

Abstract
Depending on the nucleotide sequence, the temperature, and other conditions, RNA hairpin-folding kinetics can be very complex. The complexity with a wide range of cooperative and noncooperative kinetic behaviors arises from the interplay between the formation of the loops, the disruption of the misfolded states, and the formation of the rate-limiting base stacks. With a rate constant model and a kinetic-cluster theory, we explore the broad landscape for RNA hairpin-folding kinetics. The model is validated through direct tests against several experimental measurements. The general kinetic folding mechanisms and the predicted great variety of folding kinetics are directly applicable and quantitatively testable in experiments. The results from this study suggest that 1), previous experimental findings based on the individual hairpins revealed only a small fraction of much broader and more complex RNA hairpin-folding landscapes; 2), even for structures as simple as hairpins, universal folding timescales and pathways do not exist; and 3), to treat the loop size as the sole factor to determine the hairpin-folding rate is an oversimplification.
INTRODUCTION
RNA hairpins are fundamental building blocks for complex three-dimensional RNA structures. The folding of complex RNA tertiary structures often involves the conformational change of hairpin structures (1–7). Understanding how a hairpin folds is, therefore, a prerequisite for understanding RNA tertiary structure folding. In addition, RNA functions, including splicing, translational regulation, etc., often involve the sequence-specific folding kinetics of RNA hairpins (8–10). Motivated by the significant structural and functional roles of RNA hairpins, we explore the complex sequence-dependent RNA hairpin-folding kinetics.
Since the early work of Porschke (11), there have been several experimental studies on the folding kinetics of RNA hairpin (12–16), peptide β-hairpin (17) and DNA hairpin (18–24). The early experiments focused on loop formation as the rate-limiting step. For most of the sequences studied, the folding was described by a single zipping/unzipping pathway. Moreover, depending on the loop size, most of the hairpins fold in microseconds' timescale in experiments. More recently, experiments from several groups are beginning to shed light on the complexity of RNA and DNA hairpin-folding landscapes, such as the non-Arrhenius and noncooperative kinetic behaviors (18–24). Since most of the experimental studies are limited to the kinetics specific to the sequences studied, a systematic full exploration for the sequence-dependent complexity of the folding landscape (25–27) is still missing.
Large-scale atomistic molecular dynamics simulations (28–30) enable the detailed analysis for the transition states, the kinetic intermediates, and the multiple folding pathways for the specific sequences studied. But the molecular dynamics studies are restricted to very short sequence. Monte Carlo simulations have also been used to model the RNA folding process by several authors (31–34). However, the conformational and kinetic sampling may not be complete and the method cannot give analytical results that allow for stable predictions for the long time dynamics. Recently, theoretical studies (27,35) based on statistical mechanical models were developed to study RNA folding kinetics. These statistical mechanical models are based on the master-equation (rate equation) approach (27) (see Theory and Methods, below). For short chains, the entire conformational ensemble can be exhaustively enumerated. With the complete conformational ensemble, the master equation can account for the transitions between each and every pair of the conformations and can give the information about the rate-limiting steps (36) and the population kinetics. The folding kinetics predicted from the master-equation approach is supported by the atomic simulation (29). However, the disadvantage of the master-equation approach is that it cannot give direct information about the microscopic pathways. In addition, the method is limited to short chains due to the rapid increase of the number of conformations for longer chains. More recently, aiming to study the folding mechanism at the level of microscopic pathways and to study kinetics for longer chains, we developed the kinetic-cluster model (detailed in Theory and Methods) by reducing the original large conformational ensemble into pre-equilibrated clusters (37), for which we can perform detailed microscopic kinetic analysis.
Experimental and theoretical developments suggest that the RNA hairpin-folding landscape is much more diverse and more complex than that revealed in early experiments. In this study, we develop a kinetic-cluster model for RNA hairpin folding. We go beyond the individual nucleotide sequences by focusing on the general RNA hairpin-folding scenarios as well as the specific features such as the loop and stem dependence. We first test and validate the theory through direct experimental tests for several experimentally measured duplexes and hairpins. We then employ the theory to investigate the general scenarios for RNA hairpin-folding kinetics.
THEORY AND METHODS
Transition rate model
Kinetic move set
The stability of a basepair in an RNA secondary structure (e.g., a hairpin structure) depends on the identity of the flanking basepairs and the identity of the basepair itself (38,39). This nearest-neighbor model is rationalized by the fact that RNA secondary structure is stabilized mainly by the base-stacking interactions and the hydrogen bonding—both of which are short-range local interactions and dependent primarily on the identity of adjacent basepairs (40). Adjacent basepairs form base stacks. For a given RNA sequence, we can exhaustively enumerate all the possible hairpin conformations according to the base stacks. Since a single (unstacked) basepair is not stable (38) and can quickly unfold, we define a kinetic move (equal to an elementary kinetic step for the transformation from one conformation to the other) to be the formation/disruption of a stack or a stacked basepair. Therefore, two conformations are kinetically connected if they can be interconverted through the addition or deletion of a base stack and are kinetically disconnected otherwise. Transitions between the disconnected conformations are disallowed and have rates equal to zero. Here we use the term base stack instead of basepair to define the kinetic move set because a single (unstacked) basepair is not stable. Our kinetic move model will be tested against experiments in Experimental Tests for the Rate Constant Model (see below). To compute the folding kinetics, we need a model to calculate the rate for each kinetic move.
Rate constant for a kinetic move
The transition rate for each kinetic move can be calculated from the general formula
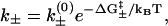 |
(1) |
with “+” for the formation and “−” for the breaking of the stack, where kB is the Boltzmann constant, T is the temperature, and 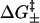 is the free energy barrier of the respective transitions, and
is the free energy barrier of the respective transitions, and 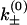 is a prefactor to be determined from the experiments.
is a prefactor to be determined from the experiments.
