

Abstract
Sample orientation relative to the static magnetic field of an NMR spectrometer allows study of membrane proteins in the lipid bilayer setting. The straightforward preparation and handling of extremely thin mica substrates with consistent surface properties has prompted us to examine oriented phospholipid bilayer and hexagonal phases on mica. The spectral characteristics of oriented lipid samples formed on mica are as good as or better than those on glass. Nine solvents with varying dielectric constants were used to cast lipid films or for vesicle spreading; film characteristics were then compared, and static solid-state 31P-NMR was used to characterize the degree of orientation of the hydrated lipid species. Lipids with four headgroup chemistries were tested: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG), 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE). Solvent affected orientation of POPG, DOPA, and DOPE, but not POPC. Film characteristics varied with solvent, with ramifications for producing homogeneous oriented lipid samples. POPC was used to optimize the amount of lipid per substrate and compare hydration methods. POPG did not orient reproducibly, whereas POPG-POPC mixtures did. DOPA showed 1–2 oriented states depending upon hydration level and deposition method. DOPE formed an oriented hexagonal phase that underwent a reversible temperature-induced phase transition to the oriented bilayer phase.
INTRODUCTION
A number of solid-state nuclear magnetic resonance (NMR) experiments probing polymers and biomolecules exploit orientation of the sample relative to the static field of the magnet (1–3). Of particular interest is the ability to study membrane proteins directly in phospholipid bilayers, rooted in pioneering studies of oriented liquid crystals and multilayers by Berendsen and co-workers (4,5). Although solid-state NMR of membrane proteins is rapidly maturing (see Refs. 3 and 6 for recent reviews), a scan of the literature shows a large variety of sample preparation methodologies. Some aspects, such as the use of a glass substrate, remain constant while others, such as bilayer deposition conditions, hydration methods, and the peptide-to-lipid ratio, may vary widely. Previous studies have examined the effects of peptide/lipid ratio (7) and levels of hydration (8). Here, we present a systematic study of a variety of aspects of oriented lipid bilayer sample preparation, with the aim of optimizing conditions for study by solid-state NMR.
The formation of planar-supported lipid bilayers on hydrophilic surfaces such as glass and mica is now quite routine. The inherently low sensitivity of the NMR signal means that relatively large amounts of sample are required; therefore, an oriented bilayer sample for solid-state NMR purposes is actually composed of numerous stacked bilayers per pair of flat substrates that is, in reality, an oriented multilayer system. In keeping with the literature, however, we refer to such an oriented multilayer sample as an oriented bilayer sample. A rough calculation for a typical sample with ∼2 mg of a phosphatidylcholine lipid (∼760 mol wt), assuming perfect packing of headgroups with area of ∼60 Å2 (9) sandwiched between a pair of 11 × 11-mm glass plates, gives ∼5000 bilayers. Therefore, layer-by-layer transfer using the Langmuir-Blodgett (10–12) and/or the Langmuir-Schaefer (13) techniques popular for single bilayers (14,15) are not generally practical. Vesicle deposition from aqueous solution (16) has been successfully used to produce solid-state NMR samples (17), allowing deposition of proteins which may not be soluble in organic solvent. Where possible, casting films from an organic solvent provides a very convenient method to form supported bilayer samples (18), and is indeed the most commonly employed method for solid-state NMR sample preparation (2,3,6).
In addition to the requirement of large sample quantities, total sample size is limited both by the bore diameter through the center of a high-field superconducting magnet and the region of the bore that experiences a homogeneous magnetic field. To maximize signal intensity, it is desirable to minimize the amount of space taken up by substrate materials versus actual sample (19). In practical terms, this means that tens of thousands of bilayers must be aligned within confined quarters while wasting the minimum possible amount of space with support substrates. Typical oriented bilayer samples for solid-state NMR are prepared on borosilicate glass plates with sides of length 5–15 mm and a thickness of <200 μm, where a 60–80-μm thickness is typically used. Glass substrates with a thickness of <∼100 μm must be chemically etched to the desired thickness using hydrofluoric acid either by the user or at considerable cost. Beyond the difficulties in production, these are hard to work with, since they are both very fragile and very difficult to further modify in terms of dimensions. From a surface chemistry standpoint, glass is a relatively heterogeneous substrate, with surface properties that are highly dependent upon the protocol used to clean it (20), making reproducibility an issue.
Muscovite mica, KAl2(Si3Al)O10(OH,F)2, provides a good alternative to glass. It is a very popular substrate in surface science since it is easily cleaved to provide a clean, molecularly flat surface with consistent chemical character (21). Furthermore, it may be readily cut into a sheet with the desired dimensions and shape, with subsequent cleavage providing a clean substrate. The normally anionic surface charge of mica may be reversed to cationic by reaction with 3-aminopropyltriethoxysilane without a loss of its surface flatness (22), adding greatly to its versatility as a substrate. Note that ruby mica contains significant amounts of iron, giving rise to an appreciable magnetic susceptibility (23). Therefore, for NMR purposes, use of high-quality Muscovite mica with minimal iron impurity is probably ideal, although good results have also been achieved with mica of a high susceptibility through signal processing (24).
Given the relative ease of substrate handling and preparation, we have decided to examine mica as a substrate for solid-state NMR of lipid bilayers. We have quantitatively examined the effect of mica mass upon 31P chemical shifts, demonstrating an increase in chemical shift with increasing substrate mass. Seul and Sammon noted a strong dependence of cast surfactant film morphology on the solvent used for deposition (18). Therefore, we examined the deposition efficacy, film characteristics, and oriented bilayer character of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) deposited from eight solvents covering a wide range of dielectric constants and by aqueous vesicle spreading. This lipid was chosen both because of its low melting temperature, allowing ready formation of bilayers at room temperature, and its popularity in solid-state NMR peptide studies. To determine the optimal amount of lipid to deposit between a pair of mica substrates, we then characterized the dependence of 31P-NMR spectral characteristics with varying amounts of lipid. The effects of hydration by direct application of a measured water aliquot and in hydration chambers at ambient temperature and elevated temperature on spectral characteristics were also compared. Similar studies are presented, for a more limited range of conditions, with a variety of other lipids with different headgroup moieties. Oriented hexagonal phase lipids were readily formed using 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE). These underwent reversible phase transitions to the oriented bilayer state after exposure to the appropriate temperature.
No one condition proved generic for all lipid headgroups. Rather, our results demonstrate that, regardless of the solid support substrate employed, consideration of nonstandard solvents should be useful for a variety of cases. Such a screening may lead to a solvent which will dissolve all lipid and protein species in a given system, improving homogeneity of the sample before hydration. This is particularly evident for all non-phosphatidylcholine headgroups that we examined, where the solvent used to form the initial lipid film on mica strongly affected the quality of the hydrated sample.
