

Abstract
Fluorescent proteins are now widely used in fluorescence microscopy as genetic tags to any protein of interest. Recently, a new fluorescent protein, Kaede, was introduced, which exhibits an irreversible color shift from green to red fluorescence after photoactivation with λ = 350–410 nm and, thus, allows for specific cellular tracking of proteins before and after exposure to the illumination light. In this work, the dynamics of this photoconversion reaction of Kaede are studied by fluorescence techniques based on single-molecule spectroscopy. By fluorescence correlation spectroscopy, fast flickering dynamics of the chromophore group were revealed. Although these dynamics on a submillisecond timescale were found to be dependent on pH for the green fluorescent Kaede chromophore, the flickering timescale of the photoconverted red chromophore was constant over a large pH range but varied with intensity of the 488-nm excitation light. These findings suggest a comprehensive reorganization of the chromophore and its close environment caused by the photoconversion reaction. To study the photoconversion in more detail, we introduced a novel experimental arrangement to perform continuous flow experiments on a single-molecule scale in a microfluidic channel. Here, the reaction in the flowing sample was induced by the focused light of a diode laser (λ = 405 nm). Original and photoconverted Kaede protein were differentiated by subsequent excitation at λ = 488 nm. By variation of flow rate and intensity of the initiating laser we found a reaction rate of 38.6 s−1 for the complete photoconversion, which is much slower than the internal dynamics of the chromophores. No fluorescent intermediate states could be revealed.
INTRODUCTION
Since the first successful cloning of the gene encoding for green fluorescent protein (GFP) was discovered in the jellyfish Aequorea victoria (1,2), a large family of fluorescent proteins has been released (3), nowadays being an invaluable tool kit for fluorescence microscopy and spectroscopy. The multitude of benefits of fluorescent proteins being tagged genetically to any protein of interest has triggered an ongoing search for proteins with new and improved properties. A wide number of GFP mutants have been introduced (4), as well as other types of fluorescent proteins, such as DsRed, the red fluorescent protein isolated from Discosoma corals (5,6). To date, fluorescent proteins with absorption and emission maxima over a broad spectral range are available with extinction coefficients between 20,000 and 160,000 M−1 cm−1 and quantum yields up to 80% (7,8). Several detrimental properties, such as long maturation times and oligomerization tendency, which have in the past restricted the use of these proteins in cell biology have been overcome successfully for several mutants (9,10).
On the basis of solved crystal structures, the origin of the specific fluorescence emitted from the amino acid chain of these proteins has been studied. The basic principle for fluorescence development is the autocatalytic and posttranslational formation of a chromophoric group by cyclization of three amino acids embedded in a barrel of a β-folded peptide chain (11,12). The extension of the formed conjugated π-system and the surrounding peptide chain give rise to the protein's typical fluorescent characteristics.
In this context, a striking feature proven for many of these fluorescent proteins—especially GFP and mutants—is the dual excitation of the chromophore at ∼395 nm and ∼475 nm, where the relative intensity of the two peaks is pH-dependent (13,14). It was proposed that the peak at shorter wavelength could be assigned to a neutral form of the chromophore, whereas the 475-nm peak is due to an ionized form. Upon excitation, the chromophore undergoes deprotonation, followed by a rearrangement of the surrounding peptide chain.
Due to variations in the sequence of the amino acid chain, i.e., mutations of amino acids involved either in the chromophoric group or in the area surrounding the chromophore, the fluorescence properties could be altered (4,15).
Recently, a novel and promising type of GFP mutant has been developed with fluorescent characteristics that emerge or change after activation by light. The possibility of “switching” the protein's emission in any temporal and spatial dimension opens a further wide gate for applications in cell biology, since it allows for specific tracking of proteins within the cytosol or on cellular membranes (16,17).
Miyawaki and co-workers (18) found a GFP-like fluorescent protein from stony coral, Trachyphyllia geoffroyi, which exhibits a spectral shift from green to red fluorescence after activation by ultraviolet light, and named it Kaede after the Japanese word for maple leaf. They could show that this photoinduced conversion is caused by a peptide cleavage in the chromophore group of the protein leading to an extension of the conjugated π-system (19) and, thus, to a spectral shift to longer wavelength. They proposed a mechanism where one of the Nα-Cα bonds in the chromophore group in the excited state is broken due to a β-elimination reaction.
Although photoconversion and spectral shifts after excitation of fluorescent proteins have been observed and described before (14,15,20), the Kaede protein investigated here exhibits an irreversible significant spectral shift of ∼60 nm and thus allows for a simple and clear differentiation of the initial and converted chromophore. The great potential of Kaede as an intracellular optical marker gives all the more reason for detailed and quantitative analysis of its photophysical characteristics, including the timescale and mechanism of the photoconversion reaction. Furthermore, understanding of the photophysics could allow the design of even more powerful fluorescent proteins.
A number of methods have been established to elucidate the photophysics of fluorescent (protein) markers. In particular, techniques based on single-molecule spectroscopy were used extensively to examine the properties of individual proteins (21,22). Provided that conformational changes are attended by variations in fluorescence parameters (such as brightness, color, or lifetime) the structural dynamics can be observed on the single-protein level.