The formation of a base stack or a loop usually involves an unfavorable entropy loss ΔS due to the accompanying restriction in the torsional angles, the desolvation, etc. We assume that the transition state is at the point where the bases have been fixed to the stacked configuration but the bases have not reacted to form the stabilizing hydrogen-bonding and base-stacking interactions. The barrier for the formation of the transition state is entropic: 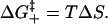
The breaking of a base stack involves an enthalpy increase ΔH due to the disruption of the hydrogen bonding and the base-stacking interactions. We assume that the transition state is at the point where the hydrogen bonding and the base-stacking interactions have been disrupted, but the torsional angles of the chain are not yet liberated from the restricted base-stack configuration. The barrier for the formation of the transition state is enthalpic: 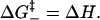
Both 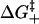 and
and 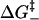 depend on the sequence identity of the bases involved. In our model, ΔH, ΔS, and
depend on the sequence identity of the bases involved. In our model, ΔH, ΔS, and 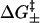 are calculated from a statistical mechanical model for RNA folding (41). Assuming
are calculated from a statistical mechanical model for RNA folding (41). Assuming 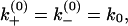 from Eq. 1, we have
from Eq. 1, we have
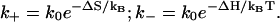 |
(2) |
The above expressions for the rate constants satisfy the detailed balance condition of 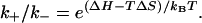 If ΔS and ΔH are T-independent, k+ would also be T-independent but k– would be strongly dependent on T and is larger for higher T.
If ΔS and ΔH are T-independent, k+ would also be T-independent but k– would be strongly dependent on T and is larger for higher T.
Rate for base-stack formation
When a base stack is formed, the increased charge density of RNA backbone would immobilize the counterions and the solvent molecules around the base stack. The reorganization of water molecules, which results in the volume contraction, increases the order of the system and yields an entropy loss (42). So the total entropic loss −ΔS (ΔS > 0) for the formation of a base stack is equal to the sum of the contributions from the loss of conformational entropy −ΔSconf (ΔSconf > 0) and the decrease in the entropy of hydration and counterions −ΔS′ (ΔS′ > 0) (42), which is ΔS = ΔSconf + ΔS′. As a result, we can write the rate k+ in Eq. 2 in the form of
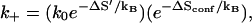 |
so k0 is larger than the effective diffusive prefactor
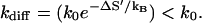 |
The rate factor 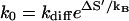 is determined not only by the solvent quality (e.g., solvent viscosity) through kdiff, but also by the hydration and the electrostatic states of the nucleotide (bases) through ΔS′. Different bases can have different hydration and ion-binding states and so can have different k0 values. For example, the GC pair and the AU pair are hydrated differently, so the GC and AU basepairs can have different k0 values.
is determined not only by the solvent quality (e.g., solvent viscosity) through kdiff, but also by the hydration and the electrostatic states of the nucleotide (bases) through ΔS′. Different bases can have different hydration and ion-binding states and so can have different k0 values. For example, the GC pair and the AU pair are hydrated differently, so the GC and AU basepairs can have different k0 values.
Rate for loop formation
According to the nearest-neighbor model, a single (unstacked) basepair is not stable (38) and has zero enthalpy (ΔH = 0). So the loop conformations closed by a single basepair are unstable and can quickly unfold (see state a in Fig. 1). As a result, because the formation of the stabilizing base stack (b → c) is much slower than the breaking of the loop (b → a),
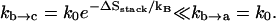 |
So the unfolded state (a in Fig. 1 A) and the (unstable) looped state (b in Fig. 1 A) can pre-equilibrate before a stable base stack (see state c in Fig. 1 A) is formed to close the loop. Here kb → c is calculated as k+ in Eq. 2, with ΔSstack equal to the entropy of the stack in state c and kb → a is calculated as k– in Eq. 2 with ΔH = 0 for the breaking of the unstacked basepair in state b.
FIGURE 1.

(A) A schematic free energy landscape for the loop formation. State a is the fully unfolded state that contains no basepair. State b contains a loop closed by a basepair. State c is a loop conformation stabilized by a closing base stack. Transitions b → c and c → b are rate-limited by an entropic barrier of TΔSstack due to the formation of the closing stack and an enthalpic barrier of ΔHstack for the breaking of the base stack, respectively. Transitions a → b and b → a are rate-limited by an entropic barrier of TΔSloop due to the formation of the loop and an enthalpic barrier of ΔHloop = 0 for the breaking of the single (unstacked) basepair, respectively. Here, according to the nearest-neighbor model, the single basepair in b does not make an enthalpic contribution (i.e., ΔHloop = 0). So the disruption of state b is very fast and the relaxation between state a and the unstable state b is fast. As a result, states a and b can have sufficient time to pre-equilibrate before the loop is stabilized through the b → c transition. (B) In the kinetic-cluster model, stabilizing the pathway conformation (in the shaded free energy landscape) concomitantly stabilizes the transition state and thus speeds up the intercluster transition. This is because the free energy difference between the transition state and the pathway conformation is equal to the barrier of the kinetic move (= TΔS and ΔH for the formation and disruption of a base stack, respectively) and is independent of the free energy of the pathway conformation. Here ΔS and ΔH are the entropy and enthalpy changes associated with the formation/disruption of the base stack.
The rate for the formation of a stabilized loop can be estimated from Fig. 1 as
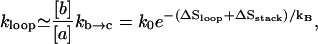 |
(3) |
where ΔSloop is the entropy loss for the loop closure in the a → b transition, and  is the relative equilibrium population between the open state a and closed state b. Eq. 3 implies that, in the present model, the rate for the formation of a stable loop is determined by not only the rate (kb → c) for the formation of the closing stack but also the stability (∼[b]/[a]) of the closed loop conformation (b in Fig. 1 A).
is the relative equilibrium population between the open state a and closed state b. Eq. 3 implies that, in the present model, the rate for the formation of a stable loop is determined by not only the rate (kb → c) for the formation of the closing stack but also the stability (∼[b]/[a]) of the closed loop conformation (b in Fig. 1 A).
Rate-limiting steps in RNA hairpin folding
The unfolding process involves the breaking of the native stacks, so the rate-limiting step of unfolding is to disrupt the slow-breaking native stacks. According to Eq. 2, the slow-breaking stacks are those with large enthalpy (ΔH).