MATERIALS AND METHODS
Materials
Egg yolk phosphatidylcholine (egg PC; >99%), 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA; >99%), and DOPE (>99%) were purchased from Avanti Polar Lipids (Alabaster, AL). POPC (>99%) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG; >99%) were purchased from Northern Lipids (Vancouver, BC, Canada). Chloroform (99.8%) and methanol (99.9%) at A.C.S. spectrophotometric grade, dichloromethane (99.6+%) and 2-propanol (99.5+%) at A.C.S. reagent grade, and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; 99+%), ethanol (200 proof), 3-pentanol (98%), and 2,2,2-trifluoroethanol (TFE; 99+%) were purchased from Sigma-Aldrich (St. Louis, MO). All reagents were used without further purification. Muscovite mica (grades V-1 and V-4, various dimensions) was purchased from Structure Probe (West Chester, PA); unless specifically noted, experiments were carried out using grade V-4 mica. Microscope cover glasses (15 × 15 × ∼0.07 mm; Marienfeld, Lauda-Koenigshofen, Germany) were sonicated in methanol for 30–45 min, rinsed with ethanol and then with deionized water, with each rinse followed by drying at 40°C, before use.
Preparation of standard samples
Standard sample cells were prepared by attaching two sheets of mica (15 × 15-mm, 30–60-μm thick) to a plastic frame (sides 15-mm long and 1-mm wide, framing a 14 × 14-mm square area, and height 1.2 mm) with either epoxy or Krazy Glue (Elmer's Products, Columbus, OH). Generally, each sheet of mica was freshly cleaved and then immediately attached to the frame. For fluid cells, one sheet was punctured with two ∼1-mm-diameter holes to allow filling of the fluid chamber by micropipette; immediately after filling, a third sheet of prewetted mica was affixed to cover the filling holes. Two 31P standard samples were prepared: one containing ∼225 μL of 1 M phosphate buffer (∼0.5 mM EDTA, ∼10 mM NaN3, 10% D2O, pH ∼4.7) and one containing ∼100 mg of egg PC.
Solvent casting of lipid samples
Powdered lipid was dissolved at ∼50 mg/mL in organic solvent. Freshly cleaved 15 × 15 mm mica substrates with a thickness of 20–60 μm were placed on microcentrifuge tube caps. Such supports served to aid in keeping low-contact-angle solvents from spreading off the edges of a mica plate. Thirty-five microliters of neat solvent were applied to prewet each substrate and allowed to dry in a fume hood. Lipid solution or suspension was deposited in a series of 20-μL aliquots, with a second 20-μL aliquot generally applied before complete drying. After each application of 2 × 20 μL, substrates were allowed to dry in a fume hood; in some cases, drying was assisted with a stream of N2 gas. After all solvent was applied, sample vials were rinsed with additional solvent sufficient to obtain application of 95% or more of the lipid. Samples were subsequently allowed to desolvate for 12–18 h in a fume hood and then under vacuum for an additional 1.5–4 h. Hydration using 0.22-μm filtered (Millipore Millex-GS, Billerica, MA) deionized water was carried out by adding an amount of ∼10 water molecules per lipid molecule by micropipette; upon hydration, mica substrates were immediately sandwiched together and tightly wrapped twice with plastic wrap. Samples were stored in this wrapped condition at ∼23°C in sealed vials. Note that comparison samples on glass were prepared identically from the prewetting step onwards.
Preparation of lipid samples by vesicle spreading
Powdered lipid was dissolved in chloroform at ∼5–10 mg/mL in a 13 × 100-mm borosilicate glass culture tube (Fisher Scientific Canada, Edmonton, Alberta). A stream of nitrogen was used to evaporate the solvent while rotating the tube to coat all sides of the tube. Any residual solvent was removed by placing the tube in a bell jar under vacuum for 1 h. A measure of 0.22-μm filtered deionized water was added to ∼10 mg/ml and the resulting solution was allowed to sit capped at room temperature for ∼1 h with periodic vortexing. After this, the cloudy solution was sonicated for 30 min at ∼35°C, producing a nearly transparent solution of small unilamellar vesicles. Vesicle formation was confirmed by solution-state 31P-NMR. The solution was applied to freshly cleaved mica substrates (no prewetting was employed) and allowed to evaporate at ∼45°C overnight. Samples were hydrated and wrapped as for solvent cast films.
Solid-state NMR
31P-NMR was carried out on a 300 MHz Varian Unity spectrometer (Varian Associates, Palo Alto, CA) equipped with a ChemMagnetics Apex DRNS Wideline static probe (Varian Associates, Fort Collins, CO) in a 7.05 T Oxford wide-bore superconducting magnet (Oxford Magnet Technology, Oxford, UK). A six-turn homebuilt rectangular coil of 22 AWG copper wire (flattened using a large, flat-tooth vise) with inner dimensions of 15.4 × 15.4 × 1.5 mm was used. In terms of tuning and matching, this resonated similarly to a standard ChemMagnetics 10-mm solenoidal coil (Varian). In some cases, the coil was used in a rotatable coil holder (25); however, all results presented here have the coil at an orientation with the normal to the substrate plane parallel to the static magnetic field of the superconducting magnet. Temperature was regulated by a steady flow of gas around the coil at 21.5°C unless otherwise noted. One-dimensional 31P-NMR spectra were recorded at 121.409 MHz with a spectral width of 80 kHz and single-pulse excitation using 90° pulses with duration between 2 and 3 μs, a field-strength equivalent between γB1/2π = 83–125 kHz. For each spectrum, 128 free induction decays were measured and fast-Fourier-transformed, with a relaxation delay of 5 s and acquisition time of 40 ms for 6396 data points. Decoupling at the 1H frequency of 299.916 MHz was applied at a field strength of γB1/2π ∼28 kHz with a Varian 1.0 kW amplifier over the duration of the 90° pulse and the acquisition period. Such 1H decoupling is not an absolute requirement, but serves to sharpen lines for the powder-type spectra being observed and improves accuracy of chemical shift measurements (26). Vesicle spectra were collected in a 5-mm solenoid coil (ChemMagnetics) in 5-mm NMR tubes cut to length; 32 transients were collected at a similar 31P field-strength with a relaxation delay of 10 s without use of the kW amplifier during 1H decoupling. Unless otherwise noted, spectra were processed using line-broadening of 50 Hz (vesicle spectrum 10 Hz).
RESULTS AND DISCUSSION
The general 31P-NMR spectral characteristics of phospholipids in oriented and nonoriented phases are first discussed. Following this is a brief discussion of shimming and referencing issues for this class of experiments. Next, the effectiveness of mica as a substrate for oriented sample solid-state NMR is examined in terms of observed spectral characteristics and chemical shift perturbation. Results of a series of experiments using POPC as a model lipid are then presented: first, deposition from a variety of solvents with different dielectric are compared; second, the effect of the amount of lipid deposited per substrate on spectral characteristics is examined; and, third, hydration of the sample under different conditions is compared. Using the optimal amount of lipid per substrate determined using POPC, the efficacy of forming oriented bilayers using POPG and DOPA and oriented hexagonal phase using DOPE is then examined, using lipid films cast from the same set of solvents. The ability to carry out the hexagonal to bilayer phase transition while retaining full planar-supported orientation is also discussed. Finally, a direct comparison is carried out between bilayers deposited on mica and those deposited on glass.
