

Abstract
An atomic force microscope (AFM) was used to visualize CWALP1923 peptides (+H3N-ACAGAWWLALALALALALALWWA-COO−) inserted in gel-phase DPPC and DSPC bilayers. The peptides assemble in stable linear structures and domains. A model for the organization of the peptides is given from AFM images and a 20 ns molecular dynamics (MD) simulation. Gold-coated AFM cantilevers were used to extract single peptides from the bilayer through covalent bonding to the cystein residue. Experimental and simulated force curves show two distinct force maxima. In the simulations these two maxima correspond to the extraction of the two pairs of tryptophan residues from the membrane. Unfolding of the peptide precedes extraction of the second distal set of tryptophans. To probe the energies involved, AFM force curves were obtained from 10 to 104 nm/s and MD force curves were simulated with 108–1011 nm/s pulling velocities (V). The velocity relationship with the force, F, was fitted to two fluctuation adhesive potential models. The first assumes the pulling produces a constant bias in the potential and predicts an F ∼ ln (V) relationship. The second takes into account the ramped bias that the linker feels as it is being driven out of the adhesion complex and scales as F ∼ (ln V)2/3.
INTRODUCTION
Biological membranes consist of a lipid bilayer with associated membrane proteins, which can traverse the bilayer either as β-barrels, helical bundles, or single α-helices. This requirement for a well-defined secondary structure is imposed by the nonpolar nature of the hydrophobic bilayer core. However, factors such as the topology and structural stability of a transmembrane protein segment will additionally be determined by interactions with the polar lipid headgroup region (1). Protein-lipid interactions also govern more global aspects of membrane organization. For example, it is increasingly recognized that the many different lipid and protein species that exist within any given membrane, rather than presenting a homogeneous mixture, segregate into distinct functional domains (2–4).
Atomic force microscopy (AFM) can resolve membrane surface features at a lateral resolution of 0.6–1 nm and a vertical resolution of 0.1 nm, under physiological conditions without the need of a crystalline system (5). Additionally, AFM can address single molecules and measure the forces required to displace, extract, and/or unfold a protein, yielding valuable dynamic and structural information (6–8). More specifically, by applying pulling forces at different velocities (dynamic force spectroscopy, DFS), the energies and forces governing protein folding and functioning can be estimated and the complex relationships between force-lifetime and chemistry can be explored (9–14).
A few membrane proteins have been imaged by AFM, including bacteriorhodopsin (bR) in the purple membrane (15), rhodopsin in disc membranes (16), aquaporin (17), and different components of several light-harvesting complexes (18–22). Pulling via an AFM cantilever, the 7 transmembrane helices of bR (8,23–25), the 12 helices of the antiporter NhaA (26), and the 8 helices of human aquaporin-1 (17) have been unfolded and extracted from the membrane. Experimental force curves have been used to identify general structural features of the unfolded membrane proteins, such as the preference of some helices to unfold in pairs, the importance of extracellular loops, and the directionality of several of the interactions (17,23,26). However, the details of the unfolding and extraction, in particular the contribution of individual amino acid residues to the process, are difficult to resolve from the unfolding pattern of a multispanning protein because force events arise from complex combinations of protein-protein, helix-helix, and helix-lipid interactions, especially in the case of native membranes.
To circumvent such issues, fundamental aspects of membrane organization and stability can be addressed with synthetic α-helical peptides, which represent a consensus sequence for transmembrane protein segments (27). WALP peptides, which were specifically designed for such studies, consist of a hydrophobic poly(leucine-alanine) stretch of variable length, which is flanked on both sides by two Trp residues. These peptides and closely related analogs with other aromatic or charged flanking residues have been extensively studied in a wide variety of model membranes (27). In gel-state lipid bilayers, WALP peptides have been shown by AFM to form highly ordered peptide-enriched domains (28) in which linear aggregates of WALP peptides are separated from each other by areas of relatively fluid phospholipids (29). Other transmembrane peptides have also been shown to form linear-type aggregates (30,31). For WALP peptides, this arrangement results from a tendency of the peptide to avoid a hydrophobic mismatch with the relatively thick gel-state lipid bilayer (32), and it is also observed when the Trp residues are replaced with tyrosines, phenylalanines, or histidines (33). WALP is a relevant model for larger integral membrane proteins because the peptides experience interactions with the lipid matrix as well as with neighboring helices. It should also be noted that several of the membrane proteins mentioned above are present in protein-rich domains or form linear aggregates and that interfacial Trp residues can modulate protein function (34,35).
The AFM unfolding experiments reported to date reveal the potential of the technique in membrane protein studies to resolve structural stability and organizational details. However, to fulfill such potential, a systematic study of the relevant individual structural elements and of the experimental conditions that determine the unfolding patterns needs to be undertaken. In this study, we use a simple CWALP1923 peptide to study several fundamental aspects of membrane protein unfolding: the role of aromatic residues, the influence of the pulling parameters, and the relevance of the different theoretical frameworks in which DFS experiments can be interpreted.
To supplement the interpretation of the experimental AFM images and DFS experiments, we have used molecular dynamics (MD) and steered molecular dynamics (SMD) simulations designed for studying force-induced reactions in biopolymers (36–38). As already demonstrated for unfolding studies of proteins and peptides in aqueous solution (7,37,39–43), our SMD simulations, to our knowledge the first to be undertaken for a membrane system, proved highly complementary to the experimental force spectroscopy measurements and highlighted the role of the Trp residues.
MATERIALS AND METHODS
Peptide synthesis
The peptide CWALP1923, with sequence 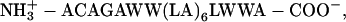 was synthesized using Fmoc chemistry as described previously for other WALP analogs (44,45), with the exception that for practical reasons the N- and C-terminus were not blocked. The identity of CWALP1923 was confirmed by MALDI-TOF mass spectrometry, and the peptide was used without further purification. The phospholipids 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) were obtained from Avanti Polar Lipids (Alabaster, AL).
was synthesized using Fmoc chemistry as described previously for other WALP analogs (44,45), with the exception that for practical reasons the N- and C-terminus were not blocked. The identity of CWALP1923 was confirmed by MALDI-TOF mass spectrometry, and the peptide was used without further purification. The phospholipids 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) were obtained from Avanti Polar Lipids (Alabaster, AL).
Vesicle preparation
The peptide was dissolved in 2,2,2-trifluoroethanol and was added to an equal volume of a mixture of chloroform and methanol (3:1 v/v) containing the lipid. The resulting solution, with a peptide/lipid molar ratio of 1:50, was dried in a rotary evaporator and subsequently under high vacuum. The solvent-free peptide/lipid film was hydrated at a temperature above the lipid main phase transition temperature with 1.25 ml of a 20 mM aqueous solution of NaCl, resulting in a lipid concentration of 0.65 mM. To obtain small unilamellar vesicles, this dispersion was briefly sonicated with an MSE sonicator (Crawley, UK) operating at an amplitude of 4 μm. The sonicated sample was centrifugated (1 h, 16000 g, 15°C) to pellet down titanium probe particles and residual multilamellar vesicles.