On the other hand, the folding process involves the formation of the native stacks and the breaking of the nonnative base stacks, so the rate-limiting steps of folding are the formation of the slow-forming native stacks and the breaking of the possible slow-breaking nonnative base stacks. According to Eq. 2, the slow-forming native stacks are those with large ΔS and the slow-breaking nonnative stacks are those with large ΔH.
The rate constants for kinetic moves can only give rates for individual transitions in the folding process. The overall folding kinetics is determined by the collective and correlated events consisting of all the possible kinetic transitions (moves) in the folding process. To treat the statistics of the kinetic transitions, we discuss the following two theories used in this study: the master-equation method and the kinetic-cluster method.
Models for folding kinetics
Master-equation method
In the master-equation approach, the populational kinetics pi(t) for the ith state (i = 1, .., Ω, where Ω is the total number of chain conformations) is described as the difference between the rates for transitions entering and leaving the state,
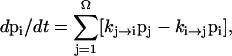 |
where kj→i and ki→j are the rate constants for the respective transitions. The above master equation has an equivalent matrix form: dp/dt = M · p, where p is the fractional populational vector col (p1, p2, … , pΩ), M is the rate matrix defined as Mij = ki→j for i ≠ j, and 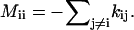 Two key issues in the master-equation method are how to compute the rate constants (ki→j and kj→i) and how to solve the master equation.
Two key issues in the master-equation method are how to compute the rate constants (ki→j and kj→i) and how to solve the master equation.
For a given initial folding condition at t = 0, by diagonalizing the rate matrix M, we have the populational kinetics p(t) for t > 0,
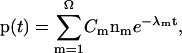 |
(4) |
where −λm and nm are the mth eigenvalue and eigenvector of the rate matrix M, and Cm is the coefficient that is dependent on the initial condition.
The eigenvalue spectrum gives the rates of the kinetic modes of the system. For a closed isolated system with the rate constants satisfying the detailed balance condition, there always exists an eigenvalue λ1 = 0 for the equilibrium mode (43). Physically, the existence of this zero eigenvalue corresponds to the fact that as t → ∞, regardless of the initial condition of the system, the system eventually relaxes to the final equilibrium state. All other λm values are negative and nonzero. The smallest nonzero |λm| gives the rate of the slowest (rate-limiting) kinetic processes. Especially, if there only one distinctively small nonzero |λm| exists in the eigenvalue spectrum, the populational kinetics p(t) would be single-exponential with the rate determined by the smallest nonzero |λm|. The eigenvectors give the basic modes of the kinetic process and are intrinsically related to the energy landscape. In fact, from the eigenvectors we can obtain the rate-limiting steps of the kinetics (36).
The great advantage of the master-equation approach is that it is based on the complete ensemble of the conformation states and accounts for the kinetic effect of each and every interconformation transition. For a given rate constant model (see Eq. 1), the master equation can give a rigorous and exact solution for the relaxation kinetics of the system. Therefore, in this study, we will use the master-equation method to test the rate constant model against the experimental data for short sequences.
The master-equation approach has its limitations. The master-equation solution can only give ensemble-averaged macroscopic kinetics and cannot give detailed information about the microscopic pathways. Moreover, RNAs are polymers, whose number of conformations (Ω) increases rapidly with the chain length (44). So the master-equation approach is limited to short sequences whose rate matrix size is not too large. Because of these reasons (and especially for the first reason), we use the kinetic-cluster method, which, as explained in the next section, can overcome the above two difficulties.
Kinetic-cluster method
The basic idea of the kinetic-cluster method is to classify the large conformational ensemble into a much reduced system of clusters (macrostates) so that the overall kinetics can be represented by the intercluster (instead of interconformation) transitions. A great advantage here is that the microscopic kinetic rates and pathways can be examined in great detail. A number of attempts have been made to model the intercluster kinetics (45–50,53). In general, there are two types of approach. In the first type of approach, the intercluster rate is computed based on the transition state with the lowest barrier (45–52). For each pair of initial and final states, the optimized pathway is used to estimate the rate constant. In the second type of approach (53), conformations that are interconvertible through barrierless transitions are classified as a cluster, and the rate constants are calculated based on all the possible intercluster kinetic pathways. Kinetic Monte Carlo simulations have also been successfully used to obtain the intercluster rate (49,54). These and other simulations (31–34) sample the kinetic paths in a stochastic way. In our present kinetic-cluster method, a collection of pre-equilibrated conformations is classified as a cluster (macrostate). Such a cluster includes the barrierless conformational cluster defined in the previous model (53) as a special subset. So the present approach is more general. From the intercluster rate constants, we construct the reduced rate matrix. From the eigenvalues and eigenvectors of the reduced rate matrix, we can analyze the rates and pathways for the folding and unfolding kinetics.
Although both the master-equation method and the present kinetic-cluster method can predict the macroscopic kinetics, and both are based on the complete conformational ensemble, the kinetic-cluster approach has the unique advantage of providing the direct information on the microscopic pathway statistics from the intercluster transitions.
The kinetic-cluster method is based on the existence of the pre-equilibrated clusters. The pre-equilibration and cluster formation have been observed in previous experiments and computer simulations (50,53,55) and the kinetic-cluster method has been rigorously validated through extensive tests against the results from the original exact master equations (37). However, the kinetic-cluster method has its limitation. The kinetic-cluster method would fail if the system does not have well-defined discrete rate-limiting steps, in which case no pre-equilibration would occur and thus no pre-equilibrated cluster would exist.
The following is a summary for the kinetic-cluster method with applications to RNA hairpin-folding kinetics.
How to classify conformations into clusters. To simplify the illustration, we use simple unfolding kinetics to show the idea. The unfolding of a hairpin involves the breaking of the native base stacks in the native hairpin structure. If the breaking of a native base stack, say, s*, is distinctively slower than the breaking of other native stacks, then the breaking of s* is the rate-limiting step for the unfolding reaction. According to the slow-breaking s*, we can define two clusters for conformations before and after the rate-limiting stack s* is broken:
 |
The existence of the rate-limiting stack s* implies that the transitions between conformations within a cluster are faster than transitions between conformations in different clusters. Therefore, conformations within a cluster may pre-equilibrate before entering a different cluster through intercluster transitions. As a result, each cluster can be treated as pre-equilibrated macrostate and the overall unfolding kinetics is determined by the slow intercluster transitions.