Spectral characteristics of an oriented bilayer sample
Before detailing our studies of lipid orientation conditions for solid-state NMR study, an outline of the features of 31P-NMR spectra that would be expected for oriented lipid samples is provided as a reference. All experiments are being carried out by static solid-state NMR spectroscopy, meaning that the orientation and position of the sample remains constant. The surface plane of a lipid bilayer may be specified by its vector normal—this will be referred to as L⊥. The second vector important for specifying spectral characteristics is the direction of the static magnetic field of the spectrometer, B0. Assuming that the planar-supported lipid bilayers will be parallel to the surface of the glass or mica substrates, the configuration that we are employing exclusively during the present study is such that L⊥ is parallel to B0. In the ideal supported bilayer sample, 100% of the lipids would be in this orientation. However, in reality, we would expect at least small components of signal to arise from other sources. First, bilayers not adsorbed to the solid supports will likely form a random distribution of orientations. Second, small liposome components will be relatively mobile compared to larger bilayer leaflets. In some instances, the lipid may also form a hexagonal phase comprising an ordered array of cylinders with lipid tailgroups on their exterior (27), as opposed to bilayers or small liposomes. Presuming an initial adsorption of hydrophilic headgroups to the substrate, the cylinders will have a long-axis lying in plane with the substrate surface. The preferred phase is temperature- and lipid-dependent.
All shifts and tensor elements in the following discussion use the chemical shift, as opposed to shielding, convention (28). Because lipids in a hydrated bilayer rotate rapidly about L⊥, the chemical shift tensor observed for the 31P atom in the phosphate headgroup will be axially symmetric, with two equal components with value δ⊥ and a third at δ∥ (29,30). For aligned bilayers, this gives rise to a signal with line-width determined both by the T2 of the 31P nucleus and by the angle (θ) between B0 and L⊥ (31) with a chemical-shift dependent upon θ (32),
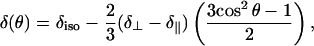 |
(1) |
where δiso is the isotropic chemical shift.
For lipids with L⊥ parallel to B0, therefore, Eq. 1 shows that the chemical shift of the signal will be at δ∥. Any unoriented lipids in bilayers will produce a powder pattern, with characteristics determined by the values δ⊥ and δ∥. Finally, any lipids in liposome impurities will be rapidly rotating, providing a time-averaged, symmetric chemical shift tensor leading to a signal at the δiso, characteristic of any 31P nucleus in a phosphate headgroup of the given lipid. Oriented hexagonal phase lipids also give rise to a single line with an angular dependence relative to B0, given by
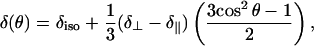 |
(2) |
where, in this case, θ is the angle between the cylinder long-axis and B0 (26). Note that the values of δ⊥ and δ∥ do not change, since these are just as applicable to a monolayer configuration of phospholipids as to a bilayer, and a hexagonal phase of phospholipids is effectively a curved monolayer. Equation 2 demonstrates that hexagonal phase lipids with the cylindrical long-axis perpendicular to B0 will have a chemical shift of 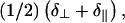 clearly distinguishable from δiso at
clearly distinguishable from δiso at 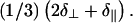
Simulation of the each of these signals is readily possible, particularly through application of software such as SIMPSON (33). A typical spectrum simulated for a nonideal oriented bilayer sample prepared using POPC is shown in Fig. 1 a. Parameters employed in this simulation, based upon the real spectrum shown in Fig. 1, were δ⊥ = −21.4 ppm and δ∥ = 25.3 ppm (for the axially symmetric tensor) and line-broadening of 170 Hz. The three components were provided using 1), Euler angles of (0,0,0) for the peak corresponding to bilayers parallel to B0; 2), a powder pattern generated using 4180 crystallite orientations for lipids having this chemical shift anisotropy tensor corresponding to randomly oriented lipid bilayers; and 3), an isotropic peak at δiso corresponding to relatively mobile liposomes. An appropriately weighted sum of the three components provides an excellent reproduction of the experimentally acquired spectrum (no line-broadening applied to the experimental data) of a nonideal POPC bilayer sample allowed to undergo dehydration for several days after preparation. Marassi and Crowell note a decrease in frequency for the oriented bilayer peak from ∼27 to 17 ppm for another PC lipid upon going from 10 water molecules to two per lipid (8). In our case, however, it appears that the bulk bilayers between the paired substrates are remaining well hydrated. Therefore, we believe that edge effects (i.e., ease of water extraction or rearrangement of lipids at edges compared to middle of sample) are more likely causing an increase in the powder pattern peak intensity at ∼−21 ppm. Note that the simulated spectrum nearly perfectly overlays the experimental data, with the exception of the small peak around 18 ppm, which can be fit by the addition of a fourth component of lipid bilayers with an angle θ = 25.2° between B0 and L⊥. This particular peak is quite commonly observed in POPC bilayer samples on both mica and glass (see, e.g., Fig. 1 c). Although headgroup hydration level could produce such a change in chemical shift (8), it is hard to envision partitioning of lipids in a bulk sample into two distinctly different hydration states. Therefore, the source of this peak is not obvious.
FIGURE 1.

(a) Simulated (orange dash) and experimental (blue) 31P-NMR spectra of nonideal oriented lipid sample (∼17.5 mg POPC on freshly cleaved mica) containing convoluted components arising from (1) oriented lipid bilayers with bilayer-normal parallel to static magnetic field of spectrometer; (2) lipid bilayers in random orientation; and (3) bilayer type liposomes. Simulated using SIMPSON (33); simulation parameters given in main text. Shown spectrum acquired after allowing sample to dehydrate for several days. (b and c) Approximately 7.6 mg POPC deposited from dichloromethane on mica (green and red) or clean, prewetted cover glass (blue). (d) Approximately 20 mg POPC deposited from methanol on two freshly cleaved, prewetted sheets of mica. Each sample desolvated under vacuum, hydrated with ∼10 equivalents of deionized water, and double-wrapped with plastic food wrap. (b) Acquired ∼4 h after hydration; (c) acquired after an additional three days at 98% relative humidity and 21°C; (d) red, initial spectrum; blue spectrum acquired immediately after removal of excess lipid and rewrapping sample with fresh plastic wrap. 31P observed at 121.4 MHz with high-power 1H-decoupling; no line-broadening applied to experimental data in a; 50 Hz line-broadening in b–d. Chemical shift of glass spectra in b and c aligned to mica spectra using peaks 2 and 3.
Shimming and referencing
Chemical shift and line-shape of nonspherical samples, such as planar-supported bilayers, are affected by the bulk magnetic susceptibility effect (34). Because all spectra presented here are acquired with the normal to the substrate plane parallel to the static magnetic field of the spectrometer (B0), the orientational dependence of the bulk magnetic susceptibility effect does not come into play, and only a single correction at this angle needs to be applied. Ideally, shimming and referencing would use a resonance from the sample itself (35). We have opted to use an external standard liquid sample as a chemical-shift reference, which was shimmed to provide an isotropic line with width at half-maximum of ∼50–70 Hz. Note that the shim-set used did not significantly affect the character of solid-state 31P-NMR spectra of phospholipid samples, but that shimming was necessary for referencing. All 31P chemical shifts we report here are referenced to a 1 M phosphate buffer at pH ∼4.7 (∼40 Hz line-width at half-maximum height; sample spectrum in Supplementary Material) at −3.6 ppm.