In this study, we first applied fluorescence correlation spectroscopy (FCS, Fig. 1). FCS has previously been used extensively to determine diffusion coefficients (23–25), e.g., to follow molecular binding or unbinding, or to map the cellular environment. Carried out in a confocal setup, FCS is a quasi-single-molecule technique allowing for the analysis of the fluctuations of the fluorescence signal of individual moving or reacting fluorophores. It enables the determination of dynamics that are accompanied by changes in brightness, such as transitions to triplet states (26) and photobleaching reactions (27). With FCS, the so-called flickering process in fluorescent proteins has been studied (20,28–31). Here, the frequent absorption-emission cycle of the chromophore is interrupted reversibly on fast timescales of microseconds to milliseconds, due to light-induced transitions from dark/dim to bright states and vice versa. It could be shown that these photophysical observations are connected to structural properties of the chromophore and the surrounding peptide chain.
FIGURE 1.

Schemes of the performed experiments. (A) Kaede proteins were observed diffusing in an aqueous droplet through the focused laser beams. The photoconversion reaction is initiated by a 405-nm diode laser, and excitation of the protein before and after reaction was performed by the 488-nm line of an Ar ion laser. Data were analyzed applying FCS. (B) For a fast time resolution, the sample solution is pumped through a microfluidic channel. Here, the proteins are passing first the focused initiating laser and second the excitation laser that is displaced for a distance d.
To use FCS for studying reversible processes, their timescale has to be clearly faster than the residence time of the molecules within the detection volume of one to a few femtoliters. In aqueous solution, accessible reaction times are typically on a microsecond timescale and faster. Irreversible reactions that are significantly slower than the time necessary for data acquisition, i.e., ∼5 s, could also be resolved.
Due to these limits of FCS, we introduced a micrometer-sized continuous-flow reaction system developed to enable fast time-resolved measurements of light-induced reactions. In a miniaturized system (so-called micro total analysis systems, μTAS), low sample (volume) consumption in combination with low sample concentration was enabled, rendering this approach extremely advantageous for efficient separation and analysis of sample mixtures (32,33). Although microflow systems were also useful for studying reactions induced by mixing of the reactants (34,35), we established in this experiment the detection of an optically triggered reaction on the single-molecule level on a polydimethylsiloxane (PDMS)/glass chip.
The sample was pumped through a microfluidic channel. After having passed the focused light of a 405-nm emitting diode laser to initiate the photoreaction, the fluorescence analysis was performed by another focused beam (λ = 488 nm) of an Ar ion laser. Contrary to conventional FCS measurements of diffusing systems, each protein molecule passing the reaction zone was detected just once. Since both educt (green Kaede) and product (red Kaede) could be excited with 488 nm and differentiated by optical filtering of the emission light, the number of green and red Kaede proteins could be counted, and thus the reaction yield could be determined. In this experimental arrangement, the time elapsing between initiation and probing of the reaction could be derived from the flow velocity, which is always recorded simultaneously by FCS. Since the time resolution of this technique is only dependent on the flow velocity, a broad temporal range is accessible. Hereby, the nature and temporal range of molecular reaction and intramolecular motility of the Kaede protein can potentially be resolved.
MATERIAL AND METHODS
Optical setup and data processing for diffusion and flow measurements
FCS and flow measurements were carried out in a confocal setup based on the experimental setup described previously (36). In the experiments described here, two light beams, i.e., the laser for activation (405 nm, blue laser diode; Laser 2000, Wessling, Germany) and the laser for excitation (488-nm line of an Ar ion laser; Lasos, Jena, Germany) are superimposed by a dichroic mirror (405 DCLP, all filters purchased from AHF, Tübingen, Germany) and adjusted into the microscope (Olympus IX 70, Tokyo, Japan). They are reflected by a dichroic mirror (495dcx, custom made) and focused by a water immersion objective with high numerical aperture (UplanApo 60×, NA 1.2, Olympus). The emitted light is collected by the same objective. The green and red part of the fluorescence spectrum is split by the dichroic mirror 560 DCLP, and the background signal is reduced by bandpass filter (525DF50 and 630DF60, see Fig. 2). The light is then collected by fiberoptics (diameter 100 μm) transferring the signal to the detectors (avalanche photodiodes CD3017, EG&G Optoelectronics, Vaudreuil, Canada). Data acquisition was performed either by a correlator card (ALV, Langen, Germany), or by the PC card AdWin light (ADWin-LD, Jäger Messtechnik, Lorsch, Germany). For the latter, a homemade program (based on TestPoint, CEC, Billerica, MA) was used (integration time of 5 ms) for data acquisition and for data saving.
FIGURE 2.

Fluorescence spectra of original (1) and photoconverted (2) Kaede protein. The dashed lines represent the transmission spectra of the optical filters used to differentiate between both chromophores (bandpass filter 525 DF 50 (3) and 630 DF 60 (5), and dichroic mirror 560 DCLP (4)).
For the flow measurements, both foci are displaced from the center of the objective in opposite directions, resulting in a slightly distorted shape of the foci. The distance of the foci centers were 15 μm determined by imaging the fluorescence from the spots on a CCD camera. The maximum light power of the beams before the objective was 13.6 mW (405 nm), and 0.38 mW (488 nm).
For acquisition of fluorescence spectra, we used a spectrometer (S2000, Ocean Optics, Dunedin, FL) coupled to glass fiber.
