

Abstract
X-ray reflectivity is used to study the interaction of C2 domains of cytosolic phospholipase A2 (cPLA2α-C2) with a Langmuir monolayer of 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC) supported on a buffered aqueous solution containing Ca2+. The reflectivity is analyzed in terms of the known crystallographic structure of cPLA2α-C2 domains and a slab model representing the lipid layer to yield an electron density profile of the lipid layer and bound C2 domains. This new method of analysis determines the angular orientation and penetration depth of the cPLA2α-C2 domains bound to the SOPC monolayer, information not available from the standard slab model analysis of x-ray reflectivity. The best-fit orientation places the protein-bound Ca2+ ions within 1 Å of the lipid phosphate group (with an accuracy of ±3 Å). Hydrophobic residues of the calcium-binding loops CBL1 and CBL3 penetrate deepest into the lipid layer, with a 2 Å penetration into the tailgroup region. X-ray measurements with and without the C2 domain indicate that there is a loss of electrons in the headgroup region of the lipid monolayer upon binding of the domains. We suggest that this is due to a loss of water molecules bound to the headgroup. Control experiments with a non-calcium buffer and with domain mutants confirm that the cPLA2α-C2 binding to the SOPC monolayer is Ca2+-dependent and that the hydrophobic residues in the calcium-binding loops are critical for membrane binding. These results indicate that an entropic component (due to water loss) as well as electrostatic and hydrophobic interactions contributes to the binding mechanism.
INTRODUCTION
It has been shown that a large number of cellular proteins reversibly translocated to cell membranes are involved in lipid-protein and protein-protein interactions. These peripheral membrane proteins play an important role in cell signaling and membrane trafficking. A large number of peripheral proteins contain one or more modular domains specialized in lipid binding. These lipid binding structural modules, also known as membrane-targeting domains, include protein kinase C (PKC) Conserved 1 (C1), PKC Conserved 2 (C2), Pleckstrin Homology (PH), Fab1, YOTB, Vac1, EEA1 (FYVE), Phox (PX), Epsin Amino-Terminal Homology (ENTH), AP180 Amino-Terminal Homology (ANTH), Bin Amphiphysin Rvs (BAR), Band 4.1, Ezrin, Radixin, Moesin (FERM), and tubby domains (1–12). The C2 domain is a Ca2+-dependent membrane-targeting domain that is present in many peripheral proteins involved in signal transduction and membrane trafficking (3,4). Structural analysis of multiple C2 domains have shown that all C2 domains share a common fold, with an eight-stranded antiparallel β-sandwich connected by variable loops (13–16). Prototype Ca2+-dependent C2 domains have multiple sites for Ca2+ ions that are composed of three Ca2+ binding loops (i.e., CBL1-3) (13–16).
Since the function and regulation of a majority of peripheral proteins depend on their interactions with membranes, extensive studies have been performed to determine the structural arrangement of peripheral proteins and membrane-targeting domains at the membrane, including their membrane-bound orientation and depth of membrane penetration. Unfortunately, high-resolution structures of bilayer-bound peripheral proteins have not been determined to date because of difficulties encountered in crystallizing peripheral proteins in the presence of lipid bilayers for x-ray diffraction studies. Other techniques, including fluorescence microscopy, electron paramagnetic resonance (EPR), and x-ray reflectivity, have been used to probe the orientation and depth of membrane penetration of these proteins (17–27). In particular, the EPR analysis of peripheral proteins with a site-specific spin probe has been used to gain structural insight into membrane-bound peripheral proteins and membrane-targeting domains, including the C2 domains of group IVA phospholipase A2 (cPLA2α), protein kinase Cα, and synaptotagmin as well as the intact MARCKS protein (19–21,28,29).
Although the EPR approach has been successfully applied to these proteins, the method requires chemical modification of the protein that may have a significant effect on its structure and membrane binding. Also, the precision of the EPR measurement greatly depends on the number of protein residues investigated by spin probe substitution, which necessitates extensive spin label incorporation. To circumvent the potentially perturbing effect of spin probe incorporation, an x-ray reflectivity analysis of an unmodified peripheral protein interacting with a lipid monolayer at the air-water interface has been developed (22–27). This technique determines a one-dimensional electron density profile along the surface normal. This electron density profile describes the lipid monolayer and the bound proteins. In the present study, we introduce the use of protein crystallographic information into the analysis of x-ray reflectivity to obtain high resolution structural information on C2 domains of cPLA2α (cPLA2α-C2) bound to a lipid monolayer of SOPC. This new method allows for determination of the angular orientation and depth of penetration of the protein bound to the lipid monolayer, as well as changes in the electron density of the lipid layer upon protein binding. This structural information provides important new clues to the membrane-binding mechanism of the domain, including the close proximity of bound Ca2+ to the lipid phosphates, the deep membrane penetration of hydrophobic residues in the Ca2+ binding loops, and the loss of lipid-bound water molecules upon protein binding. We also demonstrate that x-ray reflectivity can be used to study the interaction of proteins with monolayers of unsaturated lipids that resemble the biological membrane better than those of saturated lipids, but have been seldom used in x-ray reflectivity measurements because of their fragility (22).
MATERIALS AND METHODS
Materials
KCl, CaCl2, and HEPES from Fisher Scientific (Hampton, NH) and EGTA from Sigma (St. Louis, MO) were used as obtained. The stock solution of 1-Stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC) and 1-Palmitoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphocholine in chloroform were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further purification. Spreading solution was made by diluting the stock solution with chloroform from freshly opened bottles (Sigma). The expression and purification of the C2 domains of cPLA2α-C2 and mutants were performed as described previously (30).
Pressure, radioactive labeling, and fluorescence microscopy measurements
The change in surface pressure (π) upon protein adsorption onto the lipid monolayer was measured at constant surface area using a 10-ml circular Teflon trough and Wilhelmy plate connected to a Cahn microbalance, as described previously (30). A lipid monolayer containing SOPC was spread onto the subphase (0.1 mM KCl, 0.1 mM CaCl2, and 20 mM HEPES buffer, pH 7.0) until the desired initial surface pressure (π0) was reached. After the signal stabilized (≈30 min), cPLA2α-C2 was injected into the subphase, and the surface pressure (π) was monitored for 45 min while stirring the subphase at 60 rpm with a Teflon stir bar. Typically, π stabilized after 30 min. The maximal change in surface pressure Δπ (Δπ = π – π0) value depended on the protein concentration and reached a saturation value for a protein concentration of 6 μg/ml. Protein concentration in the subphase was maintained above this value to ensure that the observed Δπ represented a maximal value. The uncertainty on the Δπ measurement was ±0.5 mN/m.
The amount of monolayer-adsorbed protein at a given π0 was measured using radio-labeled cPLA2α-C2 domains and a hydrophobic Whatman 1PS phase-separation filter paper (Whatman, Mainstone, Kent, United Kingdom), as described previously (31,32).
Fluorescence microscopy was used to verify the absence of domains in both the SOPC monolayer and the SOPC/cPLA2α-C2 system. Using 1 mol % fluorescent dye (1-Palmitoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphocholine) in SOPC solution, no domain formation was detected before or after cPLA2α-C2 injection into the subphase.
X-ray reflectivity measurements
In this section we describe the conditions required to measure x-ray reflectivity from a Langmuir monolayer of SOPC with negligible x-ray damage for a period of up to 8 h. This is a long enough time to measure reflectivity from the SOPC, inject cPLA2α-C2 domains into the subphase, allow the domains to bind to the SOPC monolayer, and remeasure the reflectivity to determine the structural arrangement of the domains bound to the SOPC monolayer.
X-ray reflectivity experiments were conducted at beamline X19C at the National Synchrotron Light Source (Brookhaven National Laboratory, Upton, NY) with a liquid surface spectrometer described in detail elsewhere (33). Reflectivity is measured as a function of the wave vector transfer, Qz, by varying the incident angle, α (measured from the plane of the buffer surface), and measuring the intensity of scattered x rays at the reflected angle α (see Fig. 1). The wave vector transfer of the reflected x rays, 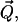 is solely in the z-direction normal to the buffer surface with its z component given by Qz = (4π/λ) sin(α), where λ = 1.54 ± 0.003 Å is the x-ray wavelength used in these measurements. Therefore, reflectivity probes variations in structure as a function of depth into the surface.
is solely in the z-direction normal to the buffer surface with its z component given by Qz = (4π/λ) sin(α), where λ = 1.54 ± 0.003 Å is the x-ray wavelength used in these measurements. Therefore, reflectivity probes variations in structure as a function of depth into the surface.
FIGURE 1.

Schematic diagram of the experimental setup at x-ray beamline X19C, National Synchrotron Light Source (Brookhaven National Laboratory): (A) focusing mirror, (B) monochromator, (C, E, H, and J) slits, (D) aluminum absorbers, (F) x-ray monitor, (G) sample trough, (I) detector absorbers, and (K) scintillation x-ray detector. The reflected angle is equal to the incident angle α and the reflected x rays are in the plane of incidence. The only nonzero component of the wave vector transfer is its z-component (z axis is normal to the buffer surface), which is given by Qz = (4π/λ) sin(α), where λ is the wavelength of the incident x rays.
The reflectivity, R(Qz), represents the scattered intensity normalized by the x-ray intensity measured immediately in front of the sample. In addition, background scattering is measured and subtracted as described elsewhere (33). To make the features of the reflectivity curve more evident, R(Qz) is divided by RF(Qz), the Fresnel reflectivity predicted for an ideal, smooth, and flat interface (34). Deviations of the measured reflectivity, R(Qz), from the Fresnel reflectivity, RF(Qz), reveal the presence of interfacial structure as a function of position along the normal to the surface. In this case, the structure is due to the lipid monolayer supported on the buffer surface and to cPLA2α-C2 bound to the lipid monolayer. Reflectivity data were analyzed using the Parratt recursive algorithm (35). The models chosen to describe the pure lipid monolayer and the lipid-protein system are discussed in the next section.
