

Abstract
Docking and fusion of single proteoliposomes reconstituted with full-length v-SNAREs (synaptobrevin) into planar lipid bilayers containing binary t-SNAREs (anchored syntaxin associated with SNAP25) was observed in real time by wide-field fluorescence microscopy. This enabled separate measurement of the docking rate kdock and the unimolecular fusion rate kfus. On low t-SNARE-density bilayers at 37°C, docking is efficient: kdock = 2.2 × 107 M−1 s−1, ∼40% of the estimated diffusion limited rate. Full vesicle fusion is observed as a prompt increase in fluorescence intensity from labeled lipids, immediately followed by outward radial diffusion (Dlipid = 0.6 μm2 s−1); ∼80% of the docked vesicles fuse promptly as a homogeneous subpopulation with kfus = 40 ± 15 s−1 (τfus = 25 ms). This is 103–104 times faster than previous in vitro fusion assays. Complete lipid mixing occurs in <15 ms. Both the v-SNARE and the t-SNARE are necessary for efficient docking and fast fusion, but Ca2+ is not. Docking and fusion were quantitatively similar on syntaxin-only bilayers lacking SNAP25. At present, in vitro fusion driven by SNARE complexes alone remains ∼40 times slower than the fastest, submillisecond presynaptic vesicle population response.
INTRODUCTION
Neurotransmitters are released by Ca2+-triggered exocytosis, in which a 50-nm diameter synaptic vesicle fuses its lipid bilayer with that of the plasma membrane and releases its contents to the cell exterior (1–3). In neurons, triggered exocytosis occurs on submillisecond timescales after the onset of Ca2+ influx, implying that the key steps must involve rearrangement of local proteins and lipids (1,4). A variety of proteins, both cytoplasmic and membrane-anchored, are known to be critical to proper function of the Ca2+-triggered fusion machinery (5,6). Fusion sites include one or more SNARE complexes (“soluble N-ethylmaleimide-sensitive factor attachment protein receptor” complexes) (7). The internal structure of the cytoplasmic domain of a SNARE complex is known from crystallographic (8) and NMR studies (9,10). Each SNARE complex comprises a four-helix bundle. One helix is carried by synaptobrevin (syb, also called VAMP-2 or the v-SNARE protein), which is anchored in the vesicle membrane. The other three are carried by a binary complex between syntaxin (syx) and SNAP25 (here the “binary t-SNARE”), with syx anchored in the plasma membrane. Accumulating evidence indicates that the Ca2+ sensor is a fourth protein called synaptotagmin (syt), which is anchored in the vesicle (5,11–15).
For the much simpler case of in vitro fusion of protein-free vesicles in solution phase, a free-energy barrier arises from the need to disrupt favorable hydrophobic lipid-lipid and hydrophilic lipid-water contacts at intermediate geometries (16,17). Certain peptides act as fusagens (18) by lowering the barrier. Recent evidence suggests that the SNARE protein anchors may create a channel through which neurotransmitters emerge (19), possibly before complete lipid mixing.
A reconstituted model system for Ca2+-triggered exocytosis in principle provides a powerful tool allowing study of the structure and dynamics of vesicle-bilayer complexes as key components are added or subtracted one by one. The first reconstitution studies involved vesicle-vesicle fusion in bulk mixtures detected by dequenching of fluorescence from lipid-based probes. In groundbreaking work, Weber, Rothman, and co-workers showed SNARE-dependent fusion between vesicles reconstituted with syb and vesicles reconstituted with preformed syx/SNAP25 complexes (20,21). Fusion occurred slowly, on a timescale of tens of minutes, and fusion was not regulated by Ca2+. Inclusion of full-length syt in the v-SNARE vesicles enhanced the fusion rate, but there was no Ca2+ dependence (22). A truncation of syx lacking the N-terminal Habc region enhanced the fusion rate moderately (23). Tucker, Weber, and Chapman demonstrated SNARE-dependent fusion that was strongly enhanced on addition of Ca2+ and the cytoplasmic domain of syt (C2A-C2B, here C2AB), but fusion remained slow (24). Evidently, the minimal Ca2+-triggered fusion system comprises SNARE complexes plus syt. In all such bulk fusion assays, the slow overall fusion rate could arise from inefficient docking of the vesicles to each other or inefficient subsequent fusion.
Throughout this article, we use docking in its generic sense of stable binding of two vesicles to each other or of a vesicle to an adsorption site on a planar bilayer. Docking or tethering of synaptic vesicles to fusion sites at the plasma membrane in vivo almost surely involves proteins in addition to the SNARE components under study here (25,26).
In the more recent vesicle-planar bilayer fusion assays (27,28), fluorescently labeled v-SNARE vesicles interact with a planar lipid bilayer supported on glass and containing preformed binary t-SNARE complexes. Wide-field fluorescence microscopy enables direct observation of individual docking and fusion events as they occur in real time. This enables independent measurement of the intrinsic docking rate constant kdock and of the unimolecular rate of fusion kfus, as first demonstrated here, to our knowledge. Two recent studies used the single-vesicle method to quantify vesicle fusion kinetics, with quite different results. Simon and co-workers formed t-SNARE planar bilayers on glass by vesicle fusion (28). The v-SNARE vesicles docked to the bilayer in the absence of Ca2+ but fused only rarely (0.35% fusion probability in 50 s). Addition of 100 μM Ca2+ (or Mg2+) stimulated fusion of ∼15% (or 4%) of the docked vesicles in 50 s, much faster than in the earlier vesicle-vesicle assays. However, there is no physiological evidence that Mg2+ should stimulate fusion. Brunger, Chu, and co-workers developed an analogous assay using bilayers containing a low number density of binary t-SNAREs or of syx alone (no SNAP25) (27). The observed fusion of docked vesicles was thermally driven by laser heating. The population decay time was ∼20 s. Surprisingly, the presence or absence of SNAP25 had little effect on the fusion probability or timescale.
This work illustrates the full power of the single-vesicle methodology to separate docking and fusion kinetics. Using the same materials as in the Tucker-Chapman vesicle-vesicle assay (24) but with low t-SNARE copy number and low v-SNARE vesicle concentration, we observe efficient, SNARE-dependent docking followed by remarkably fast, Ca2+-independent fusion. The rate constant for formation of a SNARE complex by close v-SNARE/t-SNARE encounters is kdock = (2.2 ± 0.4) × 107 M−1 s−1, only three times smaller than the estimated diffusion-limited rate constant. Using a fast camera mode (5 ms/frame), we find that 80% of the docked vesicles undergo homogeneous, unimolecular fusion with kfus = 40 ± 15 s−1 at 37°C; ∼65% of all docked vesicles fuse <25 ms after docking. This fusion rate is ∼1000 times faster than observed in the two earlier single-vesicle assays. It is ∼104 times faster than the fusion rate estimated for vesicle-vesicle assays under the assumption that fusion, not docking, is rate limiting.
Docking on t-SNARE bilayers is completely blocked by preincubation of the v-SNARE vesicles with the cytoplasmic domain of the binary t-SNARE complex and by preincubation of the binary t-SNARE bilayer with the cytoplasmic domain of the v-SNARE. Evidently, formation of trans SNARE complexes in the absence of syt and other regulatory cofactors can drive fusion on a 25-ms timescale. Syntaxin-only bilayers lacking SNAP25 yielded docking and fusion rate constants indistinguishable from the binary t-SNARE bilayers. In vitro fusion driven by ternary SNAREs in the absence of Ca2+, syt, and other auxiliary proteins at present remains ∼40 times slower than the fastest population decay of readily releasable vesicle in vivo (29).
MATERIALS AND METHODS
Protein expression and reconstitution into proteoliposomes
Proteins are expressed, purified, and reconstituted into vesicles as described previously (24). The specific proteins include: mouse synaptobrevin-2, here referred to as syb; the cytoplasmic domain of synaptobrevin syb1–94 (residues 1–94); the full-length binary t-SNARE complex composed of rat syntaxin 1A (syx) and mouse SNAP25B; the cytoplasmic domain of the binary t-SNARE complex in which syx lacks the membrane anchor (residues 1–265); and the cytoplasmic domain of syx. Note that this form of SNAP25 is not palmitoylated, unlike that in vivo. The plasmids used to generate mouse synaptobrevin 2 (pTW2; (21)), the cytoplasmic domain of syb (pET-rVAMP2CD;(21)), the t-SNARE complex composed of rat syntaxin 1A and mouse SNAP25B (pTW34;(23)), full-length syntaxin 1A alone (pTrcHis), and the cytoplasmic domain of syx (residues 1–265, pTW12; (21)) were kindly provided by J. E. Rothman (Memorial Sloan-Kettering Cancer Center, New York, NY).
Syb, the binary t-SNARE complex, and full-length syx were expressed as described previously (21,23). Bacterial pellets were resuspended (∼10 ml per liter of culture) in resuspension buffer (25 mM Hepes-KOH, pH 7.4, 400 mM KCl, 10 mM imidazole, and 5 mM β-mercaptoethanol) and incubated for 20 min on ice after addition of 0.5 mg/ml lysozyme. Protease inhibitors (1 μg/ml aprotinin, pepstatinin A and leupeptin; 1 mM phenylmethylsulfonyl fluoride) were then added and samples were sonicated in 35 ml batches on ice for 2 × 45 s (50% duty cycle). Triton X-100 was added to 2.1% (v/v) and incubated for 15 min with rotation before centrifugation of the cell lysate at 27,000 × g for 30 min in a JA-17 rotor (Beckman, Fullerton, CA). For syb purifications, the supernatant was additionally clarified by centrifugation at 35,000 rpm in a Ti45 rotor (Beckman) for 60 min. After the addition of DNase I and RNase (Sigma (St. Louis, MO), 10 μg/ml), the supernatant was then incubated for >2 h at 4°C with Ni-NTA agarose (Qiagen, Valencia, CA; 0.5 ml of a 50% slurry per liter of cell culture) equilibrated in resuspension buffer. Beads were washed extensively with resuspension buffer containing 1% Triton X-100 and then washed with OG wash buffer (25 mM Hepes-KOH [pH 7.4], 400 mM KCl, 50 mM imidazole, 10% glycerol, 5 mM β-mercaptoethanol, 1% octylglucoside). The slurry was loaded onto a column, washed with 5–10 column volumes of OG wash buffer, and step eluted with OG wash buffer containing 500 mM imidazole.
The cytoplasmic domain of syb and the soluble t-SNARE complex were purified as described above, but all detergents were omitted from the wash buffers. The purified proteins were dialyzed against 25 mM Hepes-KOH (pH 7.4), 100 mM KCl, 10% glycerol, and 1 mM DTT.
