

Abstract
After activation, many receptors and their adaptor proteins act as scaffolds displaying numerous docking sites and engaging multiple targets. The consequent assemblage of a variety of protein complexes results in a combinatorial increase in the number of feasible molecular species presenting different states of a receptor-scaffold signaling module. Tens of thousands of such microstates emerge even for the initial signal propagation events, greatly impeding a quantitative analysis of networks. Here, we demonstrate that the assumption of independence of molecular events occurring at distinct sites enables us to approximate a mechanistic picture of all possible microstates by a macrodescription of states of separate domains, i.e., macrostates that correspond to experimentally verifiable variables. This analysis dissects a highly branched network into interacting pathways originated by protein complexes assembled on different sites of receptors and scaffolds. We specify when the temporal dynamics of any given microstate can be expressed using the product of the relative concentrations of individual sites. The methods presented here are equally applicable to deterministic and stochastic calculations of the temporal dynamics. Our domain-oriented approach drastically reduces the number of states, processes, and kinetic parameters to be considered for quantification of complex signaling networks that propagate distinct physiological responses.
INTRODUCTION
Extracellular signals received by plasma membrane receptors are processed and transduced through covalent modifications of amino acid residues on receptors and their cytoplasmic substrates (1). Interaction domains of numerous adaptor proteins and signaling enzymes recognize these modified residues as binding partners. For instance, receptors that belong to the large family of receptor tyrosine kinases (RTK) modify tyrosine residues by attaching a phosphate group. Phosphotyrosine residues efficiently bind proteins with Src homology 2 (SH2) and phosphotyrosine-binding domains (2,3). Subsequent assembly of signaling complexes leads to activation of downstream enzymes and protein kinase cascades, which propagate signals down to the nucleus.
Receptors and adaptor proteins often display multiple docking sites and engage several downstream signaling proteins, thereby serving as scaffolding proteins, or scaffolds. For instance, after activation, insulin receptor (IR), insulinlike growth factor receptor-1 (IGF-1R), epidermal growth factor receptor (EGFR), and immunoreceptor tyrosine kinases, such as B-cell receptors, T-cell receptors, and Fc receptors, all bind various combinations of downstream targets generating a large number of different signaling species in highly branched networks (4–6). IR and IGF-1R, exist as dimers containing two α- and two β-subunits, whereas other RTKs are monomers that dimerize after ligand binding (7–11). Oligomerization of cell-surface receptors can generate multiple docking sites even if each receptor monomer has only one such site.
The binding partners of IR or IGF-1R include insulin receptor substrates, IRS1–IRS4, which are scaffolding proteins (4,12). Phosphorylation of various tyrosine residues on IRS-1 by IR or IGF-1R creates docking sites for several SH2 domain-containing proteins, such as growth factor receptor binding protein-2 (Grb2), the p85 subunit of phosphatidylinositol 3-kinase (PI3K), and the protein tyrosine phosphatase SHP-2 (13,14). Likewise, the Grb2-associated binders (GAB-1 and GAB-2) are scaffolding adaptors, which are phosphorylated by various RTKs (15–17) and soluble tyrosine kinases of the Src family (18,19). Phosphorylated docking sites on GABs bind numerous targets, including Shc, Grb2, p85, phospholipase Cγ, SHP-2, and the Crk adaptor protein (20,21). Different docking sites on activated receptors and scaffold proteins initiate separate signaling pathways that propagate distinct cellular responses. For instance, Grb2 binding to tyrosine phosphorylated GAB and the recruitment of the GDP/GTP exchange factor SOS to the plasma membrane enables activation of the small GTPase Ras. This leads to activation of the mitogen-activated protein kinase (MAPK) cascade that promotes mitogenesis and differentiation, whereas p85 binding to another docking site on GAB initiates the PI3K/AKT pathway implicated in glucose and lipid metabolism and cell survival (22,23).
Quantitative analysis and mathematical modeling of signaling through receptors and scaffolds is hampered by a combinatorial increase in complexity with the number of docking domains (sites) and interaction partners. A scaffolding adaptor protein can either be associated with or dissociated from a receptor. Each docking site on a scaffold can be phosphorylated or unphosphorylated, and the phosphorylated site can either be free or occupied by its binding partner. Proteins bound to a scaffold can be phosphorylated and dephosphorylated, and may associate with other signaling proteins, assembling multicomponent complexes. All these different possibilities multiply, generating tens of thousands of molecular species even for a few initial steps in signal transduction involving receptors, scaffolds, and adaptors (24,25).
An entire collection of potential molecular species corresponds to different forms of a receptor and scaffold protein and is referred to as a set of microstates. A standard mechanistic description takes into account all possible microstates and transitions between them. Such a detailed description might be necessary when binding of a protein to one docking site changes the kinetic properties of other docking sites (allosteric interactions, such as cooperative association), and/or promotes (de)phosphorylation of proteins bound to these sites (24–28). An elegant algorithm for deterministic calculations of all potential species and reactions has recently been developed (29). However, because of the enormous number of distinct microstates of regulatory complexes and a lack of knowledge of the kinetics for each possible transition, such a detailed microdescription may rapidly become impractical. Unlike deterministic algorithms, stochastic algorithms use probabilistic rules to simulate the evolution of species populations. MOLECULIZER is a stochastic simulator that employs a form of Gillespie's first reaction algorithm (30) and generates only populated states of a network, thus reducing the number of all possible states to be considered (31). An appealing stochastic approach to modeling multistate biomolecular systems was developed by Bray and colleagues (32,33). In the computer program StochSim, individual multistate molecules and complexes are represented as distinct software objects. Consequently, a combinatorial explosion of the number of microstates is circumvented by merely following stochastic changes in the states of individual, distinguishable molecules, the number of which does not increase in the course of simulations (34). The use of StochSim is especially practical for networks in small (subcellular) volumes with low numbers of molecules, where a stochastic algorithm more closely describes the physical reality than a deterministic algorithm. However, for large networks with hundreds of different proteins, the StochSim calculation time would be too slow, increasing proportionally to the number of molecules squared.
The present article demonstrates that a mechanistic description of a highly combinatorial network generated by various phosphorylation and binding forms of receptors and scaffolds may be drastically reduced using a domain-oriented approach. Provided there is a set of docking sites where molecular events are independent (i.e., allosteric interactions are absent), a signaling system can be modeled in terms of a macrodescription that follows the states of each docking domain separately, including subsequent downstream signal transduction. Compared to the combinatorial explosion of microstates and equations in a mechanistic model, for a macrodescription, the number of macrostates increases linearly, as the sum of distinct domains and binding partners. Starting with simple examples that are subsequently generalized, we demonstrate when and how the temporal behavior of a particular microstate can be expressed explicitly or approximated in terms of macrostates of distinct sites, which are determined using a significantly reduced model. Such a reconstruction of microscopic behavior is required when different microstates within the same macrostate present biologically different activities. The results presented in this article suggest a novel approach to the reduction of combinatorial complexity of multicomponent signal transduction networks.
METHODS
Kinetic description
We will analyze two signaling subnetworks, presenting a common theme in signal transduction: 1), a scaffolding adaptor protein activated by a receptor; and 2), a receptor that also acts as a scaffold. A key property that will allow us to reduce combinatorial complexity of these networks is the assumption of the absence of allosteric interactions for a subset of domains/docking sites on a receptor and/or scaffold. A quantitative description of these modules will serve as a template for constructing models of signal transduction.
Scaffold with multiple independent docking sites
We will first consider a scaffolding adaptor protein (S) that binds to an activated receptor (R) through a specific interaction domain (h) and is subsequently phosphorylated by the receptor kinase on distinct sites i (i = 1, …, n), located outside of domain h. When phosphorylated, sites i can engage several downstream signaling proteins (and their complexes) or can be dephosphorylated by phosphatases. To describe the system quantitatively, it is convenient to assign digital flags (numbers) to possible states of domain h and docking sites i (Fig. 1). For unbound S, h = 0, and for S bound to the receptor, h = 1. Each site i can be in one of (mi + 1) states denoted by ai = 0, 1, …, mi. The number ai = 0 indicates an unphosphorylated free site, 1 denotes a phosphorylated free site, 2 represents a site occupied by a binding partner (As), 3 can stand for phosphorylation of this partner (AsP) or binding a new partner (Aj), 4 can denote the binding of the complex AsPAj, and so on. Consequently, at the microlevel the state of protein S is described by a digital flag (a1, …, an; h), and the meaning of these numbers is specific for a network. The concentration of S in each state is denoted by the time-dependent function s(a1, …, an; h; t) (to simplify designations, we will omit the variable t if it does not result in misunderstanding). For any h-state, there are (m1 + 1) · (m2 + 1) · … · (mn + 1) states of S. The transitions between states are described by a graph with 2 · (m1 + 1) · (m2 + 1) · … · (mn + 1) vertices (states) presenting all feasible species. Fig. 2 illustrates such a transition graph for a scaffold with two docking sites and their partners, A1 and A2. Phosphorylation of docking sites occurs only when the scaffold is bound to the receptor (h = 1), whereas dephosphorylation may take place for both bound and unbound (h = 0) states, if docking sites are not occupied by their binding proteins (phosphatase activities are assumed to be constant parameters).
FIGURE 1.

A digital flag description of domain states of a scaffolding protein. The scaffolding adaptor protein S binds to the receptor R via a specific domain (h). For the unbound S, h = 0, and when S is bound to R, h = 1. Two docking sites on S display a variety of states: 0 indicates free unphosphorylated site (shown as a tyrosine residue Y); 1 denotes free phosphorylated site (pY); 2 represents a site occupied by a binding partner (pY-A); and 3 stands for phosphorylation of this partner (pY-pA).
FIGURE 2.

