

Abstract
Unfolding of proteins by forced stretching with atomic force microscopy or laser tweezer experiments complements more classical techniques using chemical denaturants or temperature. Forced unfolding is of particular interest for proteins that are under mechanical stress in their biological function. For β-sandwich proteins (a fibronectin type III and an immunoglobulin domain), both of which appear in the muscle protein titin, the results of stretching simulations show important differences from temperature-induced unfolding, but there are common features that point to the existence of folding cores. Intermediates detected by comparing unfolding with a biasing perturbation and a constant pulling force are not evident in temperature-induced unfolding. For an α-helical domain (α-spectrin), which forms part of the cytoskeleton, there is little commonality in the pathways from unfolding induced by stretching and temperature. Comparison of the forced unfolding of the two β-sandwich proteins and two α-helical proteins (the α-spectrin domain and an acyl-coenzyme A-binding protein) highlights important differences within and between protein classes that are related to the folding topologies and the relative stability of the various structural elements.
The mechanism of protein folding and unfolding raises one of the fundamental questions of structural biology (1, 2). Recently, atomic force and laser tweezer experiments have focused attention on unfolding individual protein molecules with external forces (3–6). This approach provides single molecule information that has not been available previously. The results are ideal for analysis by molecular dynamics simulations, which also study the behavior of individual molecules (7, 8). Structural features of the forced unfolding pathways, which are not provided by presently available measurements, are obtained from biased molecular dynamic simulation for two β-sandwich proteins (Fig. 1A) and two α-helical proteins (Fig. 1B). The differences in unfolding behavior can be correlated with the folding topology and stability of the secondary structural elements.
Figure 1.

Structures and topology of the protein studied. (A) Fn3 domain (1TEN) (32) and an Ig domain (1TIT) (33). (B) α-Spectrin domain (1AJ3) (34) and the four-helix bundle protein acyl–coenzyme A-binding protein (2ABD) (35). The N- and C-terminal atoms are represented by red and green spheres, respectively; the same colors are used to represent β-strands belonging to the N- and C-terminal sheets of the β-sandwich proteins. In the schematic topology, triangles represent β-strands, and circles represent α-helices (and also a turn of 310-helix in the Ig domain). The experimental structures are drawn with the program Molmo (36) and the folding topology, with the symbols used in the program (37).
Because unfolding in solution leads to collapsed structures (tending to a random coil at high temperature), whereas forced unfolding, as clearly demonstrated by experiments and simulations, results in an extended chain, it is important to compare the pathways that occur with the two unfolding methods. This is of particular interest because an extrapolation to zero force of atomic force microscopy experiments for a β-sandwich protein has yielded results for the unfolding rate that correlate with the denaturation in solution by Gdm Cl (6). We have used high-temperature unfolding here, because it is better suited for simulations and has been shown to produce meaningful results when compared with denaturant-induced unfolding (9, 10). In temperature-induced unfolding, the effective energy of the relatively compact denatured state is of the order of tens of kcal/mol (1 cal = 4.184 J) above the native state, whereas it is found to be more than 300 kcal/mol higher in the extended conformation induced by simulated stretching. Forced unfolding simulations (i.e., pulling the N- and C-terminal ends apart) and more classical temperature-induced unfolding simulations are reported for two β-sandwich proteins and two α-helical proteins. Comparisons of the unfolding pathways show significant differences, which can be related to the topology and energetics.