To prepare a typical sample for study by x-ray reflectivity, ∼10 μl of 1 mM SOPC in chloroform was added dropwise onto the surface of 20 mM HEPES buffer, pH 7.0, containing 0.1 mM KCl and 0.1 mM CaCl2 in a 72-mm-diameter circular Teflon trough (∼40 ml total volume). The resulting lipid monolayer (at a surface pressure π ≈ 24 mN/m) was equilibrated for 2 h and the reflectivity was measured. An amount greater than the saturated amount (i.e., >240 μg of cPLA2α-C2 or one of the mutant domains) was injected into the subphase, the system was equilibrated for 1 h with continuous slow stirring, and the reflectivity was measured. The surface pressure was monitored to ensure the integrity of the monolayer during the x-ray experiment. Typical variations in surface pressure were <1 mN/m during the period of the measurement.
Due to its unsaturated nature, SOPC lipid is prone to oxidation and radiation damage. Even without x-ray exposure, there is a small decrease of the surface pressure if monitored for a long time (on the order of 1 mN/m over 24 h). X-ray reflectivity measurements of the SOPC monolayer and the SOPC monolayer with bound cPLA2α-C2 had to be completed before the SOPC monolayer deteriorated. To decrease the total time for the experiment, the initial measurement of the SOPC monolayer can be taken with a low density of points. Our experience shows that the lipid reflectivity curve has a well-defined shape; therefore, a small number of data points at carefully chosen values of Qz determines the whole curve. Measurements of the lipid-protein system were performed with a higher density of points. To further decrease radiation exposure, we reduced the x-ray intensity by placing aluminum absorbers in the beam path in front of the sample (see Fig. 1). Since the presence of absorbers also increases the measurement time, the optimal total absorber thickness was determined. This resulted in a maximum x-ray exposure of 4 × 107 photons/(s mm2) over an x-ray footprint of ∼3 mm × 2 mm. A typical data acquisition time for the whole experiment (the lipid plus lipid-protein reflectivity curves) was 6–8 h. With the data acquisition procedure described above, no radiation damage was detected during the measurements, as indicated by the surface pressure stability and the reproducibility of the x-ray reflectivity data after repeated measurements on the same sample. An example of this reproducibility can be seen in Fig. 8, to be discussed later.
FIGURE 8.

Normalized x-ray reflectivity data for a pure SOPC layer and the SOPC/cPLA2α-C2 system with no Ca2+ present in the buffer. Solid circles indicate the data for a pure SOPC monolayer, and open diamonds represent the data for the system after the protein solution was injected into the subphase. Dashed and solid lines represent the best fits for the pure SOPC monolayer and the SOPC/cPLA2α-C2 system, respectively. The fitting parameters for the pure SOPC monolayer and the SOPC/cPLA2α-C2 system are shown in Table 3.
DATA ANALYSIS AND RESULTS
SOPC monolayer
X-ray reflectivity data from Langmuir monolayers of lipids are typically analyzed using a model for the electron density profile that consists of two slabs sandwiched between bulk aqueous buffer and bulk air (36). One slab represents the average electron density in the headgroup region of the lipids and the other slab represents the electron density in the tailgroup (acyl group) region (see dashed line in Fig. 2 b). Each slab is characterized by a constant electron density ρ throughout its thickness L. The density is smeared at the slab borders to provide a smooth crossover between slabs. This is a physical effect due to thermal fluctuations of the water surface, known as capillary waves, which result in a time-averaged smearing of the profile as measured by x-ray reflectivity. This smearing is characterized by an interfacial roughness (or width) σ. The electron density is determined by fitting the measured data to reflectivity calculated for this model profile.
FIGURE 2.

(a and b) The normalized x-ray reflectivity and the interfacial profile of the pure SOPC monolayer at π = 24 mN/m. (c and d) The normalized x-ray reflectivity and the interfacial profile of the SOPC monolayer-bound cPLA2α-C2 system. Open circles in a and c represent the experimental x-ray reflectivity data and solid lines represent the best fit of the data using models described in the text. The corresponding fitting parameters are shown in Table 1. Solid lines in b and d represent the normalized electron densities of the interfaces along the axis normal to the interface. Dashed lines in b and d represent the same electron density profiles as the solid lines with the roughness parameter, σ, set to zero for illustrative purposes.
X-ray reflectivity data for a pure SOPC monolayer at a surface pressure of 24 mN/m are shown in Fig. 2 a. Although this measurement has only a small number of data points, it is consistent with other measurements we have taken with a larger number of data points and is adequate to characterize the lipid monolayer (as described in Materials and Methods, above). The measurement shown in Fig. 2 a is the measurement that preceded the measurement of the bound protein illustrated in Fig. 2 c that will be discussed later. The data for the pure SOPC monolayer are analyzed by a two-slab model for the electron density, given by
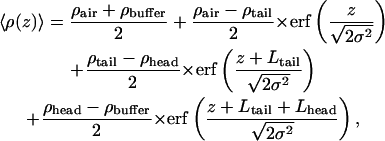 |
(1) |
where  Lhead and Ltail are the thickness of the headgroup and tailgroup; ρhead and ρtail represent the average electron densities (normalized to the electron density of the subphase, ρbuffer = 0.338 e−/Å3) of the headgroup and tailgroup; ρair, the electron density of the air, is equal to zero; and σ is the roughness of the surface calculated by capillary wave theory using the measured temperature and surface pressure.
Lhead and Ltail are the thickness of the headgroup and tailgroup; ρhead and ρtail represent the average electron densities (normalized to the electron density of the subphase, ρbuffer = 0.338 e−/Å3) of the headgroup and tailgroup; ρair, the electron density of the air, is equal to zero; and σ is the roughness of the surface calculated by capillary wave theory using the measured temperature and surface pressure.
Note that interfacial roughness used in the fits was fixed to the value calculated from the capillary wave theory using the measured temperature and surface pressure (37,38). The dependence of the fit parameters for the SOPC monolayer on the uncertainties of the surface pressure (±1 mN/m) yielded values within the quoted error bars. The contribution of the bending rigidity was not included in calculating the interfacial roughness because the bending rigidity for an SOPC monolayer has not been measured. To test the influence of including the bending rigidity, we reanalyzed our data using a typical value of 10 kT as measured for bilayers (39,40). The lipid parameters for both the pure SOPC monolayer and the SOPC/cPLA2α-C2 system changed slightly (within the quoted error bars), but there was no effect on the protein orientation and penetration.
The air-tailgroup, tailgroup-headgroup, and headgroup-buffer interfaces are located at z = 0, z = − Ltail, and z = − (Ltail + Lhead), respectively. The Parratt algorithm is used to calculate the reflectivity from the electron density profile given by Eq. 1. Fitting the data to the calculated reflectivity yields values for the fitting parameters: Lhead, Ltail, ρhead, and ρtail. The fit is shown in Fig. 2 a (solid line) and the resulting electron density profile is shown in Fig. 2 b. The fit parameters are listed in Table 1 and are very similar to parameters we measured several times for SOPC monolayers, including measurements with a much higher density of points. The thickness of the tailgroup region, Ltail, and the electron density in this region, ρtail, indicate that the acyl chains are disordered. The high electron density in the headgroup region is due to the presence of the phosphate group. The total thickness of the monolayer adds up to 21 Å, consistent with the dimensions of a phospholipid monolayer.
TABLE 1.
Parameters for pure SOPC monolayer and SOPC bound cPLA2α-C2
| SOPC/cPLA2α-C2
|
||||
|---|---|---|---|---|
| SOPC | d | a | g | |
| Ltail [Å] | 10.6 ± 0.1 | 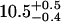 |
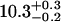 |
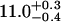 |
| Lhead [Å] | 10.5 ± 0.5 | 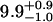 |
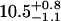 |
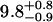 |
| CEN [Å] | 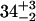 |
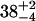 |
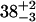 |
|
| ρtail | 0.62 ± 0.01 | 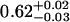 |
0.62 ± 0.03 | 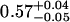 |
| ρhead | 1.25 ± 0.01 | 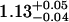 |
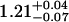 |
1.11 ± 0.04 |
| COV | 0.69 ± 0.07 | 0.9 ±0.1 | 0.66 ± 0.07 | |
| σ [Å] | 3.3 | 3.6 | 3.6 | 3.6 |
| Abox [Å2] | 1323 | 1978 | 1200 | |
| CENcryst [Å] | 43.9 | 38.2 | 39.0 | |
| L>CEN [Å] | 25.6 | 28.3 | 34.5 | |
| Aprotein [± 300 Å2] | 1900 | 2200 | 1800 | |
| PEN [± 3 Å] | 12 | 11 | 17 | |
| χ2 | 10.5 | 15.4 | 12.2 | |
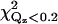 |
8.4 | 9.6 | 9.5 | |
The parameters for cPLA2α-C2 orientations shown in Fig. 7, d, a, and g, are shown. Fig. 7 d is the best fit, and Fig. 7 g is inconsistent with EPR measurements. The items Ltail, Lhead, CEN, ρtail, ρhead, and COV are fitting parameters, whereas σ is calculated from the capillary wave theory, Abox; CENcryst and L>CEN are parameters extracted from Cerius2; and Aprotein and PEN are calculated from the other parameters, as described in the text. Normalized electron densities must be multiplied by 0.338 e−/Å3 to get absolute electron densities. χ2 is the statistical measure of goodness of fit for the entire range of Qz; 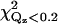 is for a smaller range of Qz (<0.2 Å−1) that is most sensitive to the protein ordering.
is for a smaller range of Qz (<0.2 Å−1) that is most sensitive to the protein ordering.