Fragment of a transition graph for a scaffolding protein with two docking sites. Species s(0,0;0), s(1,0;0), and s(2,0;0) correspond to the scaffold that is not bound to the receptor R (h = 0), unphosphorylated on docking site 2 and has docking site 1 unphosphorylated, phosphorylated, or occupied by a binding partner (A1), respectively. Species s(0,1;0), s(1,1;0), s(2,1;0) and s(0,2;0), s(1,2;0), and s(2,2;0) differ from the species above only by the state of docking site 2, which is phosphorylated (the first three) or occupied by a binding partner A2 (the last three). The terms s(i,j;1) (i,j = 0, 1, 2) are the complexes of the scaffold species s(i,j;0) with R. Reversible reactions and transitions that can occur in both directions, such as the molecule binding/dissociation, phosphorylation by the receptor kinase, and dephosphorylation by a phosphatase, are shown by lines. Arrows indicate irreversible dephosphorylation steps, which do not have phosphorylation transitions in the opposite direction, as for the scaffold that has dissociated from the receptor.
Elementary transitions between states of the scaffolding protein S are assumed to follow mass-action kinetics. Two additional assumptions are crucial. First, we assume that transitions between different states ai of each site i do not depend on states of the other sites (aj, j ≠ i), whereas these transitions may depend on the state of the h domain, which can formally be called a controlling recruitment site. For instance, no phosphorylation transitions can occur when the scaffold has dissociated from the receptor (Fig. 2). Second, transitions between different states of the recruitment site h are assumed to be independent of states ai for any site i. These kinetic properties imply a hierarchical relation between two kinds of binding sites on S.
In the notation assumed, any chemical transformation is simply a change in one of the numbers ai or h. We designate by ki(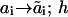 ) the pseudo-first-order rate constant for the transition from the state (a1, …, ai−1, ai, ai+1 …, an; h) to the state (
) the pseudo-first-order rate constant for the transition from the state (a1, …, ai−1, ai, ai+1 …, an; h) to the state (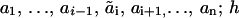 ). Upon this transition, the number ai characterizing site i changes to
). Upon this transition, the number ai characterizing site i changes to 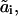 whereas states aj of other sites j do not change (as indicated by the subscript i in ki). If this is a binding reaction, the ki includes the concentration of a binding protein (Ai) as a multiplier. Similarly, the reverse transition is described by the pseudo-first order rate constant ki(
whereas states aj of other sites j do not change (as indicated by the subscript i in ki). If this is a binding reaction, the ki includes the concentration of a binding protein (Ai) as a multiplier. Similarly, the reverse transition is described by the pseudo-first order rate constant ki(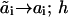 ). Association and dissociation of the scaffold S and the receptor R change only the h number and are characterized by the rate constants kr · R and k−r, respectively. For example, in Fig. 2, the rate constant for the transitions s(0, a2; 1) → s(1, a2; 1) is k1(0→1; 1) for a2 = 0, 1 or 2, and for the transitions s(a1, a2; 0)→ s(a1, a2; 1) is kr · R for any a1, a2.
). Association and dissociation of the scaffold S and the receptor R change only the h number and are characterized by the rate constants kr · R and k−r, respectively. For example, in Fig. 2, the rate constant for the transitions s(0, a2; 1) → s(1, a2; 1) is k1(0→1; 1) for a2 = 0, 1 or 2, and for the transitions s(a1, a2; 0)→ s(a1, a2; 1) is kr · R for any a1, a2.
The time evolution of the scaffold is determined by the following system of 2 · (m1+1) · (m2+1) · … · (mn+1) ordinary differential equations, which describe the chemical transformations of all feasible species as transitions between microstates of S,
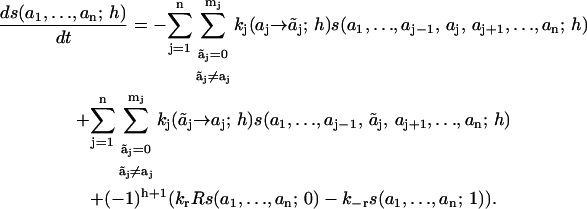 |
(1) |
This equation has a simple structure: on the right-hand side, the first and second terms describe the consumption and production of species s(a1, …, an; h; t) in reactions that occur at sites i, whereas the third term presents supply-and-demand processes arising from association (h = 0) and dissociation (h = 1) from the receptor. We will refer to this mechanistic presentation ( Eq. 1), which involves all possible microstates and transitions between them, as a microdescription of a network.
The simplest case of complete independence: sites without hierarchical control
To highlight the general principles of a domain-oriented approach to reduction in combinatorial complexity, we will also analyze an alternative, simpler model of signaling by a scaffold. In this model, chemical transformations of binding sites on the scaffold S are completely independent of states of other sites, including phosphorylation and dephosphorylation. The formal difference from the previous model is the lack of controlling recruitment site h. In this case, the state of S is characterized by the function s(a1, …, an; t). The dynamics of signaling by this scaffolding protein is described by a reduced system of (m1+1) · (m2+1) · … · (mn+1) ordinary differential equations, which is obtained from Eq. 1 by equating kr and k−r to zero.
Receptor possessing scaffolding properties
The second signaling module analyzed here is a receptor, also acting as a scaffold, of the RTK superfamily. After binding of a ligand or/and dimerization, the receptor is autophosphorylated on several docking sites, which makes them capable of binding signaling proteins with specific binding domains. We will consider two possible molecular mechanisms. The first assumes that the binding affinity of a ligand (L) for the receptor (R) does not depend on the receptor phosphorylation and dimerization states and on the states of its cytoplasmic docking sites (10). In this case, a mathematical description of the system becomes the same as the description of an activated scaffolding protein considered in Scaffold with Multiple Independent Docking Sites. States ai of docking sites are described identically for the scaffold and the receptor, and states, h = 1 and h = 0, correspond to the receptor that has or has not bound L. In fact, the transition graph is readily obtained from Fig. 2 by the replacement of S and R by R and L, respectively, and the signaling kinetics is determined by Eq. 1 after the same replacement.
In an alternative mechanism, conformational changes after receptor dimerization and/or phosphorylation preclude ligand dissociation, until the receptor is endocytosed and undergoes degradation or recycling, as for the EGF receptor (35–39). For the sake of simplicity, we will assume initially that receptors have a dimeric structure before stimulation (such as IR and IGF-1R) and undergo conformational changes and phosphorylation after the ligand binding. We designate by r0 and r(a1,…, an) the concentrations of free receptor dimer and the dimer with bound ligand(s) and docking sites (i) in states (a1,…, an), respectively. As in the previous section, ai = 0 stands for the unphosphorylated site i, ai = 1 for the free phosphorylated site, ai = 2, and so on for different combinations of adaptor proteins and their phosphorylation states bound to site i. For this signaling network, a fragment of a transition graph is given in Fig. 3. In comparison with Fig. 2, this graph is simpler, because the dissociation of ligand L is possible only when none of the receptor docking sites are phosphorylated, i.e., the flux of L occurs only for the state r(0,0).
FIGURE 3.

Fragment of a transition graph for a receptor with two docking sites for downstream interacting proteins. L, ligand; r0, free receptor; and r(0,0), ligand-receptor complex with two unphosphorylated docking sites. For all species in the network, r(i,j), i = 0, 1, or 2 indicates that the first docking site on the receptor is unphosphorylated, phosphorylated, or occupied by adaptor protein A1, respectively, and j = 0, 1, or 2 means that the second receptor docking site is unphosphorylated, phosphorylated, or occupied by adaptor protein A2. Note that phosphorylation of the receptor even on a single site locks the ligand in place.
A transition from state ai to state 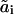 is characterized by the pseudo-first-order rate constant ki(
is characterized by the pseudo-first-order rate constant ki(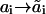 ), and the on- and off-constants for ligand and receptor association are designated by k0 and k−0, respectively. The temporal behavior of signaling species in this network is determined by
), and the on- and off-constants for ligand and receptor association are designated by k0 and k−0, respectively. The temporal behavior of signaling species in this network is determined by
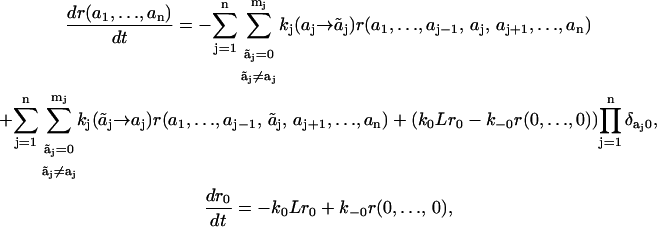 |
(2) |
where δij is the Kronecker symbol (δij = 1, if i = j, otherwise δij = 0).
RESULTS
The multiplicity of domains and binding sites on signaling molecules, such as receptors and large scaffolding adaptor proteins, is a hallmark of signal transduction networks. In this section, we apply a domain-oriented approach to combine numerous microstates into macrostates of separate domains/sites.
Dissecting signal transduction by a scaffold into signaling by separate docking sites
We assumed above that molecular processes involving a particular site of a scaffolding protein are independent of states of other docking sites, and that rate constants of receptor-scaffold association (dissociation) are the same for all states of these sites. Although there are well-characterized examples where this assumption is not valid (e.g., negative cooperativity of binding events because of steric hindrance), in other cases docking sites are located in different, distant domains of a protein, where they interact independently with their binding partners. This independence suggests that the time course of reactions involving a docking site, such as phosphorylation, dephosphorylation, and binding and release of downstream proteins, may be analyzed separately from the analysis of the entire scaffold. In fact, this approach will allow us to follow the fate of a particular docking site on a scaffold independently of reactions occurring at other sites. We will see now how this dissection of scaffold dynamics becomes feasible.
Adding up the concentrations of all forms of the adaptor protein S displaying a particular state (ai) of docking site i, we introduce the macrovariables Si(ai, h),
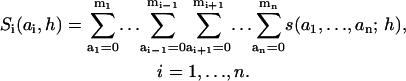 |
(3) |
Each of these macrovariables follows states of only one docking site on the scaffold and can be interpreted as the concentration of this site in a particular state. For instance, considering signaling by the adaptor protein GAB, a macrovariable may be the sum of all microstates, in which the GAB docking site for PI3K is phosphorylated and bound by PI3K, whereas sites for SHP-2, Shc (and so on) can be unphosphorylated, phosphorylated, free, or occupied by the unphosphorylated or phosphorylated binding partners.