Methods
Molecular dynamics simulations were performed with a version of the charmm program (11) modified to include an external the bias force on the protein to accelerate unfolding (for details on the simulation methodology, see ref 8). A polar hydrogen model was used for the protein (12) and an implicit Gaussian model for the solvent (13). The implicit solvent provides an adiabatic solvent response that is important for the artificially fast unfolding simulated here; i.e., the simulated unfolding takes place on a nanosecond timescale, whereas most experiments require milliseconds or longer. If water relaxation times in the nanosecond to microsecond range are involved, explicit solvent treatments may give physically less meaningful results in nanosecond simulations that mimic millisecond experiments (3, 6) than the implicit solvent method. A reaction coordinate ρ(t) leading from the initial to the final state is chosen to have the form ρ(t) = r2NC (t), where rNC (t) is the distance between the N- and C-terminal atoms at time t (8). This reaction coordinate corresponds closely to that used in the forced unfolding experiments (3–6). The time-dependent bias force is equal, in absolute value, to γ(ρ − ρa) 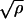 , if ρ(t) < ρa and to zero if ρ(t) ≥ ρa. The quantity ρa (t) is the maximum value of ρ reached by the reaction coordinate at times less than or equal to t. This force is parallel to the vector rNC and pulls the N- and C-terminal atoms away from each other. The method accelerates the reaction while remaining close to the minimum energy surface with normal equilibrium fluctuations; the force has little effect on the short time dynamics. The force is nonzero only when spontaneous thermal fluctuations tend to decrease the reaction coordinate; it is large when the system is crossing a barrier and small otherwise. The parameter γ is adjusted so that unfolding occurs over a nanosecond timescale (γ is a scaling factor for the perturbation, with γ = 1 corresponding to 0.0012 kcal⋅mol−1 Å−4). Different values were tried, and the general behavior was not sensitive to the choice of γ within a certain range. Each starting configuration was energy minimized, then heated for 200 ps and equilibrated at T = 300 K for 100 ps. An equilibrium simulation at least 1 ns in length was performed for each protein. Initial conditions for biased stretching simulations were taken from the equilibrium trajectory and spaced at least 100 ps apart to ensure statistical independence. Multiple simulations were calculated, as required by the chaotic nature of protein dynamics (14), to determine the common elements of the pathways for each system. All simulations were performed in the canonical ensemble using the Nosé–Hoover (15, 16) thermostat; the “mass” of the thermostat is chosen to be large enough so that local heating is negligible. A time step of 2 fs was used, and coordinates were saved every 500 steps (1 ps). During the equilibration period, all the simulated systems are relatively stable. The average Cα rms deviation from the experimental structure during the last 1 ns of equilibration was 2.9 Å for the spectrin domain [Protein Data Bank (PDB) ID 1AJ3], 3.0 Å for the acyl-coenzyme A-binding domain (PDB ID 2ABD), 2.4 Å for the immunoglobulin domain (PDB ID 1TIT), and 1.9 Å for the fibronectin type III domain (PDB ID 1TEN). Forced unfolding simulations have been made for three other Fn3 domains, the 9th and 10th repeats from human fibronectin (PDB ID 1FNF) (17), the glycosidase inhibitor (PDB ID 1HOE) (18), and two Ig domains, the titin Ig domain from human cardiac muscle (5th in the M-line region) (PDB ID 1TNM) (19) and the twitchin Ig domain from nematode muscle (18th in the A-band region) (PDB ID 1WIT) (20); they give corresponding results (ref. 8 and unpublished data). To complement these results, unfolding with a constant pulling force (8) was also studied. A constant force alters the free energy surface so that the completely stretched conformation becomes the most stable one. It accelerates unfolding by lowering the barriers between the native and the completely stretched state (21, 22). A constant force large enough that the protein unfolds on a computationally accessible timescale (∼1 ns) is likely to lead to pathways that differ more from those followed on the experimental timescale than the results obtained with the variable biasing force. Nevertheless, a constant force can be a useful way of isolating (kinetic) intermediates.