Suitability of mica as a substrate for solid-state NMR
In our experience, sheets of mica with thickness of 20–60 μm are very easy to prepare and handle. On the basis of its chemical composition, Muscovite mica would not be expected to give rise to any background signal for 31P, 13C, or 15N. One-dimensional 31P-NMR of samples sandwiched between mica sheets and observed in a homebuilt rectangular coil show standard powder pattern shapes for 15N (results not shown) and 31P (egg phosphatidylcholine; see Supplementary Material). The 31P powder pattern compares extremely well with simulation and with literature values for the principal tensor components (reviewed in Ref. 26; note the reverse-sign convention for chemical shift). Spectra of oriented lipid bilayers deposited on mica substrates demonstrate identical features to those of bilayers on glass from solvent-cast films of POPC. Fig. 1, b and c, demonstrates a direct comparison of spectral characteristics for the same amount of POPC deposited on both glass and mica. In Fig. 1 b, the characteristics are compared after hydration by direct water droplet application. At this stage, both mica samples demonstrate a higher intensity in comparison to the glass sample for the oriented bilayer peak, but very similar intensities for the isotropic and powder peaks. Such an intensity difference is common; however, Fig. 1 c demonstrates that the intensity difference is not always maintained using the same set of samples incubated for three days at 98% relative humidity and 21°C, where mica sample i maintains the intensity for the oriented bilayer peak and mica sample 2 becomes practically indistinguishable from the glass sample. It should be noted that probe tuning and matching and 90° pulse-widths were not perturbed when switching between mica and glass samples with identical dimensions. Fig. 1, b and c, is a good representation of the potential diversity in sample character, where the characteristics of mica samples tended to be as good as or better than those prepared on glass. We have been able to maintain favorable oriented peptide-lipid sample characteristics on mica by sealing the samples into plastic bags along with moist cloths (results not shown). All in all, mica provides an attractive alternative to glass as a planar support for oriented bilayers in NMR study.
Previous studies imply the possibility of chemical shift changes arising from magnetic susceptibility of the mica substrate (23,24). Since the mica that we are using is high quality Muscovite mica, containing a low amount of paramagnetic iron, the effects of additional mica upon chemical shift would be expected to be minimal. To verify this, we quantified 31P chemical shift changes for samples of lipids sandwiched between two thin sheets of mica as we introduced additional, measured quantities of mica above, below, or both. Pulse widths for 31P did not change over these sets of experiments, implying that B1 field strength is not amplified or attenuated by the presence of additional mica. A plot of the total mass of mica surrounding the sample versus observed chemical shift difference from the sample with only the support substrate pair is provided in Fig. 2. Linear regression analysis of all measured chemical shift differences (a total of 10 peaks arising from two hexagonal phase samples and three oriented bilayer samples also demonstrating powder peaks) indicates a characteristic chemical shift increase of 8.2 ± 0.3 ppm per gram of mica. A typical mass range for a 15 × 15-mm mica substrate in the thickness range of 30–60 μm is 12–35 mg; therefore, chemical shift differences for a few sheets of mica would be well under 1 ppm, while for tens of sheets it may be several ppm. We have been routinely using grade V-4 mica, defined by ASTM standards as having 25% air inclusion and minimal coloration (“good stained” visual quality) due to iron impurities. To determine the effect of the quality of the mica on chemical shift perturbation, we compared addition of type V-4 mica to the exceptionally high quality type V-1 mica, which has 0% air inclusion, and clear coloration. No significant differences in chemical shift perturbation were seen comparing grades V-1 and V-4. We are currently carrying out in-depth studies of such substrate-induced chemical shift perturbation.
FIGURE 2.

Four POPC bilayer samples and two DOPE hexagonal phase samples sandwiched between pairs of mica substrates were studied by static solid-state 31P-NMR with up to six sheets of known mass of grade V-4 and V-1 (V1 notation in legend) Muscovite mica placed above, below, or both. Chemical shift perturbation relative to the average measured shift of the indicated peak is plotted versus mass of mica added.
Solvent effects on lipid film morphology
Eight solvents with a range of dielectric constants (Table 1) were tested for cast lipid film deposition efficacy. Each solvent over the dielectric range employed from chloroform (ɛ 4.81) to methanol (ɛ 33.00) proved distinct and reproducible in terms of deposit morphology and film spreading for each lipid (detailed in the Supplementary Material). Note that a given solvent may lead to different film characteristics upon changing lipid headgroup, as might be anticipated based on differences in solubility alone. These characteristics arise from the three-phase equilibrium where the relative interfacial surface energies of the lipid solution/suspension-air, lipid solution/suspension-mica, and air-mica interactions control the degree to which the solvent-lipid mixture spreads. In cases where the lipid is not actually solubilized, but is in suspension of some description, this is an even more complicated four-phase interaction. A detailed analysis and discussion of these effects is beyond the present work (see, e.g., Ref. 36), where we carry out a much more empirical approach.
TABLE 1.
Set of solvents with varying dielectric constant (ɛ) used to test characteristics of lipid film morphology as cast on freshly cleaved Muscovite mica (experiment-dependent amounts of 1.25–10 mg of POPC, POPG, DOPA, and DOPE were deposited per 15 × 15-mm plate of mica in 20-μL aliquots of 50–75 mg/mL at 23°C)
For each type of lipid, the general trend of observed film morphologies are shown schematically in Fig. 3 and labeled types i–v. The trend is described as follows. From a solvent with a low dielectric constant, the lipid film will be constrained into a small region of the substrate (film type i). As the solvent increases in polarity the lipid film spreads increasingly from the point of application (film types ii–iii). At some crossover point, dependent upon the headgroup chemistry, the lipid becomes preferentially swept outwards from the point of application, leading to an area increasingly devoid of lipid (film types iv–v). A useful technique to avoid overflow of such rapid spreading solvents and loss of lipids is to keep the mica substrate raised above the bench, so that the substrate edges serve as a boundary against continued solvent/lipid spreading. For each of the four lipids examined, a given solvent was observed to provide maximal spreading of the lipid film (i.e., film type iii) without leaving an area at the point of application devoid of lipid. Deposition of lipid vesicles from water (ɛ 80.10) also produced reproducible film characteristics (illustrated separately in Fig. 3). These films are a bit of an amalgam between types iii and iv, and tend to display raised striations of lipid that run roughly parallel to each other over a lower coverage film of lipid. The elevated lipid around the plate edges tends to be more opaque versus the remainder, which is translucent.
FIGURE 3.

General trend observed for lipid films (types i–v) formed with POPC, POPG, DOPA, and DOPE for films cast from solvent of increasing dielectric constant (ɛ) on freshly cleaved mica substrate.
