic stability of a single tryptophan protein entrapped in a sol-gel derived glass matrix. Analyst. 123:1735–1744. [Google Scholar]
- 23.Biswas, R., and S. K. Pal. 2004. Caging enzyme function: a-chymotrypsin in reverse micelle. Chem. Phys. Lett. 387:221. [Google Scholar]
- 24.Celej, M. S., M. G. D'Andrea, P. T. Campana, G. D. Fidelio, and M. L. Bianconi. 2004. Superactivity and conformational changes on a-chymotrypsin upon interfacial binding to cationic micelles. Biochem. J. 378:1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minton, A. P. 2000. Implications of macromolecular crowding for protein assembly. Curr. Opin. Struct. Biol. 10:34–39. [DOI] [PubMed] [Google Scholar]
- 26.Ping, G., J. M. Yuan, M. Vallieres, H. Dong, Z. Sun, Y. Wei, F. Y. Li, and S. H. Lin. 2003. Effects of confinement on protein folding and protein stability. J. Chem. Phys. 118:8042–8048. [Google Scholar]
- 27.Zhou, H. X., and K. A. Dill. 2001. Stabilization of proteins in confined spaces. Biochemistry. 40:11289–11293. [DOI] [PubMed] [Google Scholar]
- 28.Brinker, A., G. Pfeifer, M. J. Kerner, D. J. Naylor, F. U. Hartl, and M. Hayer-Hartl. 2001. Dual function of protein confinement in chaperonin-assisted protein folding. Cell. 107:223–233. [DOI] [PubMed] [Google Scholar]
- 29.Klibanov, A. M. 1989. Enzymatic catalysis in anhydrous organic solvents. Trends Biosci. 14:141–144. [DOI] [PubMed] [Google Scholar]
- 30.Pal, S. K., and A. H. Zewail. 2004. Dynamics of water in biological recognition. Chem. Rev. 104:2099. [DOI] [PubMed] [Google Scholar]
- 31.Currie, E. P. K., W. Norde, and M. A. C. Stuart. 2003. Tethered polymer chains: surface chemistry and their impact on colloidal and surface properties. Adv. Colloid Interface Sci. 100:205–265. [DOI] [PubMed] [Google Scholar]
- 32.Currie, E. P. K., J. Van der Gucht, O. V. Borisov, and M. A. C. Stuart. 1999. Stuffed brushes: theory and experiment. Pure Appl. Chem. 71:1227–1241. [Google Scholar]
- 33.Fang, F., and I. Szleifer. 2002. Effect of molecular structure on the adsorption of protein on surfaces with grafted polymers. Langmuir. 18:5497–5510. [Google Scholar]
- 34.Halperin, A. 1999. Polymer brushes that resist adsorption of model proteins: design parameters. Langmuir. 15:2525–2533. [Google Scholar]
- 35.Jonsson, M., and H. O. Johansson. 2004. Effect of surface grafted polymers on the adsorption of different model proteins. Colloids Surf. B Biointerfaces. 37:71–81. [DOI] [PubMed] [Google Scholar]
- 36.Karlstrom, G., and H. O. Johansson. 2001. Lattice model calculations of interactions between proteins and surface grafted polymers with tethered affinity ligands. Colloids Surf. B Biointerfaces. 20:245–256. [DOI] [PubMed] [Google Scholar]
- 37.Kidoaki, S., Y. Nakayama, and T. Matsuda. 2001. Measurement of the interaction forces between proteins and iniferter-based graft-polymerized surfaces with an atomic force microscope in aqueous media. Langmuir. 17:1080–1087. [Google Scholar]
- 38.McPherson, T. B., S. J. Lee, and K. Park. 1995. Analysis of the prevention of protein adsorption by steric repulsion theory. In Proteins at Interfaces II. American Chemical Society, Washington, DC. 395–404.
- 39.Steels, B. M., J. Koska, and C. A. Haynes. 2000. Analysis of brush-particle interactions using self-consistent-field theory. J. Chromatogr. B. 743:41–56. [DOI] [PubMed] [Google Scholar]
- 40.Tiberg, F., C. Brink, M. Hellsten, and K. Holmberg. 1992. Immobilization of protein to surface-grafted PEO/PPO block copolymers. Colloid Polym. Sci. 270:1188–1193. [Google Scholar]
- 41.Fang, F., and I. Szleifer. 2001. Kinetics and thermodynamics of protein adsorption: a generalized molecular theoretical approach. Biophys. J. 80:2568–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hadicke, E., J. Rieger, I. U. Rau, and D. Boeckh. 1999. Molecular dynamics simulations of the incrustation inhibition by polymeric additives. Phys. Chem. Chem. Phys. 1:3891–3898. [Google Scholar]
- 43.Satulovsky, J., M. A. Carignano, and I. Szleifer. 2000. Kinetic and thermodynamic control of protein adsorption. Proc. Natl. Acad. Sci. USA. 97:9037–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Srebnik, S. 2001. Solvent effects on heteropolymer adsorption and freezing. J. Chem. Phys. 114:9179–9183. [Google Scholar]
- 45.Efremova, N. V., S. R. Sheth, and D. E. Leckband. 2001. Protein-induced changes in poly(ethylene glycol) brushes: molecular weight and temperature dependence. Langmuir. 17:7628–7636. [Google Scholar]