, if ρ(t) < ρa and to zero if ρ(t) ≥ ρa. The quantity ρa (t) is the maximum value of ρ reached by the reaction coordinate at times less than or equal to t. This force is parallel to the vector rNC and pulls the N- and C-terminal atoms away from each other. The method accelerates the reaction while remaining close to the minimum energy surface with normal equilibrium fluctuations; the force has little effect on the short time dynamics. The force is nonzero only when spontaneous thermal fluctuations tend to decrease the reaction coordinate; it is large when the system is crossing a barrier and small otherwise. The parameter γ is adjusted so that unfolding occurs over a nanosecond timescale (γ is a scaling factor for the perturbation, with γ = 1 corresponding to 0.0012 kcal⋅mol−1 Å−4). Different values were tried, and the general behavior was not sensitive to the choice of γ within a certain range. Each starting configuration was energy minimized, then heated for 200 ps and equilibrated at T = 300 K for 100 ps. An equilibrium simulation at least 1 ns in length was performed for each protein. Initial conditions for biased stretching simulations were taken from the equilibrium trajectory and spaced at least 100 ps apart to ensure statistical independence. Multiple simulations were calculated, as required by the chaotic nature of protein dynamics (14), to determine the common elements of the pathways for each system. All simulations were performed in the canonical ensemble using the Nosé–Hoover (15, 16) thermostat; the “mass” of the thermostat is chosen to be large enough so that local heating is negligible. A time step of 2 fs was used, and coordinates were saved every 500 steps (1 ps). During the equilibration period, all the simulated systems are relatively stable. The average Cα rms deviation from the experimental structure during the last 1 ns of equilibration was 2.9 Å for the spectrin domain [Protein Data Bank (PDB) ID 1AJ3], 3.0 Å for the acyl-coenzyme A-binding domain (PDB ID 2ABD), 2.4 Å for the immunoglobulin domain (PDB ID 1TIT), and 1.9 Å for the fibronectin type III domain (PDB ID 1TEN). Forced unfolding simulations have been made for three other Fn3 domains, the 9th and 10th repeats from human fibronectin (PDB ID 1FNF) (17), the glycosidase inhibitor (PDB ID 1HOE) (18), and two Ig domains, the titin Ig domain from human cardiac muscle (5th in the M-line region) (PDB ID 1TNM) (19) and the twitchin Ig domain from nematode muscle (18th in the A-band region) (PDB ID 1WIT) (20); they give corresponding results (ref. 8 and unpublished data). To complement these results, unfolding with a constant pulling force (8) was also studied. A constant force alters the free energy surface so that the completely stretched conformation becomes the most stable one. It accelerates unfolding by lowering the barriers between the native and the completely stretched state (21, 22). A constant force large enough that the protein unfolds on a computationally accessible timescale (∼1 ns) is likely to lead to pathways that differ more from those followed on the experimental timescale than the results obtained with the variable biasing force. Nevertheless, a constant force can be a useful way of isolating (kinetic) intermediates.
To compare with the results obtained by an external force directed along the reaction coordinate, high-temperature (400 K), simulations were used to obtain unfolding on the nanosecond timescale. They were started from native conformations equilibrated at room temperature by scaling the velocities and setting the Nosé–Hoover thermostat to the desired temperature. The same potential function (without the biasing term) as for the biased unfolding at 300 K was used. On a 6-ns timescale, only partial unfolding occurs at 400 K, and the unfolding is very fast (less than 500 ps) at 500 K; we report results for the 450 K simulations.
Results and Discussion
Fig. 2 shows the end-to-end distance, rNC, as a function of simulation time for the four proteins with the time-dependent external perturbation. Although individual trajectories for each protein vary as to the time of onset of unfolding, as expected, the general characteristics are preserved. Changes in the slope of the end-to-end distance as a function of time reveal bottlenecks in the induced unfolding pathway that may correspond to intermediates (see below). The two β-sandwich proteins show significant differences. The fibronectin type III (Fn3) domain (1TEN) unfolds in two major steps, whereas the Ig domain (1TIT) has only a single critical step close to the native state, although the early unfolding behavior is rather complex for both systems; a single barrier was found for both systems with a somewhat different model (7, 23). The average unfolding force as a function of rNC is shown on the right-hand side of the plots; the force peaks confirm that there are two dominant barriers for the Fn3 domain and only one for the Ig domain. The maximum local force found in the simulations is of the order of 400 pN, somewhat larger than that used in the experiments because the unfolding simulations are done on a shorter timescale (8). Results of constant force simulations indicate that forces less than 250 pN are not enough to unfold the domains on the nanosecond timescale; a larger force is required to unfold the Ig domain than the Fn3 domain, in qualitative agreement with experiments (24).