Protein treatment
Green Kaede protein was produced and purified as described before (18). It was stored at −80°C in buffer (10 mM Tris-HCl, 1 mM EDTA, 150 mM NaCl, pH 8.0). Just before the experiments, the protein was dissolved in 10 mM MOPS-buffer, pH 7.0. The concentration for diffusion measurements was between 7.8 and 39 nM, in flow measurements 390 pM. For preparation of a pure red Kaede protein solution, photoconversion was initiated by illumination of a small unit of grKaede (50 μL, 78 nM) with the unfocused light of the diode laser for 20–30 min at a laser power of 13.6 mW. All measurements were carried out at room temperature, or 22°C.
Microstructures for flow measurements
PDMS/glass microstructures were prepared as described previously (36). The dimension of the single linear channel is 50 μm horizontally and 15 μm vertically. The channels were incubated with buffer/BSA solution for ∼30 min; likewise, BSA was added to the protein solution. The flow rates were controlled by alternating droplet heights at the end reservoirs. For this, we used small tips inserted into the water reservoir covered with a foil to reduce fast evaporation of sample solution.
Calibration and fitting procedures of autocorrelation curves
Fitting of the recorded autocorrelation curves G(τ) was performed by the software Microcal Origin, version 6.0, using the following functions.
Diffusion and calibration measurements
The autocorrelation curves for fluorescent molecules with the diffusion coefficient D yielding an average time of τdiff in the detection volume with a size of ω0 and z0 in lateral and axial dimension (defined by 1/e2 – decay of the central light intensity) are fitted using the expressions (26):
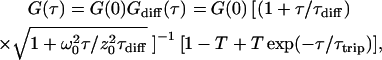 |
(1a) |
and
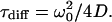 |
(1b) |
G(0) is the amplitude at τ→0, and corresponds to the inverse average number of fluorescent molecules inside the detection volume. Since most dye molecules undergo a triplet transition (37), the timescale (τtrip) and extent of triplet transition (fraction T) is also considered in calibration measurements. For simplification, it is not included in the following diffusion and flow measurements.
Autocorrelation measurements for calibration of the 488-nm laser spot were performed with a Rhodamine Green solution (diffusion coefficient of rhodamine dyes: D = 2.8 × 10−6 cm2/s; 26). Using Eqs. 1a and 1b, the dimensions of the detection volume were determined (ω0 = 0.34 μm, z0 = 2.38 μm for the green detection channel, and ω0 = 0.38 μm, z0 = 2.66 μm for the red detection channel).
Comparable calibration measurements for the 405-nm laser spot were carried out with Alexa 435. Due to the small radius and the non-Gaussian shape of the diode laser, the focus is much larger and deviant from a three-dimensional Gaussian profile. However, we estimated the lateral radius to be ω0 = ∼0.8 μm.
Diffusion with additional blinking reactions
The autocorrelation curves for reversible transitions between fluorescent states with different brightness (dark and bright states) can be approximated by additional exponential terms (20):
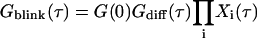 |
(2a) |
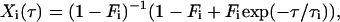 |
(2b) |
where i = 1, 2,… reflects the number of dark/bright-state pairs with the transition rate ki = 1/τi
Flow measurements
In flow measurements, the transport of fluorescent molecules through the detection volume with the flow time τflow was determined, from which the flow velocity v could be derived (38,39):
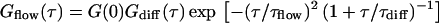 |
(3a) |
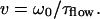 |
(3b) |
If, for high velocities, the residence time in the detection volume is mainly predefined by the directed transport (τflow ≪ τdiff), the diffusion term Gdiff can be ignored.
RESULTS AND DISCUSSION
Spectral shift
The fluorescent Kaede protein exhibits a significant shift of the emission maximum from 518 to 582 nm after illumination with 405 nm light (Fig. 2). The absolute fluorescence intensity for red Kaede decreases ∼50% for excitation with 488 nm, since the absorption maximum is also shifted. The photoconversion is irreversible. Intensive exposure with the 405-nm light to the already red converted protein causes a reduced fluorescence, which recovers partly after several minutes.
Diffusion analysis by FCS
For studies of the photophysical properties of the green and the red fluorescent chromophore (referred to henceforth as grKaede and rKaede, respectively), we applied FCS in aqueous droplets (Fig. 1 A) for a pure solution of either grKaede or rKaede. Additionally, due to the similarity of Kaede and the well-investigated fluorescent protein EGFP (enhanced green fluorescent protein) comparative measurements were carried out under the same conditions. In all experiments, the fluorophores were excited by the 488-nm line of an Ar ion laser. Fluorescence light was detected by filtering the emitted light either between 500 and 550 nm (EGFP and grKaede, emission maximum at 518 nm), or between 600 and 660 nm (rKaede, emission maximum at 582 nm).
First, we determined the average transition time, τdiff, of the proteins through the detection volume. For this, autocorrelation functions (ACF) were recorded at pH 7.0 and excitation intensity of 30 kW/cm2. The ACFs were fitted according to Eq. 1a for delay times τ > 0.1 ms to eliminate fast dynamics (see below). The diffusion time of EGFP was found to be τdiff = 0.43 ± 0.03 ms, four times smaller than the diffusion time calculated for grKaede, τdiff = 1.75± 0.04 ms, and eight times smaller than that obtained for rKaede (τdiff = 3.60 ± 0.12 ms). Using Eq. 1b, we calculated diffusion coefficients of 6.7 × 10−7 cm2/s for EGFP, 1.7 × 10−7 cm2/s for grKaede, and 1.0 × 10−7 cm2/s for rKaede, i.e., Kaede protein has a larger hydrodynamic size than EGFP, whereas the length of the amino acid chains of the primary structures are similar (18). These findings are in agreement with the observation of Ando et al. that Kaede tends to form aggregates similar to other fluorescent proteins such as DsRed (20). They determined a molecular mass of 116.0 kDa for Kaede, whereas EGFP has a molecular mass of 26.75 kDa, and suggested the formation of tetramers. However, since for globular molecules the diffusion coefficient is proportional to the cubic root of the molecular mass, we assume, from the values obtained by FCS, the formation of even larger aggregates, which are also present in freshly prepared solutions with extremely low concentration. Slightly deviating experimental protocols influencing the partial unfolding of the protein, e.g., different temperature and buffer conditions, might also contribute to the large diffusion coefficients.