These values are consistent with previous measurements (found in literature) of SOPC and similar lipids in bilayers or Langmuir monolayers. There is not much information about the structural parameters of SOPC monolayers and bilayers in the literature, but similar lipid monolayers and bilayers, such as 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were more extensively studied. Vogel et al. (40) determined the bilayer thickness of POPC to be 39 Å, corresponding to 19.5 Å single monolayer thickness, consistent with our results. Thoma et al. (41) used x-ray reflectivity to study the structure of DPPC monolayers for various lipid monolayer pressures. Their values of the headgroup thickness are in the range from 7.3 Å to 10.8 Å. Petkova et al. (42) studied DOPC bilayers by x-ray reflectivity and reported the value of 11.8 Å for the headgroup thickness. DOPC, DPPC, and POPC bilayers were studied by Liu and Nagle (43), Nagle and Tristam-Nagle (44), and Pabst et al. (45), respectively. The headgroup thickness determined by these studies was 9 Å, which is slightly smaller than the value we report here for the SOPC monolayer headgroup thickness (10.5 Å). To determine the variation of the thickness parameters with the roughness value, we fit our reflectivity data with a roughness slightly larger than the value calculated from the capillary wave theory. If the capillary wave roughness (3.3 Å) was increased to 3.5 Å, and 3.6 Å, respectively, then the headgroup thickness decreased to 9.3 Å, and 8.7 Å, respectively, and the overall monolayer thickness decreased to 20.2 Å, and 19.8 Å, respectively.
SOPC monolayer with bound cPLA2α-C2
Surface pressure data
The change in surface pressure Δπ after cPLA2α-C2 adsorption onto the SOPC monolayer is plotted in Fig. 3 as a function of the initial surface pressure π0 of the SOPC monolayer (see Materials and Methods). These data were taken on samples without prior x-ray exposure. The value of Δπ was then measured at the synchrotron, after x-ray reflectivity measurement of pure SOPC monolayer and before the x-ray reflectivity measurement of the lipid-protein system. The obtained value (Δπ = 8 mN/m) is consistent with the value measured without prior x-ray exposure.
FIGURE 3.

Change of surface pressure upon cPLA2α-C2 adsorption to the SOPC monolayer as a function of the initial monolayer pressure before adsorption. Dots indicate the data measured without x-ray exposure of SOPC monolayers as described in Materials and Methods. The open circle represents the value measured at the synchrotron immediately before measuring the x-ray reflectivity shown in Fig. 2 c.
X-ray data and reflectivity model that incorporates protein crystallography structure
X-ray reflectivity from cPLA2α-C2 domains bound to an SOPC monolayer is shown in Fig. 2 c. It is similar in form to that of the pure SOPC monolayer in Fig. 2 a, except for an additional peak located in the low Qz region. This peak is due to the C2 domain bound to the SOPC monolayer.
The standard method of analysis, previously applied to reflectivity data measured from proteins bound to Langmuir monolayers of lipids, consists of a three-slab model similar to the two-slab model we used to analyze the data from the SOPC monolayer. In this case, the third slab would represent the bound proteins. The fit to the data and resultant electron density profile from this analysis are very similar to those shown in Fig. 2, c and d. However, the fit parameters to this slab model (not shown) do not directly determine the angular orientation of the protein or the penetration depth of the protein into the region of the lipid monolayer. To extract this information, we have modified the reflectivity analysis to utilize the protein structure determined from x-ray crystallography.
The x-ray crystallography structure of cPLA2α-C2 (PDB ID:1RLW) (14) was used to model the electron density distribution of the cPLA2α-C2 domain. This structure is not a complete representation of the domain we studied because 1), there are 16 residues not resolved by x-ray diffraction; and 2), a 19-residue amino-terminal tail containing a His-tag was attached to the domain for affinity purification. These additional 35 residues are located at the end of the domain opposite to the calcium-binding loops and have no effect on the binding of this domain to the lipid monolayer. The additional residues are required to fit our data, but the results of our analysis are not sensitive to sensible variations of the conformation of these additional residues. Molecular simulations were used to model these additional residues.
This N-terminal extension was analyzed as follows: The 35-residue N-terminal extension was predicted to have random coil conformation by protein fold recognition programs (46,47). A potential structural template was detected, but the similarity was due to the inclusion of an identical His-tag. The 20 models consistent with the NMR data for this structure (PDB ID:1JDQ) (48) gave very different conformations for the N-terminal segment. Hence, no suitable structural templates for the N-terminal extension were found in the Protein Data Bank (49). An ab initio-like model was constructed with the HMMSTR/Rosetta structure prediction server (50). A composite model for the experimental construct was built with the homology-modeling protein Nest (51) and consists of two pieces: 1), the HMMSTR model for the 35-residue N-terminal portion, and 2), the experimentally determined structure of cPLA22α-C2 domain. This model predicts that the N-terminal extension is unstructured and points away from the rest of the C2 domain. Note that our initial analysis using the results of this simulation for the N-terminal extension was not fully satisfactory. A slight modification of this simulation was based upon a simple analytical form for the extra residues of the cPLA22α-C2. This modification fit the data well and is consistent with the range of conformations that can be produced by the simulation.
The cPLA2α-C2 domain studied by x-ray reflectivity is shown in Fig. 4. As will be demonstrated, the orientation shown in Fig. 4 is the best fit from our analysis and will be referred to as the basic orientation.
FIGURE 4.
Crystal structure of cPLA2α-C2. Green spheres represent calcium ions. The protein is in the basic orientation, the z axis is indicated, and the solid white line represents an xy plane separating the bottom of the lipid layer and the buffer. The part of the protein above the solid white line penetrates into the SOPC monolayer.
In this geometry the lipid monolayer is in the xy plane and the z axis is perpendicular to the monolayer. We use the same coordinate system and rotations about the same axes as described by Malmberg et al. (20) to define the basic domain orientation. This allows for a direct comparison of orientations determined by our x-ray data and those determined by EPR measurements. Three atoms were chosen as the reference points. The first and the second atoms are located in the longest β-strand of the domain and the third atom is located in the furthest strand in the same sheet. The selected atoms are the α-carbons in residues F20, A27, and K118. Two molecular vectors are then defined, a z′ vector extending from the first atom (in F20) to the second atom (in A27) and an x′ vector passing through the third atom (in K118) and perpendicular to the first vector. A molecular coordinate system is then defined, with its center at the first α-carbon, the z′ axis parallel to the z′ vector, and the x′ axis parallel to the x′ vector. The orientation of the y′ axis (perpendicular to the x′z′ plane) is defined by the requirement of a right-handedness of the x′y′z′ coordinate system.
The domain is oriented relative to an imaginary planar membrane surface in two different ways. First, the domain is placed in a common starting orientation with its z′ axis normal to the membrane. This places the calibration β-sheet perpendicular to the membrane surface. Second, the domain is placed in its final bound orientation. Last, the transformations needed to change the starting orientation into the final orientation are calculated as a rotation about the x′ axis by angle θ followed by a rotation about the z′ axis by angle φ. Our basic (and best fit) orientation is defined by angles θ = 48° and φ = 104°.
To use the structure shown in Fig. 4 in the reflectivity analysis, a one-dimensional electron density profile along the z axis, averaged over the xy directions, must be determined. This profile is referred to as 〈ρprotein_layer(zp)〉, where zp is a z axis shifted by an amount to be discussed. This protein profile, modified for the presence of buffer, is then situated between the profile for the bulk aqueous buffer and the two slabs that describe the lipid monolayer. An additional degree of freedom allows the protein profile to penetrate into the region of the monolayer. This combined profile is then used to fit the x-ray reflectivity data. The fitting determines the penetration depth of the protein into the lipid monolayer and the fraction of lipid monolayer with bound protein, as well as the lipid parameters, for the domain orientation shown in Fig. 4. The same procedure is undertaken for different angular orientations of the protein to determine if other orientations can fit the data and, if so, the fitting parameters for these orientations.
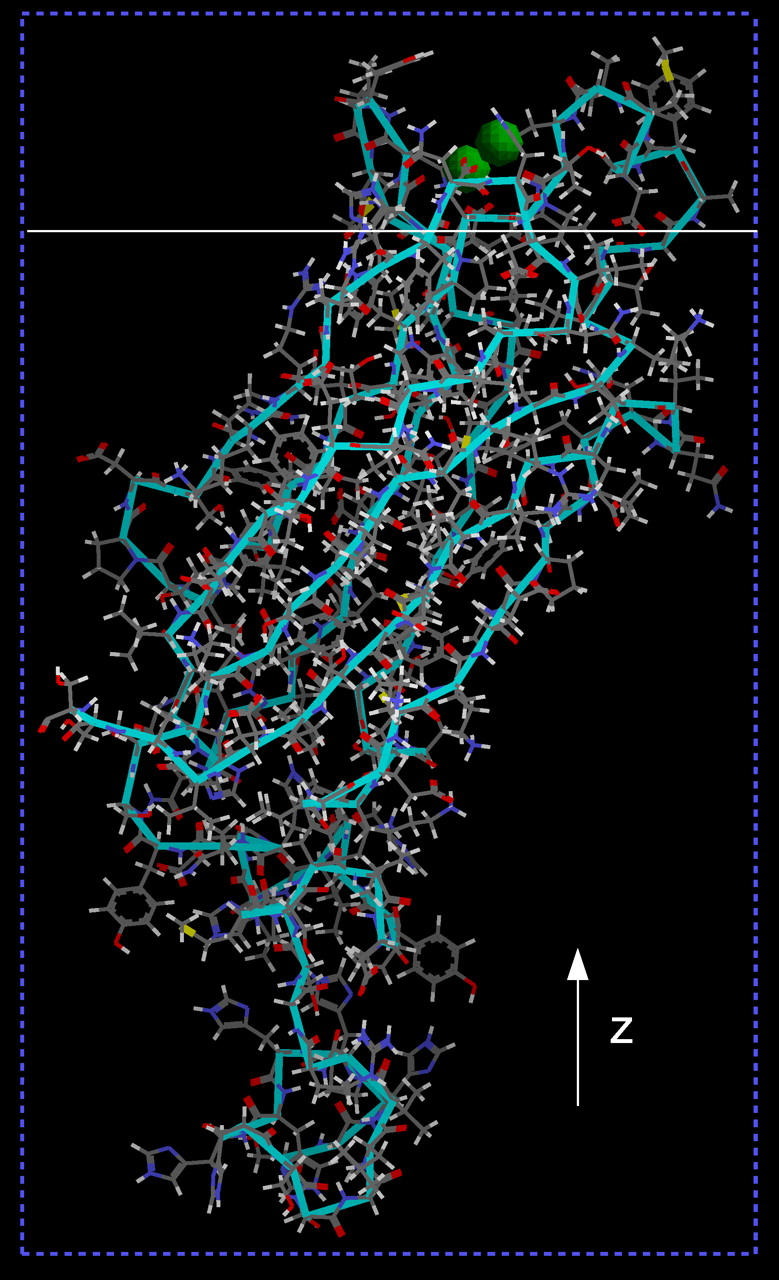
To account for the presence of buffer that surrounds the protein (i.e., buffer at the same surface depth as the protein), the electron density profile of the protein layer is modeled by enclosing a single protein in the smallest possible rectangular box. Since part of each protein is in the buffer environment, while the rest penetrates inside the lipid region that does not contain buffer, the electron density of the protein in the box 〈ρprotein_box(zp)〉 is determined for both protein in a box filled with buffer (〈ρprotein(buffer)(zp)〉) and protein in an empty box (〈ρprotein(empty)(zp)〉). The interface then consists of these boxed proteins, possibly separated by additional buffer if the protein coverage is less than 1, adjacent to the lipid monolayer (see Fig. 5).