Figure 2.

Unfolding reaction coordinate rNC (in Å) as a function of time (in picoseconds). Summary of the results for (A) the two β-sandwich domains and (B) the two α-helical domains. For a definition of the parameter γ, see Methods. The external force applied to unfold the domains is also plotted as a function of rNC; the force values are averaged over all trajectories and over 3-Å intervals of rNC.
For the Ig domain, the single large bottleneck to unfolding occurs at rather small (≈6 Å) extensions (Fig. 3), somewhat larger than the original experimental estimate of 2.5 Å (6) for the position of the transition state for unfolding; the simulation value is close to that suggested by more recent experiments (ref. 25; see also ref. 26). The critical event appears to be the breaking of the “seal” formed by the hydrogen bonds between strands A and B, and A′ and G of the two sheets (see Fig. 1); it involves pulling the N-terminal portion (strands A and A′) away from B and G (Fig. 3 E and F). After this, the domain unfolds along an essentially unique pathway with no additional bottlenecks. The two sheets now are able to slip relative to each other (Fig. 3G) and come apart, with B, E, and D lasting longer than C and F; interestingly, a nonnative hairpin forms in the late stage of unfolding (Fig. 3H) in what was the C-terminal sheet. For the Fn3 domains, the first barrier is associated with the relative “slipping” of the two sheets with respect to each other and partial disruption of the hydrophobic core (see figure 9 of ref. 8, which presents the unfolding pathways of the ninth Fn3 domain of fibronectin that are very similar to those obtained here for 1TEN). The native secondary structure is generally well preserved up to 80 Å, 2.5 times the native “length.” In the region corresponding to the second bottleneck at rNC ≃140 Å, where the molecule is stretched to approximately four times its “length,” there are two pathways (8): in one, strands A and B of the N-terminal sheet stretch completely, whereas there is little change in the C-terminal sheet; in the other, both ends (strands A and G) stretch. The bottleneck at rNC ∼140 Å is related to the simultaneous breaking of the hydrogen bonds between β-strands F and G and between C and F. The CC′ hairpin is always the last secondary structural element to disappear. Because the second barrier is significantly higher for one pathway than for the other, it is possible that partially unfolded intermediates (rNC ≃140 Å) would be observed if a low force were applied, as in constant force simulations (see Fig. 8 of ref. 8). The difference in unfolding behavior between the Fn3 and Ig domains can be understood from their folding topologies (Fig. 1A); that is, in Fn3, the N and C portions of the two β-sheets are separated, whereas they are intertwined in Ig.
Figure 3.

Pathway for the unfolding of the 1TIT domain. (A) X-ray structure, rNC = 45 Å; B–D temperature-induced unfolding pathway; (B) 0.6 ns, rNC = 34 Å, (C) 1.5 ns, rNC = 37 Å; (D) 4.8 ns, rNC = 38 Å. E–H forced unfolding pathway; (E) rNC = 54 Å; (F) rNC = 75 Å; (G) rNC = 126 Å; (H) rNC = 138 Å. Native elements of secondary structure are shown by using the same color code as in Fig. 1; nonnative are in grey.