The v-SNARE and t-SNARE vesicles were reconstituted by rapid dilution and dialysis and subsequently purified by flotation in an Accudenz (Accurate Chemical, Westbury, NY) step gradient as described previously (21). Phospholipids were from Avanti Polar Lipids (Alabaster, AL). v-SNARE vesicles were reconstituted using a lipid mix composed of 84% 1-palmitoyl, 2-oleoyl phosphatidylcholine (POPC), 15% 1,2-dioleoyl phosphatidylserine (DOPS), and 1.0% 1% head-labeled N-(tetramethylrhodamine)-1,2-diheptadecanoyl phosphatidylethanolamine (TMR-DHPE, Molecular Probes, Eugene, OR). t-SNARE vesicles were reconstituted in 85% POPC and 15% DOPS (mol/mol). The copy number of syb or binary t-SNARE complexes is varied by dilution into OG wash buffer with 500 mM imidazole before addition to the lipid film. The protein recovered in the purified vesicles was determined by an amido black protein assay. The final buffer was 25 mM Hepes-KOH (pH 7.4), 100 mM KCl. Our standard v-SNARE vesicles contain ∼100 copies/vesicle (protein/lipid ratio 1:240). Synaptic vesicles in vivo are estimated to carry ∼30 copies of syb (22,30). Our standard binary t-SNARE vesicles contain ∼0.8 copies/vesicle; ∼70–80% of the syb or t-SNARE complexes are oriented with their cytoplasmic domains facing outward. The fraction of t-SNAREs in the planar bilayer that face up into bulk solution was not determined, but this has been 50% in similar studies (27). Protein-free vesicles are reconstituted and purified as described for the SNARE-containing vesicles.
In one of the controls, the v-SNARE vesicles were treated with 0.4 μM of the botulinum neurotoxin BoNT B for 3.5 h at 37°C before addition above the t-SNARE bilayer. BoNT B cleaves most of the cytoplasmic domain of syb. The detailed protocol is published elsewhere (24).
Formation and characterization of t-SNARE bilayers
Supported lipid bilayers are formed by vesicle fusion on a clean, hydrophilic glass coverslip, a well established technique pioneered by Tamm and McConnell (31). Coverslips were cleaned by sonication in detergent for 1 h (CONTRAD 70, Decon Laboratories, King of Prussia, PA), thorough rinsing in Millipore water (Simplicity 185, Millipore, Billerica, MA), sonication in Millipore water for 30 min, and exhaustive rinsing in Millipore water. They were stored overnight at 90°C in Nano-Strip (Cyantek, Fremont, CA), a commercial mixture of H2O2 and concentrated H2SO4, and again rinsed thoroughly immediately before use. The bilayers “float” on a 1–2 nm layer of water and exhibit fast diffusion of lipids in both leaflets and of many peripheral membrane proteins (32,33). We studied a range of conditions for deposition of the t-SNARE bilayer by proteoliposome fusion on glass. The average number of t-SNAREs per liposome was either ∼0.8 (“low t-SNARE-density bilayers”, similar to the Brunger-Chu study (27) or ∼80 (“high t-SNARE-density bilayers”). The total lipid concentration was usually 25 μM, and deposition temperatures of 4°C, 25°C, and 37°C and times of 2–3 h were explored. After deposition, the bilayers were warmed to the desired temperature (either 25°C or 37°C) for 1 h and gently washed three times with buffer (60 cell volumes total) just before docking and fusion studies.
Wide-field fluorescence microscopy using 0.1% labeled lipids enabled assessment of the quality of the resulting t-SNARE bilayers on a 200 nm–20 μm scale. Bilayers formed directly at 37°C often exhibited dark, round defects of diameter ∼1 μm, evidently due to regions of bare glass. The v-SNARE vesicles docked efficiently to these defects. Deposition at 25°C led to smooth bilayers, but gave docking and fusion behavior that depended on deposition time in the range of 2–3 h. Deposition at 4°C followed by warming to 25°C or 37°C and washing minimized defects and thus became the preferred procedure.
Fluid-phase, variable temperature, tapping mode atomic force microscopy (AFM) was used to assess the quality of bilayers on a 10 nm–1 μm scale formed under conditions quite similar to those in the optical docking and fusion assay (Fig. 1). We used force-modulation etched silicon probes (FESP tips, Veeco Metrology Group, Santa Barbara, CA) in a commercial instrument (NanoScope IV, Digital Instruments, Buffalo, NY). Samples were scanned at 2 Hz. Fusion of protein-free vesicles on glass leads to the expected relatively smooth surface with few defects. “Plowing” the bilayer surface to expose glass and rescanning the same region reveals the apparent height of the deposited material as 4.5 ± 0.7 nm, consistent with the expected height of a POPC/DOPS bilayer. The apparent surface roughness is similar to that of the underlying glass surface, root-mean-square (rms) ∼0.3 nm. In contrast, t-SNARE bilayers deposited using a larger copy number of ∼80 t-SNARE/liposome and 250 μM total lipid exhibit large “mountains” of aggregated protein/lipid material above the smooth bilayer terrace that withstand the raster scan of the AFM tip. These mountains are irregular in shape, with lateral dimensions on the order of tens of μm and height ∼50 nm (not shown). Such surfaces were not studied further. Deposition using 80-copy t-SNARE vesicles at 10 times lower overall vesicle concentration (25 μM total lipid) leads to the smaller aggregates we call “mounds” (∼0.4 μm laterally and ∼10 nm taller than the bilayer itself (Fig. 1 a). We refer to these surfaces as the “high t-SNARE-density” bilayers. By direct count, the surface density of the mounds is 0.2 ± 0.1 μm−2, remarkably consistent with the estimated density of v-SNARE vesicle binding sites on the same bilayers (T0 = 0.1 μm−2, below). After deposition with the preferred t-SNARE copy number of ∼0.8 and 25 μM total lipid, AFM images (Fig. 2 b) look very similar to those of a protein-free surface. The rms roughness is again ∼0.3 nm. There is no evidence of protein-related features, probably because the AFM tip pushes small t-SNARE monomers or clusters along the surface as it rasters. We refer to these surfaces as the “low t-SNARE-density” bilayers.
FIGURE 1.

AFM images of t-SNARE bilayers. Tapping-mode AFM images of 10 μm × 10 μm patches of t-SNARE bilayer formed by deposition of vesicles on a hydrophilic glass substrate at T = 37°C. Relatively high regions are bright yellow, whereas relatively low regions are dark brown. Scans (10 μm) of apparent height along the white, horizontal lines are shown below each image (0–10 nm height scale at left). Red triangles are position markers not of interest here. (a) Deposited from vesicles with ∼80 t-SNARE copies on average at total lipid concentration 25 μM. Image at left (10-min incubation) shows ridges of lipid bilayer (yellow) with nominal height 4.3 nm above bare glass (brown). Image at right (60-min incubation) shows large regions of flat bilayer (brown). The bright yellow spots are evidently “mounds” of t-SNARE material that rise 5–10 nm above the bilayer surface and have lateral dimensions of ∼400 nm. (b) After 60-min incubation time with vesicles of ∼0.8, t-SNARE copies on average at total lipid concentration 25 μM. No mounds appear. The root mean-square vertical displacement is 0.3 nm, comparable to that of bare glass.
FIGURE 2.

Fast fusion of v-SNARE vesicles on low t-SNARE-density bilayers. (a) Field of docked, protein-free vesicles 50 s after addition to a low t-SNARE-density bilayer. Sparse docking, but no fusion, has occurred. (b) Field of v-SNARE vesicles 50 s after addition to a low t-SNARE-density bilayer. The diffuse fluorescence is due to labeled lipids that have fused into the bilayer and dispersed. (c) Sequence of images spaced by 50 ms showing fusion of a v-SNARE vesicle and outward radial diffusion of the labeled lipids. (d) Relative integrated intensity in a circle of radius 0.7 μm, I0.7μm(t), for a docked but unfused vesicle (frame a) using 40-ms frames. (e) Histogram of relative integrated fluorescence intensities for docked, protein-free vesicles (frame a). (f) Two examples of I0.7μm (t) for vesicles that undergo fast fusion as in frame b. In the lower trace, fusion occurs within one camera frame of docking, followed by diffusion out of the circle on a ∼1-s timescale. In the upper trace, the vesicle docks and waits three frames (marked by arrow) before fusion. Dashed line shows rising baseline due to leakage of labeled lipids from surrounding fusion events into the circle of integration. (g) Log plot of I0.7μm (t) for a single, well-isolated vesicle (no rising baseline). Lines are calculated by integrating Eq. 1 from r = 0 to 0.7 μm at each time t for various values of the diffusion coefficient Dlipid as shown. Dlipid = 0.6 ± 0.1 μm2 s−1 best fits the data.
Docking and fusion assay by fluorescence microscopy
A modified commercial wide-field microscope (Eclipse TE2000-U, Nikon, Melville, NY) enables excitation of fluorophores at the glass/water interface by “through the objective” total internal reflection (TIR) (34). A 0.1 mW, cw laser at 514 nm illuminates a ∼200-nm thick, 50-μm diameter cylinder including the lipid bilayer. We use a 100×, numerical aperture = 1.45 oil-immersion objective (Olympus, Melville, NY). Fluorescence from TMR-DHPE passes a 565–595 nm bandpass filter (D580/30, Chroma, Rockingham, VT) and is imaged onto a fast charge-coupled device camera (I-Pentamax, Roper Scientific, Trenton, NJ), yielding digital movies of fluorescence intensity versus time. The camera pixels are square, 15 × 15 μm2, corresponding to 150 × 150 nm2 in real space at the sample. Most movies used a 250 × 250 pixel region of interest and 40-ms frames for periods of 2–10 s. For accurate measurement of the distribution of times to fusion tfus, we restricted the region of interest to 150 × 98 pixels and used the “virtual chip” mode of the camera. This enables 5-ms or 10-ms frames that more accurately capture the time delay between firm docking and fusion.
The t-SNARE bilayer is formed on a glass coverslip that serves as the TIR window of the fluorescence microscope and also forms one wall of a small-volume flow cell. The cell volume is cylindrical, 1 mm tall × 8 mm diameter (50 μL). Quantitative measurement of docking kinetics requires a reproducible method for placing v-SNARE vesicle solution at a known bulk concentration in contact with the t-SNARE bilayer surface rapidly and uniformly. Using a pipette, we flow four times the cell volume of v-SNARE vesicle solution through the cell, fully exchanging solution in ∼2 s. Docking/fusion movies begin 10–20 s later, after manually focusing the microscope. Docking traces up to 16 min long are acquired in time-lapse mode, for which the laser illuminates the sample for 40 ms during each 2-s frame interval.