Interestingly, we found different diffusion coefficients for green and red Kaede which might be explained by an increased aggregation tendency of rKaede, or by a significant change of the protein structure during photoconversion.
Additionally, we determined the brightness per Kaede aggregate (ηmax) by FCS. The value η is calculated by multiplying the recorded photon count rate by the amplitude of the autocorrelation curve, which is the inverse average number of fluorescent molecules inside the detection volume (Eq. 1a). The value increases with the intensity of the excitation beam until photobleaching is predominant. In the setup used here, ηmax is 60 kHz (EGFP), 35 kHz (grKaede), and 20 kHz (rKaede).
Internal dynamics of green and red fluorescent Kaede
The recorded ACF for the diffusion analysis of rKaede clearly showed a “shoulder” in timescales of submilliseconds. Typically, transitions to the triplet state as commonly observed for fluorophores give rise to on/off switches in the fluorescence trace. These intersystem crossing dynamics occur usually on a faster timescale of a few microseconds (37), and are additionally present in the autocorrelation curves of Kaede. Therefore, we assumed a blinking reaction frequently observed in fluorescent proteins, i.e., a fast reversible transition from a fluorescent to a nonfluorescent state (20,28), and investigated this effect for grKaede and rKaede in detail.
In Fig. 3, the influence of protonation is demonstrated for pH values between 9 and 4.5. Although the brightness drops (to ηmax ∼5 kHz) for both grKaede and rKaede, we found a significant shift in the autocorrelation curves of grKaede with decreasing pH. The FCS curves of rKaede remain unchanged within varying proton concentration. For pH <5.5, an additional shoulder for long diffusion times was raised in the ACF of both types of Kaede that could not be prevented by further diluting the protein solution. It might be caused by (partial) unfolding of the peptide chain at low pH leading to a decreased diffusion coefficient, or by formation of even larger clusters.
FIGURE 3.

Autocorrelation functions for varying pH (solid lines represent fits according to Eqs. 2a and 2b). For grKaede (left), the curves shift to shorter residence times τ, whereas they remain almost unchanged for rKaede (right). The shoulder at 10–100 ms arises at low pH 5.0 and 4.5 for both grKaede and rKaede (pH 9, 8, 7, 6.5, 5.5, 5.0, and 4.5 in the direction of the arrow). The insets show the fit results for pH 5.5–9 obtained from Eq. 2b (transition rates τ1 (squares) and τ2 (stars, right inset) and fractions F1 (squares) and F2 (stars, right inset)).
The autocorrelation curves were fitted with one additional exponential term (Eqs. 2a and 2b, and solid lines in Fig. 3) neglecting the first part of the curves representing the triplet transitions, i.e., the fit starts for τ = 0.01 ms. Keeping τdiff = 1.74 ms constant, an increasing fraction up to 63% for decreasing τ1 from 403 μs to 235 μs (rates of 2.5–4.2 kHz) could be determined (Fig. 3 left, inset).
The timescale is comparable to values determined with the same technique for GFP (28). Here, internal protonation processes between the chromophoric group and the proton binding site close to the chromophoric group were assumed. Since grKaede protein has a sequence very similar to that of GFP, and could be found in a nonfluorescent neutral and a fluorescent protonated form (19), the fast flicker dynamics found could be assigned to the transition between these two forms. Consequently, the loss of pH dependence after photoconversion suggests the absence of a protonated form in rKaede, which is also in accordance with the proposed structure by Mizuno et al. (19).
Next, we varied the excitation light intensity (at constant pH 7.0) from 2.5 to 78 kW/cm2 (Fig. 4). For the grKaede protein, we observed a shift of the autocorrelation curves to shorter diffusion times. Again we applied the fitting function (Eqs. 2a and 2b, τdiff = 1.74 ms constant, and solid lines in Fig. 4), and also observed an increasing fraction up to 51% for rates between 0.8 and 2.2 kHz.
FIGURE 4.

Dependence of the excitation intensity on the autocorrelation curves obtained for grKaede (left) and rKaede proteins (right). Solid lines represent fits according to Eqs. 2a and 2b. The applied excitation intensities in the direction of the arrow were 2.5, 4.9, 12.4, 19.6, 39.1, and 78.0 kW/cm2. The transition rates and fractions for the blinking reaction are given in the insets (transition rates τ1 (squares) and τ2 (stars, right inset) and fractions F1 (squares) and F2 (stars, right inset)).
Likewise, the ACF of rKaede shifts for increasing excitation intensity. However, there are changes in a timescale of microseconds that are clearly different from those obtained for grKaede. The ACF could be fitted only with a two-state model (Eqs. 2a and 2b, i = 2), where we could determine flickering rates up to 32 kHz (fraction 31%) and 10 kHz (fraction 20%).