Summing up Eq. 1 for all states of docking sites j different from site i, we obtain
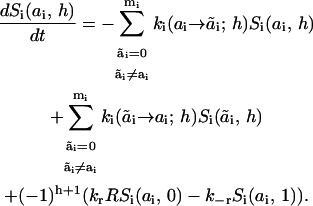 |
(4) |
This equation shows that when docking sites are independent, the time evolution of the signaling system can be analyzed in terms of macrovariables Si(ai, h), and does not require the analysis of the dynamics of microstates of the adaptor protein S. The description in terms of Si(ai, h) is referred to as a macropresentation of the signaling network that involves R and S.
A formal mathematical proof of Eq. 4 is given in Supplemental Note 1 in Supplementary Materials. In intuitive terms, recall that each microstate s(a1, …, an; h) can be presented by a vertex in an (n+1)-dimensional transition graph. Edges coming in or going out of the vertex s(a1, …, an; h) form n+1 directions that correspond to chemical transformations of any of n docking sites i and binding to/dissociation from R, which changes h (see Fig. 2 with n = 2). The macrostate Si(ai, h) is the sum of microstates over n−1 of those directions, which involve transitions that change states of n−1 docking sites other than site i and state h. In the course of summation of the rate equations (Eq. 1), terms corresponding to transitions along these n−1 directions cancel each other (each term enters twice as consumption and production with opposite signs). All transitions where ai changes have identical rate constants for each of composing microstates, and the same is true for transitions changing h. Consequently, the remaining rate terms are the products of the sums of microstates and the corresponding common rate constants. The resulting rate equations contain only macrovariables multiplied by the rate constants common for all microstates composing those macrovariables.
The separation of variables that describe different sites i's considerably reduces the number of states and equations used for a quantitative analysis of the system behavior. In a macrodescription framework, there are only 2 · (m1 + m2 + … + mn + n) states of the scaffold instead of 2 · (m1 + 1) · (m2 + 1) · … · (mn + 1) microstates (species), and the number of transitions in a transition graph decreases even more drastically. Fig. 4 illustrates this reduction for the transition graph shown in Fig. 2. Even for the simplest case of two docking sites, instead of 18 microstates, there appear to be 12 states, and the number of transitions decreases from 33 to 14. Moreover, the graph of Fig. 4 splits into two disconnected subgraphs corresponding to site 1 and site 2 transitions.
FIGURE 4.

Macrodescription of the transition graph shown in Fig. 2. Macrovariables, S1 and S2, correspond to states of docking sites 1 and 2, respectively, and are expressed in terms of microstates of Fig. 2 as follows. The upper panel: S1(0,0) = s(0,0;0) + s(0,1;0) + s(0,2;0); S1(1,0) = s(1,0;0) + s(1,1;0) + s(1,2;0); S1(2,0) = s(2,0;0) + s(2,1;0) + s(2,2;0); S1(0,1) = s(0,0;1) + s(0,1;1) + s(0,2;1), S1(1,1) = s(1,0;1) + s(1,1;1) + s(1,2;1); S1(2,1) = s(2,0;1) + s(2,1;1) + s(2,2;1). Summation goes along vertical edges of rear and front facets of the graph in Fig. 2. The lower panel: S2(0,0) = s(0,0;0) + s(1,0;0) + s(2,0;0); S2(1,0) = s(0,1;0) + s(1,1;0) + s(2,1;0); S2(2,0) = s(0,2;0) + s(1,2;0) + s(2,2;0); S2(0,1) = s(0,0;1) + s(1,0;1) + s(2,0;1); S2(1,1) = s(0,1;1) + s(1,1;1) + s(2,1;1); S2(2,1) = s(0,2;1) + s(1,2;1) + s(2,2;1). Summation goes along horizontal edges of the rear and front facets of the graph in Fig. 2. Arrows indicate one-directional transitions.
Importantly, an entire signaling network can be analyzed in terms of macrostates only, using a macrodescription of a receptor-scaffold module. In a mechanistic framework, kinetic equations that describe the dynamics of free and bound receptor forms take into account receptor interactions with each of possible forms (microstates) of the scaffold S (38,40). By contrast, if the kinetics of the scaffold protein S is described in terms of macrostates, these equations can be simplified, and the time evolution of the free receptor concentration can be determined using any of macrovariables Si (see Supplemental Note 2 in Supplementary Materials). Likewise, when there is no cross-talk between downstream signaling pathways initiated by the association of binding partners with docking sites on the scaffold, the dynamic behavior of each of these pathways can be calculated separately in terms of single macrovariables and the corresponding interacting partners, without determining the dynamics of a whole network.
The EGFR signaling network provides an instructive illustration of how a macrodescription can reduce the number of states that need to be considered in a realistic model of a signaling network. The EGFR network contains at least two major scaffolding proteins, EGFR itself and the adaptor protein GAB-1. Description of this network up to and including ERK activation requires at least 163,000 microvariables, but only ∼350 macrovariables to monitor independent and allosterically interacting domains (unpublished data). Likewise, for the IR and IGF-1R signaling networks, which involve IRS, GAB, and Grb10 adaptor proteins, the assumption of independence of some docking sites on these scaffolds results in a reduction in the number of equations from hundreds of thousands to a few hundreds, yet allows for accurate predictions of the time evolution of signaling patterns.
Retrieving a mechanistic (micro)description from the dynamics of independent docking sites
Analyzing a model of a scaffolding protein (S) with independent docking sites and receptor-binding site (Eq. 1), we found that the time evolution of each docking site (i) can be described by the dynamics of a single macrovariable Si(ai,h) without requiring monitoring of the remaining sites on S (Eq. 4). However, the concentration dynamics of a particular microstate may also be of interest—as in, for instance, when the assembly of two or more interacting proteins on S is required for activation of a downstream target. We will now show how such a microdescription can be retrieved from a macrodescription at arbitrary time t.
A scaffolding protein without controlling site h
We start with a simple model of a scaffold, formulated in The Simplest Case of Complete Independence: Sites without Hierarchical Control, above, in Methods, where there is no hierarchical control over the states of docking sites. Formally, this model does not incorporate a controlling h-site on S and assumes that phosphorylation, dephosphorylation, binding a partner, and other molecular events involving a particular docking site proceed independently of the states of any other site. In this case, the concentration of S in any microstate is characterized by the time-varying function s(a1, …, an; t), which lacks any dependence on h. This scaffold can be activated by stimulation of soluble kinases, which independently phosphorylate various docking sites. Phosphorylated motifs deliver a message to cytoplasmic binding partners of the scaffold. Alternatively, inhibition of phosphatases (e.g., as a result of oxidative stress) may shift the balance between phosphorylation and dephosphorylation of docking sites. Such perturbations will cause signaling by the scaffold.
The concentration Si(ai,t) of S in a particular macrostate (ai) is defined in Eq. 3 as the sum of all forms of the scaffold, in which docking site i is in state ai. Because of the assumed independence of docking sites, one may conjecture that the probability of finding these sites in states a1,…, an simultaneously, i.e., finding the scaffolding protein in a certain microstate s(a1,…, an), equals the product of the probabilities of the corresponding states for each docking site. The probabilities are equal to the fractional concentrations, which are obtained by dividing the concentrations s(a1,…, an) and Si(ai) by the total S concentration (Stot),
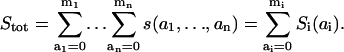 |
(5) |
Suppose that at some initial moment t0 any microprobability s(a1,…,an; t0)/Stot can be expressed as the product of the macroprobabilities Si(ai, t0)/Stot,
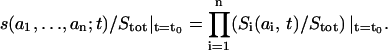 |
(6) |
Using Eqs. 1 and 4 (where both kr and k−r equal zero), it can be shown readily that Eq. 6 continues to apply for any t > t0 (Supplemental Note 3 in Supplementary Materials). Consequently, the time evolution of the concentrations s(a1, …, an; t) of the scaffold in any of its microstates can be expressed in terms of the product of the fractional concentrations Si(ai,t)/Stot of macrostates for any t > t0,
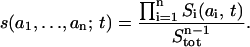 |
(7) |
It is instructive to note that in enzyme kinetics and models of ion channels, the equilibrium (or pseudo-equilibrium) concentrations of multimolecular complexes are expressed as the product of the saturation functions for distinct independent subunits or binding sites (41). However, in contrast to this earlier result of the steady-state enzyme kinetics, Eq. 7 tells us that any transient microstate of a scaffolding protein can be expressed as the product of the time-dependent probabilities (fractional concentrations) of macrostates of distinct docking sites.
Recruitment of a scaffold into a receptor-scaffold complex
In a more general case (see Eq. 1), distinct docking sites on a scaffolding protein cannot be considered completely independent. In fact, after the formation of a receptor-scaffold complex (Fig. 2), two or more docking sites can be phosphorylated while the scaffold is bound to the receptor (an alternative mechanism of entirely independent sites implies that after each phosphorylation, the scaffold dissociates from the receptor). Clearly, the fact that docking sites can be phosphorylated only if h equals 1 and not 0, imposes common constraints on otherwise independent docking sites.
Eq. 4 demonstrates that a macrostate model still applies to a receptor-scaffold system, and the time evolution of distinct domains and the initiated downstream signaling pathways can be resolved into separate dynamics of the corresponding macrovariables Si(ai, h). However, in the general case, the temporal patterns of microstates s(a1, …, an; h) cannot be exactly obtained from Si(ai, h), due to the correlation between phosphorylation of docking sites during the stage of association of S with R. Yet, as we will see, the time evolution of microstates can be accurately approximated in terms of macrostates.