POPC deposition characteristics on mica
Several general observations may be made for POPC deposition on mica. (Deposition of other headgroup types is discussed in a later section.) Upon film hydration by water droplet application and immediate sandwiching of the lipid film, films formed with each solvent and from aqueous vesicles provided practically indistinguishable oriented bilayer 31P NMR signals (e.g., Fig. 1). However, the differential spreading characteristics make certain solvents much more desirable than others. 3-Pentanol actually appears most desirable in terms of its wetting of mica (Fig. 3, type iii), but it is also slowest to evaporate. HFIP, on the other hand, spreads exceedingly fast, but leaves an area devoid of lipid at the point of aliquot application (Fig. 3, type iv). In the case of more gradual film hydration in a humidity chamber, the most desirable solvent would likely be the one that provides the best surface coverage, improving the speed of bilayer formation by decreasing the amount of diffusion required to form bilayer in areas devoid of lipid. Presumably, this would also lead to a more uniform deposition thickness over the substrate for the hydrated set of thousands of bilayers. Although commonly used, solvent mixtures should probably be avoided, since each solvent would have a different interaction with the substrate, the lipid, and the peptides, promoting heterogeneity in the initial film deposit which may or may not be removed by hydration (9,27,36). Because of its ability to form films with reasonable coverage of the mica substrate and rapid evaporation (Fig. 3, type ii with ∼70% coverage), deposition from dichloromethane was used for all remaining portions of the study involving POPC.
Optimizing lipid deposition amount
The typical protocol for oriented lipid bilayer-peptide samples utilizes stacks of tens of glass substrate plates, each with a small proportion of the total lipid-peptide sample. Each substrate plate decreases the filling factor. We found that fairly large deposits of lipid (∼20 mg) sandwiched between two mica substrates and hydrated by water droplet application provided only 35–60% of the area of the 31P signal in the regime of the spectrum consistent with an aligned bilayer sample (Fig. 1 d), with obvious “excess” lipid squeezed out at the edges of the plates. The hypothesis that this gives rise to the remainder of the signal (characteristic of a powder pattern for lipids in a bilayer configuration, as in component 2 of Fig. 1 a) was tested by removal of such excess lipid. This greatly improved the spectral characteristics, uniformly providing spectra with only a single peak arising at the parallel orientation, as can be clearly seen in Fig. 1 d. However, an optimal deposition procedure should provide such spectral characteristics without relying on the removal of sample. Therefore, we carried out a quantitative study of spectral characteristics upon the amount of lipid deposited on a pair of 15 × 15-mm mica plates.
Using POPC, a number of trials were performed where the exact amount of lipid deposited was quantified by measurement of the difference between the substrate mass immediately after cleavage and combined the substrate and lipid mass after film casting and desolvation under vacuum. A number of spectral characteristics were then examined using static solid-state 31P-NMR, and compared using the peak corresponding to the phospholipids in bilayers having L⊥ perpendicular to B0 and in the powder component with major intensity nearest to the δ⊥ component of the chemical shift tensor for lipids in the hydrated bilayer configuration. Fig. 4 shows the dependence of two such spectral characteristics upon the mass of POPC deposited: height of the peak arising from oriented bilayers (Fig. 4 a) and amount of total lipid giving rise to the area under the peak arising from the oriented bilayers (Fig. 4 b). In each case, the optimum in spectral characteristics is achieved at ∼7.5 mg of lipid deposited between two 15 × 15-mm mica substrates. For masses greater than this, both the peak-height of the oriented bilayer signal and the mass of lipid in oriented bilayers plateau. Therefore, this implies an optimum coverage for POPC of ∼17 μg of lipid per mm2 of mica substrate.
FIGURE 4.

Spectral characteristics for static solid-state 31P-NMR of POPC bilayers deposited on pairs of mica substrates and rehydrated with ∼10 water molecules per lipid for three separate sets of eight lipid depositions: trial set 1, diamonds, at 4–6 h post-hydration; trial set 2, squares, at 11–16 h post-hydration; trial set 3, triangles, at 2–4 h post-hydration; and crosses at 24–26 h post-hydration. (a) Peak-height of oriented bilayer component of spectrum normalized to the highest peak for the given data set; (b) amount of lipid in the oriented bilayer peak calculated using percent area of that peak versus the entire 31P spectrum (diagonal line, 100% of lipid oriented.)
Comparison of hydration methods
Hydration has previously been shown to be crucial in the quality of orientation for solid supported lipid bilayer samples (8,37). Spectral characteristics upon hydration by addition of a measured volume of water as shown above were compared to hydration over a period of 7–14 days in a room-temperature humidity chamber and in a humidity chamber maintained at ∼45°C. In each case, samples were prepared by depositing POPC from dichloromethane at lipid coverage levels just below the apparent critical coverage amount of ∼17 μg/mm2 seen with water droplet hydration and at approximately twice this coverage. At approximately the critical coverage level, hydration in a chamber maintained at 22°C and 98% relative humidity provided spectra that appeared to be composed practically exclusively of aligned bilayer components upon wrapping and immediate 31P-NMR analysis. Samples prepared at approximately two times the critical POPC coverage level, however, did not have significantly improved characteristics in comparison to the direct hydration method. Although a number of publications report the use of elevated temperature humidity chambers (∼45°C, ∼90% RH), we found that such an environment with POPC as the deposited lipid gave uniformly poor bilayer characteristics, with a large amount (30–60%) of lipid giving rise to a bilayer powder pattern, as opposed to oriented bilayers. These results may imply an optimum temperature for hydration of a given lipid with respect to its chain-melting phase-transition temperature where, if that temperature is greatly exceeded, the lipid is likely to form bilayers indiscriminately, rather than being driven to do so from an initial ordered layer on the solid support substrate. A systematic study of the kinetics of bilayer formation in different constant humidity-temperature environments as a function of the initial desolvated film morphology would also be useful. From our studies, hydration at room temperature in a chamber at 98% RH appeared to be optimal for POPC deposited on mica, although direct hydration by a water droplet was at least 90% as effective in forming well-oriented samples and is much less time-consuming.
Orienting phospholipids with different headgroups
The lipids POPG, DOPA, and DOPE were also dissolved in and deposited from the same set of nine solvents (Table 1) as POPC, where soluble. Characteristics of the observed film morphologies are provided in the Supplementary Material. Unlike POPC, these lipids tended to have a much stronger dependence of observed spectral characteristics upon solvent type.
POPG did not reproducibly form oriented bilayer samples on mica by solvent casting, as has been previously observed with other phosphatidylglycerol lipids (e.g., Ref. 38). Reproducibility of POPG bilayer formation would likely be facilitated by reversing the initially anionic surface charge of the mica through pretreatment by a cationic agent such as APTES or poly-L-lysine, as has been shown by atomic force microscopy (38). Vesicle deposition of POPG generally provided improved orientation, but the relative amounts of lipids in oriented (Fig. 1 a, peak 1) versus powder (Fig. 1 a, peak 2) configurations were still variable. POPG deposition at lower substrate coverage amounts improved the relative intensity of the oriented peak over the powder peak, but still showed heterogeneity in the ratio between these peaks. Mixed bilayers of POPC/POPG, commonly employed for oriented solid-state NMR of membrane proteins or peptides, were readily formed at a molar ratio of 4:1 POPC/POPG on mica from dichloromethane and HFIP, and demonstrated spectral features generally indistinguishable from those of pure POPC bilayers (results not shown). At present, we have not investigated such binary lipid systems in detail.