Supported bilayers
A 25 μl drop of unilamellar vesicle suspension was deposited onto freshly cleaved mica disks. The vesicles were allowed to adsorb to the mica for 15 h at a temperature of 4°C. After incubation the samples were gently washed with 20 mM NaCl to remove nonadsorbed vesicles and were kept above the lipid gel-to-fluid phase transition temperature (at 60°C for DPPC and at 70°C for DSPC) for 45 min. The sample was allowed to cool down to the gel phase at room temperature and was rinsed with 20 mM NaCl. Sometimes rinsing with 100 μl ultrapure water was necessary to remove unfused vesicles adsorbed on the bilayer.
AFM
The mica disks were glued to glass microscope slides and mounted on the scanner of a stand-alone MFP-3D AFM (Asylum Research, Santa Barbara, CA). The AFM has a closed loop in the x, y, and z axes. The images and pulling experiments were performed with gold-coated rectangular cantilevers CSC38/Cr-Au (Mikromasch, Tallinn, Estonia) with typical spring constants of 0.03 N/m, 0.05 N/m, and 0.08 N/m, and with “Biolevers” (Olympus, Tokyo, Japan) with a spring constant of 0.03 N/m.
Images were taken in alternate contact (AC) mode in liquid, with very low amplitudes at the primary resonance frequency that was obtained from thermal analysis of the cantilever in solution. Height, amplitude, and phase images were recorded. Heights of features in images were determined by histogram analysis. Experiments were carried out in a temperature-controlled room at 23 ± 1°C. The thermal spectrum of the cantilevers was obtained both in air and liquid, and the stiffness was estimated by fitting with the thermal noise theory (46) and compared to the Sader method for the normal spring constant of a rectangular cantilever (47). The error in calculating the spring constant is estimated to be ∼±20%. To calculate the force for peptide extraction, at least 20 single peptide extraction curves were obtained for each pulling velocity. Curves with multiple peptide extractions and many peaks were discarded.
MD simulations
MD and SMD simulations were performed using GROMACS v3.1.4 (48,49), which allows the application of forces along the z axis of the simulation box. The force field used for the peptides was GROMOS 43A2, extended to improve the simulation of the lipid components (50). All simulations were run at 300 K in an isothermal-isobaric ensemble. A Berendsen temperature and pressure-coupling scheme was chosen to keep these parameters constant. The time step for the simulation was 2 fs, and a linear constraint solver (LINCS) algorithm was used to maintain the geometry of the molecules. Long range electrostatic interactions were calculated with the particle mesh Ewald method, with up to 9 Å being treated in direct space and larger distances in Fourier space. Lennard-Jones interactions were cut off at 14 Å. Water was described with the simple point charge model (51,52).
Peptide coordinates were generated using SwissPdb-Viewer (51). The peptide was modeled as an ideal α-helix, and the side chains of the Trp residues were oriented with the N-H group pointing away from the central stretch of hydrophobic residues. Five of these CWALP1923 were assembled in a linear array, with neighboring helices in an antiparallel orientation. This row was then subjected to steepest descent energy minimization before insertion into a bilayer. The peptides were positioned manually so that their initial orientation relative to the membrane normal was distributed between 0° and 10°. All these parameters are based on experimental data (27,29).
The simulation box was constructed from a patch of fluid DPPC lipids consisting of 128 lipids with an area per lipid of 0.645 ± 0.010 nm2 and 3655 water molecules, previously equilibrated at 323 K in a 100 ns MD simulation (53,54) and available at http://www.lce.hut.fi/research/polymer/downloads.shtml. Before insertion of the row of five antiparallel CWALP1923 peptides, a cavity was created in this DPPC bilayer by removing a number of lipids. The solvent-accessible surface of the peptide row was then used as a template during an SMD simulation of 1 ns, in which the peptide-lipid interface was optimized (55). This procedure left 100 DPPC lipids and 3646 water molecules, or 17,053 atoms, in the box. The peptide-lipid system was equilibrated in six successive MD simulations of 200 ps where positional restraints, progressively decreasing to zero, were applied on the peptides. The equilibrated system was used for a 20 ns MD simulation to study the behavior of the peptide and lipid components.
Steered MD simulations
For SMD simulations, the equilibrated peptide-containing bilayer was transferred to a larger simulation box with 12,070 molecules of water. This box, containing 42,325 atoms, was first subjected to steepest-descent energy minimization. Subsequently, forces were applied through a virtual cantilever moving at a constant velocity along an axis perpendicular to the membrane plane. One extremity of the cantilever was simulated to be bound to the Cys residue of the central peptide, and the other was moved away along the pulling axis. This virtual cantilever is a harmonic spring characterized by a spring constant, and it does not have any shape nor does it undergo any hydrodynamic drag. Various cantilever spring constants and pulling velocities were simulated, ranging respectively from 0.07 to 16.67 N/m and from 1.25 to 0.0063 Å/ps. Additionally, three different peptides were extracted from the bilayer to assess the intrinsic variability of the model. The various SMD simulations ranged in length from 70 ps to 17 ns.
RESULTS
AFM images of CWALP1923 peptide in gel-state lipid bilayers
Transmembrane peptides consisting of a hydrophobic poly(leucine-alanine) core form highly ordered peptide-enriched domains in gel-state bilayers when the poly(leucine-alanine) stretch is flanked by aromatic residues, but not when it is flanked by charged residues (33). This behavior is also observed when the Trp-flanked CWALP1923 peptide is incorporated in gel-state bilayers, as shown in Fig. 1 for DPPC (A) and DSPC (B). Both these AC-mode AFM images show a bilayer region with peptide-enriched and peptide-depleted domains, as well as bilayer defects in which the mica surface is exposed. The height profile of a cross section of the DPPC system, shown in Fig. 1, reveals that the smooth surface in Fig. 1 A has a thickness of 4.9 ± 0.2 nm, as expected for an unperturbed bilayer of gel-state DPPC (28,56). The peptide-enriched domains in the DPPC system can easily be identified by their reduced height and appear either as isolated lines or as two-dimensional assemblies of these lines. In the corresponding height profile, the lines appear as 0.3 ± 0.1 nm deep depressions of decreasing width: 13 ± 3 nm at the top narrowing to 3 ± 1 nm at the bottom. As a consequence of the N- and C-terminal charges of CWALP1923, these peptide-enriched domains are somewhat less ordered than reported for other WALP analogs. In the simultaneously recorded phase images, the peptide lines and domains produce a different contrast than the flat lipid areas, implying differences in stiffness and electrostatics between peptides and peptide-depleted areas (not shown). Using a variety of techniques, Killian and co-workers have identified the lines of which these domains are composed as a linear array of antiparallel WALP peptides, one peptide wide and flanked by lipids with partly disordered acyl chains (29). This remarkable arrangement results from a combination of peptide-lipid and peptide-peptide interactions and depends on the precise composition of the peptide and of the lipid components (32).
FIGURE 1.

AFM topography images in aqueous solution of CWALP1923 peptide line domains in (A) DPPC and (B) DSPC gel-state lipid bilayers (mica, black; peptide-enriched domains, dark gray; peptide-depleted domains, light gray; unfused vesicles, white). Both images are 600 nm × 600 nm. The cross section of the AFM images depicts the bilayer thickness and the depth of the peptide-containing depressions.