FIGURE 5.
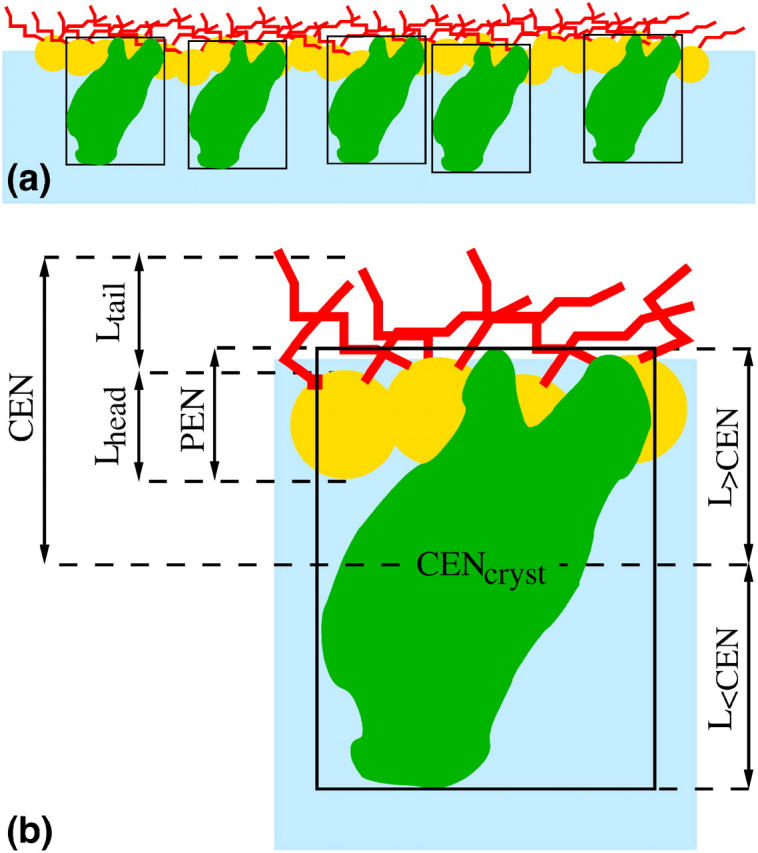
(a) Schematic view of the lipid monolayer with the adsorbed proteins enclosed in the smallest possible rectangular boxes. Yellow circles and red curves represent the lipid headgroups and tailgroups, respectively; proteins enclosed in boxes are schematically shown as green objects in black rectangles (the real structure of the protein in its smallest box is shown in Fig. 4). The parameters appearing in Eq. 6 are indicated in b, which is an enlarged version of one of the boxed proteins together with adjacent lipids.
The electron density profile of this protein in a box is determined by slicing the box into 2 Å thick slices parallel to the lipid monolayer (xy plane). The electron density of each 2 Å thick slice was calculated by dividing the number of electrons in the slice by its volume. The program Cerius2 was used to determine the number of electrons in each slice as follows. Cerius2 is capable of listing the occupancy of individual atoms in the part of the slice occupied by a protein and calculating the volume of the part of the slice occupied by a protein. The number of electrons in each slice is the sum of the number of electrons in the part of the slice occupied by the protein and the number of electrons in the part of the slice not occupied by the protein (either buffer or empty). The number of electrons in the part occupied by a protein was calculated from the occupancies of atoms obtained by Cerius2. The number of electrons in the volume not occupied by the protein (and thus filled with buffer or empty) was calculated from the known volume of this area (slice's volume minus volume of the slice occupied by protein) and the known electron density (either the electron density of the buffer in case of a box filled with buffer, or zero in case of an empty box). Resulting electron density was normalized to the electron density of the buffer.
The electron density profile of the slices for the case of protein in a box with buffer is shown in Fig. 6 (circles). It is convenient for the data-fitting algorithm to have an analytic parameterization of the discrete set of points in Fig. 6 with a (piecewise) continuous function. We found that a Gaussian function was not a good approximation to the entire 〈ρprotein_box(zp)〉 because the electron density near the calcium-binding loops drops off quickly. Therefore, we parameterized the part of 〈ρprotein_box(zp)〉 near the calcium-binding loops with a fourth-power Gaussian and the rest of 〈ρprotein_box(zp)〉 with an ordinary Gaussian. The analytic form of 〈ρprotein(buffer)(zp)〉 is given by
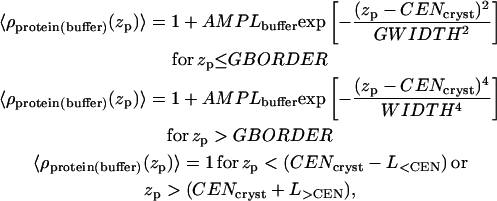 |
(2) |
where L<CEN, L>CEN, AMPLbuffer, WIDTH, GWIDTH, GBORDER, and CENcryst are described in Fig. 6. These parameters are orientation-specific and stay fixed for a given orientation. L<CEN and L>CEN characterize the protein length along the zp axis (the total protein length projected onto the zp axis is L<CEN + L>CEN). GBORDER identifies where the Gaussian switches to the fourth-power Gaussian. CENcryst is the center of the Gaussian and of the fourth-power Gaussian functions.
FIGURE 6.

Electron density along the zp axis of the protein in a box with buffer obtained by slicing the protein in the basic orientation (Fig. 4). Open circles indicate the electron density of each slice for the case of the protein in a box with buffer. The solid line represents the best fit of these electron densities using a model function described in the text. The parameter values are: L<CEN = 53.9 Å, L>CEN = 25.6 Å, CENcryst = 43.9 Å, AMPLbuffer = 0.19, AMPLempty = 0.47, WIDTH = 21.7 Å, GWIDTH = 32.4 Å, and GBORDER – CENcryst = 0 Å.
The same procedure is followed for the protein in an empty box, yielding
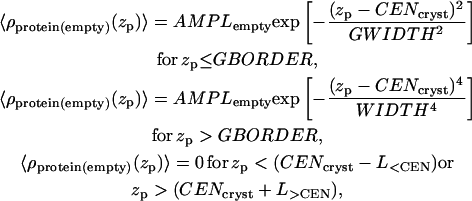 |
(3) |
where all the parameters (except AMPLempty) are the same, as in the case of the protein in a box with buffer. The electron density of a single protein in a box, 〈ρprotein_buffer(zp)〉, is then composed of two functions:
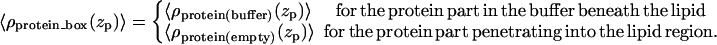 |
The electron density profile of a single protein in a box 〈ρprotein_box(zp)〉 is then used to model the electron density profile of a protein layer 〈ρprotein_layer(zp)〉 by introducing the coverage parameter COV, the fraction of surface area occupied by the protein boxes, as
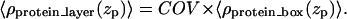 |
(4) |
Since the electron density of the buffer has been normalized to unity, this expression for 〈ρprotein_layer(zp)〉 accounts for the presence of buffer between the protein boxes if the surface is not fully covered by the protein boxes. The area per protein (Aprotein) can then be calculated as
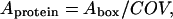 |
(5) |
where Abox is the area of the side of the box in the xy plane (parallel to the lipid monolayer).
The final model for the electron density profile of the lipid-protein system is the sum of the electron density profile of the protein layer (〈ρprotein_layer(zp)〉) and the electron density of the lipid layer described by two slabs. The coordinate system is chosen to place the air-tailgroup interface at z = 0 and the buffer-protein interface at z < 0. The position of the protein with respect to the lipid layer is given by the parameter CEN, where CEN is the distance between the protein center and the air-tailgroup interface at z = 0 (see Fig. 5). In this geometry, the penetration (PEN) of the protein into the lipid layer (measured from the headgroup/buffer interface) can be calculated as
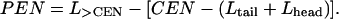 |
(6) |
Lengths appearing in Eq. 6 are indicated in Fig. 5 b, and zp is related to z via
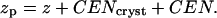 |
(7) |
Interfacial roughness is incorporated into the model by convoluting the electron density profile with a Gaussian of width σ, where σ is calculated from capillary wave theory.