DOPA reproducibly formed oriented bilayer samples with two peaks corresponding to bilayers with L⊥ parallel to B0 at a level of hydration of ∼10 equivalents of water per lipid (Fig. 5 a). Chloroform reproducibly provided samples with these spectral characteristics, indicating that the primary configuration is an oriented bilayer (Fig. 5 a). Each of the other three organic solvents in which DOPA dissolved (dichloromethane and 3-pentanol) or formed a suspension (HFIP; typically only ∼33% of the DOPA was transferred to the film) consistently showed additional peaks with increasing intensity relative to the peaks at δ∥ as solvent dielectric increased (Fig. 5 b).
FIGURE 5.

Static solid-state 31P-NMR spectra of (a and c) ∼5.7 mg DOPA deposited from chloroform; (b and d) ∼6.5 mg DOPA deposited from 3-pentanol; and (e) ∼7.2 mg DOPA (solid representation) spread from vesicle solution (shaded representation is solution-state NMR spectrum of DOPA vesicle solution) in water. Each prepared on two freshly cleaved, prewetted sheets of mica, desolvated in vacuum, hydrated with ∼10 (a, b, and e) or ∼25 (d) equivalents of water, and double-wrapped with plastic food wrap. In c, the sample was unwrapped after acquisition of the spectrum shown in a, hydrated with an additional ∼12 equivalents of water, and rewrapped.
Three possible scenarios may be envisioned which would cause a second peak corresponding to oriented bilayer phase DOPA molecules, as seen in Fig. 5 a. First, this may be indicative of different headgroup phosphatidic acid ionization states, since the headgroup may exist in an uncharged state, or in monovalently or divalently anionic states. Second, and alternately, since we are working in conditions of relatively limited water to solvate counterions, this may simply be due to the presence or absence of the sodium counterion from the initial DOPA-monosodium salt. Third, different hydration states of the DOPA headgroups may be leading to the two distinct peaks at the parallel orientation. Two strategies were employed to attempt to differentiate between these scenarios. In one, further equivalents of water were added to samples demonstrating two peaks in the region of the 31P-NMR spectrum indicating oriented bilayer samples, as in the spectrum in Fig. 5 a. In the other, samples were prepared directly with ∼25 equivalents of water per DOPA molecule. Fig. 5 c shows the same sample as in Fig. 5 a after addition of ∼12 further equivalents of water, whereas Fig. 5 d is a representative spectrum for a sample hydrated with ∼25 equivalents of water. In each case, the resulting spectra showed a single peak characteristic of an oriented bilayer of DOPA.
If the second oriented peak (Fig. 5 a) is due strictly to hydration state, then, as a sample with increased water present and showing a single oriented bilayer peak (Fig. 5, c and d) becomes dehydrated, this second peak should reappear. However, extended analysis of such samples during dehydration showed the appearance of a bilayer powder pattern peak without the reappearance of the second, upfield-oriented bilayer peak. Therefore, we can rule out different hydration states of the lipid as the cause of the pair of oriented bilayer peaks observed at ∼10 equivalents of water. Although chemical-shift anisotropy for 31P powder patterns of another lipid with the phosphatidic acid headgroup has indeed been shown to depend on ionization state (39), differentiation between a change in ionization state of the phosphatidic acid and the presence or absence of a sodium counterion for a headgroup remaining in the singly ionized state is not trivial. This is particularly hindered because the concept of pH in a system where water is so limited in amount and restricted in motion is not meaningful. Therefore, it is difficult to estimate the most favorable ionization state of the phosphatidic acid. It may be possible to differentiate between these cases by using 23Na-NMR to examine the bound state of the counterion, by carrying out high-resolution magic-angle spinning solid-state 1H-NMR on oriented bilayer samples, by attempting exchange of the Na+ ions before film deposition, or by carrying out 31P-NMR on films prepared from DOPA in different ionization states. However, these experiments are beyond the scope of this work.
Samples formed by spreading of aqueous DOPA vesicles have dramatically different character. In this case, the major peak observed at a hydration level of ∼10 equivalents of water is at the isotropic chemical shift (Fig. 5 e), implying that the vast majority of the DOPA vesicles do not actually undergo fusion to form bilayers. Note that the peak is greatly broadened in comparison to that observed for small unilamellar DOPA vesicles in solution (Fig. 5 e, gray-shaded spectrum, 10-Hz line-broadening applied). Even with increased hydration to ∼25 equivalents of water per lipid, DOPA samples prepared by vesicle fusion do not show significant improvement in orientation—further demonstrating the finicky nature of DOPA in terms of forming planar-supported samples. Although the Na+ counterion would likely be removed from the headgroup in this case, the inability to achieve well-oriented DOPA samples meant that we could not resolve the counterion issue discussed above under these conditions.
DOPE was deposited above its bilayer to hexagonal-II transition temperature (Th) of 10°C, and therefore would be expected to form oriented hexagonal phase cylinders with a reverse-phase appearance where tailgroups coat the exterior of the cylinder and headgroups are on the interior. In cases where an oriented hexagonal phase forms, the long-axis of the phospholipid cylinder would be expected to be parallel to the mica substrate plane, since this will allow PE headgroup adsorption to the hydrophilic mica substrate. Specifically, a monolayer would likely adsorb to the substrate, followed by cylinder adsorption to the exposed tailgroups of the monolayer. Such adsorption would give rise to a peak at the frequency given by Eq. 2. Note that a powder pattern of hexagonal phase lipids has half the width of a powder of bilayer phase lipids (26). It will extend upfield to δ⊥, which has a double contribution relative to the downfield edge at 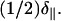 Depending upon the solvent used for deposition, a variety of cases was observed. With 3-pentanol, oriented hexagonal phase DOPE was practically exclusively observed (Fig. 6 a). The more polar solvents HFIP and TFE tended to produce samples demonstrating primarily hexagonal phase components, but with notable peak intensity (∼5–10% of the intensity of the hexagonal peak) at the δ∥-position, implying some oriented bilayer phase lipids within the sample. Conversely, the less polar solvents chloroform and dichloromethane produced samples showing a mixture of oriented and disordered hexagonal phase lipids, with small contributions from oriented bilayer phase lipids appearing in some instances, as can be seen clearly in the representative spectrum shown in Fig. 6 b. Vesicle deposition was not carried out because the required conditions would be entirely different—the steps of solvation in water and sonication would need to be carried out below the Th of 10°C (which is not readily possible with a standard sonicator), while the phase transition would then be induced during the subsequent dehydration step at elevated temperature. By carrying out orientation of DOPE entirely above Th, we avoid such perturbations in the homogeneity of the lipid phase.
Depending upon the solvent used for deposition, a variety of cases was observed. With 3-pentanol, oriented hexagonal phase DOPE was practically exclusively observed (Fig. 6 a). The more polar solvents HFIP and TFE tended to produce samples demonstrating primarily hexagonal phase components, but with notable peak intensity (∼5–10% of the intensity of the hexagonal peak) at the δ∥-position, implying some oriented bilayer phase lipids within the sample. Conversely, the less polar solvents chloroform and dichloromethane produced samples showing a mixture of oriented and disordered hexagonal phase lipids, with small contributions from oriented bilayer phase lipids appearing in some instances, as can be seen clearly in the representative spectrum shown in Fig. 6 b. Vesicle deposition was not carried out because the required conditions would be entirely different—the steps of solvation in water and sonication would need to be carried out below the Th of 10°C (which is not readily possible with a standard sonicator), while the phase transition would then be induced during the subsequent dehydration step at elevated temperature. By carrying out orientation of DOPE entirely above Th, we avoid such perturbations in the homogeneity of the lipid phase.