The mean fluorescence intensity of a docked but unfused vesicle is smaller than that of a docked and fused vesicle by a factor of 2–4, as described below. This requires a different procedure for measuring the surface density of unfused and fused vesicles. Fortunately, each experimental condition yielded essentially no fusion or essentially complete fusion of docked vesicles, so it was not necessary to blend the procedures. For docking without fusion, a centroid algorithm locates and counts vesicles in each frame using an intensity threshold. This direct count yields the total surface concentration of docked but unfused vesicles, D(t) in the kinetics model of Eq. 2 below. When fast fusion dominates, the laser seldom illuminates a docked vesicle within the 2-s frames of a time-lapse movie (laser-on duty factor of only 0.02). Instead, most vesicles are imaged only as fusion products, which appear as dim, diffuse clouds. Therefore we measure the total fluorescence intensity versus time, Itot(t), and calibrate the mean intensity per fused vesicle using 2-s long movies acquired with 40 ms frames and low vesicle density, so that single docking and fusion events are captured faithfully. A correction for slow photobleaching was also applied. The resulting absolute number density of fused vesicles is accurate to ±10%.
Enhancement of fluorescence intensity after fusion
We show below that fusion causes a sudden enhancement of a factor of 2–4 in the fluorescence intensity from the vesicle's labeled lipids. The fused vesicle's lipids lie entirely in the most intense part of the evanescent field. Before fusion, a 50-nm vesicle samples a modest range of intensities. However, this is a small effect because the 50-nm vesicle diameter is substantially smaller than the estimated 1/e penetration depth of the evanescent field into the bilayer/water layer of 150 nm. In separate experiments carried out on a bulk sample of identically labeled v-SNARE vesicles in a standard fluorimeter, we measured an enhancement of the entire fluorescence spectrum by a factor of 1.8 after complete solubilization of the lipids with detergent (data not shown). We attribute this to dequenching of TMR fluorescence, perhaps including dissociation of TMR dimers. Two polarization effects further enhance the postfusion intensity. The polarization of the TIR laser lies parallel to the plane of the bilayer (s-polarization), and both the absorption and emission transition dipole moments of TMR also preferentially orient parallel to the local bilayer plane (35). First, the TMR molecules are more efficiently excited by the laser after fusion, when all transition dipoles lie near the bilayer plane, than before fusion, when they are distributed randomly on the spherical surface of the vesicle. A geometric average of cos2θ (angle between the laser polarization axis and the transition dipole) over a random distribution of orientations on a sphere versus a disk shows that excitation could be enhanced by as much as a factor of 1.5. Second, the fluorescence collection efficiency also improves after fusion, because dipoles with horizontal polarization emit more of their intensity toward the objective than those with vertical polarization. The nearby glass surface also differentially affects fluorescence collection, further favoring postfusion emission (36,37). The combination of dequenching and the two polarization effects combine to explain the factor of 2–4 enhancement of intensity observed immediately after fusion.
RESULTS
Single-vesicle docking and fusion assay
This section describes the overall behavior of the assay, including evidence for full vesicle fusion and a qualitative discussion of various control experiments. Quantitative details of docking and fusion kinetics follow. We focus initially on the behavior of low t-SNARE-density bilayers (Materials and Methods) at 37°C. There is no Ca2+ or Mg2+ in these initial studies. The standard concentration of total lipid in the v-SNARE vesicle solution placed above the bilayer is 2.5 ± 0.8 μM, which translates to bulk vesicle concentration of V0 = (1.3 ± 0.4) × 10−10 M.
Direct observation of vesicle docking and full fusion
Docking and fusion of vesicles is described in Fig. 2. As an example of docking without fusion, Fig. 2 a shows a field of fluorescently labeled, protein-free vesicles at t = 50 s after their addition above the t-SNARE bilayer. Supplementary Movie 1 shows “swarming” behavior of protein-free vesicles that approach the surface and linger for several frames, but do not dock to form stable vesicle-planar bilayer complexes. The vesicles that do stick to the surface dock abruptly and subsequently move <70 nm rms. Repeated gentle washing with buffer does not remove any of the docked vesicles. Quantitative kinetics measurements show that compared with v-SNARE-containing vesicles, relatively few protein-free vesicles bind to low t-SNARE-density bilayers. For one such docked vesicle, Fig. 2 d shows the integrated fluorescence intensity within a circle of radius 0.7 μm centered at the docking position of one vesicle. We call this I0.7μm(t). On a 2-min timescale, we observe no evidence of either vesicle fusion or partial lipid mixing; the latter would lead to a gradual decrease in vesicle intensity and increase in background intensity over time. The distribution of fluorescence intensities of the protein-free vesicles of the species that bind to the bilayer is shown in Fig. 2 e. The long tail toward high intensity could be due to vesicle dimers, trimers, etc. as well as a broad distribution of single-vesicle sizes.
In sharp contrast, v-SNARE vesicles dock efficiently and fuse promptly on the same t-SNARE bilayers (Supplementary Movies 2 and 3). Fig. 2 b shows a snapshot of a t-SNARE bilayer the same 50 s after addition of a standard aliquot of v-SNARE vesicles. The high-intensity spots are the few remaining vesicles that have docked but not fused. The bright, hazy background is due to labeled lipids that have fused with the planar bilayer and undergone free lateral diffusion. It is clear by inspection that v-SNARE vesicles dock on the t-SNARE bilayer much more efficiently than protein-free vesicles. Movies 2 and 3 show that there is typically little or no motion away from the point of initial “contact” of the vesicle with the surface. Compared with protein-free vesicles, fewer “swarming” v-SNARE vesicles are observed.
For those vesicles that dock for at least one 40-ms camera frame before fusing, I0.7μm(t) shows a characteristic sharp increase in intensity by a factor of ∼2 that persists for 1–2 frames (Fig. 2 f), followed by a monotonic, nonexponential decay on a timescale of ∼1 s. With 5-ms frames, the intensity increase is a factor of 2–4 due to improved time resolution (see below). As seen in Movies 2 and 3 and in the sequence of images in Fig. 2 c, such events look like fast “explosions” with good circular symmetry. The sharp rise in fluorescence intensity immediately after fusion is due to dequenching of the TMR labels and to polarization effects (Materials and Methods). Fusion of other vesicles nearby causes a slowly rising baseline on most traces.
The sharp rise in fluorescence occurs in one 5-ms camera frame or less. This sets an upper limit of ∼5 ms on the timescale of lipid mixing. Evidently, the vast majority of events yield complete mixing of both vesicle leaflets with the planar bilayer. Fast hemifusion (mixing of the outer leaflet of the vesicle with the upper leaflet of the bilayer) would be readily observed as a partial “explosion” that leaves a bright core of ∼40% of the original intensity behind. The core would be clearly visible if it persisted at least 0.3 s after hemifusion. In fact, only ∼2% of the docked vesicles exhibit such behavior. These relatively rare events could be hemifusion, but they might also arise from docking of vesicle dimers or trimers (two or three v-SNARE vesicles bound together) and subsequent fusion of just one of the vesicles.
Fast diffusion of labeled lipids out of the observation circle makes the burst in I0.7μm(t) narrow in time. Assume that prompt, complete vesicle fusion instantaneously introduces a large, highly localized concentration of labeled lipids that subsequently diffuse radially outward in two dimensions with diffusion constant D. The time-dependent diffusion equation then predicts a Gaussian concentration profile that expands radially versus time (38):
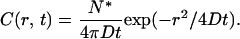 |
(1) |
Here, C has dimensions of molecules/μm2 and N* is the total number of labeled lipids released at t = 0. At each time, C(r, t) peaks at r = 0; the radius at which C(r, t) falls to half its peak value is 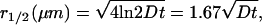 with D in μm2 s−1. In Fig. 2 g, we show a log plot of I0.7μm(t) for a single, high signal/noise, well-isolated fusion event with little baseline drift. The smooth curves are calculations of I0.7μm(t) obtained by integrating C(r,t) from r = 0 to 0.7 μm at each time, with different values of D assumed. The calculation captures the nonexponential shape of the decay and mimics the data semiquantitatively for D = 0.6 μm2 s−1. This is comparable to lipid diffusion coefficients measured by single-lipid tracking of gold-labeled phosphatidylethanolamine in a supported egg PC/cholesterol bilayer at 25°C (0.3–0.7 μm2 s−1. (39).
with D in μm2 s−1. In Fig. 2 g, we show a log plot of I0.7μm(t) for a single, high signal/noise, well-isolated fusion event with little baseline drift. The smooth curves are calculations of I0.7μm(t) obtained by integrating C(r,t) from r = 0 to 0.7 μm at each time, with different values of D assumed. The calculation captures the nonexponential shape of the decay and mimics the data semiquantitatively for D = 0.6 μm2 s−1. This is comparable to lipid diffusion coefficients measured by single-lipid tracking of gold-labeled phosphatidylethanolamine in a supported egg PC/cholesterol bilayer at 25°C (0.3–0.7 μm2 s−1. (39).
Because the vesicle's lipids promptly and completely dissolve in the bilayer in 5–10 ms or less, we infer that release of the vesicle's contents must occur at least that fast (“content-mixing” in the vesicle-vesicle literature). The overall kinetics of fusion for the population of docked vesicles is a separate question addressed in detail below.
Requirement of v-SNAREs plus binary t-SNAREs or syntaxin alone
Control studies indicate that both docking and fusion are strongly enhanced by the simultaneous presence of v-SNAREs in the vesicles and binary t-SNAREs in the planar bilayer. The control results differ from the baseline data both in the greatly reduced number density of effective vesicle binding sites and in the inability of the docked vesicles to fuse. The controls show that:
v-SNARE vesicles dock very little on protein-free bilayers and do not fuse.
Protein-free vesicles dock to a moderate extent on t-SNARE bilayers but do not fuse.
The docking of both protein-free vesicles and v-SNARE vesicles on t-SNARE bilayers is completely blocked by preincubation of the bilayer for 5 min with 5 μM of the cytoplasmic domain of syb (cd-VAMP). Presumably the cytoplasmic domain of syb binds efficiently to t-SNAREs to make cis SNARE complexes and prevent subsequent docking via trans SNAREs. The same effect evidently makes the surface less sticky toward protein-free vesicles.
The complementary control experiment preincubates the v-SNARE vesicles with cytoplasmic binary t-SNAREs (the cytoplasmic domain of syx complexed with SNAP25, 5 μM) for 5 min before addition to the t-SNARE planar bilayers. Again, docking is completely blocked.
Interestingly, we find that preincubation of the v-SNARE vesicles in 5 μM of cytoplasmic domain syx by itself also completely blocks docking of the vesicles to the t-SNARE bilayer. This suggests formation of a syb-syx complex that blocks full SNARE formation.
Bilayers formed from vesicles harboring ∼1 copy of full-length syx (without SNAP25) under otherwise identical conditions exhibit efficient docking and fast fusion of v-SNARE vesicles. The syx-only results are quantitatively indistinguishable from the binary t-SNARE results, as described below.