The nanosecond unfolding simulations of the α-helical domains occur at a somewhat smaller force than do the β-sandwich domains (Fig. 2). Moreover, the α-helical domains show more gradual unfolding. This suggests that even smaller forces would be required at the several orders of magnitude slower pulling speed used experimentally (24). The results of Rief et al. (27) for α-spectrin yield a sawtooth pattern in the force-extension curves corresponding to that observed for Ig and Fn3 domains but with smaller force peaks. As in the case of the Fn3 domains, a plateau at an extension between 110 Å and 150 Å is found in some trajectories when a constant force of 200 pN is applied, suggestive of an intermediate along one set of pathways; it consists of extended N- and C-terminal strands with all of helix B and part of helix C locked together by a nonnative β-hairpin (Fig. 4E′). Figs. 4 and 5 show representative unfolding pathways for the two α-helical domains (Fig. 1B). For the 1AJ3 domain, the initial effect of the stretching is limited to the N- and C-terminal parts of helices A and C, respectively; they tend to become longer 310 helices, with the N-terminal helix unfolding first (Fig. 4 B and C). The increase in the slope of rNC in the region with rNC between 110 Å and 150 Å (Fig. 2B) occurs when the tertiary arrangement of the helices is disrupted (Fig. 4 D to E), even though a significant fraction of the α-helical content is preserved; e.g., the conformation in Fig. 4F, which is almost four times longer than the native state, still has 59% α-helix relative to 88% in the native structure. The last helical element to unfold involves residues 82 to 89 in helix C, which, according to two different secondary structure prediction algorithms (28, 29), has the highest helical propensity. The unfolding pathway of the 2ABD is different from that of the 1AJ3 domain. It shows a large number of metastable states with rNC in the range 70–250 Å when a low (150-pN) constant force is applied. Although there is some unfolding of the N-terminal helix, the initial stages (Fig. 5 A–C) involve primarily small tertiary rearrangements of the helices. The rate of unfolding increases when the three C-terminal helices have come apart and begin to unravel (rNC ≳80 Å). A core formed by residues 32 to 54, which includes part of the B and C helices and the long loop joining them, resists most. This region has a lower helical propensity than helices A and D (29), suggesting that tertiary interactions are important in its stabilization; e.g., helices B and C are most buried (more than 80% of the surface area) in the native state and only become as exposed as helices A and D at rNC > 200 Å. Kragelund et al. (30) have concluded from solution folding studies that the interactions of helices A and D are important in the transition state. Here they separate first, because the forces are applied at the N- and C-terminal ends of helices A and D, respectively. The results for 2ABD contrast with those for the 1AJ3 domain, where a large fraction of the secondary structure survives the disruption of the native tertiary interactions.
Figure 4.

Pathway for the unfolding of the 1AJ3 domain. (A) X-ray structure, rNC = 42 Å; (B) rNC = 50 Å; (C) rNC = 70 Å; (D) rNC = 100 Å; (E) rNC = 130 Å; (E′) rNC = 142 Å; (F) rNC = 160 Å; (G) rNC = 250 Å. E is a stable intermediate found in one set of unfolding pathways.
Figure 5.

Pathway for the unfolding of 2ABD. (A) X-ray structure, rNC = 28.4 Å; (B) rNC = 30 Å; (C) rNC = 50 Å; (D) rNC = 80 Å; (E) rNC = 110 Å; (F) rNC = 150 Å; (G) rNC = 200 Å; (H) rNC = 250 Å.
To determine the thermal unfolding pathways, we consider the 6-ns simulations performed for each of the proteins at 450 K. We use the same protein and solvent model as for the forced unfolding simulations and in previous temperature-induced unfolding studies (13). At 450 K, the domains unfold smoothly, and rNC decreases from its native value, very different from the increase that occurs in rNC during forced unfolding. An essential element of temperature-induced unfolding of both β-sheet domains is that the earliest events correspond to the destruction of certain β-strands and the formation of a coil region in parts of the sequence. This is the case for strands F and C in the Ig domain (Fig. 3B) and strands A and B in the Fn3 domain. Unlike the forced unfolding, there is no tendency of the N- and C-terminal ends to become extended early and move apart. In fact, in the Ig domain, the breaking of the “seal” between the two sheets, an early event when stretching the protein, does not occur until most of the protein is unfolded at high temperature. However, in both types of unfolding simulations (i.e., with stretching and with temperature), the same β-sheet elements are unfolded last. They are the three-stranded (BED) sheet in the Ig domain and the four-stranded (C′CFG) sheet in the Fn3 domain. Moreover, in the latter, the hairpins formed by strands F and G, and, independently, C and C′, which are common to both forced unfolding pathways, are kinetically the most stable portions of the proteins during the temperature-induced unfolding.