FIGURE 6.

Static solid-state 31P-NMR spectra acquired at 21.5°C. (a) Approximately 5.8 mg DOPE deposited from 3-pentanol, and (b) ∼5.9 mg DOPE deposited from dichloromethane; each was prepared on two freshly cleaved, prewetted sheets of mica, desolvated in vacuum, hydrated with ∼10 equivalents of water, and double-wrapped with plastic food wrap.
Hexagonal to bilayer phase transitions
To test the capability of adsorbed, oriented hexagonal phase DOPE to undergo the transition to the bilayer phase and retain orientation, we examined the temperature dependence of 31P-NMR spectral characteristics. A series of experiments recorded over the range from 21.5°C to −5°C is shown in Fig. 7. As the temperature was lowered, the hexagonal phase peak at −0.4 ppm was observed to coexist with the oriented bilayer peak at ∼23.9 ppm. After incubation at −5°C, well below Th, the transition was observed to be complete. Note that we would expect the transition to occur equally fully at 5°C, but at a slower rate. The observed phase transition was fully reversible with no change in spectral characteristics observed for over several jumps between 21.5°C and −5°C. The ability to form such stable oriented hexagonal phases may be extremely useful, given the importance of so-called nonbilayer lipids such as DOPE in a variety of instances (recently reviewed in Ref. 40). Effects of the hexagonal to bilayer phase transition upon lipid-peptide interactions and membrane-associated or transmembrane peptide structure may also be conceivably studied using this type of oriented sample.
FIGURE 7.

Static solid-state 31P-NMR spectra acquired at given temperature. Approximately 7.2 mg DOPE was deposited from 3-pentanol onto two freshly cleaved, prewetted sheets of mica, desolvated in vacuum, hydrated with ∼10 equivalents of water, and double-wrapped with plastic food wrap. Note that chemical shifts are not corrected for temperature.
SUMMARY
Oriented samples of a variety of phospholipids were prepared on mica substrates. Mica was found to be a very convenient alternative to glass, with comparable or better solid-state 31P-NMR spectral characteristics for the phospholipid headgroup of oriented, planar-supported POPC bilayer samples. It should be noted that mica provides chemically identical surface characteristics upon cleavage, unlike a glass surface, which has characteristics dependent upon the cleaning method. The effects of the amount of mica present upon 31P chemical shift were quantified, with a downfield shift noted for increasing mica mass. This had a linear dependence upon the mass of mica added, however, so can be easily compensated for.
A set of eight solvents of varying dielectric constant was used for lipid film deposition upon pairs of mica substrates. POPC film characteristics on mica varied over the set of solvents, but oriented bilayer characteristics were indistinguishable by 31P-NMR upon drying under vacuum and subsequent hydration with ∼10 equivalents of water. DOPA and DOPE, on the other hand, not only differed in film characteristics from solvent to solvent, but also demonstrated definite preferences for certain solvents in the quality of oriented bilayer or hexagonal phase lipid samples formed, respectively. POPG did not consistently form well-oriented supported bilayers on its own, but readily formed oriented bilayers on mica from various solvents when deposited in binary mixtures with POPC.
Deposition of POPC was used to search for an optimal amount of lipid per pair of solid support substrates. Quantification of spectral features indicated an asymptotic effect for the amount of lipid per substrate, beyond which the formation of oriented bilayer relative to disordered powder from bilayers was not favored. Hydration of POPC films by direct addition of water versus in a hydration chamber at 98% relative humidity and 22°C did not provide significantly different oriented bilayer quality, whereas incubation at 90% relative humidity at 45°C actually greatly decreased the quality of bilayer orientation, as judged by 31P-NMR spectra.
DOPA formed oriented bilayers by solvent casting with two peaks corresponding to different oriented lipid species at ∼10 water molecules per lipid. Addition of further water to >20 molecules per lipid caused the disappearance of the more upfield of these peaks, giving rise to a well-oriented bilayer sample with a single peak. Since dehydration causes a bilayer phase lipid powder pattern peak to appear, rather than reappearance of the upfield peak, we do not attribute the two peaks to different headgroup hydration states. Rather, the second peak is likely due either to the presence of some sodium counterions which are displaced by additional water or to different ionization states. In the case of aqueous vesicle spreading, spectral characteristics indicated that the majority of lipids remain in liposomes.
DOPE formed extremely well-oriented hexagonal phase lipids at room temperature, which could be converted within hours to the oriented bilayer phase by incubation at −5°C. This phase transition was fully reversible, and could be carried out several times in each direction for a given sample.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
Thanks to Jeff DeVries for flat-coil fabrication and spectrometer maintenance and to Sam Graziano at the Faculty of Medicine Workshop for helpful discussions and expert machining. Thanks also to Dr. Steffen Graether at PENCE and Drs. Carla Franzin and Francesca Marassi at The Burnham Institute for valuable discussions. We are grateful to Dr. Marassi for providing us with the glass substrates employed.
J.K.R. is supported by Postdoctoral Fellowships from the Natural Sciences and Engineering Research Council of Canada and Alberta Heritage Foundation for Medical Research. B.D.S. receives support as a Canada Research Chair in Structural Biology. This work has been funded by the Canadian Protein Engineering Network of Centres of Excellence.
References
- 1.Schmidt-Rohr, K., and H. W. Spiess. 1994. Multidimensional Solid-State NMR and Polymers. Academic Press, NY.
- 2.Marassi, F. M. 2002. NMR of peptides and proteins in oriented membranes. Concepts Magn. Reson. 14:212–224. [Google Scholar]
- 3.Watts, A., S. K. Straus, S. L. Grage, M. Kamihira, Y. H. Lam, and X. Zhao. 2004. Membrane protein structure determination using solid-state NMR. Methods Mol. Biol. 278:403–474. [DOI] [PubMed] [Google Scholar]
- 4.de Vries, J. J., and H. J. C. Berendsen. 1969. Nuclear magnetic resonance measurements on a macroscopically ordered smectic liquid crystalline phase. Nature. 221:1139–1140. [Google Scholar]
- 5.Hemminga, M. A., and H. J. Berendsen. 1972. Magnetic resonance in ordered lecithin-cholesterol multilayers. J. Magn. Reson. 8:133–143. [Google Scholar]
- 6.Opella, S. J., and F. M. Marassi. 2004. Structure determination of membrane proteins by NMR spectroscopy. Chem. Rev. 104:3587–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moll 3rd, F., and T. A. Cross. 1990. Optimizing and characterizing alignment of oriented lipid bilayers containing gramicidin D. Biophys. J. 57:351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marassi, F. M., and K. J. Crowell. 2003. Hydration-optimized oriented phospholipid bilayer samples for solid-state NMR structural studies of membrane proteins. J. Magn. Reson. 161:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanford, C. 1980. The Hydrophobic Effect. John Wiley & Sons, New York.