Effects of laser intensity, Ca2+ and Mg2+, temperature, and t-SNARE density
Fusion in our system is fast in the absence or presence of dipositive cations. Preincubation of v-SNARE vesicles in 1 mM Ca2+ or Mg2+ changes the docking kinetics on low t-SNARE-density bilayers somewhat, but does not affect the fusion rate within experimental uncertainty. Quantitative details are given below. To test for possible thermal effects on fusion, we varied the laser intensity up and down by a factor of two, but observed no qualitative change in the fast fusion behavior. In addition, the degree of fusion observed on a particular bilayer is independent of the time delay between addition of the v-SNARE vesicles and the onset of laser illumination.
Docking and fusion are comparably efficient at 25°C and 37°C, although we carried out careful quantitative work only at 37°C. In several experiments, we deposited the low t-SNARE-density bilayer at 4°C for 2.5 h, warmed the bilayer to 25°C for 1 h, washed, and studied the docking and fusion at 25°C. As at 37°C, the v-SNARE vesicles dock efficiently and the vast majority fuse promptly at 25°C. However, after fusion, the labeled lipids often disperse into irregular, asymmetric patterns that suggest confinement in distinct lipid domains. The bilayer may segregate into domains at 4°C, with the t-SNAREs preferring one domain type over the other (40). Evidently the domains do not completely disperse in 1 h at 25°C, whereas the bilayer behaves homogeneously after 1 h at 37°C.
Finally, when the number of t-SNAREs per proteoliposome is increased from ∼0.8 to ∼80 to form the high t-SNARE-density bilayers (Materials and Methods), we observe much less efficient docking of v-SNARE vesicles and no fusion. These are the same surfaces that exhibit large mounds of t-SNARE protein in AFM images (Fig. 1 a). This strongly suggests that the t-SNAREs on the high protein density surface are somehow entangled and unable to form good trans SNARE complexes with syb. Addition of Ca2+ to the high t-SNARE density surfaces enhances the docking somewhat, but does not induce fusion on a 1-min timescale.
Quantitative docking kinetics
The simplest adsorption kinetics mechanism assumes that diffusion of solution-phase v-SNARE vesicles is sufficiently fast to maintain a concentration at the surface equal to the bulk v-SNARE vesicle concentration V0. This is equivalent to the “well-stirred reactor” model described by the simple kinetics mechanism:
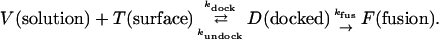 |
(2) |
Here V represents the solution-phase v-SNARE vesicles; T represents the effective surface t-SNARE binding sites; D represents docked but unfused vesicles on the surface; kdock and kundock are adsorption and desorption rate constants; F represents fusion products on the surface; and kfus is the unimolecular rate of fusion of docked vesicles. We use surface concentrations in vesicles-μm−2 and bulk concentrations in molar units, so kdock has units M−1s−1, and kundock and kfus have units s−1. The sum D(t) + F(t) includes all vesicles that have docked up to time t, whether or not they have subsequently fused. The instantaneous total vesicle docking rate is the sum Rdock(t) = dD/dt + dF/dt in vesicles μm−2 s−1.
Under the assumptions that: 1), the bulk vesicle concentration V = V0 is independent of time (many more vesicles than docking sites, which is true by design); 2), kundock is effectively zero on the 16-min timescale of the experiments (as observed); and 3), diffusion rapidly fills the boundary layer just above the surface as adsorption occurs (which is only approximately true); the sum of docked and fused vesicles is a rising single exponential:
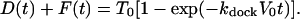 |
(3) |
Here T0 is the initial concentration of active surface sites and V0 is the bulk vesicle concentration. Within the model, D(t) + F(t) has initial slope Rdock(t = 0) = kdockT0V0 and an asymptotic limit of T0.
Examples of docking kinetics data are shown in Fig. 3. In Fig. 3 b, we show docking-plus-fusion kinetics plots for v-SNARE vesicles added to a bilayer formed with 0.8 t-SNARE/vesicle (the low t-SNARE-density bilayer) and to a bilayer formed with 80 t-SNARE/vesicle (the high t-SNARE-density bilayer). In both cases, the nominal concentration of total lipid was 2.5 ± 0.8 μM, which translates to bulk vesicle concentration of V0 = (1.3 ± 0.4) × 10−10 M, assuming 2.0 × 104 lipids/vesicle (outer leaflet of 50 nm diameter, inner leaflet of 40 nm diameter, 0.65 nm2 per vesicle head). Clearly the initial docking rate Rdock and the effective total density of binding sites T0 are at least 20 times larger for the low t-SNARE-density bilayer than for the high t-SNARE-density bilayer. Fig. 3 c shows examples of control measurements for protein-free vesicles docking to the low t-SNARE-density bilayer and for v-SNARE vesicles docking to the protein-free bilayer. Compared with v-SNARE vesicles on low t-SNARE-density bilayers (Fig. 3 b), T0 is evidently ∼10 times smaller for protein-free vesicles and ∼100 times smaller for the protein-free bilayer. Quantitative fits will confirm these visual estimates. In several of these cases, the intrinsic docking rate constant will prove to be substantial; the primary effect of the controls is to drastically reduce the density of effective binding sites.
FIGURE 3.

Docking kinetics. (a) Schematic of flow cell for vesicle docking kinetics experiments. (b) Plots of total surface density of docked vesicles versus time, whether fused or unfused (Materials and Methods). (□) v-SNARE vesicles on low t-SNARE-density bilayer. Red line is least-squares fit to simple adsorption model (Eq. 3), yielding kdock = 1.1 × 107 M−1 s−1 and adsorption site density T0 = 4.4 μm−2. Black line is hand-adjusted best fit to the diffusion-adsorption model (see text and Supplementary Material), with kdock = 2.0 × 107 M−1 s−1 and T0 = 4.7 μm−2. The values of Dves = 3.3 μm2 s−1 and V0 = 1.7 × 10−10 M were held fixed. The dot-dash line shows the prediction of the simple kinetics model (Eq. 3) using the same values of kdock and T0. (Red circles) v-SNARE vesicles on high t-SNARE-density bilayer. Best-fit value is T0 = 0.10 μm−2, 40 times lower than the effective site density on the low t-SNARE-density bilayer. (Blue triangles) Protein-free vesicles on low t-SNARE-density bilayer. These data are repeated in frame c to emphasize the change of vertical scale. (c) Docking of v-SNARE vesicles on low t-SNARE-density bilayers as in frame b for two controls. Note vertical scale change from frame b. Lines are best fits to the diffusion-adsorption model. (Blue inverted triangles) Protein-free vesicles on low t-SNARE-density bilayer; T0 = 0.4 μm−2. (Red circles) v-SNARE vesicles on protein-free bilayer; T0 = 0.03 μm−2. (See Table 1 for all fitting results.) (d) Summary of absolute docking site density for v-SNARE vesicles on low t-SNARE-density bilayer (leftmost bar) and for all controls as indicated. Blue bars refer to left-hand scale, red bars to right-hand scale. “v-SN ves” means v-SNARE vesicle; “t-SN” means binary t-SNARE; “PF” means protein-free. Preincubations of the binary t-SNARE bilayer with cytoplasmic domain syb, and of the v-SNARE vesicles with the cytoplasmic domain of syx and with the cytoplasmic domain of the binary t-SNARE as shown. (Below) Percent of fusion-active vesicles, defined as the number of vesicles that fuse in 4 s divided by the total number of vesicles that dock in 4 s. (e) Docking curves for v-SNARE vesicle on low t-SNARE-density bilayers with addition of 1 mM Mg2+ and Ca2+, compared with standard buffer as indicated.
For v-SNARE vesicles docking to the low t-SNARE-density bilayer, a least-squares fit of the data to the equation D(t) + F(t) = T0 [1 – exp(–kexptt)] is shown as the thin red line in Fig. 3 b. The best-fit values are T0 = 4.4 μm−2 and kexpt = 0.00182 s−1, yielding kdock = kexpt/V0 = 1.1 × 107 M−1 s−1 (with V0 = 1.7 × 10−10 M as below). The reproducibility across different samples is ∼±25% in kdock and ±40% in T0. The fit is reasonably good, but the simple model cannot accurately capture the detailed curvature of the data. This arises primarily from depletion of the vesicle concentration at the surface by adsorption, as shown below. In addition, the binding sites likely vary in t-SNARE cluster size and in the probability that a vesicle docks irreversibly when it touches a site. The resulting heterogeneity in kdock could give rise to faster than average docking initially, as the more favorable sites would be filled first.
We further tested the model by doubling the bulk vesicle concentration to 2V0. The resulting data (not shown) could not be fit by the simple model with values of kdock and T0 at all comparable to the best-fit values derived from the data taken at V0. This led us to investigate a more accurate “diffusion-adsorption” model that explicitly includes the effects of depletion of the concentration of v-SNARE vesicles at the surface. This standard “unstirred reactor” problem must be solved numerically with assumed input values of T0, V0, kdock, and Dves, the diffusion coefficient of v-SNARE vesicles in bulk buffer solution (41). The details are given in the Supplementary Material. In practice, we fixed Dves = 3.3 μm2 s−1, as directly measured previously for comparable 50-nm diameter vesicles (42). We treated T0 and kdock as manually adjustable parameters and V0 as a mildly adjustable parameter within its estimated ±30% accuracy. Extensive exploration showed that the data can be very well fit, including the details of curvature, using the best estimate of V0 = 1.3 × 10−10 M. However, other data sets, especially the docking of v-SNARE vesicles in the presence of Ca2+ and Mg2+, were not well fit unless V0 was adjusted mildly upward to 1.7 × 10−10 M. For consistency, we chose to fix V0 at this latter value in all of the fitting, leaving only kdock and T0 as adjustable parameters.
As shown in Fig. 3 b, the diffusion-adsorption model fits the curvature of the kinetics plot significantly better than the simple model. In the example in Fig. 3 b, the best-fit values are kdock = 2.0 × 107 M−1 s−1 and T0 = 4.7 μm−2. In conditions for which adsorption is efficient, like the v-SNARE plus t-SNARE data shown, the main effect of the diffusion-adsorption model compared with the simple model is to increase kdock by a factor of ∼2; T0 remains essentially the same. As expected, the effect on kdock is more modest for the controls with smaller T0, since diffusion is then better able to keep up with adsorption. The need to include diffusion explicitly is underscored by the dot-dash curve in Fig. 3 b, which shows the prediction of the simple adsorption model using the best-fit kinetic parameters from the diffusion-adsorption model. The diffusion-adsorption model also shows clearly why the approach to the asymptote T0 is so slow. According to the model, it would take 2000 s = 33 min for the surface density to reach 95% of its asymptote. In practice, we found that photobleaching and defocusing of the microscope limited our ability to obtain accurate data beyond ∼1000 s.