Two types of conformations: Pathway conformation and nonpathway conformations. For an intercluster transition, a conformation Ni in cluster N is transformed to a corresponding conformation Ui in cluster U through the breaking of the rate-limiting stack s*. There exist many such intercluster pathways between the clusters. We define pathway conformations as conformations (such as Ni in cluster N) that are directly connected to the other clusters through kinetic movement. They are called pathway conformations because they form the intercluster pathways. All the other conformations are called nonpathway conformations. We distinguish these two types of conformations because only the pathway conformations directly contribute to the intercluster kinetics.
Intercluster transition rates and the dominant pathways. The intercluster transition rate is given by the sum over all the possible microscopic pathways Ui ↔ Ni between pathway conformations in the respective clusters,
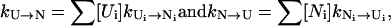 |
(5) |
where [Ui] and [Ni] are the equilibrium fractional populations of Ui and Ni in the respective clusters,
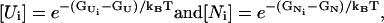 |
(6) |
where  and
and 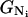 are the free energies of conformations Ui and Ni, respectively, and GU and GN are the free energies of clusters U and N, respectively,
are the free energies of conformations Ui and Ni, respectively, and GU and GN are the free energies of clusters U and N, respectively,
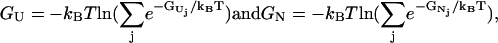 |
(7) |
where the summations in Eq. 7 are for all the (pathway and nonpathway) conformations in the respective clusters.
From the above equations for the intercluster rates, we find that the intercluster transition rates are determined by two factors: the stabilities of the pathway conformations (e.g., [Ui] and [Ni]) and the rates (e.g., 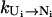 and
and 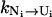 ) for each intercluster pathway. The interplay between these two factors leads to the following general conclusions for the intercluster kinetics:
) for each intercluster pathway. The interplay between these two factors leads to the following general conclusions for the intercluster kinetics:
Pathway conformations versus nonpathway conformations. For given rate constants (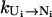 ), stabilizing the pathway conformations (versus the nonpathway conformations) speeds up the intercluster transition. As illustrated in Fig. 1 B, stabilizing the pathway conformation would concomitantly stabilize the transition state and thus speed up the transition. This is because the free energy difference between the transition state and the pathway conformation is determined by the barrier of the kinetic move, which is independent of the stability of the pathway conformation.
), stabilizing the pathway conformations (versus the nonpathway conformations) speeds up the intercluster transition. As illustrated in Fig. 1 B, stabilizing the pathway conformation would concomitantly stabilize the transition state and thus speed up the transition. This is because the free energy difference between the transition state and the pathway conformation is determined by the barrier of the kinetic move, which is independent of the stability of the pathway conformation.
Fast versus slow pathway conformations. Some pathway conformations have large transition rate 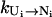 and are thus called fast pathway conformations and others are the slow pathway conformations. Stabilizing the fast pathway conformations (versus the slow pathway conformations) leads to faster intercluster transitions.
and are thus called fast pathway conformations and others are the slow pathway conformations. Stabilizing the fast pathway conformations (versus the slow pathway conformations) leads to faster intercluster transitions.
Dominant pathways. The probability for the molecule to take a specific pathway, say, Ui → Ni, is determined by its fractional contribution to the total rate:
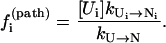 |
(8) |
We call 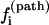 the kinetic partitioning factor or pathway partitioning probability. The pathways that have the largest
the kinetic partitioning factor or pathway partitioning probability. The pathways that have the largest 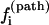 are the dominant pathways. Due to the temperature and sequence-dependence of the rate constants and of the stabilities of the pathway conformations, the dominant pathways can be quite sensitive to the temperature and the sequence. Moreover, the folding and unfolding reactions can involve different rate-limiting base stacks and can thus be described by different clusters. As a result, folding and unfolding reactions can have quite different dominant pathways.
are the dominant pathways. Due to the temperature and sequence-dependence of the rate constants and of the stabilities of the pathway conformations, the dominant pathways can be quite sensitive to the temperature and the sequence. Moreover, the folding and unfolding reactions can involve different rate-limiting base stacks and can thus be described by different clusters. As a result, folding and unfolding reactions can have quite different dominant pathways.
EXPERIMENTAL TESTS FOR THE RATE CONSTANT MODEL
There are two key issues of the present theory: the rate constant model and the kinetic-cluster method. Although the latter has been extensively validated (37), the former requires rigorous tests. The rate constant model includes two ingredients: 1), the definition of the kinetic move; and 2), the transition rate for a kinetic move (see Eq. 2). In this section, we aim to test the rate constant model through experimental comparisons. In addition, we can fit the prefactor k0 in the rate constant equation (Eq. 2) from experiments.
We use the experimentally measured parameters (ΔH, ΔS) for the base stacks (56). To compute the transition rates from Eq. 2, we need to know the prefactor k0. Due to the different energetics of the GC and AU basepairs, we assign different k0 values for stacks with GC and AU basepairs. We determine k0 by fitting the experimental data. Specifically, we use the relaxation rates for the (AnUn)2 duplex (57) and for the (A4GCU4)2 duplex (58) for AU and GC, respectively. After the prefactors are determined, we would further validate the model by applying the model to predict the relaxation rates for a wide temperature range for other hairpin-folding experiments; see Table 1.
TABLE 1.
A summary for the experiments used to test the theory
It is important to note that for simple sequences such as (AnUn)2, the k0 value serves as a global scaling factor for the overall rates. So for simple sequences (AnUn)2, k0 plays no role in determining the physical nature of the kinetics, namely, the pathways, the transition states, and temperature-dependence of the rates. For all the experimentally measured sequences that we tested, the predicted shapes of the rate versus temperature curves (equal to the Arrhenius plot) agree with the experimental results. Furthermore, the k0 parameters fitted from one experiment can give good predictions for other experiments, i.e., the fitted k0 parameters are transferable between different experiments. These results suggest that the model, including the definition of the kinetic moves and the rate constants for the kinetic moves may be valid and reliable.