For the α-helical domains, the stretched and temperature-induced unfolding pathways are very different: the helices become frayed and fragmented and unfold much earlier at high temperature than in the stretching pathways, where the unfolding of helices is more cooperative, and, once helices are broken, they do not reform. In temperature-induced unfolding, there is a tendency to form transient nonnative β-sheets, which stabilize the collapsed form.
Conclusion
Comparison of the forced unfolding of two protein classes (all-β-sandwich proteins and all-α-helix bundle proteins) have shown significantly different behavior, both within a protein class and for the different classes. Important differences are also found between the unfolding induced by high temperature and by external pulling forces. The result is interpreted in terms of the type of perturbation, the folding topologies, the nature of the secondary and tertiary interactions, and the relative stability of the various structural elements. The complex behavior observed in the simulations contrasts with the simple sawtooth profile observed in the first generation of atomic force microscopy (AFM) unfolding experiments; they revealed only the onset of the unfolding of individual domains in multidomain proteins. It is encouraging that recent forced unfolding experiments of an engineered polyprotein made of identical Ig domains (25) show a previously undetected component, which is attributed to an intermediate. Thus, it is likely that improved AFM experiments will show more of the features observed in the simulation of four different proteins. When combined with protein engineering experiments of the same proteins (30) and simulations for the mutated systems, a detailed microscopic description of the unfolding reaction under different conditions will become available.
Acknowledgments
We thank Jane Clarke for comments on the manuscript. Support by the Ministère de l'Education Nationale de la Recherche et de la Technologie and by the Centre National de la Recherche Scientifique to ESA 7006 (Strasbourg) and by the National Science Foundation (Harvard University) is acknowledged. The computer time used for this work was provided by the Informatique et Calcul Parallèle de Strasbourg. E.P. was supported by the European Union through a Marie Curie fellowship.
Abbreviation
- PDB
Protein Data Bank
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.100124597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.100124597
References
- 1.Dobson C M, S̆ali A, Karplus M. Angew Chem Int Ed Eng. 1998;37:868–893. doi: 10.1002/(SICI)1521-3773(19980420)37:7<868::AID-ANIE868>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 2.Fersht A R. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman; 1999. [Google Scholar]
- 3.Rief M, Gautel M, Oesterhelt F, Fernandez J M, Gaub H E. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 4.Kellermayer M S, Smith S B, Granzier H L, Bustamante C. Science. 1997;276:1112–1116. doi: 10.1126/science.276.5315.1112. [DOI] [PubMed] [Google Scholar]
- 5.Mehta A D, Rief M, Spudich J A, Smith D A, Simmons R M. Science. 1999;283:1689–1695. doi: 10.1126/science.283.5408.1689. [DOI] [PubMed] [Google Scholar]
- 6.Carrion-Vasquez M, Oberhauser A F, Fowler S B, Marszalek P E, Broedel S E, Clarke J, Fernandez J M. Proc Natl Acad Sci USA. 1999;96:3694–3699. doi: 10.1073/pnas.96.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu H, Isralewitz B, Krammer A, Vogel V, Schulten K. Biophys J. 1998;75:662–671. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paci E, Karplus M. J Mol Biol. 1999;288:441–459. doi: 10.1006/jmbi.1999.2670. [DOI] [PubMed] [Google Scholar]