The “best-fit” diffusion-adsorption result in Fig. 3 b is not a least-squares fit, but rather the result of trial and error. The calculated adsorption curve is highly sensitive to the combination of input parameters. For fixed Dves, the product kdockT0 controls the initial slope (as in the simple model); T0 controls the long-time asymptote; and kdock and V0 affect the curvature of the approach to saturation of the surface sites. The most important derived parameters kdock and T0 separate reasonably well, but we are unable to set statistically valid error limits on the results. Our manual fitting procedure consistently locks on to very similar “best-fit” values for a given data set, and kdock is reproducible across nominally identical samples to ∼±25%. The best-fit T0 varies by ±50% across nominally identical samples. Much of the variability is probably due to real differences in the quality of the t-SNARE binding sites from sample to sample.
Averages of the resulting model parameters from fits to the diffusion-adsorption model under various experimental conditions are collected in Table 1. Each entry is the mean of 3–4 independent determinations on separate bilayers. For v-SNARE vesicles docking to the low t-SNARE-density bilayer, we find kdock = (2.2 ± 0.4) × 107 M−1 s−1 and an effective density of binding sites T0 = 4 ± 1 μm−2. Error estimates indicate the range of “best-fit” values across triplicate experiments. In contrast, the high t-SNARE-density bilayer shows efficient docking to a much smaller density of binding sites: kdock = (1.2 ± 0.7) × 107 M−1 s−1 and T0 = 0.10 ± 0.05 μm−2. The low t-SNARE-density bilayer contains 100 times fewer t-SNAREs but evidently exhibits 40 times as many effective docking sites. We argue below that the binding sites on the high t-SNARE-density bilayer are the large domains of aggregated t-SNAREs directly observed by AFM (Fig. 1 a).
TABLE 1.
Summary of docking and fusion kinetics parameters
| Conditions* | kdock (M−1 s−1)† | T0 (site μm−2) † | kfus (s−1) |
|---|---|---|---|
| v-SN ves on t-SN bilayer (0.8 copy) | (2.2 ± 0.4) × 107 | 4 ± 1 | 40 ± 15‡ |
| Protein-free ves on t-SN bilayer (0.8 copy) | (2.8 ± 0.5) × 107 | 0.4 ± 0.1 | <0.0002§ |
| v-SN ves on protein-free bilayer | (8 ± 4) × 106 | 0.03 ± 0.01 | <0.0002§ |
| v-SN ves + 1 mM Mg2+ on t-SN bilayer (0.8 copy) | (1.4 ± 0.4) × 107 | 8 ± 1 | >15¶ |
| v-SN ves + 1 mM Ca2+ on t-SN bilayer (0.8 copy) | (5 ± 3) × 106 | 25 ± 10 | >15¶ |
| v-SN ves on t-SN bilayer (80 copy) | (1.2 ± 0.7) × 107 | 0.10 ± 0.05 | <0.0002§ |
t-SN bilayer (0.8 copy) and (80 copy) denote the low and high t-SNARE-density bilayers (Materials and Methods). See Fig. 1 for AFM images of these surfaces. Other abbreviations as in Fig. 3. Refer to Fig. 3 for additional control results.
Results of least-squares fits to the diffusion-adsorption model (see text and Supplementary Material), with Dves fixed at 3.3 μm2 s−1 (42) and V0 fixed at 1.7 × 10−10 M (see text). Each entry is the mean of 3–4 independent determinations on separate bilayers. Uncertainties span the range of results.
From fit of histogram of tfus shown in Fig. 4 b.
Conservative upper bound from the absence of vesicle fusion in 500 s movies.
Lower bound from histogram of tfus with 40-ms bins; 5-ms movies were not taken with Ca2+ and Mg2+.
To test the effect on docking of removal of SNAP25 from the binary t-SNARE complex, we formed low syx-density bilayers in exactly the same way and added v-SNARE vesicles as before. Three bilayers of each type were directly compared on the same day. These resulting docking curves for low density binary t-SNARE bilayers and low density syx-only bilayers coincide within ±20% (data not shown). Inclusion of SNAP25 had no measurable effect on docking within our experimental reproducibility.
The quantitative analyses of the other controls reveal 10–40 times smaller effective binding site density T0 (Fig. 3 d and Table 1). All of the intrinsic docking rate constants lie in the range 0.5 × 107–2.8 × 107 M−1 s−1. Docking to the few available adsorption sites is fairly efficient in all the control experiments. All values of kdock lie within a factor of 10 of theoretical estimates of the diffusion-limited docking rate constant to surface sites of effective radius R ∼ 7 nm (Discussion). For very low T0, surface imperfections may provide the bulk of the binding sites. However, the t-SNARE bilayers bind protein-free vesicles 10% as efficiently as they bind v-SNARE vesicles, suggesting a significant nonspecific interaction that cannot lead to fusion.
Fig. 3 e compares representative docking kinetics plots for v-SNARE vesicles on the low t-SNARE-density bilayer in plain buffer, in the presence of 1 mM Mg2+, and in the presence of 1 mM Ca2+. The solid lines are best fits to the diffusion-adsorption model. The three curves are qualitatively similar, but they differ substantially in their curvature. The plain buffer experiments rise more rapidly at early time. This effect requires additional study, but the diffusion-adsorption model consistently finds significantly lower values of kdock and higher values of T0 in the presence of Mg2+ or Ca2+ (Table 1). In future investigations, it will be important to improve the accuracy of V0 and Dves if subtle variations in kdock and T0 are to be interpreted with confidence.
In a final qualitative control, the v-SNARE vesicles were pretreated with BoNT B (as described in Materials and Methods), which acts by cleavage of the helix-forming cytoplasmic domain of syb. On the standard, low t-SNARE-density bilayer, docking of the pretreated vesicles is inhibited by a factor of three at all times relative to the untreated vesicles (data not shown). As judged by movies with 40-ms frames, those vesicles that dock proceed to fuse on a similar timescale to the normal v-SNARE vesicles, suggesting they have sufficient remaining intact syb to enable ternary SNARE-complex formation.
Quantitative fusion kinetics
In this section, we describe the fusion of v-SNARE vesicles on the low t-SNARE-density bilayer in quantitative detail. The single-vesicle assay enables measurement of the distribution of tfus, the residence time of firmly docked vesicles on the surface before fusion. From the usual 40 ms/frame movies, which give good signal/noise ratio, we found that the time between docking and fusion is usually <80 ms, i.e., two frames or less (Fig. 2 f). This made it possible to set a lower limit kfus > 15 s−1 but difficult to determine the actual decay rate. Using the fast camera mode enabled us to obtain docking and fusion movies at 10 ms/frame and 5 ms/frame (Supplementary Movie 3), albeit with low signal/noise ratio. Examples of traces of I0.7μm(t) from these fast movies (Fig. 4 a) usually show a distinct “hesitation” of 1–10 camera frames between docking and fusion, with fusion signaled by a sharp increase in intensity by a factor of 2–4 in one frame. We assign each fusion event to a 5-ms bin (bin 1: 0 < tfus ≤ 5 ms; bin 2: 5 ms < tfus ≤ 10 ms; etc.). Both the I0.7μm(t) plot and visual inspection of each frame just before fusion are necessary for proper binning. The noisy intensity plot does not always distinguish a firmly docked vesicle from a vesicle that is “searching” the surface in the vicinity of its eventual docking site. However, the onset of the expansion of labeled lipids is often detected visually two frames later than the sharp jump in I0.7μm (t), due to the 150-nm equivalent size of the pixels and the optical resolution limit.
FIGURE 4.

Fast fusion kinetics. (a) Examples of integrated intensity in a 0.7-μm radius circle, I0.7μm (t), for fast-fusing vesicles obtained with 5-ms and 10-ms camera frames on binary t-SNARE bilayers. The points within ovals are frames in which the vesicle was stationary (docked) before fusion. In the 5-ms trace, the three points preceding the oval correspond to frames in which the vesicle visits the circle of integration but is not yet firmly docked. See text for details. (b) Histogram of tfus combining 62 events with tfus < 0.1 s taken from 10 movies on five different binary t-SNARE bilayers. “Sim” traces show the results of averaging an exponential decay with kfus as indicated over a uniform distribution of docking times relative to the camera frames, normalized to 62 total events. See text. (c) Histogram of tfus combining 47 events with tfus < 0.1 s taken from 11 movies on six different syx-only bilayers. “Sim” traces as in b but normalized to 47 total events.
A histogram of 62 events with tfus < 100 ms acquired in 10 movies using five different surfaces is shown in Fig. 4 b. This represents 77% of all the docking events observed during the 10-s movies. An additional 18 vesicles (23%) fused in the longer time range 0.1–4 s. For homogeneous fusion kinetics and sufficient camera speed, meaning that all vesicles and docking/fusion sites have the same kfus and 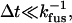 the histogram would peak in the first time bin and decay exponentially:
the histogram would peak in the first time bin and decay exponentially:
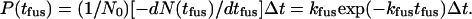 |
(4) |
Here N(tfus) is the number of docked vesicles that have survived to time tfus, N0 is the total number of fusion events analyzed, P(tfus) is the probability that a given vesicle fuses in the time bin centered at tfus, and Δt is the bin width.
Even for Δt = 5 ms, many fusion events occur in two frames or less. The camera's time-averaging broadens the cusp of the exponential decay. A particular docking event may begin at any time within the first 5-ms camera frame. This significantly attenuates the probability that fusion is observed in the first camera frame, since many vesicles dock late in the frame. The lines in Fig. 4 b show simulations of this effect for different assumed values of kfus, obtained by averaging Eq. 4 over a uniform distribution of docking times within the first camera frame. All simulations are normalized to 62 total events. The fast fusion events (those 77% of docked vesicles for which fusion occurs in 100 ms or less) are well modeled with kfus = 40 ± 15 s−1 (τfus = 25 ± 15 ms). The χ2 statistic computed with Poisson-like weighting factors (variance of each nonzero channel estimated as the number of counts; each channel with no counts weighted as one) indicates that kfus = 40 s−1 gives the best fit. The reduced χ-squared statistic 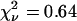 , indicating the model is adequate. That is, we find no statistically significant heterogeneity in the population of fast-fusing vesicles or their docking sites. Values of kfus > 50 s−1 place too many events at short times, whereas values of kfus < 30 s−1 place too few events at short times and too many at long times. The 23% population of more slowly fusing vesicles (100 ms < τfus < 4 s) clearly indicates significant kinetic heterogeneity within the entire vesicle population or among their docking/fusion sites.
, indicating the model is adequate. That is, we find no statistically significant heterogeneity in the population of fast-fusing vesicles or their docking sites. Values of kfus > 50 s−1 place too many events at short times, whereas values of kfus < 30 s−1 place too few events at short times and too many at long times. The 23% population of more slowly fusing vesicles (100 ms < τfus < 4 s) clearly indicates significant kinetic heterogeneity within the entire vesicle population or among their docking/fusion sites.