Supplemental Note 4 in Supplementary Materials shows that although the products of the fractional concentrations (probabilities) of macrostates may coincide with the microstate concentrations at the initial time point (e.g., before receptor activation; compare to Eq. 6), they start to deviate from each other, as time progresses. This deviation is brought about by the terms that involve the rate constants of the association and dissociation of the scaffold and receptor, kr and k−r (see Eqs. S4.3 and S4.4, Supplemental Note 4 in Supplementary Materials). This finding suggests that it may be helpful to analyze cases, where these rates are much faster or much slower than the rates that change the states of docking sites i. Under the condition where the association/dissociation steps changing h are much faster than reactions changing states ai of docking sites, the ratio of the concentrations of the receptor-bound forms of the scaffold (h = 1) and the free forms (h = 0) with the same states of docking sites are fixed by the following rapid-equilibrium relations (42,43):
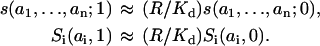 |
(8) |
Using Eq. 8, we express Eqs. 1 and 4 in terms of so-called slow variables, which equal the overall concentrations of bound and unbound scaffold forms with the same states ai and, thus, do not change in the fast association/dissociation reactions of the scaffold and receptor (see Supplemental Note 5 in Supplementary Materials). The resulting equations for slow variables are parallel to the equations that describe a scaffold with completely independent sites, and similarly to Eq. 7, the concentrations of microstates for both the receptor-bound (h = 1) and unbound (h = 0) scaffold forms can be found as follows (Supplemental Note 5 in Supplementary Materials):
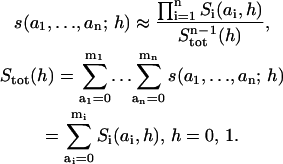 |
(9) |
Importantly, this equation suggests the use of an alternative scaling of the fractional concentrations of micro- and macrostates exploiting the normalizing factor, Stot(h), which is the total concentration of all scaffold forms with a certain h. With this normalization, the fractional concentrations s(a1, …, an; h)/Stot(h) and Si(ai, h)/Stot(h) present the conditional probabilities of finding the scaffold in that particular micro or macrostate, under the condition when it is either associated with (h = 1) or dissociated from the receptor (h = 0). Equation 9 shows that when the association and dissociation reactions are fast, all microstates (i.e., all network species) can be approximated by the product of the conditional probabilities (alternatively normalized fractional concentrations) of macrostates. However, even for this alternative scaling of concentrations, Eq. 9 cannot be considered an exact relationship for all feasible values of the kinetic constants (see Supplemental Note 6 in Supplementary Materials).
In the other extreme case, when binding/association of the receptor and scaffold is much slower than processes that change states ai of docking sites, Eq. 9 continues to apply. In fact, because changes in h are very slow, one may consider the pseudo-equilibrium for the microstates of the scaffold bound to the receptor, h = 1 (in Fig. 2, these microstates are at the front facet of the graph), separately from the pseudo-equilibrium for the free scaffold forms, h = 0 (the rear facet of the graph in Fig. 2). Since at constant h, transitions between states of any docking site do not depend on other sites, the equilibrium values of s(a1, …, an; h)/Stot(h) equal the product of Si(ai, h)/Stot(h) for the same h. Because of the rapid-equilibrium condition, the exact values of microstate concentrations will not differ significantly from the equilibrium concentrations at all times.
Numerical experiments demonstrate that over a wide range of parameters the concentrations of network species (microstates of the scaffold) are well approximated by Eq. 9. Fig. 5 illustrates this by covering three cases, where the apparent forward (krR) and reverse (k−r) rate constants of binding of the scaffold to the receptor are much greater, in the same range, or much smaller than the rate constants of reactions involving scaffold docking sites. The active receptor concentration at t = 0 was set to be 100 nM (a typical range for EGFR activated with saturating [EGF] in liver cells; see Ref. 37), and the total abundance of the scaffold, and the scaffold's binding partners A1 and A2 (see Fig. 2), was set to be 50 nM. To simulate a transient temporal pattern of receptor activation, the concentration of active free receptor was assumed to decrease with the first-order rate constant of 0.01 s−1. The precise concentrations of all micro- and macrostates were calculated according to Eqs. 1 and 4. The approximate values of microstate concentrations were obtained as the product of macrostate concentrations according to Eq. 9 (Fig. 5 illustrates the exact and approximate solutions for microstates s(2,2;1) and s(2,2;0) that correspond to the receptor-bound and unbound scaffold with docking sites occupied by their partners A1 and A2).
FIGURE 5.

Time-course of receptor-bound and unbound scaffold forms with docking sites occupied by their partners A1 and A2. A–C illustrate three cases, where the scaffold-receptor association/dissociation reactions are much faster, comparable, or slower than the reactions involving scaffold docking sites. The left and right panels (marked by numbers 1 and 2) present the short and extended time windows. Exact concentrations are calculated according to Eq. 1 (microdescription) and marked with ○ and □ for scaffold forms bound and unbound to the receptor, i.e., s(2,2;1) and s(2,2;0), respectively. The approximate concentrations of microstates are obtained by solving Eq. 4 using Eq. 9 and marked with + and × for forms bound and unbound to the receptor, respectively. The rate constants are the following (see Fig. 2): kr = 0.05 nM−1 · s−1, 5 · 10−3 nM−1 · s−1 and 5 · 10−4 nM−1 · s−1 and k−r = 5 s−1, 0.5 s−1, and 0.05 s−1 for A–C, respectively (Kd = 100 nM for all cases); k1(0→1;1) = 0.2 s−1, k1(0→1;0) = 0, k1(1→0;1) = k1(1→0;0) = 0.8 s−1, k1(1→2;1) = k1(1→2;0) = 0.02 nM−1 · s−1, k1(2→1;1) = k1(2→1;0) = 0.8 s−1; k2(0→1;1) = 0.8 s−1, k2(0→1;0) = 0, k2(1→0;1) = k2(1→0;0) = 0.2 s−1, k2(1→2;1) = k2(1→2;0) = 0.02 nM−1 · s−1, k2(2→1;1) = k2(2→1;0) = 0.2 s−1; R = R0 · exp(−kdeac · t), R0 = 100 nM, kdeac = 0.01 s−1; Stot = (A1)tot = (A2)tot = 50 nM. The initial conditions for Eq. 1 and 4 were set as follows: s(0,0;0) = 50 nM, s(0,0;1) = 1 · 10−10 nM, whereas all other s(a1,a2;h) = 0; S1(0,0) = S2(0,0) = 50 nM, S1(0,1) = S2(0,1) = 1 · 10−10 nM, and all other Si(ai,h) = 0. The freely available Jarnac software package was used for simulations (56).
The time courses for precise and approximate concentrations of scaffold microstates shows that the best approximation corresponds to the fast receptor-scaffold association/dissociation reactions (Fig. 5, A1 and A2). If krR and k−r have the same magnitude as the other rate constants in Eq. 1, the deviation of the exact solution from Eq. 9 increases, but remains reasonably small (Fig. 5, B1 and B2). When the association/dissociation of the receptor and scaffold is much slower, microstates that correspond to phosphorylated forms can be approximated well only in the case where the scaffold is bound to the receptor (Fig. 5, C1 and C2). Indeed, for the free scaffold the pseudo-equilibrium solution for phosphorylated forms is equal to zero, since only the phosphatase reactions carry on when the scaffold has dissociated from the receptor.
Macrodescription of receptor possessing scaffolding properties
If ligand binding and dissociation are independent of the conformation and phosphorylation states of the cytoplasmic receptor tail including its independent docking sites, the macrodescription developed here for scaffold signaling is applicable to this receptor. As shown in Methods, all that is needed is the replacement of the word receptor with ligand, and scaffold with receptor in that description (for simplicity, it is assumed that the receptor is a dimer before stimulation with the ligand, such as IR or IGF-1R).
In an alternative model, the dissociation of the ligand from the receptor is possible only when all docking sites of the receptor are unphosphorylated (i.e., in microstate r(0,…,0), Fig. 3). Adding up the concentrations of all receptor forms that display a particular state (ai) of docking site i, we introduce the macrovariables Ri(ai),
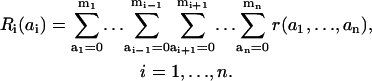 |
(10) |
These macrovariables, also referred to as the receptor macrostates, are merely the concentrations of receptor docking sites in particular states for the receptor that has bound the ligand.