In this section, where the goal is to test the rate constant model, we use the original master equation instead of the kinetic-cluster method to remove the possibility of any error caused by the kinetic-cluster method. So the tests would be exclusively focused on the rate constant model. For each sequence tested, we consider the complete ensemble of all the possible conformations and compute the interconformation rate-matrix from Eq. 2. By diagonalizing the rate matrix, we obtain the relaxation rates from the eigenvalues of the rate matrix for different temperatures.
Duplex formation
For the duplex formation, we use the zipper model. The model assumes that all the base stacks occur contiguously in a region. We use base stacks to describe the duplex structure because stacking is the major stabilizing force. Previous approach to the duplex formation is based on a prior assumption on the two states of the transition (57). A steady-state approximation was used. In the present study, we do not make a prior assumption about the two-state of the kinetics. So the model can account for any possible kinetic intermediates. The formation of the first stack of the duplex from the single strand is a second-order chemical reaction process, while the helix growth is a first-order (linear) process. Let [A0] and [AN] be the concentration of the single and the fully zipped (native) duplex with N basepairs, respectively. When we use 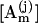 to denote the concentration of the jth state with m stacks, the rate equation can be written as
to denote the concentration of the jth state with m stacks, the rate equation can be written as
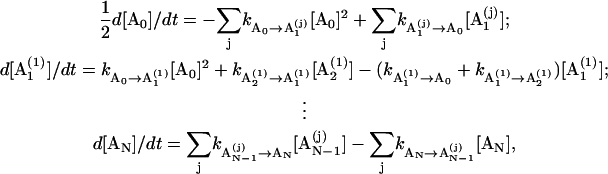 |
where ka → b denotes the a → b transition rate. Furthermore, because the temperature jump in the experiment is small (≃3 − 4°) (57), the concentration deviation from the equilibrium level is small. Therefore, we can use the following linear approximation for the concentration deviation x0 = [A0] – [A0]eq from the equilibrium value 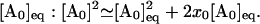 With this approximation, the above equation becomes a linear master equation for the concentration deviations of A0, AN, and
With this approximation, the above equation becomes a linear master equation for the concentration deviations of A0, AN, and 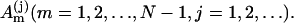
The rate for the formation of the first base stack in a contiguous helix region 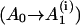 is equal to
is equal to 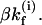 Here β is the nucleation probability, i denotes the position of the base stack, and
Here β is the nucleation probability, i denotes the position of the base stack, and 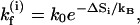 where ΔSi is entropic change for the formation of the base stack. After the first stack is formed, the growth rate (e.g.,
where ΔSi is entropic change for the formation of the base stack. After the first stack is formed, the growth rate (e.g., 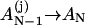 ) of a base stack j is equal to
) of a base stack j is equal to  The breaking rate for a base stack j (e.g.,
The breaking rate for a base stack j (e.g., 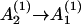 ) is equal to
) is equal to 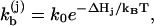 where ΔHj is enthalpic change for the formation of the base stack.
where ΔHj is enthalpic change for the formation of the base stack.
The nucleation parameter β is the probability that two bases in different strands will approach each other. The value β can be determined from the measured equilibrium constant K, which, in the zipper model, is given by
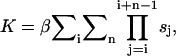 |
where i denotes the nucleating base stack, n is the length of the helix, and 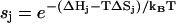 is the stability of the jth stack.
is the stability of the jth stack.
The ΔH and ΔS parameters are typically measured under 1 M NaCl condition. However, many of the experiments that we use for comparison were performed under other ionic conditions (see Table 1). For example, the measurements for the (AnUn)2 kinetics is under 0.25 M Na+ condition (57). So we need to know the ΔH and ΔS parameters under arbitrary ionic conditions. We use the following empirical ion concentration-dependent parameters, which are reliable for ion concentrations not too low (≥mM) (59):
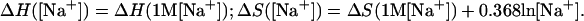 |
The lowest nonzero eigenvalue of the above linear rate equation gives the relaxation rate and the relaxation time τ = (relaxation rate)−1. Comparison between our model prediction and the (AnUn)2 experimental data gives the prefactor k0 = 6.6 × 1012 s−1 for AU stacks.
The relaxation time τ as a function of the reciprocal of temperature is shown in Fig. 2 a. The results show that the theoretical predictions are in good agreement with the experimental data for a wide temperature range and for different chain lengths. The agreement is better for longer chains. For short chains (small n), the theory-experiment difference comes from the finite size effects of the oligomers. For example, the electrostatic effect can cause nonadditivity in the base-stack stabilities (57). Moreover, the rates are overestimated for short chains due to the ignored conformations in the zipper model and the possible inaccuracy of the ion-dependence of the energy parameters used in the model.
FIGURE 2.

The temperature (T in Kelvin) dependence of the relaxation time for the duplex formation. (Symbol, experiment; line, model.) (a) (AnUn)2 (from bottom to top, n = 4, 5, 6, 7) (0.25 M Na+). (b) (A4GCU4)2 (1.05 M Na+). The experimental relaxation time are determined by 1/τ = 4k1C0 + k–1, where k1 and k–1 and C0 are from the experiment (58).
We can determine the association rate constant k1 and the dissociation rate constant k–1 for the two-state transition through the relaxation time τ and the equilibrium constant K,
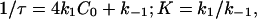 |
where 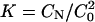 is the equilibrium constant, and CN and C0 are the measured concentrations of the duplex and monomer, respectively. Consistent with the experimental results, our model predicts that k1 is only weakly dependent on temperature T and the chain length n, and k–1 decreases as temperature T decreases or duplex length n increases (data not shown).
is the equilibrium constant, and CN and C0 are the measured concentrations of the duplex and monomer, respectively. Consistent with the experimental results, our model predicts that k1 is only weakly dependent on temperature T and the chain length n, and k–1 decreases as temperature T decreases or duplex length n increases (data not shown).