For protein-free vesicles docking to the low t-SNARE-density bilayers, we never observed a fusion event among 500 vesicles during time-lapse movies lasting 500 s. The conservative assumption of <10% fusion sets the upper limit kfus < 0.0002 s−1, which we include in Table 1. Analogous limits are given for the other control conditions, none of which showed fusion on a comparable timescale to the v-SNARE vesicles on the low t-SNARE-density bilayer.
In several experiments, we preincubated the v-SNARE vesicles in 1 mM Ca2+ or Mg2+ before addition to the low t-SNARE-density bilayers. We obtained fusion movies only with the slower, 40-ms frames. The 40-ms histograms of tfus look identical using normal buffer, on addition of 1 mM Ca2+, and on addition of 1 mM Mg2+ (data not shown). This establishes the lower bounds kfus > 15 s−1 in Table 1.
Finally, we tested for fusion on syx-only bilayers prepared in the same way as the low t-SNARE-density bilayers but without expressing SNAP25. The histogram of tfus for 47 fusion events occurring in 100 ms or less after firm docking as observed in three separate movies with 5-ms frames is shown in Fig. 4 c. These represent ∼80% of all the vesicles that docked on the syx-only surface. Clearly, the fusion is comparably efficient and comparably fast on the syx-only bilayers and the binary t-SNARE bilayers (Fig. 4 b). More data are needed to improve the statistics, test for heterogeneity, and determine kfus accurately, so we do not include these results in Table 1. Both docking and fusion results are very similar with and without SNAP25.
DISCUSSION
Nature of fast fusion sites
The high absolute docking efficiency (see below), the absence of vesicle undocking, the preponderance of fast v-SNARE vesicle fusion events, and the success of the various control experiments all strongly suggest that t-SNAREs within the active docking sites on the low t-SNARE-density bilayer interact freely and specifically with v-SNAREs to form ternary SNARE complexes that induce fast fusion. Quantitative modeling of the outgoing wave of labeled lipids (Fig. 2 g) shows that the sites also permit free release of the vesicle's lipids after fusion. However, the exact nature and stoichiometry of the docking/fast fusion sites is unknown. There is presumably a distribution of t-SNARE cluster numbers in the docking sites, potentially including monomers, dimers, and small multimers. The fusion assay finds remarkably little kinetic heterogeneity among the fast fusion sites.
The low t-SNARE-density bilayer is sparse in overall protein, with average lateral density ∼100 t-SNARE/μm2. This is 25 times larger than the estimated density of effective surface sites for v-SNARE binding (T0 = 4 μm−2). Each binding site may be a t-SNARE multimer. In addition, some t-SNAREs may be inactive (e.g., lying face-down). In the movies at 5 ms/frame, v-SNARE vesicles are observed to “search” the surface briefly before docking firmly; they do not move perceptibly in the dwell time just before fusion. One possibility consistent with our observations is that most t-SNAREs are initially mobile monomers, but they diffuse and combine to form immobile clusters on the surface during the 3.5 h incubation/annealing time. Such immobile t-SNARE clusters are probably the primary docking and fast fusion sites. There is precedent for clustering of binary t-SNAREs in the literature (43). It is significant that the docked vesicles become immobile within 10–20 ms of first touching the surface. Especially in the 5-ms movies, we see evidence of a brief “search” of the local surface before firm docking. A vesicle docked to a freely mobile t-SNARE cluster would not seriously impede its diffusion. The diffusion coefficient of a 50-nm vesicle in buffer is 3.3 μm2 s−1 (42), significantly larger than Dlipid. In future work, it is important to probe diffusion of the t-SNAREs and to better characterize the structure of the purported t-SNARE cluster sites using fluorescence resonance energy transfer, as demonstrated earlier (27,44,45).
Absolute docking efficiency
The “intrinsic” docking rate constant kdock can be interpreted as the product of a diffusion-limited rate constant kdiff for vesicle docking site encounters and the probability of docking per encounter: kdock = kdiff pdock. We estimate kdiff from the theoretical expression for the diffusive flux that impinges on sparse, perfectly sticky, circular surface binding sites of radius R. The flux per binding site is given by: kdiffV0 = 4DvesRV0 (vesicles/s), where V0 is the concentration of v-SNARE vesicles and Dves is the vesicle diffusion coefficient (38). We must assume a value for R, the effective radius of a docking site. The ∼7-nm length of a SNARE complex is smaller than the ∼25-nm radius of the vesicle. For the low t-SNARE-density bilayer, we take R ∼ 7 nm to approximate the lateral reach of a single v-SNARE or t-SNARE (8). Inserting Dves = 3.3 × 10−8 cm2 s−1 then yields kdiff = 5.6 × 107 M−1 s−1, ∼2.5 times the experimental value kdock = 2.2 × 107 M−1 s−1 (i.e., pdock ∼0.4). The t-SNARE sites on the low-density surface capture v-SNARE vesicles with high probability per encounter.
For v-SNARE vesicles adsorbing to the high t-SNARE-density bilayer, the primary docking sites are probably the large t-SNARE aggregates observed by AFM (Fig. 1 a). The relatively small density of effective docking sites on that bilayer (T0 = 0.1 ± 0.05 μm−2) is essentially the same as the density of aggregated t-SNARE mounds obtained by direct count on the AFM images (0.2 ± 0.1 μm−2). Such sites are larger than the approaching vesicle, so we take R to be the “radius” of an aggregate: R ∼ 200 nm. This gives kdiff = 1.6 × 109 M−1 s−1, ∼60 times larger than kdock = 1.2 × 107 M−1 s−1 (i.e., pdock ∼ 0.02). If the aggregates are indeed the binding sites, then the binding probability per encounter is very low. The t-SNAREs must be in very different condition on the low and high t-SNARE density surfaces.
Recent evidence suggests the possibility that binding of the SNARE-forming, cytoplasmic segment of syb to the vesicle bilayer itself provides one level of regulation of synaptic response. Electron paramagnetic resonance data indicate that insertion of the 7–8 residues nearest the membrane anchor inhibits formation of a ternary SNARE complex among anchored syb and the cytoplasmic domains of syx (lacking the Habc domain) and SNAP25 (46). A protein-binding assay using reconstituted proteoliposomes, harvested chromaffin granules, or harvested synaptic vesicles reached the same conclusion (47,48). Our study provides direct evidence that ternary SNARE formation is reasonably efficient between anchored syb and anchored binary t-SNAREs that include the Habc domain. Evidently anchored, binary t-SNAREs are better able than cytoplasmic domains to pry loose the anchor-proximal segment of syb and form full ternary SNARE complexes.
Comparison of in vitro fusion assays
The behavior of reconstituted fusion systems has varied widely across laboratories. A detailed comparison may help guide future improvements. In the vesicle-vesicle geometry, both bilayers are curved. The vesicle/planar-bilayer geometry more closely approximates that found in nature. All else being equal, we would expect a stronger driving force toward fusion (more negative ΔG) and thus a smaller barrier to fusion in the vesicle-vesicle assays than in the vesicle-planar bilayer assays. Yet the single-vesicle studies have consistently found substantially faster fusion.
Vesicle-vesicle assays
In the vesicle-vesicle assays, fusion is detected by fluorescence dequenching of labeled lipid components, with the intensity calibrated in “rounds of fusion”. Weber, Rothman, and co-workers first demonstrated SNARE-dependent fusion; in the optimized system, the time to the first round of fusion was ∼20 min (20,21). Fusion was not regulated by Ca2+, even in the presence of syt. Subsequent studies have all found slow fusion on a timescale of tens of minutes (22,42). Tucker, Weber, and Chapman demonstrated a fourfold enhancement of the fusion rate in the presence of Ca2+ and the cytoplasmic domain of syt, but fusion remained slow (24).
We can use our measured values of kdock to estimate the time to one round of fusion in a hypothetical vesicle-vesicle assay whose proteins' docking behavior mimicked ours. In three-dimensional solution, the diffusion-limited reaction rate constant for vesicle-vesicle collisions is kdiff = 4πN0(RA + RB)(DA + DB), where N0 is Avogadro's number, A and B label the two types of vesicles, RA = RB is the common vesicle radius, and DA = DB is the common vesicle diffusion constant. If we take RA = RB = 25 nm and DA = DB = 3.3 × 10−8 cm2 s−1 from the literature (42), then kdiff = 2.5 × 109 M−1 s−1. Typical bulk fusion assays use vesicle concentrations Cv-SNARE ∼ 12 nM ≪ Ct-SNARE ∼ 110 nM. Under these pseudo-first-order conditions, a diffusion-limited fusion reaction (diffusion-limited docking followed by prompt fusion) would initially show exponential decay of the unfused v-SNARE vesicle population with pseudo-first-order rate constant keff = kdiff Ct-SNARE ∼ 280 s−1 (τeff ∼ 4 ms). One full round of fusion would occur on a timescale of ∼2τeff ∼ 8 ms. If pdock = 0.4, as estimated for our low t-SNARE-density planar bilayer, the timescale for the first round of fusion lengthens to ∼20 ms. If the t-SNAREs on the vesicles were aggregated and relatively inert like the mounds on the high t-SNARE-density bilayer, pdock = 0.02 predicts ∼0.4 s to the first round of fusion.
In fact, the observed time to one round of fusion is 20–50 min, ∼5000 times slower than the longer of these estimates. For the fusion rate to be controlled by inefficient docking, the docking probability per encounter would have to be ∼10−5, much smaller than anything directly observed in our assay. Therefore, we strongly suspect that the fusion step itself is the bottleneck. Jahn and co-workers reached a similar conclusion (42). If so, then the time to the first round of fusion provides the rough estimate kfus ∼ 0.001 s−1. This is ∼104 times slower than in our vesicle-bilayer assay using the fusion-efficient low t-SNARE-density bilayer. It is consistent with the estimated upper bound kfus < 0.002 s−1 for fusion of v-SNARE vesicles on the high t-SNARE-density bilayer in our work.
Significantly, our vesicle-planar bilayer assay uses the same materials and procedures as the Tucker-Chapman assay to make both the v-SNARE and t-SNARE vesicles. Evidently, the t-SNARE vesicles are the impediment to faster fusion in the vesicle-vesicle assay. We tentatively conclude that all vesicle-vesicle assays to date are fusion-rate limited. The high barrier to fusion may well arise from the entangled or aggregated state of the t-SNARE complexes on the vesicle surface, although we lack direct evidence of this. If our 80-copy t-SNARE vesicles contain “preaggregated” t-SNAREs, deposition of these vesicles might nucleate formation of large aggregated mounds on the glass substrate. In contrast, for average copy number ∼0.8 t-SNARE/vesicle, such “preaggregation” is essentially impossible. The resulting low t-SNARE-density bilayers induce fast fusion of v-SNARE vesicles.