Unfortunately, Eq. 2 cannot be transformed into a system of differential equations that include the macrostates Ri only. In fact, the rate of dissociation of the ligand from the receptor equals k−0 · r(0,…,0), whereas any of the variables Ri(0) is the sum of microstate r(0,…,0) and various other microstates r(a1,…,ai−1,0,ai+1,…,an). Summing up Eq. 2 for all states of docking sites j ≠ i and docking site i in state ai, we obtain the equation system that explicitly involves the microstate r(0,…,0):
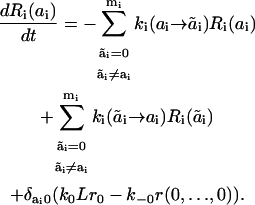 |
(11) |
The above analysis of signaling by a scaffold suggests that microstate concentrations can often be well approximated in terms of scaled macrostate concentrations. To scale the concentrations, we introduce the total concentration (RL) of the receptor-ligand complexes:
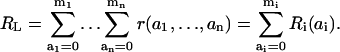 |
(12) |
Normalizing by RL, we obtain the fractional concentrations (conditional probabilities) of micro- and macrostates of the receptor that have bound the ligand L, r(a1,…,an)/RL and Ri(ai)/RL. Suppose the microstate concentration r(0,…,0) can be approximated by the product of the scaled macrostate concentrations Ri(0), as
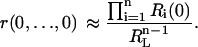 |
(13) |
Substituting this estimate in Eq. 11, we arrive at the following approximate description of receptor kinetics in terms of macrostates (the superscripted asterisk indicates the approximate concentrations),
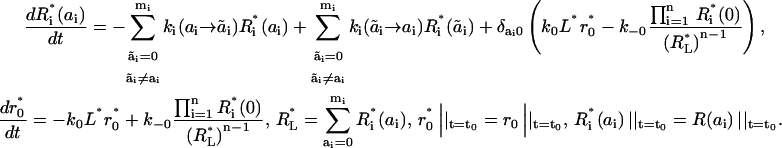 |
(14) |
Solving Eq. 14, we can approximate the microstate concentrations by the product of scaled macrostate concentrations (Supplemental Note 7 in Supplementary Materials shows that this relationship is not exact),
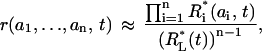 |
(15) |
The remaining central question is what the difference (ϕ) between exact and approximate microstate concentrations amounts to:
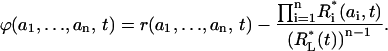 |
(16) |
Before the stimulation, at the initial moment t = 0, both r(a1,…,an) and Ri* (ai) equal zero, and therefore ϕ = 0. Remarkably, it appears that at t→∞, when all variables are approaching their steady-state values, the difference ϕ again tends to zero. The steady-state concentrations of micro- and macrostates of the receptor are obtained from Eqs. 2 and 14, respectively, by equating the time derivatives to zero. At steady state, the terms multiplied by the Kronecker symbols become zero, and equations for r(a1,…,an) and Ri* (ai) become identical to the steady-state equations for micro- and macrostates s(a1,…,an) and Si(ai) of a scaffold without controlling site h (these equations are derived from Eqs. 1 and 4, respectively, by equating kr, k−r, and the time derivatives to zero). Since we proved that for a scaffold with totally independent sites, the steady-state microstate concentrations are equal to the products of the scaled macrostate concentrations (see Eq. 7), the same is true for the steady-state concentrations of micro- and macrostates of the receptor, and, therefore, ϕ→0 when t→∞. To understand this result in intuitive terms, note that the graph, which describes transitions between different receptor forms (Fig. 3), is almost identical to the graph for a scaffold lacking controlling site h (see the front facet of Fig. 2). The only difference between the two graphs is the dead-end edge attached to the vertex (0,0) and describing receptor-ligand association/dissociation in Fig. 3. At the steady state, the flux along this edge equals zero, and the relationships between the concentrations of micro- and macrostates become identical for both graphs. Note that results similar to Eqs. 10–16 continue to apply for a receptor model that includes the dimerization of monomers upon ligand binding (Supplemental Note 8 in Supplementary Materials).
Numerical experiments demonstrate that, within a wide parameter range, the difference ϕ between the exact microstate concentrations and their approximation in terms of the products of scaled macrostate concentrations remains small for receptor signaling. Fig. 6 illustrates this for a variety of cases where receptor-ligand interactions are fast and have either low or high affinity (Fig. 6, A and B), slower (Fig. 6 C), or comparable affinity (Fig. 6 D) with the rates of the chemical reactions at receptor docking sites. In addition, the total ligand concentration is assumed to be either conserved (Fig. 6, A–C) or free ligand concentration is considered as an external variable that exponentially decreases with time after the initial burst (Fig. 6 D). The precise values of microstate concentrations were calculated according to Eq. 2 (marked by open circles in Fig. 6), and the approximate values were calculated according to Eqs. 14 and 15 (marked by plus symbols). Note that Fig. 6 D shows the temporal dynamics on the timescale of hours, because the ligand degradation is a slow process, whereas all other panels depict the time courses on the scale of seconds. In all cases considered, the approximations for the microstate r(2,2) that corresponds to the receptor form bound to both adaptor proteins A1 and A2 (Fig. 3), were very close to the exact solutions, although the precision of the approximation could depend on the affinity of the ligand for the receptor (compare Fig. 6, A and B).
FIGURE 6.

Time-course of the receptor forms with docking sites occupied by adaptor proteins A1 and A2. A and B illustrate cases of fast receptor-ligand binding/dissociation with low and high affinity, respectively. C corresponds to slow receptor-ligand binding/dissociation with low affinity. The total ligand concentration (Ltot) is assumed constant (A–C). D illustrates the case of an exponential decrease of the free ligand concentration (L). Exact values of the microstate concentration r(2,2) are calculated according to Eq. 2 (microdescription) and marked with ○. Approximate values are determined by solving Eq. 14 and using Eq. 15 and marked with +. The rate constants for the model are the following: k0 = 0.05 nM−1 · s−1, 1.667 nM−1 · s−1, 5 · 10−4 nM−1 · s−1, and 5 · 10−3 nM−1 · s−1 for A–D, respectively; k−0 = 5 s−1, 5 s−1, 0.05 s−1, and 0.5 s−1 for A–D, respectively; Kd = 100 nM for A–D and 3 nM for B; k1(0→1;1) = 0.2 s−1, k1(0→1;0) = 0, k1(1→0;1) = k1(1→0;0) = 0.8 s−1, k1(1→2;1) = k1(1→2;0) = 0.02 nM−1 · s−1, k1(2→1;1) = k1(2→1;0) = 0.8 s−1; k2(0→1;1) = 0.8 s−1, k2(0→1;0) = 0, k2(1→0;1) = k2(1→0;0) = 0.2 s−1, k2(1→2;1) = k2(1→2;0) = 0.02 nM−1 · s−1, k2(2→1;1) = k2(2→1;0) = 0.2 s−1; total abundances Rtot = 100 nM; (A1)tot = (A2)tot = 50 nM; Ltot = 100 nM for A–C, L = L0 · exp(−kdeg · t), L0 = 100 nM, kdeg = 1 · 10−3 s−1 for D. The initial conditions for Eqs. 2 and 18 were set as follows: r0 = 100 nM, r(0,0) = 1 · 10−10 nM, whereas all other r(a1,a2) = 0; 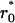 = 100 nM,
= 100 nM, 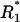 (0) =
(0) = 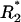 (0) = 1 · 10−10 nM, and all other
(0) = 1 · 10−10 nM, and all other 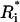 (ai) = 0.
(ai) = 0.
Interestingly, for the general case of scaffold signaling, the difference between the exact solution to Eq. 1 and its approximation (obtained from Eqs. 4 and 9) does not tend to zero at t→∞. In fact, there are uncompensated fluxes between vertices at the front and rear facets, since phosphorylation reactions occur only at the front facet, when a scaffold is bound to a receptor (Fig. 2). This observation explains why, in the case of signaling by a receptor described by Eq. 2, the quality of the approximation of microstates is generally better than in the case of a scaffold described by Eq. 1 (compare Figs. 5 and 6).
General principles of reducing combinatorial complexity: which kinetic properties are critical?
Table 1 summarizes the feasibility of applying a macrodescription to different classes of signaling modules considered in this article. These results indicate which conditions need to be satisfied for the reduction of a combinatorial, mechanistic description to a macromodel of a signaling system. A key prerequisite is that a signaling protein contains domains/sites that do not influence other sites, allosterically or through interactions with bound partners. We refer to these sites as a-type docking sites; their states are characterized by a set of numbers, a = (a1, …, an). If, in addition, a-type sites do not depend on the state of any other domain on a receptor or scaffold, the dynamics of these sites can be modeled separately without requiring any information on the states of other sites. Obviously, this is the simplest protein system, which we refer to as a scaffolding protein with kinetically independent docking sites and with no hierarchy in the control of these sites (Table 1). In the general case, the chemical transformations of docking sites may depend on the states of a second group of sites, called controlling sites. We refer to these controlling domains (regions) as h-type sites that may influence the transitions between the states of all other sites on a signaling molecule. Two examples of such signaling systems include: 1), a receptor-scaffold module, where the transformation of scaffold docking sites depends on whether the scaffold is bound to, or dissociated from the receptor (characterized by the digital flag of the h-site); and 2), a receptor that acts as a scaffold having independent docking sites, the transformations of which depend on whether the ligand is bound to the receptor (Table 1). Importantly, the controlling hierarchy does not allow for the reverse interaction, in which a-sites would influence the transformations of h-sites (for instance, ligand-receptor interactions are assumed to be independent of the states of receptor docking sites in the example above; see Table 1, fifth row).
TABLE 1.