For CWALP1923, this is highlighted by incorporation in bilayers of DSPC, a lipid which has acyl chains with 18 carbon atoms, only two ethylene moieties more than in the case of DPPC. As can be seen in Fig. 1 B, this small change in the lipid component has a pronounced influence on the morphology of the peptide-enriched domains: isolated peptide lines rather than extended two-dimensional arrays are now the dominant feature. The lines in DSPC also vary in thickness. The thinner line-shaped depressions have a width of ∼11 nm near the bilayer surface, which is reduced to ∼4 nm at the bottom of the depression, and are thus similar in width to the isolated lines in the DPPC system. The wider lines can be up to 36 nm wide at the bilayer surface, but in this case the bottom of the depression displays a height fluctuation of ∼0.4 nm, suggesting that these wider depressions consist of multiple linear arrays of peptide which are still separated by lipids. In all cases, the depressions are typically 1.4 ± 0.6 nm deep, ∼1 nm deeper than the lines observed in the DPPC system. Since the peptide-depleted DSPC domains were ∼1 nm thicker than the corresponding DPPC domains, it appears that the CWALP1923 peptides, which are the core component of the lines, have not changed their conformation in response to the different lipid environment. WALP analogs have indeed been shown to form a stable well-defined α-helix in different lipid matrices (57).
CWALP1923 unfolding and extraction from lipid bilayers at different velocities by AFM
The extraction of peptides with the gold-coated AFM tip was preceded by AFM imaging of the samples, after which the tip was shifted to the area where the peptides were visible (Fig. 2 A). Taking great care in not damaging the bilayer, the tip was moved toward the sample in 50–100 nm approach-retraction cycles. When cantilever deflection was detected and the tip touched the surface, the cantilever was moved 1 nm upwards so that the point of maximum proximity with the sample during a cycle was set above the bilayer, without touching it. Subsequently, approach-retraction cycles were carried out until a peptide-binding event could be detected. This procedure increased the probability of a single molecule-binding event. To increase the accessibility to the peptide, the Cys residue had been placed some residues away from the tryptophans, which are expected to be situated relatively deep in the lipid headgroup region (58). After the pulling experiments, the bilayer was imaged again to verify that the bilayer was undamaged and to validate the pulling data. In Fig. 2 B, it can be seen that some peptides have been extracted and that lipids have diffused into the space left by the missing peptides. During the 6 min between the acquisition of the two images, lipid mobility is also evident in peptide-free areas: in the upper part of Fig. 2 A some lipids had been extracted, and in Fig. 2 B the damage has healed. The rates of lipid diffusion involved here are within the typical lateral diffusion coefficient for a lipid molecule in the gel phase, estimated to be ∼104 nm2/s (59). However transfer of lipids previously adhered to the tip back into the bilayer defect could also explain the healing (I. Reviakine, Technical University Clausthal, personal communication, 2005). When the tip was forced to pierce completely through the bilayer, an indentation and a change of slope were detected in the approach curve. The retraction curves showed 500 pN–2 nN deep adhesion peaks followed by a random number of weaker peaks (not shown). Images show that the cantilever has removed a whole piece of bilayer and that this defect does not heal afterwards. After this, all the retraction curves showed many peaks, as if bilayer material had adhered to the tip, rendering it inadequate for further pulling.
FIGURE 2.

AFM topography images of peptide domains in a DPPC bilayer before (A) and after (B) peptide extraction. The arrow in A points to a place where lipids have been extracted. In B, the bilayer has healed. The arrow in B points to the space left by the extracted peptides, which has been filled by lipids.
When the Cys residue of a CWALP1923 peptide sticks to the gold tip, the retraction curves consistently show two adhesion peaks (Fig. 3). As the peptide is pulled, the force measured by the bending of the cantilever increases, reaches a maximum, and then briefly relaxes. Subsequently the force increases again, as the last part of the peptide clings to the lipids just before it is completely extracted, and the force returns to zero. Extraction force curves from the DPPC bilayer were obtained with hard (0.15 N/m) and soft (0.02 N/m) cantilevers (Fig. 3, A and B) with retraction velocities from 10 nm/s to 10,000 nm/s. The characteristic double step in the pulling curves appears at all velocities measured and with all the cantilevers used. Only at very low velocity, where small forces in the range of 10 pN are sufficient to extract the peptide, did the two step feature disappear. This may be due to rearrangement and rebinding of the peptide as it is being extracted as well as to the sensitivity of the system.
FIGURE 3.

Experimental peptide unfolding force curves from DPPC with (A) a 0.15 N/m cantilever, (B) a 0.02 N/m cantilever, and (C) from DSPC with a 0.07 N/m cantilever at different retraction velocities.
The unfolding curves obtained with a softer cantilever are much longer. Fig. 4 A compares the time needed for extraction of the peptides at the same retraction velocity for the soft and hard cantilevers. The softer cantilever needs more time to pull the peptide out since it takes longer to transmit the retraction of the piezo.
FIGURE 4.

(A) Experimental force curves for the extraction of CWALP1923 from a DPPC bilayer at 4000 nm/s retraction velocity with 0.02 N/m and 0.15 N/m cantilevers. (B) Force maxima as a function of log (V), for peptide extraction from a DPPC bilayer with 0.02 N/m and 0.15 N/m cantilevers, and from a DSPC bilayer with a 0.07 N/m cantilever. The superposed lines show the linear fit of the forces versus log (V).
For both soft and hard cantilevers, the length of the force curve increases with the pulling velocity (V). The maximum rupture forces needed for peptide extraction were obtained at different velocities. In Fig. 4 B, the rupture forces are plotted against V for the two cantilevers used. The cantilever calibration error (see Materials and Methods) has not been included in the calculation of the forces. As predicted by the theory of kinetics under force (10,11), the forces scale quasilogarithmically with the velocity. At higher velocities the rupture-force distribution broadens (note the larger error bars), as different extraction paths are available for the peptide (60). The distribution of forces at very low velocity retraction cannot be accurate. The force sensitivity of our system is limited by the Brownian motion of the cantilever. Forces below 10 pN cannot enter the statistics and therefore they are shifted toward high forces.
CWALP1923 peptides were also extracted from DSPC bilayers (Fig. 3 C). The double peak feature of the force curves is more pronounced than in the extraction from DPPC. The maximum rupture forces are plotted against V in Fig. 4 B, showing a quasilogarithmic scaling but with a smaller slope.
MD simulation of CWALP1923 in a DPPC bilayer
For a small segment of a peptide-enriched CWALP1923 domain, a molecular model was constructed consisting of a row of five peptides with alternating orientation which is flanked by several layers of relatively fluid lipids (see Materials and Methods). During a 20 ns MD simulation of this system, the temperature average was constant at 299.85 ± 1.64 K. As depicted in Fig. 5 C, the root mean-square deviation (RMSD) of the position of the peptide atoms compared to their initial position shows an increase from 0 to 0.3 nm before oscillating between 0.25 and 0.35 nm, and the secondary structure of all the peptides remained unchanged. This MD simulation thus demonstrates that the peptide-lipid system is stable. A side view of the simulation box after the MD run is shown in Fig. 5 A. The average tilt angle of the peptides has evolved to 10.7 ± 3.8°, which compares well to 2H NMR studies on similar peptides in fluid bilayers (61). The depth of the peptide row, calculated by subtracting the coordinates of the lipid phosphate group from the coordinates of the peptides extremities as defined by the center of mass of the last residues, is on average 1.67 ± 0.71 Å. Alternatively, the density profile of the modeled system, depicted in Fig. 5 B, indicates that the distance between the membrane surface and the peptide termini is between 2 and 8 Å. These values are compatible with the depth of 3 ± 1 Å as observed by AFM (see above).