Vesicle-planar bilayer assays
The three single-vesicle studies themselves exhibit a wide range of behavior. The Simon study (28) found docking but little or no fusion in the absence of dipositive cations. Only 0.35% of the docked vesicles fused in 50 s. This corresponds to kfus ∼ 7 × 10−5 s−1. (This is our calculation, aimed at placing all studies approximately on the same quantitative scale.) Either Ca2+ (or Mg2+) induced fusion of 15% (or 4%) of the docked vesicles within 50 s. Half of these “competent” vesicles fused within 10 s of Ca2+ addition (kfus ∼ 0.07 s−1). On removal of the regulatory Habc domain of syx and without Ca2+, 10% of docked vesicles fused in 50 s, and 70% of the competent vesicles fused within 20 s of docking (kfus ∼ 0.06 s−1). Evidently, Ca2+ and Mg2+ enhance both the number density of fusion-active sites and kfus itself, whereas removal of Habc primarily increases the number density of fusion-active sites. The presence of Habc endows each t-SNARE with two spatially separate helical bundles, which we suggest can “cross-link” pairs of t-SNAREs and thus enhance t-SNARE aggregation.
One significant difference between our work and the Simon study may be the density of t-SNAREs in the planar bilayer. They used average copy number ∼8 t-SNARE/vesicle (lipid/protein 3000:1), 10 times smaller than the ∼80-copy vesicles we used to form the high t-SNARE-density bilayers that exhibit large mounds of aggregated protein (Fig. 1 a). Both resulting bilayers exhibit very low fusion rates. We did not observe the triggering of fusion on addition of Ca2+ or Mg2+ observed earlier, perhaps suggesting that the t-SNARE bilayers in the Simon study are less entangled than in our 80-copy bilayers. It may also be significant that in the Simon study the t-SNAREs were reconstituted into 100% POPC vesicles.
Our low t-SNARE-density conditions have protein content quite similar to that in the Brunger-Chu study (27). They formed a low syx-density planar bilayer (0.1–100 syx/μm2) by vesicle deposition using 100% eggPC (no PS) and subsequently added SNAP25 to form binary t-SNARE complexes in situ. The experimental conditions would prevent observation of prompt fusion events in the dark. Under laser illumination, ∼50% of the vesicles fuse on a 10–20 s timescale at 37°C (kfus ∼ 0.07 s−1, comparable to the Simon study with added Ca2+ or Mg2+). Evidently the fusion is thermally activated by laser heating. Individual fusion events were abrupt (sub-100 ms). At relatively high syx concentration (270 μm−2, ∼3 times higher than our low-density conditions), the mean number of SNARE complexes per docked vesicle was ∼12, consistent with the suggestion that our fast fusion sites are t-SNARE clusters. As few as 1–2 SNAREs were sufficient to cause docking and thermally induced fusion. There was no Ca2+ effect.
The obvious differences between our work and the Brunger-Chu study lie in the lipid composition (synthetic 85% POPC/15% DOPS for us, 100% eggPC for them) and in the method of formation of anchored t-SNARE complexes. Our t-SNAREs are fully formed in vivo and harvested as a single entity, whereas Brunger-Chu added SNAP25 to syx preanchored in the planar bilayer. Although the fusion rate constants are very different in the two studies, in both cases the rate is remarkably insensitive to replacement of the binary t-SNARE by syntaxin alone, i.e., to the presence or absence of SNAP25. Equally remarkably, the measured docking kinetics in our study are indistinguishable with or without SNAP25.
All four helices in the full SNARE bundle are amphipathic, with hydrophobic sides facing inward in the bundle (8). This may explain why anchored syb is able to bind to anchored syx in both studies, and why preincubation of the v-SNARE vesicles in cytoplasmic-domain syx completely blocks docking to t-SNARE bilayers in our study. Such “imperfect SNAREs” might comprise various combinations of four helices (e.g., two from syx, none from SNAP25, and two from syb), only three helices, etc. Evidently, such imperfect SNAREs are able to form stable complexes and to drive fusion. There is some precedent for this idea in a recent study (49) demonstrating that large dense-core neurosecretory granules isolated from the bovine neurohypophysis spontaneously fuse with a planar lipid bilayer containing syntaxin 1A but no SNAP25.
Why are the docking and fusion rates indistinguishable in our assay with or without SNAP25, whereas in the recent vesicle-vesicle fusion assay truncations of SNAP25 that mimic the actions of BoNT A and E suppressed fusion? We already argued that because our assay and the Tucker-Chapman assay use the same v-SNARE vesicles, the bottleneck causing ∼10-min fusion in the vesicle-vesicle assay must be due to the entangled condition of the t-SNAREs. Truncation of SNAP25 might somehow enhance the entanglement of the t-SNARE binding sites, rendering them more inert. In our assay, the 25-ms fusion remains slow on the molecular timescale, i.e., there is still a bottleneck to our fusion process as well. The results suggest that our bottleneck (and that in the Brunger-Chu assay) arises from the condition of the v-SNAREs or from the dynamics of trans SNARE formation after the v-SNARE vesicle has docked, not from the condition of the t-SNAREs. This may be related to the observation that insertion into the cis bilayer of the 7–8 residues of syb nearest the membrane anchor inhibits formation of a ternary SNARE complex among anchored syb and the cytoplasmic domains of syx and SNAP25 (45–47). Future experimental work will shed more light on this issue.
CONCLUSIONS AND PROGNOSIS
In summary, we have demonstrated how to use the single-vesicle methodology to independently measure v-SNARE vesicle docking and fusion rate constants on planar t-SNARE bilayers. Both rates are informative. On low t-SNARE-density bilayers, v-SNARE vesicles dock with ∼40% efficiency per encounter with a binding site; ∼77% of the docked vesicles achieve fast fusion with kfus = 40 ± 15 s−1 in the absence of Ca2+ and of syt. Complete lipid mixing (and thus content release) occurs within 5–10 ms. The fusion rate constant is ∼1000 times faster than observed in previous single-vesicle assays and ∼104 times faster than the combined docking/fusion rate observed in vesicle-vesicle assays. Our results indicate that the condition of the binary t-SNAREs on a vesicle or bilayer surface controls both the effective surface binding site density and also kfus.
The role of SNARE complexes in presynaptic vesicle fusion remains controversial. Indeed, some workers (50) have raised legitimate questions as to whether the early vesicle-vesicle fusion assays (21) carried out at high protein/lipid ratio ∼1:20 truly prove that ternary SNAREs are fusagens. Our new data demonstrate that trans SNARE complexes can drive fusion of v-SNARE vesicles having protein/lipid ratio of ∼1:240 on a timescale of 25 ms at 37°C. The vesicle-planar bilayer geometry has more realistic curvature than the vesicle-vesicle geometry, and fast fusion occurs with or without Ca2+ and in the absence of all auxiliary proteins. We have not yet fully optimized the protein concentrations, method of deposition, lipid mixtures, or vesicle size in our assay. Measurement of the intrinsic free energy barrier to vesicle-plus-bilayer fusion for the optimal number of well-formed trans SNARE complexes now becomes an important goal. It will be technically feasible to measure fusion rate constants of 500 s−1 or faster using higher laser intensity and the new generation of charge-coupled device cameras.
Clearly SNARE complexes alone can drive vesicle fusion much more rapidly than previously observed. Is the current value kfus = 40 s−1 sufficiently fast to explain the submillisecond synaptic response time of specific systems to the Ca2+ trigger event in vivo? Seemingly not. It is important to distinguish the time to the first presynaptic response (release of the first few vesicles) and the kinetic response time of the entire population of “readily releasable” vesicles (51). The presynaptic response time τsynapse depends on the number of readily releasable vesicles N and the kinetic time constant 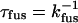 as τsynapse ∼ τfus/N. In goldfish bipolar neurons (52), the population of readily releasable vesicles is ∼2000 per neuron and τfus ∼ 120 ms, so the presynaptic response time can be submillisecond. In contrast, in calyx of Held nerve terminals, the entire population of ∼4000 readily releasable vesicles exocytoses with τfus ∼ 0.6 ms at 40 μM Ca2+ (29). The corresponding kfus ∼ 1700 s−1 is ∼40 times faster than our kfus = 40 s−1.
as τsynapse ∼ τfus/N. In goldfish bipolar neurons (52), the population of readily releasable vesicles is ∼2000 per neuron and τfus ∼ 120 ms, so the presynaptic response time can be submillisecond. In contrast, in calyx of Held nerve terminals, the entire population of ∼4000 readily releasable vesicles exocytoses with τfus ∼ 0.6 ms at 40 μM Ca2+ (29). The corresponding kfus ∼ 1700 s−1 is ∼40 times faster than our kfus = 40 s−1.
It remains to be seen just how fast an optimized trans SNARE system alone can drive fusion. We currently have no information about the nature of the bottleneck in our system. The starting point for our measurement of tfus is the time at which the vesicle firmly docks, i.e., when perceptible lateral motion ceases on a ∼70 nm length scale. We do not know what the SNARE components are doing in the time between firm docking and fusion. Suppose one ternary SNARE can cause firm docking, but multiple SNAREs must form before fusion. In that case, kfus may measure the time for complementary proteins on the trans bilayers to find each other. Alternatively, a sufficient number of ternary SNAREs may form very quickly and contents may release rapidly, but our assay is limited in response time by an activation barrier to lipid mixing that must be overcome by thermal energy. In comparison with our docking-fusion study, the starting configuration in synaptic vesicle exocytosis just upstream of the Ca2+ trigger is presumably highly specific. For example, preoriented and prefolded SNARE helices may be poised to assemble the ternary SNARE. Alternatively, a partially formed ternary SNARE complex may be poised to zipper rapidly. In future work, simultaneous study of contents release and lipid mixing combined with fluorescence resonance energy transfer studies of interprotein distances versus time will begin to elucidate the slow steps of SNARE-induced fusion in vitro. Addition of auxiliary proteins such as Ca2+/syt and complexin may further enhance kfus.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
J.C.W. thanks the National Science Foundation (CHE-0071458 and CHE-0452375), the University of Wisconsin Alumni Research Foundation, and the University of Wisconsin-Madison Department of Chemistry for support of this research. This study was further supported by grants to E.R.C. from the National Institutes of Health (NIGMS GM 56827 and NIMH MH61876) and the American Heart Association (0440168N). W.C.T. thanks the National Institutes of Health for a postgraduate fellowship. We thank Dr. Min Dong for materials and help with the BoNT B treatment of v-SNARE vesicles.
References
- 1.Katz, B. 1969. The Release of Neural Transmitter Substances. Charles C. Thomas, Springfield, IL.