To determine the prefactor k0 for base stacks with the GC basepair, we study the kinetics for the duplex (A4GCU4)2 (58). The duplex involves both AU and GC basepairs. Given the k0 value for the AU pairs, we find that k0 is equal to k0 = 6.6 × 1013 s−1 for the GC pair. The k0 for the GC pair is larger than that for AU because a GC pair involves stronger bonding than an AU pair. In Fig. 2 b, we show the temperature-dependence of the relaxation rate for (A4GCU4)2, and we find good agreement between theory and experiment for a wide range of temperature.
Hairpin formation
Our purpose here is to have further tests on the rate constant model through experimental comparisons for the temperature-rate dependence. We use our model to make predictions for two RNA hairpin-folding experiments (58,60) that showed quite different temperature-dependence of the relaxation rate. In Fig. 3, a and b, we show the results for sequences A6C6U6 (58) at 1 M NaCl and r(AUCCUAUT4UAGGAU) (60) at 0.2 M NaCl, respectively. We find reasonably good theory-experiment agreements.
FIGURE 3.

The temperature (T in °C) dependence of the relaxation time for hairpin disruption. (Symbol, experiment; dashed line, model.) (a) A6C6U6 ((11);1 M NaCl). (b) r(AUCCUAUT4UAGGAU) ((60); 0.2 M Na+).
The eigenvalues of the rate matrix in the master equation gives the relaxation rate kr. For a two-state transition, from kr, we can obtain the folding rate kf and the unfolding rate ku separately from the following two equations: kr = kf + ku and K = kf/ku, where K is the equilibrium constant, which can be determined from our statistical thermodynamic model. At unfolding temperatures T > Tm (Tm is the folding-unfolding transition temperature), the relaxation process is predominantly an unfolding process, so 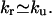 At folding temperatures T < Tm, the relaxation process is mainly a folding process, so
At folding temperatures T < Tm, the relaxation process is mainly a folding process, so 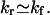
As shown in Fig. 3 a for sequence A6C6U6, the value kf increases as temperature increases, indicating a positive activation enthalpy for the folding. In our model, such positive activation enthalpy arises from the breaking of the misfolded states, which can be formed through sliding of one or more bases from the native basepair positions. Another possibility for the positive activation enthalpy of folding is for the breaking of the single-strand stacking, which is not considered in the model. In fact, neglecting the breaking of the single-strand stacking may be a reason why our predicted rate is larger than the experiment result in Fig. 3 a.
For sequence r(AUCCUAUT4UAGGAU), the T4 loop was treated as the RNA loop U4. Due to the different chain stiffness, the entropy of T4 loop may be smaller than that of U4. As a result, the rate for loop closure may be underestimated by the model; see Fig. 3 b.
GENERAL THEORY OF HAIRPIN-FOLDING KINETICS
In this section, using the kinetic-cluster method, we present a general theory for RNA hairpin-folding kinetics. We first discuss the possible slow processes of hairpin folding and the different folding kinetics scenarios with different slow processes as the rate-limiting steps for the overall folding. We then discuss the kinetic clusters and the energy landscapes for each different scenario.
Rate-limiting steps
To fold from a fully unfolded state, the hairpin forming chain undergoes the following four types of the possible slow processes (see Fig. 4):
FIGURE 4.

The four types of possible slow folding processes: (a) loop formation, (b) formation of a rate-limiting stack, (c) direct folding, the loop is closed by a rate-limiting stack (shaded) from the fully unfolded state, and (d) detrapping through disruption of the nonnative base stack (pattern-fill).
Loop nucleation (Fig. 4 a)
The formation of the first base stack can be rate-limiting because it concurrently causes the closure of a loop, and has a rate constant of
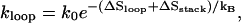 |
(9) |
where ΔSstack and ΔSloop are the entropic losses associated with the formation of the base stack and the corresponding loop. The process is slow if the total entropic loss (ΔSloop + ΔSstack) is large.
Formation of the rate-limiting stack (Fig. 4 b)
If a native base stack s* exists, whose entropy ΔS* is exceedingly larger than that of other native stacks, according to Eq. 2, the formation of s* would be exceedingly slower than the formation of other native stacks. As a result, the formation of s*, which has a rate constant of
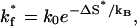 |
(10) |
is a rate-limiting step for the overall folding process.
Direct folding (Fig. 4 c)
In the loop nucleation process, if the loop is closed by a rate-limiting stack s* from the fully unfolded state, the loop closing process would be extremely slow with a rate constant of
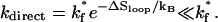 |
(11) |
The contribution from such process to the overall folding kinetics can often be ignored due to the extremely slow rate.
Detrapping (Fig. 4 d)
In the folding reaction, nonnative base stacks formed in the process need to be disrupted for the folding process to proceed. The disruption of nonnative base stacks (detrapping) has a rate constant of
 |
(12) |
where ΔHnn is the enthalpy cost to break the nonnative base stack. The detrapping process can be slow for large ΔHnn or low temperature T. The process can become a rate-limiting step if kdetrap is small.
Kinetic cluster
According to the above possible slow steps, we can classify the conformational ensemble into five clusters, C, In, Inn, Nn, and Nnn, such that
 |
(13) |
In and Inn are the conformations in cluster I without and with nonnative base stacks, respectively, and Nn and Nnn are the conformations in cluster N with and without nonnative base stacks, respectively. Fig. 5 summarizes the kinetic-cluster classifications. In Fig. 5 a, F = I + N is the cluster for all the folded and partially folded conformations and U = C + I is the cluster for all the conformations with the rate-limiting stack s* disrupted.
FIGURE 5.

Summary of the two types of kinetic-cluster classifications.
By defining the nonnative clusters Inn and Nnn, we neglect the heterogeneity of the non-native stacks by grouping different nonnative states into the same cluster. This assumption may cause an overestimation for the folding rate due to the neglected transition time between different nonnative conformations within clusters Inn and Nnn. The overestimation for the rate is negligible for larger kdetrap when the disruption of the nonnative stacks is fast and can become important if kdetrap is small. Nevertheless, the separation of nonnative Inn from the nativelike In and nonnative Nnn from native Nn can, to the lowest-order approximation, account for the trapping effect in the folding process.