FIGURE 5.

(A) Simulation box used for equilibration of the peptides in the DPPC bilayer (DPPC lipids, green; peptide, cyan; tryptophan residues, purple; cystein residues, yellow; water, red and white). (B) Density profile of the simulated membrane system: the total peptide density is shown in cyan, and the other curves represent various lipid moieties as indicated. (C) RMSD of the position of the peptide atoms during the 20 ns MD simulation with respect to their initial position.
Simulation of the AFM extraction curves
Structural analysis
An 8 ns SMD simulation of the extraction of a CWALP1923 peptide from a DPPC bilayer was performed to correlate the shape of the AFM force curves with molecular events during the peptide extraction. The SMD simulation was carried out with a 0.167 N/m virtual spring attached to the Cys residue, moving at a constant speed of 0.0125 Å/ps (see Materials and Methods). Fig. 6 A displays a side view of the system before and near the end of the SMD simulation, the RMSD analysis of the extracted peptide is shown in Fig. 6 C, and the evolution of the secondary structure of all the peptides during the simulation is given in Fig. 6 D. The force experienced by the two extremities of the virtual spring was calculated, and the two different force profiles are presented in Fig. 6 B, where the force experienced by the Cys residue is shown in red and the force experienced by the end of the cantilever is shown in black. Some key molecular events which define the shape of the force curves were identified by visual inspection of the SMD trajectory and are given in detail in the figure legend. In summary, the helix is partially unfolded until further unfolding requires extraction of Trp6 and Trp7 from the bilayer, which gives rise to the first distinct force maximum. After removal of the first set of tryptophan residues, the unfolding rate increases: many residues unfold simultaneously, and the peptide chain rapidly becomes more extended. When the poly(leucine-alanine) chain is fully extended, the second set of tryptophan residues is still anchored into the opposite side of the bilayer. The displacement of Trp21 and Trp22 gives rise to the second distinct force peak. Once these tryptophans start moving, the entire peptide is extracted from the bilayer. Thus, the two main force peaks observed experimentally (Fig. 3) are also evident from the SMD simulation, where they correspond to the displacement of the first and second set of Trp residues. Less distinct features of the SMD force profile represent either unfolding events or collisions with side chains of neighboring peptides.
FIGURE 6.

(A) Side view of the simulation box before and after the SMD pulling (0.167 N/m cantilever, 0.0125 Å/ps pulling velocity, 7870 ps simulation length) of the central peptide. (DPPC lipids, green; peptide, cyan; tryptophan residues, purple; cystein residues, yellow; water, red and white). (B) The simulated force profile is displayed for the side of the virtual spring attached to the Cys residue (red) and for the other end of the spring (black), which is set to move at constant velocity. (C) RMSD of the peptide during the SMD simulation. (D) The secondary structure evolution of the five peptides shows four stable α-helical peptides, whereas the pulled peptide (represented from N- to C-terminus by residues 47–69) is progressively unfolded. Selected collision, unfolding, and stretching events: 1 (720 ps) the hydrogen bond of residue Ala3 breaks and subsequently the residue unfolds. 2 (1200 ps) unfolding of Gly4. 3 (1450 ps) unfolding of Ala5. 4 (1600–1700 ps) the unwound residues become completely extended. 5 (1950 ps) unfolding of Trp6. 6 (2000–2950 ps) Trp6 collides with a Trp side chain of a neighboring peptide, preventing unfolding, the helix then unfolds at Trp7, while its side chain still interacts with the lipids. 7 (4050 ps) Leu8 unfolds and the side chain of Trp6 moves out of the bilayer. 8 (4070–4800 ps) residues Ala9 to Leu12 unfold. 9 (5050 ps) the side chain of Trp7 moves out of the bilayer and the rate of unfolding increases, several residues unfold simultaneously. 10 (5200–5350 ps) the remainder of the helix collapses and the peptide chain rapidly becomes extended. 11 (5600 ps) the peptide chain moves slowly through the membrane, with the side chains of Trp21 and Trp22 colliding with the Leu side chains of the neighboring peptides, and at the same time the chain becomes further extended. 12 (7000 ps) the side chain of Trp21 reaches the middle of the bilayer and the peptide chain becomes fully extended. 13 (7500 ps) collision between the side chain of Trp22 and a Trp side chain of a neighboring peptide. 14 (7700 ps) after Trp22 has passed this obstruction, the entire peptide moves rapidly out of the membrane. 15 (7870 ps) the peptide has been completely extracted.
Systematic study of the AFM pulling parameters
A series of 12 SMD simulations ranging from 70 ps to 16.5 ns with different spring constants (0.06, 0.167, 1.667, 16.67 N/m) at different velocities (1.25, 0.125, 0.0125, and 0.00625 Å/ps) were performed. In Fig. 7 A, the maximum force observed at the different loading rates (force/ps) is shown and compared with the experimental forces for the extraction of the peptide from DPPC with a 0.16 N/m cantilever. The distribution of maximum forces is broader for higher forces, as in the case of the AFM experiment. Fig. 7, D–F, shows the structural variation in force profiles simulated for three of the five peptides, with the same velocity and force constant. For each peptide, the force was applied on the N-terminus, and due to their antiparallel orientation, the peptides were pulled from different sides of the membrane. The three peptides correspond to the helices defined by the residues 24–46, 47–69, and 70–92, as described in Fig. 6 D.
FIGURE 7.

(A) Effect of the loading rate on the maximum force required for extraction of CWALP1923 from a DPPC bilayer, for experiments and simulations. (B–C) Simulated force-extension profiles with a 1.667 N/m cantilever (B) and a 0.167 N/m cantilever (C). (D–F) Simulated force curves with a 1.667 N/m cantilever at three different pulling velocities: 1.25 (D), 0.125 (E), and 0.0125 Å/ps (F) for three different peptides.
Fig. 7, B and C, shows the effect of the spring constant and pulling velocity on the calculated force profile. As in the experiment (Fig. 3), the softer cantilever produces longer force versus distance curves and results in a longer extraction time (Table 1).
TABLE 1.
Relation between pulling time, spring constant, and pulling velocity in the simulations
| Spring constant, N/m
|
|||||
|---|---|---|---|---|---|
| Pulling time, ps | 0.06 | 0.167 | 1.667 | 16.67 | |
| Pulling velocity, Å/ps | 1.25 | 310 | 182 | 80 | 70 |
| 0.125 | 1960 | 1135 | 680 | 670 | |
| 0.0125 | N.A. | 7870 | 6000 | 5630 | |
Both in simulations (Fig. 7) and experiments (Fig. 3), the length of the force curves increases with the velocity. A comparison between position of the Cys residue and the AFM force curves in the simulations indicates that this effect is mainly due to the cantilever deflection (62). As shown in Fig. 6 B, a force curve at 0.0125 Å/ps with a 0.1667 N/m cantilever reproduces within 2 Å the actual distance between the Trp extraction peaks. At higher velocities the cantilever deflects more, and consequently, the AFM force curves deviate further from the actual trajectory of the Cys residue. Softer cantilevers bend more and produce longer force curves which carry less structural information. At a high pulling velocity and a low spring constant, the force versus distance plot becomes an almost featureless straight line (Fig. 7 C).This increase in length cannot be due to membrane deformations.