- 10.Langmuir, I. 1917. The constitution and fundamental properties of solids and liquids. II. Liquids. J. Am. Chem. Soc. 39:1848–1906. [Google Scholar]
- 11.Blodgett, K. B. 1934. Monomolecular films of fatty acids on glass. J. Am. Chem. Soc. 56:495. [Google Scholar]
- 12.Blodgett, K. B. 1935. Films built by depositing successive monomolecular layers on a solid surface. J. Am. Chem. Soc. 57:1007–1022. [Google Scholar]
- 13.Langmuir, I., and V. J. Schaefer. 1938. Activities of urease and pepsin monolayers. J. Am. Chem. Soc. 60:1351–1360. [Google Scholar]
- 14.Steinem, C., A. Janshoff, W.-P. Ulrich, M. Sieber, and H.-J. Galla. 1996. Impedance analysis of supported lipid bilayer membranes: a scrutiny of different preparation techniques. Biochim. Biophys. Acta. 1279:169–180. [DOI] [PubMed] [Google Scholar]
- 15.Tamm, L. K., and H. M. McConnell. 1985. Supported phospholipid bilayers. Biophys. J. 47:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brian, A. A., and H. M. McConnell. 1984. Allogeneic stimulation of cytotoxic T-cells by supported planar membranes. Proc. Natl. Acad. Sci. USA. 81:6159–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong, X. M., J. Choi, C. M. Franzin, D. Zhai, J. C. Reed, and F. M. Marassi. 2004. Conformation of membrane-associated proapoptotic tBid. J. Biol. Chem. 279:28954–28960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seul, M., and M. J. Sammon. 1990. Preparation of surfactant multilayer films on solid substrates by deposition from organic solution. Thin Solid Films. 185:287–305. [Google Scholar]
- 19.Freeman, R. 1997. A Handbook of Nuclear Magnetic Resonance. Addison-Wesley Longman, Harrow, Essex, England.
- 20.Cras, J. J., C. A. Rowe-Taitt, D. A. Nivens, and F. S. Ligler. 1999. Comparison of chemical cleaning methods of glass in preparation for silanization. Biosens. Bioelectron. 14:683–688. [Google Scholar]
- 21.Tabor, D. 1992. Mica in the service of surface science. Colloids Surf. 65:ix–xiii. [Google Scholar]
- 22.Lindsay, S. M., Y. L. Lyubchenko, N. J. Tao, Y. Q. Li, P. I. Oden, J. A. Derose, and J. Pan. 1993. Scanning tunneling microscopy and atomic force microscopy studies of biomaterials at a liquid-solid interface. J. Vac. Sci. Technol. A. 11:808–815. [Google Scholar]
- 23.Kendall, J. T., and D. Yeo. 1951. Magnetic susceptibility and anisotropy of mica. Proc. Phys. Soc. London B. 64:135–142. [Google Scholar]
- 24.Oyler, N. A., and R. Tycko. 2004. Absolute structural constraints on amyloid fibrils from solid-state NMR spectroscopy of partially oriented samples. J. Am. Chem. Soc. 126:4478–4479. [DOI] [PubMed] [Google Scholar]
- 25.Rainey, J. K., J. S. DeVries, and B. D. Sykes. 2005. A rotatable flat-coil for static solid-state nuclear magnetic resonance spectroscopy. Rev. Sci. Instr. 76:086102. [Google Scholar]
- 26.Seelig, J. 1978. 31P nuclear magnetic resonance and head group structure of phospholipids in membranes. Biochim. Biophys. Acta. 515:105–140. [DOI] [PubMed] [Google Scholar]
- 27.Israelachvili, J. N. 1992. Intermolecular and Surface Forces, 2nd ed. Academic Press, San Diego, CA.
- 28.Mason, J. 1993. Conventions for the reporting of nuclear magnetic shielding (or shift) tensors suggested by participants in the NATO ARW on NMR shielding constants at the University of Maryland, College Park, July 1992. Solid State Nucl. Magn. Reson. 2:285–288. [DOI] [PubMed] [Google Scholar]
- 29.Mehring, M., R. G. Griffin, and J. S. Waugh. 1971. 19F shielding tensors from coherently narrowed NMR powder spectra. J. Chem. Phys. 55:746–755. [Google Scholar]
- 30.Kohler, S. J., and M. P. Klein. 1976. 31P nuclear magnetic resonance chemical shielding tensors of phosphorylethanolamine, lecithin, and related compounds—applications to head-group motion in model membranes. Biochemistry. 15:967–973. [DOI] [PubMed] [Google Scholar]
- 31.Hemminga, M. A., and P. R. Cullis. 1982. 31P NMR studies of oriented phospholipid multilayers. J. Magn. Reson. 47:307–323. [Google Scholar]
- 32.McLaughlin, A. C., P. R. Cullis, M. A. Hemminga, D. I. Hoult, G. K. Radda, G. A. Ritchie, P. J. Seeley, and R. E. Richards. 1975. Application of 31P NMR to model and biological membrane systems. FEBS Lett. 57:213–218. [DOI] [PubMed] [Google Scholar]
- 33.Bak, M., J. T. Rasmussen, and N. C. Nielsen. 2000. SIMPSON: a general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 147:296–330. [DOI] [PubMed] [Google Scholar]
- 34.Ulrich, R., R. W. Glaser, and A. S. Ulrich. 2003. Susceptibility corrections in solid state NMR experiments with oriented membrane samples. II. Theory. J. Magn. Reson. 164:115–127. [DOI] [PubMed] [Google Scholar]
- 35.Soubias, O., O. Saurel, V. Reat, and A. Milon. 2002. High resolution 13C NMR spectra on oriented lipid bilayers: from quantifying the various sources of line broadening to performing 2D experiments with 0.2–0.3 ppm resolution in the carbon dimension. J. Biomol. NMR. 24:15–30. [DOI] [PubMed] [Google Scholar]
- 36.Rowlinson, J. S., and B. Widom. 1982. Molecular Theory of Capillarity. Clarendon Press, Oxford, UK.
- 37.Hallock, K. J., K. Henzler Wildman, D. K. Lee, and A. Ramamoorthy. 2002. An innovative procedure using a sublimable solid to align lipid bilayers for solid-state NMR studies. Biophys. J. 82:2499–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi, E. J., and E. K. Dimitriadis. 2004. Cytochrome-c adsorption to supported, anionic lipid bilayers studied via atomic force microscopy. Biophys. J. 87:3234–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pott, T., J. C. Maillet, and E. J. Dufourc. 1995. Effects of pH and cholesterol on DMPA membranes: a solid state 2H- and 31P-NMR study. Biophys. J. 69:1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van den Brink-van der Laan, E., J. A. Killian, and B. de Kruijff. 2004. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochim. Biophys. Acta. 1666:275–288. [DOI] [PubMed] [Google Scholar]
- 41.Lide, D. R., editor. 2001. CRC Handbook of Chemistry and Physics, 82nd Ed. CRC Press, Boca Raton, FL.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.