For interpretation of the ACFs, photobleaching reactions have to be considered. Photobleaching reactions are irreversible, and cause a complete loss of the fluorescent properties. Since they are induced by light, they occur during the transition of the fluorescent protein through the laser spot. The dwell time of the diffusing molecules after photobleaching is beyond registration and, thus, the autocorrelation curve is shifted to a shorter diffusion time τdiff (27). In the fitting function, these reactions could be approximated by an additional exponential term (27), i.e., in the same way as the blinking reactions. Thus, the exact differentiation between blinking and photobleaching reactions is only possible by FCS as long as the timescales are different.
Despite the difficulty in accounting for photobleaching, we followed the discussion in Malvezzi-Campeggi et al. (20) where a similar light-induced flickering process for DsRed could be observed by FCS. Thus, we can assume at least one spectrally distinguishable state in rKaede in addition to the basic fluorescent form that was not present before the photoconversion reaction.
It should be mentioned that we transferred results from similar studies of different fluorescent proteins (EGFP and DsRed) to interpret the ACFs for the Kaede protein, which seems to be justified due to the structural similarity of these proteins. Because of the specific characteristics of Kaede, particularly aggregation, the observed changes in the ACFs for varying pH and excitation light intensity could also be caused by internal dynamics of the single chromophore, or by interactions of chromophores within single protein aggregates. Furthermore, alteration of the equilibrium of different-sized protein aggregates cannot be fully excluded.
Nevertheless, for both pH and light-intensity dependence, the FCS studies clearly demonstrate the change of fluorescent characteristics during the photoconversion reaction, reflecting indeed the formation of a new chromophore.
Flow measurements
Since the photoconversion reaction itself was beyond the time resolution of FCS, a different experimental setup was designed with a flowing system to increase the time resolution of the measurements. Furthermore, in a flowing system, every single protein molecule would be analyzed once.
The microfluidic structures used here were prepared by casting PDMS from a silicon master. Covering it with a glass plate, it could be adapted to the optical setup. Choosing a miniaturized channel with dimensions of 25 × 15 μm2 over a length of 5 mm, the sample consumption of ∼15 μl in subnanomolar concentration was indeed extremely low.
For the flow experiments, the spots of the excitation laser and the laser that initiates photoconversion were created by the same objective, but were slightly displaced (distance: 15 μm, see illustration in Fig. 1 B). Proper alignment of the channel to the flow direction allowed for subsequent initiation of the reaction and probing by the excitation laser. Green and red fluorescence was recorded simultaneously by two detectors (“red” and “green”). From the fluorescence traces (Fig. 5 A), we could observe signal bursts corresponding to passing protein molecules. For a pure buffer solution pumped through the microchannels, almost no fluorescence bursts were recorded. However, stray light from the diode laser triggering the photochemical conversion, and from the Ar/Kr laser probing the emission behavior could not be suppressed completely by optical filtering. By subtracting the additional signal at high intensities of the diode laser, the background signal in all experiments was equalized to ∼20 kHz. For data evaluation, fluorescence bursts with intensity >50 kHz on either the green or red detector were assigned to a passing molecule. This value corresponds to the background signal plus double the standard deviation of the average fluctuation of the background signal (15 kHz). Detected photons above the threshold were added until the signal dropped again below the threshold. The ratio of red to green signal of a fluorescence burst was calculated. From measurements with pure grKaede and rKaede solutions, we could guarantee the clear differentiation between both forms, i.e., a ratio <1 could be directly assigned to grKaede, and analogously a ratio >1 corresponded to rKaede.
FIGURE 5.

Single-molecule detection in a microfluidic channel. (A) Out of the fluorescence trace of the green and red detection channel, the fluorescence bursts above a set threshold (50 kHz) are analyzed. Histograms of the bursts for low (B) and high (C) intensity of the initiation laser (0.07 and 427 kW/cm2 at 405 nm). The ratio of red to green intensity per burst corresponds to the color of detected protein. For grKaede, Ired/Igreen < 1; for rKaede, Ired/Igreen > 1.
In Fig. 5, the histograms shown were obtained by the analysis described before. By raising the intensity of the 405-nm initiation laser beam (from 0.07 to 427 kW/cm2 in Fig. 5, B and C), we observed the expected increase of red Kaede protein.
For very high intensities of the blue diode laser, a tremendous decrease of the total number of protein molecules as well as of rKaede protein was found, which could either be due to molecules being in a nonfluorescent intermediate state, or to quenching and bleaching of the protein before or after photoconversion. For the latter, we could directly verify quenching of rKaede fluorescence in a flowing sample solution of pure rKaede when irradiated with 405 nm and subsequently with 488 nm light. From these calibration measurements, a linear reduction of rKaede with intensity of the 405 nm laser of up to 30% was determined, which was considered in the following calculations.
Taking these corrections into account, the increase of rKaede versus the applied intensity is nonlinear, i.e., the slope in a double-log plot (graph not shown) is <1. This could be a result of saturation effects of the 405-nm laser spot, as well as of competing reactions, such as photobleaching or photophysical transitions that are likely to be induced by 405-nm light preventing the green-to-red-photoconversion. In this context, the findings by Jung et al. (40) should be mentioned: Those authors observed an influence of 405-nm light on the population of the fluorescent bright state for GFP and mutants (depopulation of the neutral form), which results in an increase of the green fluorescence. Since these chromophores are similar to the ones investigated here, their findings could probably be adapted to explain the reduced quantum yield of the photoconversion. For the Kaede protein, depopulation of the excited neutral form would result in a reduction of the photoreaction.