The x-ray reflectivity data are fit with the reflectivity calculated by the Parratt algorithm applied to the above model. There are six fitting parameters—four for the lipid layer (Lhead, Ltail, ρhead, and ρtail) and two for the protein layer (CEN and COV). The fit is shown in Fig. 2 c, the electron density profile in Fig. 2 d, and fit parameters are listed in Table 1, column d. The fit to the reflectivity data is good throughout the entire range of Qz. Note that the region of low Qz is most sensitive to the protein and the region of higher Qz is sensitive to the lipid structure. This indicates that the protein orientation shown in Fig. 4 is consistent with our data, though it does not imply that other orientations are inconsistent with our data. Although this protein orientation provides the best fit to our data, fits to the data for a variety of other orientations are discussed in the next section to illustrate the dependence upon orientation.
The parameters describing the SOPC monolayer are similar to those of the monolayer without bound protein with the exception that the electron density in the headgroup region is lower. This indicates that the number of electrons attributable to the headgroup region of the lipid monolayer changed upon protein binding. We speculate that some water molecules hydrated to the headgroup are removed upon protein binding.
The amount of water can be estimated by counting the electrons in the lipid region as follows. The total number of electrons in the lipid region (per lipid) will be the total number of electrons in SOPC (436 electrons) plus the number of electrons in hydrated water molecules (10 nw, where nw is the number of hydrated water molecules per lipid). The area per lipid, A, can be written in terms of this total number of electrons as A = (436 +10 nw)/[(Lhead × ρhead × ρbuffer) + (Ltail × ρtail × ρbuffer)], where ρbuffer = 0.338 e−/Å3. For the measurement of the SOPC monolayer without protein (see Table 1), A = 65.5 + 1.5 nw. Similarly, for the measurement of cPLA2α-C2 bound to SOPC (Table 1, analysis d), 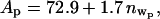 where the subscript p refers to the presence of the bound protein. Since the tail parameters do not change upon protein binding, it is sensible to assume that the area per lipid does not change significantly with protein binding. Therefore, Ap = A and
where the subscript p refers to the presence of the bound protein. Since the tail parameters do not change upon protein binding, it is sensible to assume that the area per lipid does not change significantly with protein binding. Therefore, Ap = A and 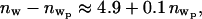 where
where 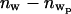 represents the number of water molecules lost from the headgroup region upon protein binding. The amount of lost water depends upon the value of
represents the number of water molecules lost from the headgroup region upon protein binding. The amount of lost water depends upon the value of 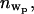 which we do not measure accurately. However, this analysis indicates that a minimum of five water molecules per lipid is lost upon protein binding. Literature values of ∼30 hydrated waters (nw = 30) lead to a slightly larger value of eight water molecules per lipid lost upon protein binding (43,44,52).
which we do not measure accurately. However, this analysis indicates that a minimum of five water molecules per lipid is lost upon protein binding. Literature values of ∼30 hydrated waters (nw = 30) lead to a slightly larger value of eight water molecules per lipid lost upon protein binding (43,44,52).
Approximately two-thirds of the lipid layer is covered by protein (COV = 0.69). Equation 5 yields the area per protein as 1900 Å2, similar to the result of the radioactive labeling method (see Materials and Methods) that yielded the value 2110 Å2. Equation 6 yields a value of 12 ± 3 Å for the penetration of the protein, in this orientation, into the lipid layer. This places the Ca2+ ions within 1 Å of the lipid phosphate groups.
Protein orientation and penetration
To investigate other protein orientations that may be consistent with the x-ray data, the protein in the orientation shown in Fig. 4 was rotated to eight additional orientations. Since rotations around the z axis change only the viewing angle, but not the orientation with respect to the membrane, only rotations around the x and y axes can give new orientations. The orientations chosen are shown in Fig. 7. These orientations were analyzed by repeating the procedure described in the previous section. This determined new electron density profiles 〈ρprotein_layer(zp)〉, which were then used to fit the reflectivity data. Only the two orientations shown in Fig. 7, a and g, in addition to that shown in Fig. 4 (or, equivalently, Fig. 7 d), provide adequate fits to the data. The protein orientation in Fig. 7 a is obtained by rotating the protein by 25° around the y axis from the orientation in Fig. 7 d and the orientation in Fig. 7 g is obtained by rotating the protein by −25° around the y axis and 25° around the x axis from the orientation in Fig. 7 a. The fitting parameters for the three orientations that fit the data are listed in Table 1. Two of the three rotations (d and a) that provide adequate fits to the data can be compared by the statistical measure of goodness of fit χ2. Two different values of χ2 were considered, one that measures the goodness of fit over the entire range of Qz (denoted χ2) and the other that measures the goodness of fit over a smaller range of Qz (<0.2 Å−1, denoted 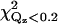 ) that is most sensitive to the protein ordering. The values of χ2 and
) that is most sensitive to the protein ordering. The values of χ2 and 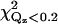 for nine orientations shown in Fig. 7 and for orientations predicted from earlier EPR studies (19,20) are shown in Table 2. Orientation d has the lowest values of χ2 and
for nine orientations shown in Fig. 7 and for orientations predicted from earlier EPR studies (19,20) are shown in Table 2. Orientation d has the lowest values of χ2 and 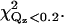 The statistical significance of the difference between these values for orientations d and a is ∼2−sigma. Although orientation g also provides an adequate fit to the data, we do not consider this orientation further because it is inconsistent with the depth parameters from the EPR measurements determined by Malmberg et al. (20) and because it places hydrophilic residues of the CBL1 as deep or deeper into the lipid monolayer as hydrophobic residues of the CBL3.
The statistical significance of the difference between these values for orientations d and a is ∼2−sigma. Although orientation g also provides an adequate fit to the data, we do not consider this orientation further because it is inconsistent with the depth parameters from the EPR measurements determined by Malmberg et al. (20) and because it places hydrophilic residues of the CBL1 as deep or deeper into the lipid monolayer as hydrophobic residues of the CBL3.
FIGURE 7.

Protein orientations selected for analysis. The basic orientation is shown on d. The orientation obtained from the basic orientation (d) by rotating it by 25° around the y axis is shown on a. Orientations obtained from the orientation (a) by rotating it by (b) −25° around the x axis; (c) 25° around the x axis; (e) 25° around the y axis; (f) −25° around the y axis and −25° around the x axis; (g) −25° around the y axis and 25° around the x axis; (h) 25° around the y axis and −25° around the x axis; and (i) 25° around the y axis and 25° around the x axis, are shown. Shaded spheres represent two calcium ions. The white solid lines in a, d, and g indicate the border between the lipid headgroups and the buffer. The positions of the white lines were calculated for each orientation via Eq. 6. The part of the protein above the lines penetrates into the SOPC monolayer.
TABLE 2.
Comparison of χ2 and 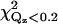
| Orientations shown in Fig. 7
|
EPR studies
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | d | e | f | g | h | i | EPR1 | EPR2 | |
| θ [°] | 56 | 79 | 36 | 48 | 70 | 73 | 23 | 90 | 52 | 68 | 52 |
| φ [°] | 74 | 86 | 54 | 104 | 51 | 108 | 94 | 66 | 31 | 66 | 53 |
| χ2 | 15.4 | 27.9 | 13.0 | 10.5 | 46.2 | 18.6 | 12.2 | 60.1 | 24.3 | 23.8 | 17.1 |
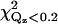 |
9.6 | 24.9 | 19.6 | 8.4 | 39.9 | 13.8 | 9.5 | 60.5 | 36.1 | 17.0 | 14.3 |
The values of χ2 and 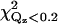 for orientations shown in Fig. 7 and orientations predicted by Frazier et al. (19) (denoted by EPR1) and Malmberg et al. (20) (denoted by EPR2) are given. Orientations a, d, and g provide adequate fits to the data. Orientation d has the lowest values of χ2 and
for orientations shown in Fig. 7 and orientations predicted by Frazier et al. (19) (denoted by EPR1) and Malmberg et al. (20) (denoted by EPR2) are given. Orientations a, d, and g provide adequate fits to the data. Orientation d has the lowest values of χ2 and 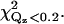 The statistical significance of the difference between these values for orientations d and a is ∼2−sigma. Orientation g is inconsistent with the depth parameters from the EPR measurements determined by Malmberg et al. (20) and it places hydrophilic residues of the CBL1 as deep or deeper into the lipid monolayer as hydrophobic residues of the CBL3.
The statistical significance of the difference between these values for orientations d and a is ∼2−sigma. Orientation g is inconsistent with the depth parameters from the EPR measurements determined by Malmberg et al. (20) and it places hydrophilic residues of the CBL1 as deep or deeper into the lipid monolayer as hydrophobic residues of the CBL3.
The fits to orientations d and a indicate that the thickness and electron density of the SOPC tailgroup layer did not change upon binding of the cPLA2α-C2 (Table 1). The thickness of the headgroup layer did not change either, but the electron density of the headgroup decreased upon binding of the cPLA2α-C2 to the SOPC monolayer. The area per protein for these orientations is within error bars of the value of 2110 Å2 from radioactive labeling (Materials and Methods). The orientation in Fig. 7 a determines a penetration of 11 Å, similar to the 12 Å determined for the orientation in Fig. 7 d. Although the orientation in Fig. 7 g determines a much higher penetration, 17 Å, there are good reasons to disregard this fit, as just discussed.