Energy landscapes
The competition between the loop nucleation (kloop), the formation of the rate-limiting (native) stack (kf*), and the detrapping of the nonnative stacks (kdetrap) results in a great variety of different scenarios for the energy landscapes and folding kinetics (see Fig. 6). We can classify the folding landscapes and folding kinetics into the following different scenarios; see Table 2 for a summary.
FIGURE 6.

Schematic free energy landscapes for different folding scenarios. The shaded line shows the formation of the native-like looped structures In from the unfolded state C. Inn represents the non-native intermediate state. (a) Scenario 1: The rate-limiting process is the loop formation. The folding is a two-state (C → F = I + N). (b) Scenario 2: The rate-limiting process is the formation of a rate-limiting base stack. In and Inn equilibrate quickly and form a pre-equilibrate macrostate (cluster) I. The folding is a two-state process U (= C + I) → N. (c) Scenario 3: Weakly noncooperative folding with a rate-limiting stack and a (or few) slow-breaking misfolded nonnative base stacks. The kinetics is multi-state (five clusters: C, In, Inn, Nn, Nnn). N = Nn + Nnn (Nn and Nnn not shown). (d) Scenario 4: Strongly noncooperative folding. The disruption of an average nonnative stack is very slow and nearly every non-native state can form a kinetic trap. See also Table 2.
TABLE 2.
A summary for the different scenarios of the folding kinetics
| Scenario | Energy landscape | Rate-limiting step | Cooperativity | Temperature |
|---|---|---|---|---|
| 1 | Fig. 5 a | Loop formation | Two-state C → F (= I + N) | T > Tr |
| 2 | Fig. 5 b | Formation of the rate-limiting native base stack s* | Two-state U (= C + I) → N | T > Tr |
| 3 | Fig. 5 c | Formation of s* and detrapping of the nonnative states | Multi-state C, In, Inn, Nn, Nnn | T ≤ Tr |
| 4 | Fig. 5 d | Rate-limiting steps not discrete | Glassy | T < Tr |
Cooperative folding (if detrapping is fast)
The folding is cooperative (two-state) and single-exponential if the detrapping is much faster than both the loop nucleation and the formation of the rate-limiting stack:
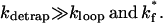 |
(14) |
If kdetrap is large, detrapping of nonnative base stacks is not a slow process. As a result, In and Inn, which are separated by the slow-breaking nonnative stacks, can equilibrate quickly. As a result, they can merge to form larger pre-equilibrated cluster I. For the same reason, Nn and Nnn can be merged into a larger cluster N. The overall conformation ensemble can be classified into three clusters: C, I, and N (see Fig. 5 b). According to the competition between the loop nucleation and the formation of the rate-limiting stack, we can further distinguish two different folding scenarios:
Scenario 1: Cooperative folding through loop formation (if the formation of the native base stacks is fast). If the formation of the native base stacks is faster than the loop formation,
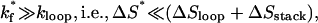 |
(15) |
no rate-limiting (native) base-stack s* exists. As a result, cluster I and N, which are separated by the possible slow-forming stacks s*, would quickly pre-equilibrate and form cluster F = I + N (see Fig. 5 a). The overall conformational ensemble is classified into two clusters, C and F, and the folding is a two-state transition: C → F corresponding to the loop formation (see Fig. 6 a). In fact, for most of the previous RNA and DNA hairpin-folding experiments (11), there is no rate-limiting (native) base stack, and the folding reaction C → F is rate-limited by the loop nucleation.
Microscopically, the transition C → F can be understood as the nonspecific formation of the loops. Once the looped conformations are formed, they can quickly interconvert to form the stable native structure through the fast breaking/formation of base stacks and the accompanying changes of the loops (e.g., through the chain sliding modes). Since the loop entropy is a logarithmic function of the loop size, which is not sensitive to the loop size change, the transitions between different looped structures within cluster F are not rate-limited by the loop changes, and are thus, fast.
Scenario 2: Cooperative folding through formation of the rate-limiting base stack (if loop formation is fast). If the slowest kinetic move is to form native base stack s*,
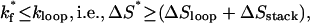 |
(16) |
then stack s* is major divider to separate different clusters and the conformational ensemble is reduced to a two-cluster system: U (= C + I) and N (see Fig. 5 b). Cluster U is the collection of conformations without the slow-forming s* (see Fig. 6 b), and the folding kinetics is two-state: U → N corresponding to the slow formation of s*.
Starting from the fully unfolded conformation C, the chain initially undergoes transition C → I to form cluster U. Because many different ways exist to form the nonspecific loops from the fully unfolded state, the C → I process is fast even if kloop is not much larger than kf*.
After initial fast formation of cluster U, the chain undergoes slow transition U → N to form the rate-limiting stack s*. Some conformations in U may be transiently populated at this stage because they have large (fractional) equilibrium population in U but not in the final overall conformational ensemble U + N. These conformations are kinetic intermediates. How do the emergence of the intermediate states and the two-state cooperativity compromise with each other? Since the different conformations, including the kinetic intermediates, in the cluster U have already reached equilibrium, transitions between conformations in U do not contribute to the folding time. Equivalently speaking, conformations in U act as an effective single state for the folding kinetics. Therefore, regardless of the existence of the kinetic intermediates, the resultant folding kinetics is two-state (U → N) and single-exponential.
Noncooperative folding (if detrapping is slow)
Scenario 3: Weakly noncooperative folding kinetics. If one or only a few nonnative stacks exist whose disruption rate kdetrap is slow (i.e., comparable to kf*), the folding would be (weakly) noncooperative. For example, if one slow-disruption nonnative base stack exists, we can describe the kinetics using five clusters (C, In, Inn, Nn, Nnn in Fig. 5 b; see Fig. 6 c for a schematic plot of the free energy landscape). The resultant kinetics would be multi-state and multi-exponential.
The kinetic traps would form kinetic intermediates in the folding process. These intermediates are different from the intermediates that emerged in the two-state cooperative folding process. The kinetic traps in the noncooperative kinetics prevent the formation of the pre-equilibrated cluster I (I = In + Inn), while the intermediates in the cooperative kinetics are the result of the pre-equilibration of the cluster U(= C + I).