In contrast, photobleaching by the probe beam (488 nm) should be very small in flowing systems and negligible for the data analysis, as long as the measured burst size (i.e., detected photons before photobleaching) is clearly above the threshold. Indeed, increasing the excitation intensity of the 488-nm laser beam did not result in a significant reduction of counted proteins, and was set to the highest available intensity of 78 kW/cm2.
Since the photoreaction is initiated and probed individually for each fluorescent molecule, it is possible, with this technique, to reveal fluorescent intermediates during the reaction. However, we frequently found fluorescence bursts either with high green or high red fluorescence, i.e., there was no indication for an intermediate fluorescent state with equal green and red fluorescence intensity. That observation is particularly surprising, since Kaede protein is most probably aggregated, i.e., comprises several individual chromophores. We suggest an efficient energy transfer from green to red chromophore in which the green fluorescence is almost completely suppressed after photoconversion of only one chromophore inside a single aggregate. This is in agreement with the measurements of immobilized proteins, where an alternating emission pattern with intensity either in the green or red detection channel was recorded (data not shown), and is a subject for further study.
Determination of the reaction rate
In the experiments described next, the flow speed of the sample solution was varied. While recording the fluorescence trace for passing proteins, the autocorrelation curves were obtained simultaneously (Fig. 6 B). Using Eq. 3a, autocorrelation curves were fitted. From the results, we evaluated the flow velocities determined at the central position of the microchannel that correspond to the time delay between initiation of the reaction and probing of the reaction progress (t = 15 μm/v). By changing the flow velocity from 1.50 to 0.23 mm/s, we varied the delay time between 10 and 65 ms.
FIGURE 6.

(A) Reaction yield (counted rKaede) for various flow velocities and intensities of the 405-nm diode laser. The intensity of the initiating 405-nm laser diode was corrected taking into account the time the proteins are exposed to the light while passing the focus. The time given for each graph corresponds to the delay between initiation and probing of the reaction. (B) The applied flow velocities in A at the channel center were determined with FCS (measured data and fit). The arrow indicates increasing flow velocity. (C) Determination of the reaction rate: From A, the reaction yield was determined for a constant intensity (value 4.7 of the corrected intensity) for different delay times. The solid line represents the monoexponential fit with the increase time of 25.9 ms and amplitude of 2% at delay time of 0 ms, corresponding to the amount of rKaede frequently observed in a pure grKaede solution.
During this time, the molecules were transported directly from the initiating laser to the probing laser. The flow rates inside the PDMS/glass structures used here could in principle be increased up to a few mL/h, corresponding to a flow speed at the center of the microfluidic channel of a few hundred mm/s. This makes the technique adaptable to study any fast light-induced reactions in time ranges up to 105/s.
However, although the flow speed could be enhanced easily, deceleration to the range of diffusion will induce more complexity in data analysis. Diffusion is a random movement in all directions. Thus, movement of molecules out of focus of the probing laser after having passed the initiation laser, as well as the movement of molecules into the probing laser focus without passing the initiation laser, cannot be ignored. The maximum distance d that is passed by a molecule with the diffusion coefficient D (here: 1.7 × 10−7 cm2/s for grKaede) during the time t can be calculated by the expression d2 = 2Dt for the applied delay times, d = 0.6–1.5 μm. For slow velocities, these distances are in the range of the size of the laser spot, and we omit them in our calculations to avoid complexity of data analysis. In this context, a channel with smaller dimensions or a sheath flow might be helpful and would additionally prevent the protein from adsorption to the channel walls.
For analysis of the reaction progress, it must be considered that the flow velocity influences the integrated intensity of the initiating laser light: A fast flow velocity results in a short exposure time of the initiation light to grKaede protein. Therefore, the intensity was corrected by multiplying the applied intensity with the duration time of the proteins inside the detection volume: Istandardized = Idetermined × τfl.
Fig. 6 A gives an overview of the reaction yield, i.e., counted rKaede protein in relation to the total number of protein molecules, for different flow speed and intensity of the initiation laser. For slow velocities, the reaction yield of rKaede increases up to 73%, whereas fast flow velocities produce just a few detectable photoconverted rKaede molecules of <10% as a result of the short time remaining from initiation to probing of the reaction. For calculation of the reaction rate, the reaction yield was plotted against the time lag (Fig. 6 C) for the same standardized intensity (Istandardized = 4.5). The data were adjusted with a monoexponential function, and an exponential increase of 25.9 ms was found, which corresponded to a reaction rate of 38.6 s−1 for the irreversible photoreaction.
The determined reaction rate is in a range that is much slower than reaction rates typically observed for photoinduced reactions, such as photoisomerization (10−8–10−11 s−1). However, the photoconversion reaction described here is most likely a cleavage reaction (19) that is coupled with a rearrangement of the peptide chain surrounding the chromophore, which is very likely a much slower process. Mizuno et al. (19) proposed that the photoreaction occurs after excitation of the neutral chromophore and deprotonation in the excited state. Without relaxation, the cleavage reaction is induced. The reaction rate determined using the method described here covers the complete process from loss of green fluorescence to the formation of the red chromophore.