Control experiments: buffer without Ca2+ and mutants
An experiment with non-Ca2+ buffer was conducted to investigate the effect of Ca2+ on cPLA2α-C2 binding. A 20-mM HEPES buffer, pH 7.0, containing 0.1 mM KCl and 0.1 mM EGTA, was used instead of the Ca2+ buffer previously described. Protein injection under the SOPC monolayer did not result in any change of the 27 mN/m initial lipid pressure. X-ray reflectivity from the pure SOPC monolayer supported on the non-Ca2+ buffer and the reflectivity after the protein injection are the same (see Fig. 8). Two-slab-model fits to both sets of data have nearly identical fitting parameters (Table 3) that model a lipid layer without cPLA2α-C2 adsorption. This measurement confirms earlier proposals that Ca2+ is required for binding cPLA2α-C2 domains to lipids (30,53–59). Importantly, this measurement illustrates our ability to measure a Langmuir monolayer of SOPC over an extended period of time (8 h from the beginning of x-ray exposure) without noticeable deterioration from radiation damage or other effects.
TABLE 3.
Fitting parameters for experiment with non-calcium buffer
| SOPC | SOPC/cPLA2α-C2 | |
|---|---|---|
| Ltail [Å] | 10.43 ± 0.04 | 10.2 ± 0.1 |
| Lhead [Å] | 11.1 ± 0.4 | 11.1 ± 0.5 |
| ρtail | 0.63 ± 0.01 | 0.63 ± 0.02 |
| ρhead | 1.246 ± 0.007 | 1.257 ± 0.009 |
| σ [Å] | 3.4 | 3.4 |
Fitting parameters for the SOPC monolayer before and after addition of protein solution to the non-calcium buffer. Normalized electron densities must be multiplied by 0.338 e−/Å3 to get absolute electron densities.
The role of the hydrophobic residues on the putative binding sites was studied by measuring x-ray reflectivity from the system with mutated cPLA2α-C2. Two calcium-binding loops of cPLA2α-C2, CBL1, and CBL3, have a cluster of hydrophobic residues on the tips of their structure (see Fig. 9). According to our best-fit model (Fig. 7, d), the two residues L39 and V97 penetrate most deeply into the lipid layer by penetrating 2 Å into the tailgroup region of the lipids (see Table 1). It has been proposed that these residues are involved in membrane binding (18,30,58). To test this idea, we performed two experiments, in which we mutated two hydrophobic residues: L39 in CBL1 and V97 in CBL3 to alanine (L39A and V97A). The initial surface pressures for the experiments with L39A and V97A mutants were 25.3 mN/m and 25.8 mN/m, respectively; and the corresponding pressure changes after the mutant injections were Δπ = 0.6 ± 0.5 mN/m and Δπ = 1.0 ± 0.5 mN/m, respectively. The reflectivity data before and after injecting the mutants are similar (see Fig. 10); therefore, these data do not indicate any binding of the mutants onto the SOPC monolayer at these pressures. The reflectivity data were fit with a two-slab model, with fitting parameters very similar to those of pure lipid monolayers (Table 4).
FIGURE 9.
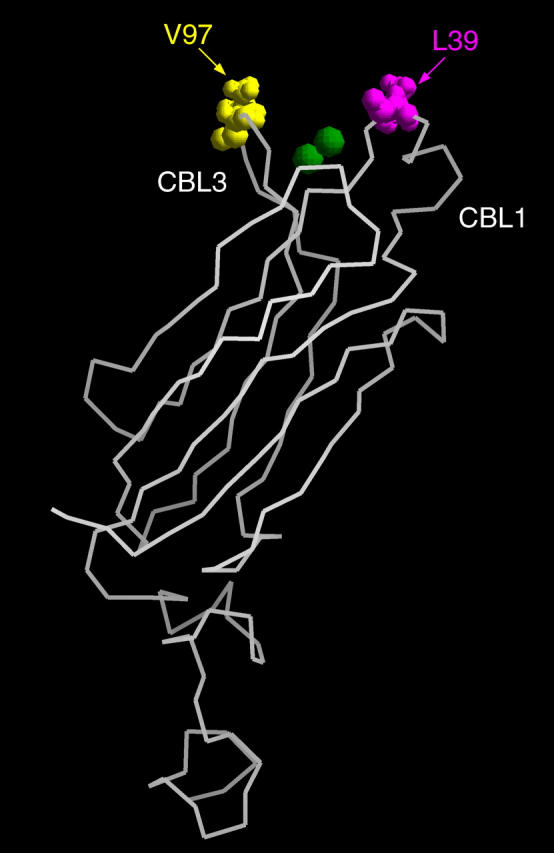
Crystal structure of cPLA2α-C2. Two calcium ions are shown as green spheres. Mutated hydrophobic residues L39 in CBL1 and V97 in CBL3 are represented by magenta and yellow spheres, respectively.
FIGURE 10.

Normalized x-ray reflectivity data for pure SOPC layers and SOPC/cPLA2α-C2 systems with mutations (a) L39A in CBL1, and (b) V97A in CBL3. Solid circles indicate the data for a pure SOPC monolayer, and open diamonds represent the data for the system after the protein mutants (a) L39A and (b) V97A were injected into the subphase. Dashed and solid lines represent the best fits for the pure SOPC monolayer and the mutant-SOPC system, respectively. The corresponding fitting parameters are shown in Table 4.
TABLE 4.
Fitting parameters for experiments with mutants
| L39A mutant
|
V97A mutant
|
|||
|---|---|---|---|---|
| SOPC | SOPC/cPLA2α-C2 | SOPC | SOPC/cPLA2α-C2 | |
| Ltail [Å] | 10.49 ± 0.08 | 10.2 ± 0.1 | 10.78 ± 0.08 | 10.4 ± 0.1 |
| Lhead [Å] | 10.6 ± 0.5 | 10.4 ± 0.5 | 10.3 ± 0.4 | 10.8 ± 0.5 |
| ρtail | 0.58 ± 0.01 | 0.62 ± 0.02 | 0.62 ± 0.01 | 0.61 ± 0.02 |
| ρhead | 1.217 ± 0.009 | 1.23 ± 0.01 | 1.248 ± 0.008 | 1.222 ± 0.009 |
| σ [Å] | 3.3 | 3.3 | 3.3 | 3.4 |
Fitting parameters for the SOPC monolayer before and after addition of mutated protein solution to the calcium buffer. Normalized electron densities must be multiplied by 0.338 e−/Å3 to get absolute electron densities.
These results are in a good agreement with studies by Bittova et al. (30) on the effects of mutations in the membrane-binding residues on the membrane binding affinity and monolayer penetration. They reported that the binding affinities of L39A and V97A mutants were four times lower than that of the wild-type. They also measured the critical surface pressure (πc), which specifies an upper limit of π0 for protein penetration into a monolayer. They reported critical surface pressures, 27 mN/m and 26 mN/m for L39A and V97A mutants, respectively, that are significantly lower, than the 34 mN/m measured for the wild-type cPLA2α-C2. Based on mutational effects of hydrophobic residues L39 and V97 on activity such as membrane binding and monolayer penetration, Bittova et al. (30) reported that L39 and V97 are directly involved in membrane penetration and hydrophobic interaction, and should be fully inserted into the hydrophobic core of the membrane.
DISCUSSION
In this work, synchrotron x-ray reflectivity was used to investigate the orientation and position of cPLA2α-C2 domain bound to an SOPC monolayer at the air-water interface. X-ray reflectivity from the monolayer was measured before and after injecting protein solution into the subphase. The SOPC layer is modeled by a two-slab model with one slab corresponding to the headgroup region and the other to the tailgroup region of the SOPC. Although a third slab can be used to model cPLA2α-C2 domains bound to the SOPC monolayer, this simple model is incapable of directly determining the orientation and penetration of the domains. To determine this information from the reflectivity measurements, a model that utilizes the reported crystal structure of cPLA2α-C2 was developed. The use of the crystal structure for the modeling of SOPC monolayer-bound cPLA2α-C2 is justified by the following observations. First, the crystal structure of cPLA2α-C2 (PDB ID:1RLW) (14) is similar to the solution structure determined by NMR (PDB ID:1BCI) (17). Second, EPR measurements indicate that the average conformations of the calcium-binding loops undergo only subtle changes upon membrane binding (20).
Four fitting parameters characterized the SOPC layer, and an additional two parameters were used to model the protein domain. These two parameters are CEN, which determines the cPLA2α-C2 penetration depth into the SOPC monolayer and COV, which characterizes the fraction of the SOPC monolayer covered by proteins. The orientation shown in Fig. 4 (also Fig. 7 d) provided the best fit to the reflectivity data. Eight additional orientations, obtained by 25° rotations around the y and/or x axes, were investigated. Of these eight orientations, six resulted in significantly worse fits than the basic orientation and one led to an unrealistic penetration. An additional orientation, in Fig. 7 a, provided an adequate fit though not as good as Fig. 7 d. The protein penetration value of 12 ± 3 Å, that indicates the distance from the headgroup-buffer plane to the residue that penetrates most deeply, places two hydrophobic residues, L39 and V97, just within the lipid tailgroups.