In the experiment, the distance between the two peaks is variable (Fig. 3). This is possibly caused by the natural oscillation of the peptide (Fig. 5 C) and the different pathways in which it can be extracted. In the simulations, shifts in the position of the two force maxima (Fig. 7, D–F) are due to small differences between the conformation and relative position of the peptides, leading to differences in the way side chains collide with each other. The maximum force in the simulations is ∼700 pN, which is the same order of magnitude as experimentally observed, although the pulling velocity is 5 to 6 orders of magnitude higher.
DISCUSSION
Unfolding and extraction: Trp anchoring determines the unfolding pattern
DFS studies of the extraction of lipids from membranes revealed energy barriers that are consistent with the hydrophobic half thickness of the studied membrane (63). It was concluded that the main lipid anchoring force is hydrophobic interaction and that the lipids are extracted without major structural changes. However in the case of peptides and proteins, it is not clear in what order unfolding and extraction occur (17). A recently proposed experimental model for unfolding of bR by force spectroscopy (24,64) suggests that transmembrane helices unfold within the membrane rather than being first extracted from the hydrophobic membrane core and then unfolded.
The molecular events underlying the shape of the simulated force curves (Fig. 6 B) highlight the importance of the bulky Trp side chains in the extraction pathway. The first set of Trp residues exits the bilayer first, marking the first peak in the force curve. As the cantilever keeps pulling, the peptide unfolds inside the bilayer, whereas the tryptophans at the opposite end of the peptide resist displacement. The eventual extraction of this second set of Trp residues coincides with the second maximum in the force curve. Thus, in contrast to the model proposed by Ganchev et al. for the extraction of SH-WALP23 (58), our data imply that the tryptophans are more important for CWALP1923 anchoring into the bilayer than the hydrophobic interaction of the poly(leucine-alanine) chain.
The first set of tryptophans is able to delay extraction through van der Waals interactions with the side chains from neighboring peptides and also through electrostatic interactions with polar lipid moieties and with the Trp side chains of neighboring peptides, as described in the legend of Fig. 6 B. Also the second set of tryptophans initially resists displacement in this way. Once enough force has been built up to overcome this resistance, the extraction of the peptide is slowed down by interactions and collisions of these Trp side chains with Trp and Leu side chains from the neighboring peptides. The tryptophan side chains can be pulled through the bilayer either by allowing time for structural rearrangements which minimize side chain clashes (as in event 6 in Fig. 6 B) or by applying a stronger force and thereby forcing obstacles to move away, as observed in the shorter simulations where stronger forces were applied.
Our results suggest that Trp may also play an important role in the unfolding of membrane proteins in general. In this context bR is the best characterized protein, and it shows a well-defined unfolding pattern. Although Müller et al. (23) propose that bulky residues such as tyrosine are responsible for the force profile, a close inspection of the unfolding pattern of bR reveals a striking coincidence of some of the unfolding peaks with the position of tryptohan residues in particular. The indole group of Trp is the bulkiest amino acid side chain; therefore one can expect a stronger resistance to displacement across the membrane. The energy barriers can also be interpreted by Trp interactions (64). All the Trp residues in bR are situated in the extracellular leaflet of the bilayer, and indeed it has been observed that extracellular loops particularly resist unfolding (24). Recent studies have shown the stiffness asymmetry of bR (K. Voïtchovsky, M. Kamihira, S. Antoranz, A. Watts, and J. Ryan, unpublished results), and highlight the specific bR-lipid interactions in the extracellular side of the protein. Indeed tryptophans play a singularly important role in membrane anchoring (65). Unfolding experiments and simulations with model peptides mixing different flanking residues may provide more information about their relative relevance.
Force distributions
DFS experiments show that the width of the distribution of maximum forces increases with the pulling velocity (Fig. 4 B). It has been argued that this is caused by experimental uncertainty (10). However, the MD simulations also show a wider distribution of forces for high pulling velocities (Fig. 7). Theoretical calculations for thermally assisted bond rupture predict that the width of the force distribution increases with the velocity, since the variance is set by the thermal force and it is broadened by kinetics (11,60). The experiments and simulations presented here support this interpretation.
Validity of the loading rate concept
In previous protein-unfolding studies, the energy barriers have been obtained from the slopes of forces versus loading rates (force/time) plots (11,58). This procedure pools together data obtained with different spring constants. However the unfolding depends on the cantilever stiffness as can be seen in Figs. 3 and 4. In this respect, the MD simulations coincide with the experimental results. In the simulations, softer cantilevers produce longer unfolding curves and take a longer time in producing them (Fig. 7). Fig. 8 shows the differences in the unfolding paths for two simulations with the same loading rate but with different cantilever stiffness and pulling velocity. This result questions the use of the loading rate for calculating energy barriers of protein unfolding. Indeed, using different combinations of cantilever stiffness and pulling rate can lead to the same loading rate values, but the force gradient applied on the peptide will not be the same. The experimental force curves for two cantilevers of different stiffness have different slopes resulting in different energy barriers (Fig. 4). Theoretical calculations taking into account viscous dissipation do predict differences in the unfolding for different spring constants (60). Experimentally, the large error in calculating the cantilever force constant presents an additional problem when mixing data obtained with different cantilevers.
FIGURE 8.

Peptide secondary structure evolution for two simulations with the same loading rate: (A) with a 0.166 N/m cantilever at 0.125 Å/ps pulling velocity, and (B) with a 1.667 N/m cantilever at 0.0125 Å/ps pulling velocity. The time required to unfold and extract the peptide is different, as is the peptide unfolding path.
Thermally assisted bond rupture: “creep models”
In DFS experiments an adhesion bond is driven away from its equilibrium position at a given velocity. The rupture of the bond occurs by a thermally assisted escape from the bound state across an activation barrier. In previous experimental studies of bond rupture and unfolding of biological molecules, DFS has been used to extract information about the energy barriers traversed along the force-driven pathway (11) with a simple linear creep model which assumes that the pulling force produces a small constant bias which reduces the height of the potential barrier. Eventually, the barrier is crossed at the maximum force (F), which is taken from experimental force profiles such as those in Fig. 3. This model predicts a linear dependence of F versus ln (V), in accordance with the solution of the Langevin equation, where the effect of the thermal fluctuation is given by a random force and viscous dissipation is taken into account. The position of the barrier can be obtained from the slope of the F versus ln (V) plots, given in Fig. 4 B.
When fitting our data to this theory for the extraction of CWALP1923 from DPPC with a 0.02 N/m cantilever, the regression of the linear fit (R) is 0.99004. The forces for extracting CWALP1923 from DPPC with a 0.15 N/m cantilever fit with R = 0.98732. The fitting of the forces measured for CWALP1923 in DSPC with a 0.07 N/m cantilever leads to R = 0.97232. In Fig. 7 A the forces observed in the experiments and the MD simulations have been plotted together as a function of the loading rate. An asymptotic behavior with a rapid increase of the unfolding force is observed, as previously described for the unfolding of a soluble protein (7).
It has been argued that the linear creep description of the bond dissociation by force is an unlikely regime (60,66–68). As the linker is driven out of the adhesion complex, the bias is ramped up and a bond rupture happens when a potential barrier almost disappears. The close relationship between adhesion, stick-slip, and friction, which ultimately involve the making and breaking of bonds, is at the heart of recent theoretical studies that suggest a universal ramped creep model, which predicts an F ∼ (ln V)2/3 relationship (60,66–68). Interestingly, a more rigorous theoretical analysis such as the generalized Fokker-Plank approach produces a force equivalent to the solution of the Langevin equation used in the “creep models” (66), confirming its generality. Both creep models are predicted to fail at low pulling velocities, when rebinding effects become important (60).