- 46.Huang, Y. B., W. Leobandung, A. Foss, and N. A. Peppas. 2000. Molecular aspects of muco- and bioadhesion: tethered structures and site-specific surfaces. J. Control. Release. 65:63–71. [DOI] [PubMed] [Google Scholar]
- 47.Woodle, M. C. 1998. Controlling liposome blood clearance by surface-grafted polymers. Adv. Drug Deliv. Rev. 32:139–152. [DOI] [PubMed] [Google Scholar]
- 48.Moskovitz, Y., and S. Srebnik. 2004. Stabilization of surface-immobilized enzymes using grafted polymers. Phys. Rev. E. 70:032902-1–032902-4. [DOI] [PubMed] [Google Scholar]
- 49.Scheutjens, J. M. H. M., and G. J. Fleer. 1979. Statistical theory of the adsorption of interacting chain molecules. 1. Partition function, segment density distribution, and adsorption isotherms. J. Phys. Chem. 83:1619–1635. [Google Scholar]
- 50.Wijmans, C. M., J. Scheutjens, and E. B. Zhulina. 1992. Self-consistent field-theories for polymer brushes—lattice calculations and an asymptotic analytical description. Macromolecules. 25:2657–2665. [Google Scholar]
- 51.Engel, M. F. M., A. J. W. G. Visser, and C. P. M. Van Mierlo. 2004. Conformation and orientation of a protein folding intermediate trapped by adsorption. Proc. Natl. Acad. Sci. USA. 101:11316–11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gray, J. J. 2004. The interaction of proteins with solid surfaces. Curr. Opin. Struct. Biol. 14:110–115. [DOI] [PubMed] [Google Scholar]
- 53.Henry, M., C. Dupont-Gillain, and P. Bertrand. 2003. Conformation change of albumin adsorbed on polycarbonate membranes as revealed by ToF-SIMS. Langmuir. 19:6271–6276. [Google Scholar]
- 54.Lensun, L., T. A. Smith, and M. L. Gee. 2002. Partial denaturation of silica-adsorbed bovine serum albumin determined by time-resolved evanescent wave-induced fluorescence spectroscopy. Langmuir. 18:9924–9931. [Google Scholar]
- 55.Michael, K. E., V. N. Vernekar, B. G. Keselowsky, J. C. Meredith, R. A. Latour, and A. J. Garcia. 2003. Adsorption-induced conformational changes in fibronectin due to interactions with well-defined surface chemistries. Langmuir. 19:8033–8040. [Google Scholar]
- 56.Srebnik, S., A. K. Chakraborty, and D. Bratko. 1998. Random heteropolymer adsorption on disordered multifunctional surfaces: effect of specific intersegment interactions. J. Chem. Phys. 109:6415–6419. [Google Scholar]
- 57.Hoffman, A. S., G. Schmer, C. Harris, and W. G. Kraft. 1972. Covalent binding of biomolecules to radiation-grafted hydrogels on inert polymer surfaces. Trans. Am. Soc. Artif. Intern. Organs. 18:10–18. [DOI] [PubMed] [Google Scholar]
- 58.Park, K. D., T. Okano, C. Nojiri, and S. W. Kim. 1988. Heparin immobilization onto segmented polyurethane-urea surfaces—effect of hydrophilic spacers. J. Biomed. Mater. Res. 22:977–992. [DOI] [PubMed] [Google Scholar]
- 59.Jeon, S. I., J. H. Lee, J. D. Andrade, and P. G. Degennes. 1991. Protein surface interactions in the presence of polyethylene oxide. 1. Simplified theory. J. Colloid Interface Sci. 142:149–158. [Google Scholar]
- 60.Sofia, S. J., V. Premnath, and E. W. Merrill. 1998. Poly(ethylene oxide) grafted to silicon surfaces: grafting density and protein adsorption. Macromolecules. 31:5059–5070. [DOI] [PubMed] [Google Scholar]
- 61.Szleifer, I. 1997. Protein adsorption on tethered polymer layers: effect of polymer chain architecture and composition. Phys. A (Amsterdam). 244:370–388. [Google Scholar]
- 62.Wijmans, C. M., F. A. M. Leermakers, and G. J. Fleer. 1994. Chain stiffness and bond correlations in polymer brushes. J. Chem. Phys. 101:8214–8223. [Google Scholar]
- 63.Dill, K. A., S. Bromberg, K. Z. Yue, K. M. Fiebig, D. P. Yee, P. D. Thomas, and H. S. Chan. 1995. Principles of protein folding—a perspective from simple exact models. Protein Sci. 4:561–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scopes, R. K. 1994. Protein Purification: Principles and Practice. Springer-Verlag, New York. 95–101.
- 65.Tang, H., and I. Szleifer. 1994. Phase behavior of grafted polymers in poor solvents. Europhys. Lett. 28:19–24. [Google Scholar]
- 66.Yeung, C., A. C. Balazs, and D. Jasnowt. 1993. Lateral instabilities in a grafted layer in a poor solvent. Macromolecules. 26:1914–1921. [Google Scholar]
- 67.Holm, C., J. F. Joanny, K. Kremer, R. R. Netz, P. Reineker, C. Seidel, T. A. Vilgis, and R. G. Winkler. 2004. Polyelectrolyte theory. In Polyelectrolytes with Defined Molecular Architecture II. Springer-Verlag, Berlin. 67–111.
- 68.Ruhe, J., M. Ballauff, M. Biesalski, P. Dziezok, F. Grohn, D. Johannsmann, N. Houbenov, N. Hugenberg, R. Konradi, S. Minko, et al. 2004. Polyelectrolyte brushes. In Polyelectrolytes with Defined Molecular Architecture I. Springer-Verlag, Berlin. 79–150.