Macrodescription of signaling modules
| Estimation of microstates in terms of macrostates
|
||||
|---|---|---|---|---|
| System properties | Definition of macrostates | Is macrostate description exact? | During the time- evolution of the system | At the steady-states |
| A scaffold with kinetically independent docking sites. No controlling hierarchy, no allosteric interactions or interactions through bound partners. | A macrostate is the sum of all microstates having a particular state of a docking site. | Yes. | Exact. | Exact. |
| A scaffold-receptor module. Docking sites on the scaffold do not interact allosterically or through bound partners. The state of a controlling site (h) on the scaffold influences the chemical transformations of docking sites. | A macrostate is the sum of all microstates having a particular state of a docking site and a certain state h. | Yes. | Approximate. The accuracy of the approximation is higher if the changes in h-states occur much faster than the transformations of docking sites. | Approximate. The accuracy of the approximation is higher if the changes in h-states occur much faster or much slower than the transformations of docking sites. |
| A receptor acting as a scaffold with independent docking sites. Ligand-receptor interactions are independent of the states of receptor docking sites. The state of a controlling site (h) is determined by ligand binding and affects the docking sites. | A macrostate is the sum of all microstates having a particular state of a docking site and a certain state h. | Yes. | Approximate. The accuracy of the approximation is higher if the changes in h-states occur much faster than the transformations of docking sites. | Approximate. The accuracy of the approximation is higher if the changes in h-states occur much faster or much slower than the transformations of docking sites. |
| A receptor that acts as a scaffold with independent sites. No controlling hierarchy (no h-sites), but ligand dissociation occurs only if all docking sites on the receptor are unphosphorylated. | A macrostate is the sum of all microstates of the receptor with bound ligand and a particular state of a docking site. | No. Only an approximate macro-description. | Approximate, usually with high accuracy. | Exact. |
| Each site influences any other site, either directly or indirectly (via a bound partner). | Macrodescription is not applicable. | |||
Although so far we explicitly considered proteins with a single controlling domain, the same principles apply to systems with multiple controlling sites. General rules for developing macromodels of systems containing proteins with these two types of domains, a-type and h-type, are given by Eqs. 3 and 4, where the variable h is considered the vector-variable, h = (h1,…, hr). An example of a system with two controlling sites is a scaffold that displays a receptor-binding site h1, a different phosphatase-binding site h2, and a number of docking sites i that do not interact with each other. One or more of these docking sites can be phosphorylated or dephosphorylated while the scaffold is associated with the receptor (h1 = 1) and/or with the phosphatase (h2 = 1), respectively. The microstates of a scaffold protein are determined as s(a1,…, an; h1, h2). The rate constants for transitions between states (ai) of docking sites i may depend on both h1 and h2; e.g., dephosphorylation occurs only if h2 = 1. The differential equations for microstate concentrations are similar to Eq. 1, but include an extra term that describes the association/dissociation of the scaffold and phosphatase. A macromodel of this receptor-scaffold-phosphatase signaling module is constructed as follows. The macrovariables Si(ai,h1,h2) depend on the states of controlling sites h1 and h2 and are defined by Eq. 3, where h = (h1, h2). The differential equations for macrostate concentrations are given by Eq. 4 with an extra term that accounts for the scaffold-phosphatase interaction. Remarkably, not only the macromodel, but also the approximation of the microstate concentrations by the product of the scaled macrostate concentrations continues to apply. The latter is given by Eq. 9, where the normalizing factor Stot(h) should be determined for each of four combinations of the states of controlling sites (h1, h2). If the association/dissociation of the scaffold with the receptor and the phosphatase is much faster than the changes in the states of docking sites, the normalizing factors and scaffold forms that differ only by states h1, h2 (i.e., bound to or unbound from the receptor and the phosphatase) are related by rapid-equilibrium relationships, and the accuracy of the approximation is very high. For arbitrary rate constants, numerical experiments suggest that the accuracy is generally good (the approximation errors are similar to those illustrated in Fig. 5 for similar values of kinetic parameters; data not shown).
Importantly, model reduction is still possible even if some of docking sites on a scaffold interact allosterically or through their bound proteins. We refer to these sites as b-type docking sites. We assume that the states (bj) of b-type site j can influence the transformations between the states (bl) of any b-type sites, but do not influence the transformations of a-type docking sites and controlling h-sites. For instance, the tyrosine phosphatase SHP2 that binds to GAB-1 was shown to dephosphorylate specific p85 binding sites, negatively regulating EGF-dependent PI3K activation without significant effects on binding of other partners to GAB-1 (44). Likewise, SHP2 that binds to the EGFR-GAB1 complex specifically dephosphorylates Tyr992 on EGFR, whereas the dephosphorylation of other docking sites is not significantly changed (45). Tyr992 is a specific binding site for the GTPase activating protein RasGAP; the dephosphorylation of this site increases Ras-GTP levels and thereby positively influences the MAPK activation. A general approach to circumvent the combinatorial explosion of the number of microstates, s(a1,…, an; b1,…, bq; h1, …, hr), for signaling proteins with these three types of sites (independent a-type and allosterically interacting b-type docking sites and controlling h-domains) is the following. We introduce a mesoscopic description with two sets of macrovariables. One set, S(ai, h), is analogous to the macrovariables considered above (Eq. 3), and is determined by summing up the microconcentrations of all the states (b) of allosterically interacting sites and all a-type docking sites except site i. These variables merely follow the states of each independent docking site at certain states of controlling h-sites. The second set of variables S(b, h) is determined by the sum of microconcentrations of all the states (a) of independent docking sites. These variables characterize the states of all allosterically interacting sites simultaneously at certain states of controlling h-sites. The differential equations obtained by the summation of the corresponding equations for a mechanistic description constitute a macromodel of a signaling module. The conditions necessary for the applicability of a macromodel, including the possible influences and dependences between different types of sites and the requirements for the rate constants of the corresponding transformations are shown in Table 2.
TABLE 2.
Types of sites and imposed conditions that allow for a macromodel reduction
| Sites | Influence: | Depend on: | Transformation | Rate constants may depend on: |
|---|---|---|---|---|
| Controlling (h-sites). | All types of sites. | Other controlling sites. | 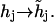 |
The states (h) of controlling sites. |
| Independent docking sites (a-sites). | No effect on other sites. | Controlling sites. | 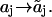 |
The states aj and 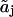 and the states (h) of controlling sites. and the states (h) of controlling sites. |
| Interacting allosterically or via bound partners (b-sites). | Other allosterically interacting sites only. | Controlling and other allosterically interacting sites. | 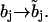 |
All states (b) of allosterically interacting sites and the states (h) of controlling sites. |
Experimental verification of a macrodescription
The importance of a macrodescription goes beyond the reduction of a mechanistic model of signal transduction, providing a direct connection to experimentally observable variables. In fact, macrovariables are quantified in experimental studies by Western blot analysis using site-specific antibodies, whereas microstates of a scaffold protein cannot be assessed readily at the present state-of-the-art. When the structural and kinetic information suggests that allosteric interactions are absent (at least for some protein domains), a macrostate model can be developed. Such a macro (or mesoscopic) description of a signaling system has the advantage of providing a direct test of the model against the experiment using current techniques to quantify posttranslational protein modifications (46,47).
It is instructive to ask how we can proceed from standard cell biological experiments determining the functional states of signaling molecules using Western blots to assess if a macropresentation of a system is applicable. Answering this question requires a modification to the commonly used experimental designs. An analysis is outlined here for a simple example of a scaffold (S) that has two docking sites. Suppose proteins A and B were detected by Western blot in immunoprecipitates of total cell lysates obtained with an antibody against S. These data do not show 1), whether proteins A and B can bind simultaneously to the same molecule of S; and 2), if they do, whether docking sites for A and B can be considered independent, or whether allosteric interactions occur that would impede a macrodescription of this system. These issues can be addressed as follows. First, separate immunoprecipitates of S, B, and A are prepared at several time-points after cell stimulation. Using an antibody against A, we quantify the intensity of the A-band in the immunoprecipitates of S and in the total cell lysates on the same membrane, thereby determining the relative amount, AS/Atot = r1. Assuming that A binds B only through the ternary complex ASB and quantifying the A-band in the immunoprecipitates of B and in the total cell lysates on the same membrane, we determine the relative amount, ASB/Atot = r2. Likewise, using an antibody against B for a detection and the immunoprecipitates of S and A, we determine SB/Btot = r3 and ASB/Btot = r4. Finally, using an antibody against S for detection and the immunoprecipitates of A and B, we find AS/Stot = r5 and SB/Stot = r6. Using these data, we can determine the relative abundance of proteins Atot/Btot/Stot, and relative amount of ASB/Stot = r2 · r5/r1 = r4 · r6/r3. This equation shows that there is the relationship between these data (r2 · r3 · r5 = r1 · r4 · r6), and only five measurements are needed to determine any unknown fraction at any given time. However, the additional measurement is useful for the statistical estimates and tests for the consistency of data (including the possible presence of other complexes containing A, B, and S).
Now recall that the relative amounts quantified in these experiments are the macrovariables, S(A)/Stot = r5 and S(B)/Stot = r6, and the microvariable S(A,B)/Stot = r2 · r5/r1 (for the states of the scaffold S, mnemonic notations are used, which are readily comparable to the notation utilized in Eq. 7). If the data show that S(A,B)/Stot is not significantly different from the product of S(A)/Stot and S(B)/Stot (i.e., r5 = r4/r3, or equivalently, r6 = r2/r1) there is no allosteric interaction between docking sites for proteins A and B (see Eq. 7). Hence, a macromodel of this system can be constructed where the temporal dynamics of each docking site is determined separately. A substantial deviation of the microvariable from the product of the macrovariables tells us that there is a positive or negative cooperativity between two docking sites. If r5 ≪ r4/r3, there is positive cooperativity (binding a partner to one docking site facilitates binding to the other site), and if the opposite inequality holds, negative cooperativity is predicted. In such cases, it is necessary to monitor the states of two docking sites simultaneously in the framework of a mechanistic model.
DISCUSSION
Many cellular proteins participate in the transfer and processing of information, rather than catalyzing the chemical transformation of metabolic intermediates or building cellular structures. Information is received in the form of growth factors, hormones, and other environmental signals that stimulate plasma membrane receptors. After stimulation, activated receptors and large adaptor proteins often act as scaffolds that assemble multiprotein complexes on their docking sites (Fig. 1). For instance, activated RTKs, such as growth factor receptors and IR, and scaffolding/adaptor proteins, such as GAB 1/2, IRS 1–4, and Grb10, gather a variety of complexes with downstream partners, initiating diverse signaling branches and pathways (2,3,7,48,49). The multiplicity of docking sites, phosphorylation, dephosphorylation, subsequent binding and dissociation, and the resulting random assemblage of a battery of protein complexes leads to a combinatorial increase in the number of feasible molecular species that present different states of a receptor/scaffold signaling system.
In this article, we show a way to circumvent this combinatorial complexity so as to facilitate modeling of signaling networks. Using a domain-oriented analysis, we dissect the dynamics of a highly multidimensional system into separate trajectories of individual docking sites on receptors and scaffolds. Our analysis effectively separates a highly branched signaling network into interacting pathways originated by protein complexes assembled on different docking sites, dramatically reducing the number of states and differential equations to be considered. Closely related approaches were successfully applied to model the kinetics of bivalent ligand-receptor interactions that lead to the formation of multireceptor aggregates (50,51).