The experimental data given in Fig. 4 were also fitted to the ramped creep model. The fit, R, of the ramped creep model was marginally worse for soft cantilevers (extraction from DPPC with a 0.02 N/m cantilever gives R = 0.98767, and extraction from DSPC with a 0.07 N/m cantilever gives R = 0.94956). However the fit is slightly better for the harder cantilever (extraction from DPPC with a 0.15 N/m cantilever gives R = 0.98824). The fit quality of the ramped creep model is expected to increase for a system that is less overdamped, i.e., a stiffer spring constant, and with measurements over larger velocity ranges (60,67).
The creep models that incorporate simple adhesive potentials with thermal fluctuations fit the pulling data reasonably well. This proves that the physics behind peptide extraction is similar to the recently developed models for the effect of thermal fluctuations in atomic friction (66,67,69). The ramped creep model has been shown to give slightly better fits in friction experiments with glassy polymers (69). However the linear creep model seems to better describe the barrier hopping when peptides are extracted with soft cantilevers. This implies that the potential barrier is not too high, i.e., the rate of thermal fluctuation over the barrier is proportional to the pulling velocity. Indeed the forces needed for extracting the peptides from the bilayer are much lower than the friction forces fitting the ramped creep model (69), reflecting the relative weakness of the forces involved in the peptide extraction.
The reasonable fitting of our data with the creep models stresses the conceptual equivalence of atomic friction and membrane protein unfolding. The Trp-induced force peaks correspond to slip-stick motion, which can be suppressed using very slow velocities (Fig. 3). This enables side-chain rearrangements to occur, resulting in low friction sliding. The extraction peak distance is variable as a result of the temporal irregularity of the peptide-peptide and peptide-lipid interaction potential.
Energy barrier calculations
We can extract information about the energy barriers by fitting our data to the linear creep theory. The force has the form F = const + (kBT/Δx) ln (VKΔx/(k0kBT)), where k0 is the spontaneous rate of bond dissociation, Δx the distance from the minimum to the activation barrier of the reaction potential U(x), K the cantilever spring constant, T the temperature, and kB the Boltzmann constant. For the extraction of CWALP1923 from DPPC with a 0.02 N/m cantilever, a barrier is crossed at 0.23 ± 0.02 nm from the equilibrium position. When extracting CWALP1923 from DPPC with a 0.15 N/m cantilever, the barrier is at 0.29 ± 0.02 nm. As expected, the position of the transition state is different from the extraction of lipids (63). The CWALP1923 peptide extraction barriers are much smaller than the hydrophobic thickness of fluid DPPC and DSPC bilayers, suggesting that breakage of intermolecular bonds that stabilize the structure starts the unfolding as in the case of bR (64) and similar WALP peptides (58). The force profile obtained by the SMD simulation shows that the movement and extraction of the Trp residues mark the maxima in the pulling curve. Therefore an energy barrier of ∼2 Å should correlate with the extraction of the first set of Trp residues. In the simulation, these Trp side chains are anchored in the bilayer by electrostatic interactions with polar lipid moieties and with the Trp side chains of neighboring peptides. A displacement of ∼2 Å would be sufficient to disrupt these interactions (Fig. 5 B). Indeed, displacement of Trp side chains from the lipid carbonyl region is unfavorable (65). The fitting of the pulling of CWALP1923 from DSPC with a 0.07 N/m cantilever leads to a barrier at 0.52 ± 0.07 nm, reflecting the increased mismatch with the thicker bilayer.
From the experiments (Fig. 4 A) and the SMD simulations, we know that a softer cantilever unfolds the peptide at a slower rate (Table 1), giving more time for rebinding events to happen as transient capture wells are created (60,70). This may give rise to an energy barrier at a shorter distance from the equilibrium position than when the peptide is pulled with a harder cantilever.
CONCLUSIONS
We have demonstrated the importance of Trp residues in membrane protein unfolding with a very simple model peptide. Both in the experiments and in the simulations, extraction of the Trp residues produces the force maxima and the main energy barriers. The resistance of the tryptophan side chains to displacement causes the peptide to unfold before it can be completely extracted. This result is meaningful to interpret the unfolding pattern of real membrane proteins such as bR, where a correlation between the positions of force peaks and the location of Trp residues in the structure can be made.
Additionally, the simulations and the experimental force curves demonstrate the influence of velocity and stiffness of the cantilever on the unfolding path of the peptides. This questions the validity of using “loading rates” for calculating energy barriers with different cantilevers for membrane protein unfolding. The relationship of forces and velocities agrees with the creep models that consider barrier-hopping fluctuations in adhesive potentials, with slips occurring at lower energy values than determined by the energy barriers. This agreement underscores the theoretical and conceptual equivalence of membrane protein unfolding and atomic friction; in this study, the Trp peaks correspond to stick-slip motion, and the average force scales quasilogarithmically with the velocity.
Acknowledgments
The authors thank Mark Samson, Andrew Hung, and Kislon Voïtchovsky for fruitful discussions, Gabriel Mendes for assistance with the sample preparation, Jiayu Li and Yuen-Han Lam for peptide synthesis, and Antoinette Killian for kindly providing a preprint of the Ganchev et al. manuscript.
This work was supported by the Biotechnology and Biological Sciences Research Council, the Engineering and Physical Sciences Research Council, the Medical Research Council, and the Ministry of Defence through the Bionanotechnology IRC. M.R.R.dP. acknowledges support from the European Molecular Biology Organization.