CONCLUSION AND OUTLOOK
Green fluorescent Kaede protein undergoes a photoreaction when exposed to 405 nm light that results in a broad red shift of the fluorescence spectrum accompanied by altered fluorescence properties. The spectroscopic techniques presented here allowed for a comprehensive study of fast reversible dynamics (in the microsecond time range) of the protein before and after photoreaction, as well as the irreversible dynamics of the photoreaction itself (millisecond time range) on a single-molecule scale. The study revealed temporal information that could be assigned to the data determined by structural analysis in former studies by Mizuno et al. (19).
Using FCS, internal blinking reactions of the chromophore group could be observed. In particular, the reconstruction of the chromophore after the photoreaction was apparent, accompanied by the loss of pH-dependent flicker processes on the one hand, and the formation of light-induced flicker dynamics on the other hand.
The dynamics during the reaction was resolved, introducing a microflow continuous reaction system. Accessible reaction rates up to 105 s−1 are exclusively dependent on the distance between two focused laser spots (limited by the size of the objective from 0 to 15 μm), and the applied flow speed that is determined simultaneously by FCS. Using this technique, any photoinduced reaction could be directly observed on a single-molecule scale. Of course, the formation of reaction educts, products, and probable intermediates must be accompanied by changes in the fluorescence characteristics such as the spectral shift from green to red described here.
The remaining uncertainties regarding the determined reaction rate are due to the presumably complex photophysics and the aggregation tendency of Kaede, and will be investigated in detail in upcoming studies. They will certainly limit the usefulness of the protein for cellular applications. Some modifications in the amino acid sequence, as done for similar fluorescent proteins, might introduce improved characteristics, and less aggregation, which opens the way to switching and tracking proteins at any time and place.
Acknowledgments
We thank A. Miyawaki and H. Mizuno for kindly providing the Kaede protein.
Financial support by the Bundesministerium für Bildung und Forschung (Biofuture grant to P.S.), the Deutsche Forschungsgemeinschaft for support (Graduiertenkolleg 723 to S.S.), and Europäischen Fonds für regionale Entwicklung (grant No. 4-0123.55-20-0370-03/3) is gratefully acknowledged.
P. S. Dittrich's present address is Dept. of Miniaturization, ISAS—Institute for Analytical Sciences, Bunsen-Kirchhoff-Str. 11, D-44139 Dortmund, Germany.
S. P. Schäfer's present address is Institute of Biophysics/BIOTEC, Technische Universität Dresden, Tatzberg 47-51, D-01307 Dresden, Germany.
References
- 1.Prasher, D. C., V. K. Eckenrode, W. W. Ward, and F. G. Prendergast. 1992. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 111:229–233. [DOI] [PubMed] [Google Scholar]
- 2.Chalfie, M., Y. Tu, G. Euskirchen, W. W. Ward, and D. C. Prasher. 1994. Green fluorescent protein as a marker for gene expression. Science. 263:802–805. [DOI] [PubMed] [Google Scholar]
- 3.Zhang, J., R. E. Campbell, A. Y. Ting, and R. Y. Tsien. 2002. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 3:906–918. [DOI] [PubMed] [Google Scholar]
- 4.Tsien, R. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- 5.Matz, M. V., A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L. Markelov, and S. A. Lukyanov. 1999. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 17:969–973. [DOI] [PubMed] [Google Scholar]
- 6.Baird, G. S., D. A. Zacharias, and R. Y. Tsien. 2000. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA. 97:11984–11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyawaki, A. 2002. Green fluorescent protein-like proteins in reef Anthozoa animals. Cell Struct. Funct. 27:343–347. [DOI] [PubMed] [Google Scholar]
- 8.Kohl, T., and P. Schwille. 2004. Fluorescence correlation spectroscopy with autofluorescent proteins. Adv. Biochem. Biotechnol. In press. [DOI] [PubMed]
- 9.Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiedenmann, J., A. Schenk, C. Röcker, A. Girod, K. D. Spindler, and G. U. Nienhaus. 2002. A far-red fluorescent protein with fast maturation and reduced oligomerization tendency from Entacmaea quadricolor (Anthozoa, Actinaria). Proc. Natl. Acad. Sci. USA. 99:11646–11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heim, R., D. C. Prasher, and R. Y. Tsien. 1994. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA. 91:12501–12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ormö, M., A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien, and S. J. Remington. 1996. Crystal structure of the Aequorea Victoria green fluorescent protein. Science. 273:1392–1395. [DOI] [PubMed] [Google Scholar]
- 13.Lossau, H., A. Kummer, R. Heinecke, F. Pöllinger-Dammer, C. Kompa, G. Bieser, T. Jonsson, C. M. Silva, M. M. Yang, D. C. Youvan, and M. E. Michel-Beyerle. 1996. Time-resolved spectroscopy of wild-type and mutant green fluorescent proteins reveals excited state deprotonation consistent with fluorophore-protein interactions. Chem. Phys. 213:1–16. [Google Scholar]