Two previous EPR measurements of the penetration and orientation of the cPLA2α-C2 domain bound to lipid vesicles were compared to our results by fitting our x-ray reflectivity data with the predictions for the orientations from these earlier studies (see Table 5) (19,20). Although our results generally agree with two previous EPR studies on the lipid vesicle-bound cPLA2α-C2 domain, some important differences have been noticed (19,20). The orientation proposed by Frazier et al. (19) does not fit our data well, and yields values of χ2 and 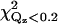 that are approximately twice as large as our best-fit χ2 values (Fig. 7 d, and see Table 2). The orientation proposed by Malmberg et al. (20) is similar to the orientation shown in Fig. 7 a; however, Malmberg's orientation also yields a value of χ2 ∼60% larger and a value of
that are approximately twice as large as our best-fit χ2 values (Fig. 7 d, and see Table 2). The orientation proposed by Malmberg et al. (20) is similar to the orientation shown in Fig. 7 a; however, Malmberg's orientation also yields a value of χ2 ∼60% larger and a value of 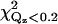 approximately twice as large as our best fit values (Fig. 7 d and see Table 2). In addition, our best fit orientation of cPLA2α-C2 (Fig. 7 d) places the Ca2+ ions 4.4 Å above the headgroup-buffer interface, i.e., within 1 Å of the lipid phosphates (see Ref. 51 for a determination of the distance between the phosphates and the headgroup). This is in agreement with the Malmberg's model, which places the Ca2+ ions at the depth of the membrane phosphates (20). However, the x-ray data reveal an inconsistency between the orientation and penetration proposed by Malmberg et al. If we analyze our x-ray data with the orientation proposed by Malmberg et al. (20), the Ca2+ ions are positioned just above the headgroup-buffer interface, ∼5 Å away from the phosphates and, therefore, 5 Å away from the position determined by Malmberg et al. (see Table 5).
approximately twice as large as our best fit values (Fig. 7 d and see Table 2). In addition, our best fit orientation of cPLA2α-C2 (Fig. 7 d) places the Ca2+ ions 4.4 Å above the headgroup-buffer interface, i.e., within 1 Å of the lipid phosphates (see Ref. 51 for a determination of the distance between the phosphates and the headgroup). This is in agreement with the Malmberg's model, which places the Ca2+ ions at the depth of the membrane phosphates (20). However, the x-ray data reveal an inconsistency between the orientation and penetration proposed by Malmberg et al. If we analyze our x-ray data with the orientation proposed by Malmberg et al. (20), the Ca2+ ions are positioned just above the headgroup-buffer interface, ∼5 Å away from the phosphates and, therefore, 5 Å away from the position determined by Malmberg et al. (see Table 5).
TABLE 5.
Comparison of various models
| Model | θ [°] | φ [°] | Ca2+ [Å] | Deepest residue | Cα [Å] |
|---|---|---|---|---|---|
| Fig. 7 d, best fit | 48 | 104 | 4.4 | L39 | 8.2 |
| Fig. 7 a | 56 | 74 | 0.7 | V97 | 6.4 |
| (20) | 52 | 53 | 5.3 (−0.1) | L39 | 12.0 (6.6) |
| (19) | 68 | 66 | 8.1 | V97 | 14.8 |
A comparison of four models for the cPLA2α-C2 domain orientation is shown. Three atoms were used to define a starting position in which a calibration β-strand is perpendicular to the membrane. The first rotation (θ) is about the molecular x′ axis, which is perpendicular to the calibration β-strand and lies in the plane of the calibration β-sheet. The second rotation (φ) is about the molecular z′ axis, co-linear with the calibration β-strand. Also shown for each model is the average depth of the Ca2+ ions, and the depth of its most deeply buried α-carbon. The depths are given relative to the headgroup-buffer interface. The depths determined by Malmberg et al. (20) and Frazier et al. (19), originally given relative to the headgroup phosphate plane, were converted to distances from the headgroup-buffer interface by adding 5 Å to them. The depths determined by our analysis applied to the orientation determined by Malmberg et al. (20) are shown in the parentheses.
Recent electrostatic potential calculations have indicated that nonspecific electrostatic interactions can explain the calcium-induced binding of C2 domains such as Sytl-C2A, PKCβ-C2, and PLCδ1 that preferentially bind to membranes containing anionic phospholipids in the presence of Ca2+ (60). These calculations were less successful in explaining the binding of cPLA2α-C2 domains that can bind to membranes containing only zwitterionic phospholipids. In this case, it was shown that the electrostatic free energy of interaction between cPLA2α-C2 domains and the surface of phospholipid bilayers is always unfavorable and does not explain the binding. Nevertheless, these calculations demonstrated that the presence of Ca2+ can enhance the membrane-binding affinity of the domains.
Our results display several structural features not accounted for in the electrostatic potential calculations because these calculations model the interior of the membrane as a structureless dielectric continuum (60). The electron density in the region of the SOPC lipids reveals a reduction in the electron density of the headgroup upon binding of the cPLA2α-C2 domain whereas the thickness of both headgroup and tailgroup and the electron density of tailgroup remain unchanged. This indicates that the number of electrons attributable to the headgroup region of the lipid changes upon cPLA2α-C2 domain binding. Calculations from our electron density profiles of the total number of electrons per lipid lead to the speculative conclusion that at least five water molecules previously hydrated to the headgroup leave the headgroup upon protein binding in the best-fit orientation d.
This will provide an entropic contribution to the free energy that favors protein binding. Furthermore, the near vicinity of the C2 domain-bound Ca2+ to the phosphate groups of the lipids and the location of the hydrophobic residues L39 and V97 just within the region of the lipid tailgroups will lead to favorable enthalpic contributions to the free energy of binding. In conclusion, our new method of analysis of x-ray reflectivity leads to detailed structural information that lends new insight into the mechanism and energetics of cPLA2α-C2 membrane interactions.
Acknowledgments
We acknowledge Professor Wai-Yee Keung (University of Illinois at Chicago) for suggesting the use of the fourth-power Gaussian, and Aleksey Tikhonov, the X19C beamline scientist.
This work was supported by National Institutes of Health grants (Nos. GM53987, GM52598, and GM53987 to W.C.) and National Science Foundation grants (Nos. DMR0092469 and CHE0315691 to M.L.S.). The Brookhaven National Laboratory is supported by the Department of Energy.
References
- 1.Cho, W. 2001. Membrane targeting by C1 and C2 domains. J. Biol. Chem. 276:32407–32410. [DOI] [PubMed] [Google Scholar]
- 2.Yang, C., and M. G. Kazanietz. 2003. Divergence and complexities in DAG signaling: looking beyond PKC. Trends Pharmacol. Sci. 24:602–608. [DOI] [PubMed] [Google Scholar]
- 3.Nalefski, E. A., and J. J. Falke. 1996. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 5:2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizo, J., and T. C. Sudhof. 1998. C2 domains, structure and function of a universal Ca2+ binding domain. J. Biol. Chem. 273:15879–15882. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson, K. M., J. M. Kavran, V. G. Sankaran, E. Fournier, S. J. Isakoff, E. Y. Skolnik, and M. A. Lemmon. 2000. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol. Cell. 6:373–384. [DOI] [PubMed] [Google Scholar]
- 6.Lemmon, M. A., and K. M. Ferguson. 2000. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem. J. 350:1–18. [PMC free article] [PubMed] [Google Scholar]
- 7.Stenmark, H., R. Aasland, and P. C. Driscoll. 2002. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett. 513:77–84. [DOI] [PubMed] [Google Scholar]
- 8.Xu, Y., L. F. Seet, B. Hanson, and W. Hong. 2001. The Phox homology (PX) domain, a new player in phosphoinositide signaling. Biochem. J. 360:513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Camilli, P., H. Chen, J. Hyman, E. Panepucci, A. Bateman, and A. T. Brunger. 2002. The ENTH domain. FEBS Lett. 513:11–18. [DOI] [PubMed] [Google Scholar]
- 10.Habermann, B. 2004. The BAR-domain family of proteins: a case of bending and binding? EMBO Rep. 5:250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bretscher, A., K. Edwards, and R. G. Fehon. 2002. ERM proteins and Merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3:586–599. [DOI] [PubMed] [Google Scholar]
- 12.Carroll, K., C. Gomez, and L. Shapiro. 2004. Tubby proteins: the plot thickens. Nat. Rev. Mol. Cell Biol. 5:55–63. [DOI] [PubMed] [Google Scholar]
- 13.Essen, L. O., O. Perisic, D. E. Lynch, M. Katan, and R. L. Williams. 1997. A ternary metal binding site in the C2 domain of phosphoinositide-specific phospholipase C-δ1. Biochemistry. 36:2753–2762. [DOI] [PubMed] [Google Scholar]
- 14.Perisic, O., S. Fong, D. E. Lynch, M. Bycroft, and R. L. Williams. 1998. Crystal structure of a calcium-phospholipid binding domain from cytosolic phospholipase A2. J. Biol. Chem. 273:1596–1604. [DOI] [PubMed] [Google Scholar]
- 15.Sutton, R. B., B. A. Davletov, A. M. Berghuis, T. C. Sudhof, and S. R. Sprang. 1995. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell. 80:929–938. [DOI] [PubMed] [Google Scholar]
- 16.Sutton, R. B., and S. R. Sprang. 1998. Structure of the protein kinase Cβ phospholipid-binding C2 domain complexed with Ca2+. Structure. 6:1395–1405. [DOI] [PubMed] [Google Scholar]