We analyzed molecular scenarios that are commonly present in RTK signaling networks: a scaffold activated by a receptor or soluble kinases, and a receptor that can bind several target proteins simultaneously, thus displaying scaffolding properties. In the simplest model of a scaffold, the chemical transformations of any docking site, including its phosphorylation and dephosphorylation, are assumed to be completely independent of the states of other sites. In a more complex case, the scaffold-receptor association keeps a tight rein on otherwise independent docking sites, since their phosphorylation occurs only when the scaffold is bound to the receptor. To account for this effect, we set the notation of a controlling site (h) on the scaffold. When two or more different docking sites are phosphorylated while the scaffold is bound to the receptor (h = 1), the reaction mechanism is called processive. A model of completely independent sites assumes that docking sites are phosphorylated and dephosphorylated in a distributive mechanism, where phosphorylation (dephosphorylation) of more than one site requires a new binding interaction between soluble kinases (phosphatases) and their target proteins (52,53). Fig. 2 shows that in the model of a receptor-scaffold signaling module analyzed here, scaffold phosphorylation includes elements of both processive and distributive mechanisms. We demonstrated that regardless of the mechanism of phosphorylation, signaling by a scaffold and its binding partners can be analyzed in terms of macrostates of the system (Eqs. 3 and 4). The macrostates Si(ai, h) represent separate states (ai) of distinct docking sites i on a scaffold protein (S) and may depend on h for a receptor-scaffold signaling module. Even for two docking sites, a macrodescription brings about a considerable reduction in the number of states and transitions, as illustrated by Figs. 4 and 2.
Using a macrodescription, the dynamics of microstates can be retrieved for a scaffold protein with totally independent sites. In this case, the microstate concentrations are exactly expressed as the product of fractional concentrations of macrostates (Eq. 7). For a receptor-scaffold signaling module, i.e., for a scaffold with a controlling site, a similar relationship (Eq. 9) that expresses the microstate dynamics is merely an approximation. Furthermore, the macrostate concentrations are no longer normalized by the total concentration of the scaffold protein. Depending on whether a particular macrostate corresponds to the form associated with or dissociated from the receptor, the normalizing factor is the total concentration of receptor-bound or receptor-unbound scaffold forms (Eq. 9). Both these total concentrations can be measured in co-immunoprecipitation experiments (37,46). We showed that the approximation of the dynamics of microstates in terms of the product of the scaled macrostate concentrations holds well within a wide range of kinetic parameters, as illustrated in Fig. 5. These results imply that by using antibodies that specifically recognize phosphorylation of individual residues/sites on a scaffold protein, we can predict the temporal dynamics of any given microstate, e.g., the state where all docking sites are phosphorylated and occupied by their binding partners, in terms of the product of scaled concentrations of macrostates.
We analyzed two models of a receptor that possesses scaffolding proteins: 1), a model where the ligand affinity does not depend on receptor dimerization and phosphorylation state; and 2), a model where the affinity of the ligand drastically increases after the receptor undergoes dimerization, activating conformational change, or phosphorylation. In the extreme case, the dissociation of ligand is possible only from the unphosphorylated receptor or receptor monomer (see kinetic schemes in Fig. 3 and see Supplemental Note 8, Fig. S8.1, in Supplementary Materials). Whereas the first model is mathematically equivalent to a scaffold-receptor signaling module, where both mechanistic and macrodescription can be applied, the second model does not allow a precise quantitation in terms of only macrovariables. In fact, in contrast to signaling by a scaffold protein, the differential equations for the macrovariables (Eq. 11) include a separate microstate that corresponds to the active, but unphosphorylated receptor. This unique microstate serves as the input into a transition graph (Fig. 3) for receptor docking sites, and cannot be eliminated from Eq. 11 by any linear transformation of variables. We derived a nonlinear approximation for the concentrations of macrostates (Eq. 14) and showed how the microstate concentrations are estimated (Eq. 15). Interestingly, the quality of the approximation of a mechanistic description by macrovariables appears to be even better for a receptor with independent docking sites than for a scaffold-receptor signaling system (Figs. 5 and 6). In fact, only for signaling by a receptor that possesses scaffolding properties does the difference ϕ between the exact microstate concentrations and their approximations in terms of the concentrations of macrostates tend to zero when the system approaches the steady state, whereas ϕ does not tend to zero for a scaffold-receptor signaling module.
The notion of multiple controlling sites is also useful for the analysis of more complex models of receptor activation. For instance, it is known that following insulin binding, IR undergoes autophosphorylation at three neighboring tyrosine residues in the activation loop of the intracellular kinase domain (54). Phosphorylation of tyrosine residues in the activation loop causes a 36-fold increase in the kinase activity of IR (54). Assuming that there are several independent docking sites on the receptor with the phosphorylation state of the activation loop determining the kinase activity, we can describe all microstates of this receptor by a set of numbers (a1, …, an; h1, h2), i.e., by a model with two controlling sites, where h1 = 1 or 0 indicates that the ligand is associated with or dissociated from the receptor, and h2 = 1 or 0 stands for the phosphorylated or unphosphorylated activation loop. Using our approach, the kinetic behavior of this system can be analyzed at the macrostate level and, in addition, the microstate concentrations can be approximated by the product of the scaled concentrations of macrostates, with generally good quality.
In this article, we applied our analysis to signaling networks described by deterministic differential equations. Signaling events that occur in small subcellular volumes involve small numbers of molecules. These signaling systems are intrinsically noisy and should be analyzed by stochastic methods (30,55). Importantly, our results appear to be equally applicable to exact stochastic calculations of the temporal dynamics of noisy systems. Indeed, the stochastic kinetic equation, referred to as the master equation, uniquely corresponds to ordinary differential equations, which are derived for the probabilities of numbers of molecules in certain molecular states. For instance, the reaction parameters of the master equation are equal to the first-order rate constants of ordinary differential equations for unimolecular reactions and, for bimolecular reactions, to the second-order rate constants divided by the reaction volume (30). The absence of allosteric interactions implies that the kinetic parameters of reactions that occur at one docking site are independent of the states of the other docking sites on a signaling protein. Under this condition, the probabilities of transformations of macrostates are the sums of the probabilities of transformations of microstates and are calculated in a Gillespie-type exact stochastic algorithm that is related to the master equation (30). Therefore, the implications of the independence of the reaction parameters for the summation of the microstates appear to be similar for deterministic and stochastic systems. Employing a macroscopic, domain-oriented approach significantly reduces the number of distinct states and reactions and the required calculation time even when the pathway kinetics is simulated according to exact stochastic methods. In conclusion, the methods proposed here make it possible to exploit reduced, tractable models for a quantitative description of complex multiprotein signaling networks.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
This work was supported by grants No. GM59570 and No. AA08714 from the National Institutes of Health.
References
- 1.Hunter, T. 2000. Signaling—2000 and beyond. Cell. 100:113–127. [DOI] [PubMed] [Google Scholar]
- 2.Schlessinger, J. 2000. Cell signaling by receptor tyrosine kinases. Cell. 103:211–225. [DOI] [PubMed] [Google Scholar]
- 3.Pawson, T., G. D. Gish, and P. Nash. 2001. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 11:504–511. [DOI] [PubMed] [Google Scholar]
- 4.White, M. F. 2002. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 283:E413–E422. [DOI] [PubMed] [Google Scholar]
- 5.Liu, P., P. Wang, P. Michaely, M. Zhu, and R. G. Anderson. 2000. Presence of oxidized cholesterol in caveolae uncouples active platelet-derived growth factor receptors from tyrosine kinase substrates. J. Biol. Chem. 275:31648–31654. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein, B., J. R. Faeder, and W. S. Hlavacek. 2004. Mathematical and computational models of immune-receptor signalling. Nat. Rev. Immunol. 4:445–456. [DOI] [PubMed] [Google Scholar]
- 7.Paz, K., H. Voliovitch, Y. R. Hadari, C. T. Roberts, Jr., D. LeRoith, and Y. Zick. 1996. Interaction between the insulin receptor and its downstream effectors. Use of individually expressed receptor domains for structure/function analysis. J. Biol. Chem. 271:6998–7003. [DOI] [PubMed] [Google Scholar]
- 8.Hammond, B. J., J. Tikerpae, and G. D. Smith. 1997. An evaluation of the cross-linking model for the interaction of insulin with its receptor. Am. J. Physiol. 272:E1136–E1144. [DOI] [PubMed] [Google Scholar]
- 9.Ottensmeyer, F. P., D. R. Beniac, R. Z. Luo, and C. C. Yip. 2000. Mechanism of transmembrane signaling: insulin binding and the insulin receptor. Biochemistry. 39:12103–12112. [DOI] [PubMed] [Google Scholar]