References
- 1.Killian, J. A., and G. von Heijne. 2000. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 25:429–434. [DOI] [PubMed] [Google Scholar]
- 2.Mukherjee, S., and F. R. Maxfield. 2004. Membrane domains. Annu. Rev. Cell Dev. Biol. 20:839–866. [DOI] [PubMed] [Google Scholar]
- 3.Wisniewska, A., J. Draus, and W. K. Subczynski. 2003. Is a fluid-mosaic model of biological membranes fully relevant? Studies on lipid organization in model and biological membranes. Cell. Mol. Biol. Lett. 8:147–159. [PubMed] [Google Scholar]
- 4.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- 5.Engel, A., and D. Müller. 2000. Observing single biomolecules at work with the atomic force microscope. Nat. Struct. Biol. 7:715–718. [DOI] [PubMed] [Google Scholar]
- 6.Fisher, T., P. Marszalek, and J. Fernandez. 2000. Stretching single molecules into novel conformations using the atomic force microscope. Nat. Struct. Biol. 7:719–724. [DOI] [PubMed] [Google Scholar]
- 7.Rief, M., and H. Grubmüller. 2002. Force spectroscopy of single biomolecules. ChemPhysChem. 3:255–261. [DOI] [PubMed] [Google Scholar]
- 8.Müller, D. J., W. Baumeister, and A. Engel. 1999. Controlled unzipping of a bacterial surface layer with atomic force microscopy. Proc. Natl. Acad. Sci. USA. 96:13170–13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heymann, B., and H. Grubmüller. 2000. Dynamic force spectroscopy of molecular adhesion bonds. Phys. Rev. Lett. 84:6126–6129. [DOI] [PubMed] [Google Scholar]
- 10.Evans, E. 1999. Energy landscapes of biomolecular adhesion and receptor anchoring at interfaces explored with dynamic force spectroscopy. Faraday Discuss. 111:1–16. [DOI] [PubMed]
- 11.Evans, E. 2001. Probing the relation between force—lifetime—and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct. 30:105–128. [DOI] [PubMed] [Google Scholar]
- 12.Evans, E., and K. Ritchie. 1997. Dynamic strength of molecular adhesion bonds. Biophys. J. 72:1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans, E. B. 1999. Looking inside molecular bonds at biological interfaces with dynamic force spectroscopy. Biophys. Chem. 82:83–97. [DOI] [PubMed] [Google Scholar]
- 14.Bell, G. I. 1978. Models for the specific adhesion of cells to cells. Science. 200:618–627. [DOI] [PubMed] [Google Scholar]
- 15.Müller, D., H. Sass, S. Muller, G. Buldt, and A. Engel. 1999. Surface structures of native bacteriorhodopsin depend on the molecular packing arrangement in the membrane. J. Mol. Biol. 285:1903–1909. [DOI] [PubMed] [Google Scholar]
- 16.Fotiadis, D., Y. Liang, S. Filipek, D. A. Saperstein, A. Engel, and K. Palczewski. 2004. The G protein-coupled receptor rhodopsin in the native membrane. FEBS Lett. 564:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Möller, C., D. Fotiadis, K. Suda, A. Engel, M. Kessler, and D. Müller. 2003. Determining molecular forces that stabilize human aquaporin-1. J. Struct. Biol. 142:369–378. [DOI] [PubMed] [Google Scholar]
- 18.Bahatyrova, S., R. N. Frese, C. A. Siebert, J. D. Olsen, K. O. van der Werf, R. van Grondelle, R. A. Niederman, P. A. Bullough, C. Otto, and C. N. Hunter. 2004. The native architecture of a photosynthetic membrane. Nature. 430:1058–1062. [DOI] [PubMed] [Google Scholar]
- 19.Scheuring, S., F. Reiss-Husson, A. Engel, J.-L. Rigaud, and J.-L. Ranck. 2001. High-resolution AFM topographs of Rubrivivax gelatinous light-harvesting complex LH2. EMBO J. 20:3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheuring, S., J. Seguin, S. Marco, D. Levy, B. Robert, and J.-L. Rigaud. 2003. Nanodissection and high-resolution imaging of the Rhodopseudomonas viridis photosynthetic core complex in native membranes by AFM. Proc. Natl. Acad. Sci. USA. 100:1690–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheuring, S., J. N. Sturgis, V. Prima, A. Bernadac, D. Levy, and J.-L. Rigaud. 2004. Watching the photosynthetic apparatus in native membranes. Proc. Natl. Acad. Sci. USA. 101:11293–11297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fotiadis, D., P. Qian, A. Philippsen, P. A. Bullough, A. Engel, and C. N. Hunter. 2004. Structural analysis of the reaction center light-harvesting complex I photosynthetic core complex of Rhodospirillum rubrum using atomic force microscopy. J. Biol. Chem. 279:2063–2068. [DOI] [PubMed] [Google Scholar]
- 23.Müller, D. J., M. Kessler, F. Oesterhelt, C. Möller, D. Oesterhelt, and H. Gaub. 2002. Stability of bacteriorhodopsin alpha-helices and loops analyzed by single-molecule force spectroscopy. Biophys. J. 83:3578–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janovjak, H., M. Kessler, D. Oesterhelt, H. Gaub, and D. J. Muller. 2003. Unfolding pathways of native bacteriorhodopsin depend on temperature. EMBO J. 22:5220–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oesterhelt, F., D. Oesterhelt, M. Pfeiffer, A. Engel, H. E. Gaub, and D. J. Muller. 2000. Unfolding pathways of individual bacteriorhodopsins. Science. 288:143–146. [DOI] [PubMed] [Google Scholar]
- 26.Kedrov, A., C. Ziegler, H. Janovjak, W. Kuhlbrandt, and D. J. Muller. 2004. Controlled unfolding and refolding of a single sodium-proton antiporter using atomic force microscopy. J. Mol. Biol. 340:1143–1152. [DOI] [PubMed] [Google Scholar]
- 27.de Planque, M. R., and J. A. Killian. 2003. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 20:271–284. [DOI] [PubMed] [Google Scholar]
- 28.Rinia, H., R. Kik, R. Demel, M. Snel, J. Killian, J. van Der Eerden, and B. de Kruijff. 2000. Visualization of highly ordered striated domains induced by transmembrane peptides in supported phosphatidylcholine bilayers. Biochemistry. 39:5852–5858. [DOI] [PubMed] [Google Scholar]
- 29.Sparr, E., D. N. Ganchev, M. M. E. Snel, A. Ridder, L. M. J. Kroon-Batenburg, V. Chupin, D. T. S. Rijkers, J. A. Killian, and B. de Kruijff. 2005. Molecular organization in striated domains induced by transmembrane alpha-helical peptides in dipalmitoyl phosphatidylcholine bilayers. Biochemistry. 44:2–10. [DOI] [PubMed] [Google Scholar]
- 30.Amrein, M., A. von Nahmen, and M. Sieber. 1997. A scanning force and fluorescence light microscopy study of the structure and function of a model pulmonary surfactant. Eur. Biophys. J. 26:349–357. [DOI] [PubMed] [Google Scholar]
- 31.Van Gorkom, L. C. M., L. I. Horvath, M. A. Hemminga, B. Sternberg, and A. Watts. 1990. Identification of trapped and boundary lipid binding sites in M13 coat protein/lipid complexes by deuterium NMR spectroscopy. Biochemistry. 29:3828–3834. [DOI] [PubMed] [Google Scholar]
- 32.Rinia, H., and B. de Kruijff. 2001. Imaging domains in model membranes with atomic force microscopy. FEBS Lett. 504:194–199. [DOI] [PubMed] [Google Scholar]
- 33.Rinia, H., J. Boots, D. Rijkers, R. Kik, M. Snel, R. Demel, J. Killian, J. van der Eerden, and B. de Kruijff. 2002. Domain formation in phosphatidylcholine bilayers containing transmembrane peptides: specific effects of flanking residues. Biochemistry. 41:2814–2824. [DOI] [PubMed] [Google Scholar]
- 34.Draheim, R., A. Bormans, R. Lai, and M. Manson. 2005. Tryptophan residues flanking the second transmembrane helix (TM2) set the signaling state of the Tar chemoreceptor. Biochemistry. 44:1268–1277. [DOI] [PubMed] [Google Scholar]