- 2.Augustine, G. J. 2001. How does calcium trigger neurotransmitter release? Curr. Opin. Neurobiol. 11:320–326. [DOI] [PubMed] [Google Scholar]
- 3.Sollner, T. H. 2003. Regulated exocytosis and SNARE function (Review). Mol. Membr. Biol. 20:209–220. [DOI] [PubMed] [Google Scholar]
- 4.Llinas, R., I. Z. Steinberg, and K. Walton. 1981. Relationship between presynaptic calcium current and postsynaptic potential in squid giant synapse. Biophys. J. 33:323–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai, J., and E. R. Chapman. 2004. The C2 domains of synaptotagmin—partners in exocytosis. Trends Biochem. Sci. 29:143–151. [DOI] [PubMed] [Google Scholar]
- 6.Whiteheart, S. W., and E. A. Matveeva. 2004. Multiple binding proteins suggest diverse functions for the N-ethylmaleimide sensitive factor. J. Struct. Biol. 146:32–43. [DOI] [PubMed] [Google Scholar]
- 7.Sollner, T., M. K. Bennett, S. W. Whiteheart, R. H. Scheller, and J. E. Rothman. 1993. A protein assembly-disassembly pathway in-vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 75:409–418. [DOI] [PubMed] [Google Scholar]
- 8.Sutton, R. B., D. Fasshauer, R. Jahn, and A. T. Brunger. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 395:347–353. [DOI] [PubMed] [Google Scholar]
- 9.Chen, X., D. R. Tomchick, E. Kovrigin, D. Arac, M. Machius, T. C. Sudhof, and J. Rizo. 2002. Three-dimensional structure of the complexin/SNARE complex. Neuron. 33:397–409. [DOI] [PubMed] [Google Scholar]
- 10.Hazzard, J., T. C. Sudhof, and J. Rizo. 1999. NMR analysis of the structure of synaptobrevin and of its interaction with syntaxin. J. Biomol. NMR. 14:203–207. [DOI] [PubMed] [Google Scholar]
- 11.Matthew, W. D., L. Tsavaler, and L. F. Reichardt. 1981. Identification of a synaptic vesicle-specific membrane protein with a wide distribution in neuronal and neurosecretory tissue. J. Cell Biol. 91:257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perin, M. S., N. Brose, R. Jahn, and T. C. Sudhof. 1991. Domain structure of synaptotagmin (p65). J. Biol. Chem. 266:623–629. [PubMed] [Google Scholar]
- 13.Brose, N., A. G. Petrenko, T. C. Sudhof, and R. Jahn. 1992. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 256:1021–1025. [DOI] [PubMed] [Google Scholar]
- 14.Chapman, E. R. 2002. Synaptotagmin: a Ca2+ sensor that triggers exocytosis? Nat. Rev. Mol. Cell Biol. 3:498–508. [DOI] [PubMed] [Google Scholar]
- 15.Tucker, W. C., and E. R. Chapman. 2002. Role of synaptotagmin in Ca2+-triggered exocytosis. Biochem. J. 366:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haque, M. E., and B. R. Lentz. 2004. Roles of curvature and hydrophobic interstice energy in fusion: studies of lipid perturbant effects. Biochemistry. 43:3507–3517. [DOI] [PubMed] [Google Scholar]
- 17.Malinin, V. S., and B. R. Lentz. 2004. Energetics of vesicle fusion intermediates: comparison of calculations with observed effects of osmotic and curvature stresses. Biophys. J. 86:2951–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, Y., X. Han, and L. K. Tamm. 2003. Thermodynamics of fusion peptide-membrane interactions. Biochemistry. 42:7245–7251. [DOI] [PubMed] [Google Scholar]
- 19.Han, X., C. T. Wang, J. Bai, E. R. Chapman, and M. B. Jackson. 2004. Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science. 304:289–292. [DOI] [PubMed] [Google Scholar]
- 20.Nickel, W., T. Weber, J. A. McNew, F. Parlati, T. H. Sollner, and J. E. Rothman. 1999. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc. Natl. Acad. Sci. USA. 96:12571–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weber, T., B. V. Zemelman, J. A. McNew, B. Westermann, M. Gmachl, F. Parlati, T. H. Sollner, and J. E. Rothman. 1998. SNAREpins: minimal machinery for membrane fusion. Cell. 92:759–772. [DOI] [PubMed] [Google Scholar]
- 22.Mahal, L. K., S. M. Sequeira, J. M. Gureasko, and T. H. Sollner. 2002. Calcium-independent stimulation of membrane fusion and SNAREpin formation by synaptotagmin I. J. Cell Biol. 158:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parlati, F., T. Weber, J. A. McNew, B. Westermann, T. H. Sollner, and J. E. Rothman. 1999. Rapid and efficient fusion of phospholipid vesicles by the alpha-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc. Natl. Acad. Sci. USA. 96:12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tucker, W. C., T. Weber, and E. R. Chapman. 2004. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 304:435–438. [DOI] [PubMed] [Google Scholar]
- 25.Coppola, T., S. Magnin-Luthi, V. Perret-Menoud, S. Gattesco, G. Schiavo, and R. Regazzi. 2001. Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin. J. Biol. Chem. 276:32756–32762. [DOI] [PubMed] [Google Scholar]
- 26.Peters, C., M. J. Bayer, S. Buhler, J. S. Andersen, M. Mann, and A. Mayer. 2001. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature. 409:581–588. [DOI] [PubMed] [Google Scholar]
- 27.Bowen, M. E., K. Weninger, A. T. Brunger, and S. Chu. 2004. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys. J. 87:3569–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fix, M., T. J. Melia, J. K. Jaiswal, J. Z. Rappoport, D. You, T. H. Sollner, J. E. Rothman, and S. M. Simon. 2004. Imaging single membrane fusion events mediated by SNARE proteins. Proc. Natl. Acad. Sci. USA. 101:7311–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolfel, M., and R. Schneggenburger. 2003. Presynaptic capacitance measurements and Ca2+ uncaging reveal submillisecond exocytosis kinetics and characterize the Ca2+ sensitivity of vesicle pool depletion at a fast CNS synapse. J. Neurosci. 23:7059–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jahn, R., and T. C. Sudhof. 1994. Synaptic vesicles and exocytosis. Annu. Rev. Neurosci. 17:219–246. [DOI] [PubMed] [Google Scholar]
- 31.Tamm, L. K., and H. M. McConnell. 1985. Supported phospholipid bilayers. Biophys. J. 47:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson, S. J., T. M. Bayerl, D. C. McDermott, G. W. Adam, A. R. Rennie, R. K. Thomas, and E. Sackmann. 1991. Structure of an adsorbed dimyristoylphosphatidylcholine bilayer measured with specular reflection of neutrons. Biophys. J. 59:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiessling, V., and L. K. Tamm. 2003. Measuring distances in supported bilayers by fluorescence interference-contrast microscopy: polymer supports and SNARE proteins. Biophys. J. 84:408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Axelrod, D. 2001. Selective imaging of surface fluorescence with very high aperture microscope objectives. J. Biomed. Opt. 6:6–13. [DOI] [PubMed] [Google Scholar]
- 35.Harms, G. S., M. Sonnleitner, G. J. Schutz, H. J. Gruber, and T. Schmidt. 1999. Single-molecule anisotropy imaging. Biophys. J. 77:2864–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson, N. L., K. H. Pearce, and H. V. Hsieh. 1993. Total internal reflection fluorescence microscopy: application to substrate-supported planar membranes. Eur. Biophys. J. 22:367–378. [DOI] [PubMed] [Google Scholar]
- 37.Abney, J. R., B. A. Scalettar, and N. L. Thompson. 1992. Evanescent interference patterns for fluorescence microscopy. Biophys. J. 61:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berg, H. C. 1993. Random Walks in Biology. Princeton, N.J.: Princeton University Press.
- 39.Lee, G. M., A. Ishihara, and K. A. Jacobson. 1991. Direct observation of Brownian motion of lipids in a membrane. Proc. Natl. Acad. Sci. USA. 88:6274–6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bacia, K., C. G. Schuette, N. Kahya, R. Jahn, and P. Schwille. 2004. SNAREs prefer liquid-disordered over “raft” (liquid-ordered) domains when reconstituted into giant unilamellar vesicles. J. Biol. Chem. 279:37951–37955. [DOI] [PubMed] [Google Scholar]
- 41.Vijayendran, R. A., F. S. Ligler, and D. E. Keckband. 1999. A computational reaction-diffusion model for the analysis of transport-limited kinetics. Anal. Chem. 71:5405–5412. [DOI] [PubMed] [Google Scholar]
- 42.Schuette, C. G., K. Hatsuzawa, M. Margittai, A. Stein, D. Riedel, P. Kuster, M. Konig, C. Seidel, and R. Jahn. 2004. Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc. Natl. Acad. Sci. USA. 101:2858–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laage, R., J. Rohde, B. Brosig, and D. Langosch. 2000. A conserved membrane-spanning amino acid motif drives homomeric and supports heteromeric assembly of presynaptic SNARE proteins. J. Biol. Chem. 275:17481–17487. [DOI] [PubMed] [Google Scholar]
- 44.Weninger, K., M. E. Bowen, S. Chu, and A. T. Brunger. 2003. Single-molecule studies of SNARE complex assembly reveal parallel and antiparallel configurations. Proc. Natl. Acad. Sci. USA. 100:14800–14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagner, M. L., and L. K. Tamm. 2001. Reconstituted syntaxin1A/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys. J. 81:266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kweon, D. H., C. S. Kim, and Y. K. Shin. 2003. Regulation of neuronal SNARE assembly by the membrane. Nat. Struct. Biol. 10:440–447. [DOI] [PubMed] [Google Scholar]
- 47.Hu, K., C. Rickman, J. Carroll, and B. Davletov. 2004. A common mechanism for the regulation of vesicular SNAREs on phospholipid membranes. Biochem. J. 377:781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu, K., J. Carroll, S. Fedorovich, C. Rickman, A. Sukhodub, and B. Davletov. 2002. Vesicular restriction of synaptobrevin suggests a role for calcium in membrane fusion. Nature. 415:646–650. [DOI] [PubMed] [Google Scholar]
- 49.McNally, J. M., D. J. Woodbury, and J. R. Lemos. 2004. Syntaxin 1A drives fusion of large dense-core neurosecretory granules into a planar lipid bilayer. Cell Biochem. Biophys. 41:11–24. [DOI] [PubMed] [Google Scholar]
- 50.Rizo, J. 2003. SNARE function revisited. Nat. Struct. Biol. 10:417–419. [DOI] [PubMed] [Google Scholar]
- 51.Almers, W. 1994. Synapses. How fast can you get? Nature. 367:682–683. [DOI] [PubMed] [Google Scholar]
- 52.von Gersdorff, H., and G. Matthews. 1994. Dynamics of synaptic vesicle fusion and membrane retrieval in synaptic terminals. Nature. 367:735–739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.