- 9.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 10.Ladurner A G, Itzhaki L S, Daggett V, Fersht A R. Proc Natl Acad Sci USA. 1998;95:8473–8478. doi: 10.1073/pnas.95.15.8473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brooks B R, Bruccoleri R E, Olafson B D, States D J, Swaminathan S, Karplus M. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 12.Neria E, Fischer S, Karplus M. J Chem Phys. 1996;105:1902–1921. [Google Scholar]
- 13.Lazaridis T, Karplus M. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 14.Braxenthaler M, Unger R, Auerbach D, Given J A, Moult J. Proteins. 1997;29:417–425. [PubMed] [Google Scholar]
- 15.Nosé S. Mol Phys. 1984;52:255–268. [Google Scholar]
- 16.Hoover W G. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 17.Leahy D J, Aukhil I, Erickson H P. Cell. 1996;84:155–164. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- 18.Pflugrath J W, Wiegand G, Huber R, Vertesy L. J Mol Biol. 1986;189:383–386. doi: 10.1016/0022-2836(86)90520-6. [DOI] [PubMed] [Google Scholar]
- 19.Pfuhl M, Pastore A. Structure (London) 1995;3:391–401. doi: 10.1016/s0969-2126(01)00170-8. [DOI] [PubMed] [Google Scholar]
- 20.Fong S, Hamill S J, Proctor M, Freund S M, Benian G M, Chothia C, Bycroft M, Clarke J. J Mol Biol. 1996;264:624–639. doi: 10.1006/jmbi.1996.0665. [DOI] [PubMed] [Google Scholar]
- 21.Evans E, Ritchie K. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans E. Faraday Discuss Chem Soc. 1999;111:1–16. [Google Scholar]
- 23.Krammer A, Lu H, Isralewitz B, Schulten K, Vogel V. Proc Natl Acad Sci USA. 1999;96:1351–1356. doi: 10.1073/pnas.96.4.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rief M, Gautel M, Schemmel A, Gaub H E. Biophys J. 1998;75:3008–3014. doi: 10.1016/S0006-3495(98)77741-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marszalek P E, Lu H, Li H, Carrion-Vazquez M, Oberhauser A F, Schulten K, Fernandez J M. Nature (London) 1999;402:100–103. doi: 10.1038/47083. [DOI] [PubMed] [Google Scholar]
- 26.Lu M, Schulton K. Chem Phys. 1999;247:141–153. [Google Scholar]
- 27.Rief M, Pascual J, Saraste M, Gaub H E. J Mol Biol. 1999;286:553–561. doi: 10.1006/jmbi.1998.2466. [DOI] [PubMed] [Google Scholar]
- 28.Muñoz V, Serrano L. Biopolymers. 1997;41:495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 29.Chandonia J-M, Karplus M. Proteins. 1999;35:293–306. [PubMed] [Google Scholar]
- 30.Kragelund B B, Osmark P, Neergaard T B, Schiødt J, Kristiansen K, Knudsen J, Poulsen F M. Nat Struct Biol. 1999;6:594–601. doi: 10.1038/9384. [DOI] [PubMed] [Google Scholar]
- 31.Hamill S J, Steward A, Clarke J. J Mol Biol. 2000;297:165–178. doi: 10.1006/jmbi.2000.3517. [DOI] [PubMed] [Google Scholar]
- 32.Leahy D J, Hendrickson W A, Aukhil I, Erickson H P. Science. 1992;258:987–991. doi: 10.1126/science.1279805. [DOI] [PubMed] [Google Scholar]
- 33.Improta S, Politou A S, Pastore A. Structure (London) 1996;4:323–337. doi: 10.1016/s0969-2126(96)00036-6. [DOI] [PubMed] [Google Scholar]
- 34.Pascual J, Pfuhl M, Walther D, Saraste M, Nilges M. J Mol Biol. 1997;273:740–751. doi: 10.1006/jmbi.1997.1344. [DOI] [PubMed] [Google Scholar]
- 35.Andersen K V, Poulsen F M. J Biomol NMR. 1993;3:271–284. doi: 10.1007/BF00212514. [DOI] [PubMed] [Google Scholar]
- 36.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 37.Flores T P, Moss D S, Thornton J M. Protein Eng. 1997;7:31–37. doi: 10.1093/protein/7.1.31. [DOI] [PubMed] [Google Scholar]