- 35.Clark, E., J. East, and A. Lee. 2003. The role of tryptophan residues in an integral membrane protein: diacylglycerol kinase. Biochemistry. 42:11065–11073. [DOI] [PubMed] [Google Scholar]
- 36.Grubmüller, H., B. Heymann, and P. Tavan. 1996. Ligand binding: molecular mechanics calculation of the streptavidin biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- 37.Lu, H., B. Isralewitz, A. Krammer, V. Vogel, and K. Schulten. 1998. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys. J. 75:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Izrailev, S., A. R. Crofts, E. A. Berry, and K. Schulten. 1999. Steered molecular dynamics simulation of the Rieske subunit motion in the cytochrome bc(1) complex. Biophys. J. 77:1753–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paci, E., and M. Karplus. 2000. Unfolding proteins by external forces and temperature: the importance of topology and energetics. Proc. Natl. Acad. Sci. USA. 97:6521–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marszalek, P. E., H. Lu, H. B. Li, M. Carrion-Vazquez, A. F. Oberhauser, K. Schulten, and J. M. Fernandez. 1999. Mechanical unfolding intermediates in titin modules. Nature. 402:100–103. [DOI] [PubMed] [Google Scholar]
- 41.Krammer, A., H. Lu, B. Isralewitz, K. Schulten, and V. Vogel. 1999. Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch. Proc. Natl. Acad. Sci. USA. 96:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu, H., and K. Schulten. 1999. Steered molecular dynamics simulations of force-induced protein domain unfolding. Proteins. 35:453–463. [PubMed] [Google Scholar]
- 43.Paci, E., and M. Karplus. 1999. Forced unfolding of fibronectin type 3 modules: an analysis by biased molecular dynamics simulations. J. Mol. Biol. 288:441–459. [DOI] [PubMed] [Google Scholar]
- 44.de Planque, M. R. R., J. W. P. Boots, D. T. S. Rijkers, R. M. J. Liskamp, D. V. Greathouse, and J. A. Killian. 2002. The effects of hydrophobic mismatch between phosphatidylcholine bilayers and transmembrane alpha-helical peptides depend on the nature of interfacially exposed aromatic and charged residues. Biochemistry. 41:8396–8404. [DOI] [PubMed] [Google Scholar]
- 45.de Planque, M. R. R., J. A. W. Kruijtzer, R. M. J. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe, B. de Kruijff, and J. A. Killian. 1999. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane alpha-helical peptides. J. Biol. Chem. 274:20839–20846. [DOI] [PubMed] [Google Scholar]
- 46.Hutter, J. L., and J. Bechhoefer. 1993. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 64:1868–1873. [Google Scholar]
- 47.Sader, J. E., J. W. M. Chon, and P. Mulvaney. 1999. Calibration of rectangular atomic force microscope cantilevers. Rev. Sci. Instrum. 70:3967–3969. [Google Scholar]
- 48.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. (Online). 7:306–317. [Google Scholar]
- 49.Berendsen, H. J. C., D. Vanderspoel, and R. Vandrunen. 1995. Gromacs: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91:43–56. [Google Scholar]
- 50.Berger, O., O. Edholm, and F. Jahnig. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72:2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- 52.Berendsen, H. J. C., J. R. Grigera, and T. P. Straatsma. 1987. The missing term in effective pair potentials. J. Phys. Chem. 91:6269–6271. [Google Scholar]
- 53.Patra, M., M. Karttunen, M. T. Hyvonen, E. Falck, P. Lindqvist, and I. Vattulainen. 2003. Molecular dynamics simulations of lipid bilayers: major artifacts due to truncating electrostatic interactions. Biophys. J. 84:3636–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patra, M., M. Karttunen, M. T. Hyvonen, E. Falck, and I. Vattulainen. 2004. Lipid bilayers driven to a wrong lane in molecular dynamics simulations by subtle changes in long-range electrostatic interactions. J. Phys. Chem. B. 108:4485–4494. [Google Scholar]
- 55.Faraldo-Gomez, J. D., G. R. Smith, and M. S. P. Sansom. 2002. Setting up and optimization of membrane protein simulations. Eur. Biophys. J. 31:217–227. [DOI] [PubMed] [Google Scholar]
- 56.Marsh, D. 1990. Handbook of Lipid Bilayers. CRC Press, Boca Raton, FL.
- 57.de Planque, M. R. R., E. Goormaghtigh, D. V. Greathouse, R. E. Koeppe, J. A. W. Kruijtzer, R. M. J. Liskamp, B. de Kruijff, and J. A. Killian. 2001. Sensitivity of single membrane-spanning alpha-helical peptides to hydrophobic mismatch with a lipid bilayer: effects on backbone structure, orientation, and extent of membrane incorporation. Biochemistry. 40:5000–5010. [DOI] [PubMed] [Google Scholar]
- 58.Ganchev, D., D. Rijkers, M. Snel, J. Killian, and B. de Kruijff. 2004. Strength of integration of transmembrane alpha-helical peptides in lipid bilayers as determined by atomic force spectroscopy. Biochemistry. 43:14987–14993. [DOI] [PubMed] [Google Scholar]
- 59.Cevc, G., and D. Marsh. 1987. Phospholipid Bilayers. Wiley-Interscience, New York.
- 60.Dudko, O. K., A. E. Filippov, J. Klafter, and M. Urbakh. 2003. Beyond the conventional description of dynamic force spectroscopy of adhesion bonds. Proc. Natl. Acad. Sci. USA. 100:11378–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strandberg, E., S. Ozdirekcan, D. T. S. Rijkers, P. C. A. van der Wel, R. E. Koeppe II, R. M. J. Liskamp, and J. A. Killian. 2004. Tilt angles of transmembrane model peptides in oriented and non-oriented lipid bilayers as determined by 2H solid-state NMR. Biophys. J. 86:3709–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cappella, B., and G. Dietler. 1999. Force-distance curves by atomic force microscopy. Surf. Sci. Rep. 34:1–104. [Google Scholar]
- 63.Evans, E., and F. Ludwig. 2000. Dynamic strengths of molecular anchoring and material cohesion in fluid biomembranes. J. Physics: Condens. Matter. 12:A315–A320. [Google Scholar]
- 64.Janovjak, H., J. Struckmeier, M. Hubain, A. Kedrov, M. Kessler, and D. J. Muller. 2004. Probing the energy landscape of the membrane protein bacteriorhodopsin. Structure. 12:871–879. [DOI] [PubMed] [Google Scholar]
- 65.de Planque, M. R., B. B. Bonev, J. A. Demmers, D. V. Greathouse, R. E. Koeppe, F. Separovic, A. Watts, and J. A. Killian. 2003. Interfacial anchor properties of tryptophan residues in transmembrane peptides can dominate over hydrophobic matching effects in peptide-lipid interactions. Biochemistry. 42:5341–5348. [DOI] [PubMed] [Google Scholar]
- 66.Dudko, O. K., A. E. Filippov, J. Klafter, and M. Urbakh. 2002. Dynamic force spectroscopy: a Fokker-Planck approach. Chem. Phys. Lett. 352:499–504. [Google Scholar]
- 67.Sang, Y., M. Dube, and M. Grant. 2001. Thermal effects on atomic friction. Phys. Rev. Lett. 8717:1743011–1743014. [DOI] [PubMed] [Google Scholar]
- 68.Urbakh, M., J. Klafter, D. Gourdon, and J. Israelachvili. 2004. The nonlinear nature of friction. Nature. 430:525–528. [DOI] [PubMed] [Google Scholar]
- 69.Sills, S., and R. M. Overney. 2003. Creeping friction dynamics and molecular dissipation mechanisms in glassy polymers. Phys. Rev. Lett. 91:0955011–0955014. [DOI] [PubMed] [Google Scholar]
- 70.Evans, E., A. Leung, D. Hammer, and S. Simon. 2001. Chemically distinct transition states govern rapid dissociation of single L-selectin bonds under force. Proc. Natl. Acad. Sci. USA. 98:3784–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]