- 17.Xu, G. Y., T. McDonagh, H. A. Yu, E. A. Nalefski, J. D. Clark, and D. A. Cumming. 1998. Solution structure and membrane interactions of the C2 domain of cytosolic phospholipase A2. J. Mol. Biol. 280:485–500. [DOI] [PubMed] [Google Scholar]
- 18.Nalefski, E. A., and J. J. Falke. 1998. Location of the membrane-docking face on the Ca2+-activated C2 domain of cytosolic phospholipase A2. Biochemistry. 37:17642–17650. [DOI] [PubMed] [Google Scholar]
- 19.Frazier, A. A., M. A. Wisner, N. J. Malmberg, K. G. Victor, G. E. Fanucci, E. A. Nalefski, J. J. Falke, and D. S. Cafiso. 2002. Membrane orientation and position of the C2 domain from cPLA2 by site-directed spin labeling. Biochemistry. 41:6282–6292. [DOI] [PubMed] [Google Scholar]
- 20.Malmberg, N. J., D. R. Van Buskirk, and J. J. Falke. 2003. Membrane-docking loops of the cPLA2 C2 domain: detailed structural analysis of the protein-membrane interface via site-directed spin-labeling. Biochemistry. 42:13227–13240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frazier, A. A., C. R. Roller, J. J. Havelka, A. Hinderliter, and D. S. Cafiso. 2003. Membrane-bound orientation and position of the synaptotagmin I C2A domain by site-directed spin labeling. Biochemistry. 42:96–105. [DOI] [PubMed] [Google Scholar]
- 22.Bitto, E., M. Li, A. M. Tikhonov, M. L. Schlossman, and W. Cho. 2000. Mechanism of annexin I-mediated membrane aggregation. Biochemistry. 39:13469–13477. [DOI] [PubMed] [Google Scholar]
- 23.Lee, K. Y. C., J. Majewski, T. L. Kuhl, P. B. Howes, K. Kjaer, M. M. Lipp, A. J. Waring, J. A. Zasadzinski, and G. S. Smith. 2001. Synchrotron x-ray study of lung surfactant-specific protein SP-B in lipid monolayers. Biophys. J. 81:572–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weygand, M., B. Wetzer, D. Pum, U. B. Sleytr, N. Cuvillier, K. Kjaer, P. B. Howes, and M. Lösche. 1999. Bacterial S-layer protein coupling to lipids: x-ray reflectivity and grazing incidence diffraction studies. Biophys. J. 76:458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Möhwald, H., H. Balts, M. Schwendler, C. A. Helm, G. Brezesinski, and H. Haas. 1995. Phospholipid and protein monolayers. Jpn. J. Appl. Phys. 34:3906–3913. [Google Scholar]
- 26.Gidalevitz, D., Z. Huang, and S. A. Rice. 1999. Protein folding at the air-water interface studied with x-ray reflectivity. Proc. Natl. Acad. Sci. USA. 96:2608–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gidalevitz, D., Y. Ishitsuka, A. S. Muresan, O. Konovalov, A. J. Waring, R. I. Lehrer, and K. Y. C. Lee. 2003. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA. 100:6302–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohout, S. C., S. Corbalan-Garcia, J. C. Gomez-Fernandez, and J. J. Falke. 2003. C2 domain of protein kinase Cα: elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry. 42:1254–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin, Z., and D. S. Cafiso. 1996. Membrane structure of protein kinase C and calmodulin binding domain of myristoylated alanine rich C kinase substrate determined by site-directed spin labeling. Biochemistry. 35:2917–2925. [DOI] [PubMed] [Google Scholar]
- 30.Bittova, L., M. Sumandea, and W. Cho. 1999. A structure-function study of the C2 domain of cytosolic phospholipase A2. J. Biol. Chem. 274:9665–9672. [DOI] [PubMed] [Google Scholar]
- 31.Momsen, W. E., and H. L. Brockman. 1997. Recovery of monomolecular films in studies of lipolysis. Methods Enzymol. 286:292–305. [DOI] [PubMed] [Google Scholar]
- 32.Stahelin, R. V., F. Long, K. Diraviyam, K. S. Bruzik, D. Murray, and W. Cho. 2002. Phosphatidylinositol 3-phosphate induces the membrane penetration of the FYVE domains of Vps27p and Hrs. J. Biol. Chem. 277:26379–26388. [DOI] [PubMed] [Google Scholar]
- 33.Schlossman, M. L., D. Synal, Y. Guan, M. Meron, G. Shea-McGarthy, Z. Huang, A. Acero, S. M. Williams, S. A. Rice, and P. J. Viccaro. 1997. A synchrotron x-ray liquid surface spectrometer. Rev. Sci. Instrum. 68:4372–4384. [Google Scholar]
- 34.Born, M., and E. Wolf. 1980. Principles of Optics. Pergamon Press, Oxford, UK.
- 35.Parratt, L. G. 1954. Surface studies of solids by total reflection of x-rays. Phys. Rev. 95:359–369. [Google Scholar]
- 36.Nielsen, J., and D. McMorrow. 2000. Elements of Modern X-Ray Physics. John Wiley and Sons, Chichester, UK.
- 37.Buff, F. P., R. A. Lovett, and F. H. Stillinger. 1965. Interfacial density profile for fluids in the critical region. Phys. Rev. Lett. 15:621–623. [Google Scholar]
- 38.Mitrinović, D. M., A. M. Tikhonov, M. Li, Z. Huang, and M. L. Schlossman. 2000. Noncapillary-wave structure at the water-alkane interface. Phys. Rev. Lett. 85:582–585. [DOI] [PubMed] [Google Scholar]
- 39.Niggemann, G., M. Kummrow, and W. Helfrich. 1995. The bending rigidity of phosphatidylcholine bilayers. Dependence on experimental methods, sample cell sealing and temperature. J. Phys. II. 5:413–425. [Google Scholar]
- 40.Vogel, M., C. Münster, W. Fenzl, and T. Salditt. 1999. Thermal unbinding of highly oriented phospholipid membranes. Phys. Rev. Lett. 84:390–393. [DOI] [PubMed] [Google Scholar]
- 41.Thoma, M., M. Schwendler, H. Baltes, C. A. Helm, T. Pfohl, H. Riegler, and H. Möhwald. 1996. Ellipsometry and x-ray reflectivity studies on monolayers of phosphatidylethanolamine and phosphatidylcholine in contact with n-dodecane, n-hexadecane, and bicyclohexyl. Langmuir. 12:1722–1728. [Google Scholar]
- 42.Petkova, V., J. J. Benattar, and M. Nedyalkov. 2002. How to control the molecular architecture of a monolayer of proteins supported by a lipid bilayer. Biophys. J. 82:541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu, Y., and J. F. Nagle. 2004. Diffuse scattering provides material parameters and electron density profiles of biomembranes. Phys. Rev. E. 69:040901-1–040901-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagle, J. F., and S. Tristam-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pabst, G., M. Rappolt, H. Amenitsch, and P. Laggner. 2000. Structural information from multilamellar liposomes at full hydration: full q-range fitting with high quality x-ray data. Phys. Rev. E. 62:4000–4009. [DOI] [PubMed] [Google Scholar]
- 46.Shi, J., T. J. Blundell, and K. Mizuguchi. 2001. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310:243–257. [DOI] [PubMed] [Google Scholar]
- 47.Kelley, L. A., R. M. MacCallum, and M. J. Sternberg. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Membr. Biol. 299:499–520. [DOI] [PubMed] [Google Scholar]
- 48.Yee, A., X. Chang, A. Pineda-Lucena, B. Wu, A. Semesi, B. Le, T. Ramelot, G. M. Lee, S. Bhattacharyya, P. Gutierrez, A. Denisov, C. H. Lee, J. R. Cort, G. Kozlov, J. Liao, G. Finak, L. Chen, D. Wishart, W. Lee, L. P. McIntosh, K. Gehring, M. A. Kennedy, A. M. Edwards, and C. H. Arrowsmith. 2002. An NMR approach to structural proteomics. Proc. Natl. Acad. Sci. USA. 99:1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bystroff, C., V. Thorsson, and D. Baker. 2000. HMMSTR: a hidden Markov model for local sequence-structure correlations in proteins. J. Mol. Biol. 301:173–190. [DOI] [PubMed] [Google Scholar]
- 51.Petrey, D., Z. Xiang, C. L. Tang, L. Xie, M. Gimpelev, T. Mitros, C. S. Soto, S. Goldsmith-Fischman, A. Kernytsky, A. Schlessinger, I. Y. Koh, E. Alexov, and B. Honig. 2003. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins. 6(Suppl.):430–435. [DOI] [PubMed] [Google Scholar]
- 52.Wiener, M. C., and S. H. White. 1992. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J. 61:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nalefski, E. A., L. A. Sultzman, D. M. Martin, R. W. Kriz, P. S. Towler, J. L. Knopf, and J. D. Clark. 1994. Delineation of two functionally distinct domains of cytosolic phospholipase A2, a regulatory Ca2+-dependent lipid-binding domain and a Ca2+-independent catalytic domain. J. Biol. Chem. 269:18239–18249. [PubMed] [Google Scholar]
- 54.Clark, J. D., L. L. Lin, R. W. Kriz, C. S. Ramesha, L. A. Sultzman, A. Y. Lin, N. Milona, and J. L. Knopf. 1991. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell. 65:1043–1051. [DOI] [PubMed] [Google Scholar]
- 55.Gijón, M. A., D. M. Spencer, A. L. Kaiser, and C. C. Leslie. 1999. Role of phosphorylation sites and the C2 domain in regulation of cytosolic phospholipase A2. J. Cell Biol. 145:1219–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davletov, B., O. Perisic, and R. L. Williams. 1998. Calcium-dependent membrane penetration is a hallmark of the C2 domain of cytosolic phospholipase A2 whereas the C2A domain of synaptotagmin binds membranes electrostatically. J. Biol. Chem. 273:19093–19096. [DOI] [PubMed] [Google Scholar]
- 57.Lichtenbergova, L., E. T. Yoon, and W. Cho. 1998. Membrane penetration of cytosolic phospholipase A2 is necessary for its interfacial catalysis and arachidonate specificity. Biochemistry. 37:14128–14136. [DOI] [PubMed] [Google Scholar]
- 58.Perisic, O., H. F. Paterson, G. Mosedale, S. Lara-Gonzáles, and R. L. Williams. 1999. Mapping the phospholipid-binding surface and translocation determinants of the C2 domain from cytosolic phospholipase A2. J. Biol. Chem. 274:14979–14987. [DOI] [PubMed] [Google Scholar]
- 59.Evans, J. H., D. M. Spencer, A. Zweifach, and C. C. Leslie. 2001. Intracellular calcium signals regulating cytosolic phospholipase A2 translocation to internal membranes. J. Biol. Chem. 276:30150–30160. [DOI] [PubMed] [Google Scholar]
- 60.Murray, D., and B. Honig. 2002. Electrostatic control of the membrane targeting of C2 domains. Mol. Cell. 9:145–154. [DOI] [PubMed] [Google Scholar]