- 10.De Meyts, P., and J. Whittaker. 2002. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat. Rev. Drug Discov. 1:769–783. [DOI] [PubMed] [Google Scholar]
- 11.Harmer, N. J., D. Chirgadze, K. Hyun Kim, L. Pellegrini, and T. L. Blundell. 2003. The structural biology of growth factor receptor activation. Biophys. Chem. 100:545–553. [DOI] [PubMed] [Google Scholar]
- 12.Saltiel, A. R., and J. E. Pessin. 2002. Insulin signaling pathways in time and space. Trends Cell Biol. 12:65–71. [DOI] [PubMed] [Google Scholar]
- 13.Myers, M. G., Jr., Y. Zhang, G. A. Aldaz, T. Grammer, E. M. Glasheen, L. Yenush, L. M. Wang, X. J. Sun, J. Blenis, J. H. Pierce, and M. F. White. 1996. YMXM motifs and signaling by an insulin receptor substrate 1 molecule without tyrosine phosphorylation sites. Mol. Cell. Biol. 16:4147–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White, M. F. 1998. The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol. Cell. Biochem. 182:3–11. [PubMed] [Google Scholar]
- 15.Ingham, R. J., M. Holgado-Madruga, C. Siu, A. J. Wong, and M. R. Gold. 1998. The GAB1 protein is a docking site for multiple proteins involved in signaling by the B cell antigen receptor. J. Biol. Chem. 273:30630–30637. [DOI] [PubMed] [Google Scholar]
- 16.Lehr, S., J. Kotzka, A. Herkner, A. Sikmann, H. E. Meyer, W. Krone, and D. Muller-Wieland. 2000. Identification of major tyrosine phosphorylation sites in the human insulin receptor substrate GAB-1 by insulin receptor kinase in vitro. Biochemistry. 39:10898–10907. [DOI] [PubMed] [Google Scholar]
- 17.Rodrigues, G. A., M. Falasca, Z. Zhang, S. H. Ong, and J. Schlessinger. 2000. A novel positive feedback loop mediated by the docking protein GAB1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol. Cell. Biol. 20:1448–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong, M., C. Mounier, V. Dumas, and B. I. Posner. 2003. Epidermal growth factor-induced DNA synthesis. J. Biol. Chem. 278:5837–5844. [DOI] [PubMed] [Google Scholar]
- 19.Chan, P. C., Y. L. Chen, C. H. Cheng, K. C. Yu, L. A. Cary, K. H. Shu, W. L. Ho, and H. C. Chen. 2003. Src phosphorylates Grb2-associated binder 1 upon hepatocyte growth factor stimulation. J. Biol. Chem. 278:44075–44082. [DOI] [PubMed] [Google Scholar]
- 20.Nishida, K., and T. Hirano. 2003. The role of GAB family scaffolding adapter proteins in the signal transduction of cytokine and growth factor receptors. Cancer Sci. 94:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamasaki, S., K. Nishida, Y. Yoshida, M. Itoh, M. Hibi, and T. Hirano. 2003. GAB1 is required for EGF receptor signaling and the transformation by activated ErbB2. Oncogene. 22:1546–1556. [DOI] [PubMed] [Google Scholar]
- 22.Shepherd, P. R., D. J. Withers, and K. Siddle. 1998. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem. J. 333:471–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo, H. R., H. Hattori, M. A. Hossain, L. Hester, Y. Huang, W. Lee-Kwon, M. Donowitz, E. Nagata, and S. H. Snyder. 2003. AKT as a mediator of cell death. Proc. Natl. Acad. Sci. USA. 100:11712–11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faeder, J. R., W. S. Hlavacek, I. Reischl, M. L. Blinov, H. Metzger, A. Redondo, C. Wofsy, and B. Goldstein. 2003. Investigation of early events in Fc-ɛ RI-mediated signaling using a detailed mathematical model. J. Immunol. 170:3769–3781. [DOI] [PubMed] [Google Scholar]
- 25.Hlavacek, W. S., J. R. Faeder, M. L. Blinov, A. S. Perelson, and B. Goldstein. 2003. The complexity of complexes in signal transduction. Biotechnol. Bioeng. 84:783–794. [DOI] [PubMed] [Google Scholar]
- 26.Levchenko, A., J. Bruck, and P. W. Sternberg. 2000. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. USA. 97:5818–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ekman, S., A. Kallin, U. Engstrom, C. H. Heldin, and L. Ronnstrand. 2002. SHP-2 is involved in heterodimer specific loss of phosphorylation of Tyr771 in the PDGF β-receptor. Oncogene. 21:1870–1875. [DOI] [PubMed] [Google Scholar]
- 28.Markevich, N. I., J. B. Hoek, and B. N. Kholodenko. 2004. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J. Cell Biol. 164:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blinov, M. L., J. R. Faeder, B. Goldstein, and W. S. Hlavacek. 2004. BioNetGen: software for rule-based modeling of signal transduction based on the interactions of molecular domains. Bioinformatics. [DOI] [PubMed]
- 30.Gillespie, D. T. 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81:2340–2361. [Google Scholar]
- 31.Lok, L., and R. Brent. 2005. Automatic generation of cellular reaction networks with MOLECULIZER 1.0. Nat. Biotechnol. 23:131–136. [DOI] [PubMed] [Google Scholar]
- 32.Morton-Firth, C. J., and D. Bray. 1998. Predicting temporal fluctuations in an intracellular signalling pathway. J. Theor. Biol. 192:117–128. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu, T. S., N. Le Novere, M. D. Levin, A. J. Beavil, B. J. Sutton, and D. Bray. 2000. Molecular model of a lattice of signalling proteins involved in bacterial chemotaxis. Nat. Cell Biol. 2:792–796. [DOI] [PubMed] [Google Scholar]
- 34.Le Novere, N., and T. S. Shimizu. 2001. STOCHSIM: modelling of stochastic biomolecular processes. Bioinformatics. 17:575–576. [DOI] [PubMed] [Google Scholar]
- 35.Haugh, J. M., K. Schooler, A. Wells, H. S. Wiley, and D. A. Lauffenburger. 1999. Effect of epidermal growth factor receptor internalization on regulation of the phospholipase C-γ1 signaling pathway. J. Biol. Chem. 274:8958–8965. [DOI] [PubMed] [Google Scholar]
- 36.Haugh, J. M., A. C. Huang, H. S. Wiley, A. Wells, and D. A. Lauffenburger. 1999. Internalized epidermal growth factor receptors participate in the activation of p21(ras) in fibroblasts. J. Biol. Chem. 274:34350–34360. [DOI] [PubMed] [Google Scholar]
- 37.Kholodenko, B. N., O. V. Demin, G. Moehren, and J. B. Hoek. 1999. Quantification of short term signaling by the epidermal growth factor receptor. J. Biol. Chem. 274:30169–30181. [DOI] [PubMed] [Google Scholar]
- 38.Schoeberl, B., C. Eichler-Jonsson, E. D. Gilles, and G. Muller. 2002. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat. Biotechnol. 20:370–375. [DOI] [PubMed] [Google Scholar]
- 39.Hatakeyama, M., S. Kimura, T. Naka, T. Kawasaki, N. Yumoto, M. Ichikawa, J. H. Kim, K. Saito, M. Saeki, M. Shirouzu, S. Yokoyama, and A. Konagaya. 2003. A computational model on the modulation of mitogen-activated protein kinase (MAPK) and AKT pathways in heregulin-induced ErbB signalling. Biochem. J. 373:451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kholodenko, B. N., M. Cascante, and H. V. Westerhoff. 1995. Control theory of metabolic channelling. Mol. Cell. Biochem. 143:151–168. [DOI] [PubMed] [Google Scholar]
- 41.Cornish-Bowden, A. 1995. Fundamentals of Enzyme Kinetics. Portland Press, London, UK.
- 42.Heinrich, R., S. M. Rapoport, and T. A. Rapoport. 1977. Metabolic regulation and mathematical models. Prog. Biophys. Mol. Biol. 32:1–82. [PubMed] [Google Scholar]
- 43.Kholodenko, B. N., S. Schuster, J. Garcia, H. V. Westerhoff, and M. Cascante. 1998. Control analysis of metabolic systems involving quasi-equilibrium reactions. Biochim. Biophys. Acta. 1379:337–352. [DOI] [PubMed] [Google Scholar]
- 44.Zhang, S. Q., W. G. Tsiaras, T. Araki, G. Wen, L. Minichiello, R. Klein, and B. G. Neel. 2002. Receptor-specific regulation of phosphatidylinositol 3′-kinase activation by the protein tyrosine phosphatase SHP2. Mol. Cell. Biol. 22:4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agazie, Y. M., and M. J. Hayman. 2003. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell. Biol. 23:7875–7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moehren, G., N. Markevich, O. Demin, A. Kiyatkin, I. Goryanin, J. B. Hoek, and B. N. Kholodenko. 2002. Temperature dependence of the epidermal growth factor receptor signaling network can be accounted for by a kinetic model. Biochemistry. 41:306–320. [DOI] [PubMed] [Google Scholar]
- 47.Markevich, N. I., G. Moehren, O. Demin, A. Kiyatkin, J. B. Hoek, and B. N. Kholodenko. 2004. Signal processing at the Ras circuit: what shapes Ras activation patterns? IEE Sys. Biol. 1:104–113. [DOI] [PubMed] [Google Scholar]
- 48.Staubs, P. A., D. R. Reichart, A. R. Saltiel, K. L. Milarski, H. Maegawa, P. Berhanu, J. M. Olefsky, and B. L. Seely. 1994. Localization of the insulin receptor binding sites for the SH2 domain proteins p85, SYP, and GAP. J. Biol. Chem. 269:27186–27192. [PubMed] [Google Scholar]
- 49.Deng, Y., S. Bhattacharya, O. R. Swamy, R. Tandon, Y. Wang, R. Janda, and H. Riedel. 2003. Growth factor receptor-binding protein 10 (Grb10) as a partner of phosphatidylinositol 3-kinase in metabolic insulin action. J. Biol. Chem. 278:39311–39322. [DOI] [PubMed] [Google Scholar]
- 50.Perelson, A. S., and C. DeLisi. 1980. Receptor clustering on a cell surface. Math. Biosci. 48:71–110. [Google Scholar]
- 51.Posner, R. G., C. Wofsy, and B. Goldstein. 1995. The kinetics of bivalent ligand-bivalent receptor aggregation: ring formation and the breakdown of the equivalent site approximation. Math. Biosci. 126:171–190. [DOI] [PubMed] [Google Scholar]
- 52.Ferrell, J. E., Jr., and R. R. Bhatt. 1997. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J. Biol. Chem. 272:19008–19016. [DOI] [PubMed] [Google Scholar]
- 53.Zhao, Y., and Z. Y. Zhang. 2001. The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J. Biol. Chem. 276:32382–32391. [DOI] [PubMed] [Google Scholar]
- 54.Ablooglu, A. J., and R. A. Kohanski. 2001. Activation of the insulin receptor's kinase domain changes the rate-determining step of substrate phosphorylation. Biochemistry. 40:504–513. [DOI] [PubMed] [Google Scholar]
- 55.Rao, C. V., D. M. Wolf, and A. P. Arkin. 2002. Control, exploitation and tolerance of intracellular noise. Nature. 420:231–237. [DOI] [PubMed] [Google Scholar]
- 56.Sauro, H. M., M. Hucka, A. Finney, C. Wellock, H. Bolouri, J. Doyle, and H. Kitano. 2003. Next generation simulation tools: the Systems Biology Workbench and BioSPICE integration. OMICS. 7:355–372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.