- 14.Chattoraj, M., B. A. King, G. U. Bublitz, and S. G. Boxer. 1996. Ultra-fast excited state dynamics in green fluorescent protein: multiple states and proton transfer. Proc. Natl. Acad. Sci. USA. 93:8362–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brejc, K., T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormö, and S. J. Remington. 1997. Structural basis for dual excitation and photoisomerization of the Aequorea Victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA. 94:2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ando, R., H. Mizuno, and A. Miyawaki. 2004. Regulated fast nucleocytoplamic shuttling observed by reversible protein highlighting. Science. 306:1370–1373. [DOI] [PubMed] [Google Scholar]
- 17.Patterson, G. H., and J. Lippincott-Schwartz. 2002. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 297:1873–1877. [DOI] [PubMed] [Google Scholar]
- 18.Ando, R., H. Hama, M. Yamamoto-Hino, H. Mizuno, and A. Miyawaki. 2002. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:12651–12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizuno, H., T. K. Mal, K. I. Tong, R. Ando, T. Furuta, M. Ikura, and A. Miyawaki. 2003. Photo-induced peptide cleavage in the green-to-red conversion of a fluorescent protein. Mol. Cell. 12:1051–1058. [DOI] [PubMed] [Google Scholar]
- 20.Malvezzi-Campeggi, F., M. Jahnz, K. G. Heinze, P. Dittrich, and P. Schwille. 2001. Light-induced flickering of DsRed provides evidence for distinct and interconvertible fluroescent states. Biophys. J. 81:1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie, X. S., and J. K. Trautman. 1998. Optical studies of single molecules at room temperature. Annu. Rev. Phys. Chem. 49:441–480. [DOI] [PubMed] [Google Scholar]
- 22.Weiss, S. 1999. Fluorescence spectroscopy of single biomolecules. Science. 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- 23.Rigler, R., Ü. Mets, J. Widengren, and P. Kask. 1993. Fluorescence correlation spectroscopy with high count rate and low background: analysis of translational diffusion. Eur. Biophys. J. 22:169–175. [Google Scholar]
- 24.Schwille, P., J. Bieschke, and F. Oehlenschläger. 1997. Kinetic investigations by fluorescence correlation spectroscopy: the analytical and diagnostic potential of diffusion studies. Biophys. Chem. 66:211–228. [DOI] [PubMed] [Google Scholar]
- 25.Schwille, P., U. Haupts, S. Maiti, and W. W. Webb. 1999. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 77:2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Widengren, J., R. Rigler, and Ü. Mets. 1994. Triplet-state monitoring by fluorescence correlation spectroscopy. J. Fluoresc. 4:255–258. [DOI] [PubMed] [Google Scholar]
- 27.Dittrich, P. S., and P. Schwille. 2001. Photobleaching and stabilization of fluorophores used for single-molecule analysis with one- and two-photon excitation. Appl. Phys. B. 73:829–837. [Google Scholar]
- 28.Haupts, U., S. Maiti, P. Schwille, and W. W. Webb. 1998. Dynamics of fluorescent fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 95:13573–13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwille, P., S. Kummer, A. A. Heikal, W. E. Moerner, and W. W. Webb. 2000. Fluorescence correlation spectroscopy reveals fast optical excitation-driven intramolecular dynamics of yellow fluorescent proteins. Proc. Natl. Acad. Sci. USA. 97:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heikal, A. A., S. T. Hess, G. S. Baird, R. Y. Tsien, and W. W. Webb. 2000. Molecular spectroscopy and dynamics of intrinsically fluorescent proteins: Coral red (dsRed) and yellow (Citrine). Proc. Natl. Acad. Sci. USA. 97:11996–12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schenk, A., S. Ivanchenko, C. Röcker, J. Wiedenmann, and G. U. Nienhaus. 2004. Photodynamics of red fluorescent proteins studied by fluorescence correlation spectroscopy. Biophys. J. 86:384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reyes, D. R., D. Iossifidis, P. Auroux, and A. Manz. 2002. Micro total analysis systems. 1. Introduction, theory, and technology. Anal. Chem. 74:2623–2636. [DOI] [PubMed] [Google Scholar]
- 33.Auroux, P., D. Iossifidis, D. Reyes, and A. Manz. 2002. Micro total analysis systems. 2. Analytical standard operations and applications. Anal. Chem. 74:2637–2652. [DOI] [PubMed] [Google Scholar]
- 34.Lipman, E. A., B. Schuler, O. Bakajin, and W. A. Eaton. 2003. Single-molecule measurement of protein folding kinetics. Science. 301:1233–1235. [DOI] [PubMed] [Google Scholar]
- 35.Dittrich, P. S., B. Müller, and P. Schwille. 2004. Studying reaction kinetics by simultaneous FRET and cross-correlation analysis in a miniaturized continuous flow reactor. Phys. Chem. Chem. Phys. 6:4416–4420. [Google Scholar]
- 36.Dittrich, P. S., and P. Schwille. 2003. An integrated microfluidic system for reaction, high-sensitivity detection and sorting of fluorescent cells and particles. Anal. Chem. 75:5767–5774. [DOI] [PubMed] [Google Scholar]
- 37.Mets, Ü., and R. Rigler. 1994. Submillisecond detection of single rhodamine molecules in water. J. Fluoresc. 4:259–264. [DOI] [PubMed] [Google Scholar]
- 38.Magde, D., W. W. Webb, and E. L. Elson. 1978. Fluorescence correlation spectroscopy. III. Uniform translation and laminar flow. Biopolymers. 17:361–376. [Google Scholar]
- 39.Dittrich, P. S., and P. Schwille. 2002. Spatial two-photon fluorescence cross-correlation spectroscopy for controlling molecular transport in microfluidic structures. Anal. Chem. 74:4472–4479. [DOI] [PubMed] [Google Scholar]
- 40.Jung, G., S. Mais, A. Zumbusch, and C. Bräuchle. 2000. The role of dark states in the photodynamics of the green fluorescent protein examined with two-color fluorescence excitation spectroscopy. J. Phys. Chem. 104:873–877. [Google Scholar]