Scenario 4: Strongly noncooperative folding kinetics. The folding kinetics becomes strongly noncooperative if the detrapping from an average nonnative stack is slow: 〈kdetrap〉 ≤ kf*, where 〈…〉 denotes the average over all the possible nonnative base stacks. See Fig. 6 d for a schematic free energy landscape. When the above condition is satisfied, on average, each individual nonnative state should be treated as a separate cluster. As a result, the use of In, Inn, Nn, Nnn becomes invalid and the resultant folding kinetics is strongly multi-state (noncooperative) and strongly multiple-exponential.
Folding rate-temperature dependence
In this and the next section, we use a 21-nt sequence UAUAUCGC7CGAUAUA as an example to illustrate the complex hairpin-folding kinetics using the kinetic-cluster theory. From the previously reported statistical thermodynamic statistical model (41), we find the folding/unfolding melting temperature Tm≃50°C for this sequence.
As shown in Fig. 7 b, from the large (ΔS, ΔH) parameters, the formation and disruption of the native base stack s* = (5,6,16,17) = (U,C,G,A) are rate-limiting for the folding and unfolding kinetics due to large ΔS = 35.5 eu and ΔH = 13.3 kcal/mol, respectively. The rate constants for the formation and disruption of s* are given by Eq. 2: 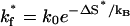 =1.29×106s−1and
=1.29×106s−1and which is equal to 3.19 × 104 s−1 at T = 37°C.
which is equal to 3.19 × 104 s−1 at T = 37°C.
FIGURE 7.

(a) Temperature (T in °C) dependence of the relaxation rate for a hairpin-forming sequence UAUAUC- GC7CGAUAUA. (Solid line) From the original complete conformational ensemble (master-equation method); (dashed line) from the two-cluster model (Scenario 2); and (dotted line) from the five-cluster model (Scenario 3). (b) The native structure and energy parameters of the native state. The shaded stack is the rate-limiting stack. (c) The pathway conformations Ui and Ni (i = 1, 2, … , 20) in the respective clusters U and N and the corresponding intercluster pathways and rates. U1 → N1 is the direct folding pathway through the loop closing by the rate-limiting stack. N19 → U19 is a double-breaking pathway that involves the simultaneous breaking of two stacks (one of which is the rate-limiting stack). U4 is a nonnative pathway conformation because it contains a nonnative base stack.
Plotted in Fig. 7 a is the temperature-dependence of the relaxation rate kr for the sequence. The predicted kr-T relationship (Arrhenius plot) is consistent with the experimental findings for some DNA hairpins (21). The relaxation rate kr is related to the folding rate kf and the unfolding rate ku through the equation kr = kf + ku for a two-state transition. The kinetics is dominated by the folding reaction for T < Tm and kr≃kf and by the unfolding reaction for T > Tm and kr≃ku. The Arrhenius plot in Fig. 7 a has two characteristic temperatures: Tm≃50°C and a rollover temperature Tr≃10°CC. According to Tm and Tr, the kr-T relationship can be classified into three regimes:
Unfolding at T > Tm: kr ∼ ku increases as T is increased because the rate for breaking the stack ∼kb* is larger for higher T.
Folding at temperature T between Tr and Tm: kr ∼ kf increases as T decreases, indicating a negative apparent activation energy.
Folding at low temperature T < Tr: kr ∼ kf decreases as T decreases, indicating a positive activation energy.
What causes the rollover of the rate-temperature dependence plot? How to predict the rollover temperature Tr from the microscopic energetics? In this section, we explore the connections between the macroscopic Arrhenius plot and the microscopic kinetic clusters.
Since the formation of s* is the sole dominant rate-limiting step, we can classify the conformational ensemble into two clusters U and N; see Fig. 5 b. For this 21-nt sequence, there are a total of 20 hairpin conformations in cluster U to which the rate-limiting stack s* can be added through a kinetic move. These 20 conformations (Ui, i = 1, 2, …20; see Fig. 7 c) are the pathway conformations in cluster U. There are 20 corresponding intercluster pathways (see Fig. 7 c). Among the 20 pathways, U1 (= C) → N1 is through the direct folding and is extremely slow (see Eq. 11). The rest of the 19 pathways are fast-folding pathways (see Fig. 7 c).
The temperature-dependent competition between the stabilities of the following three types of conformations leads to the complex temperature-dependence of the folding rate: 1), the fast-folding (U2, …, U20); 2), the slow-folding (U1) pathway conformations; and 3), the nonpathway conformations in cluster U. For the given sequence, as shown in Fig. 8 a, a decrease in temperature causes two competing effects:
The fast-folding pathway conformations U2, …, U20 are stabilized and the slow-folding conformation U1 are relatively destabilized, causing a faster folding rate.
The nonpathway (nonnative) states in U are stabilized, causing a decrease in the total population of the pathway conformations in cluster U and hence a slower folding rate.
FIGURE 8.

(a) The populational distribution of the pathway conformations Ui → Ni for i = 1, 2, …, 20 (solid for T = 30°C and shaded for T = 90°C). The total faction population of the nonpathway conformation is listed as the state 21 in the figure. (b) The probability 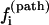 for the molecule to fold along Ui → Ni (i = 1, 2, …20) (solid for T = 30°C and shaded for T = 90°C). As temperature T is increased from 30°C to 90°C, the populational distribution is shifted from the fast-folding conformation (i = 20) to the slow-folding conformation (i = 1), causing a slow folding process.
for the molecule to fold along Ui → Ni (i = 1, 2, …20) (solid for T = 30°C and shaded for T = 90°C). As temperature T is increased from 30°C to 90°C, the populational distribution is shifted from the fast-folding conformation (i = 20) to the slow-folding conformation (i = 1), causing a slow folding process.
For the given sequence, the former effect dominates and thus the folding is accelerated at lower temperatures.
As the temperature is further lowered, at a critical temperature Tr, the Arrhenius plot shows a rollover behavior, i.e., the rate decreases as temperature is decreased. Two possible mechanisms for the rollover exist:
In the cooperative folding regime (Scenario 2), the rollover can be caused by the stabilization of the nonpathway conformations or the slow-folding pathway conformations in cluster U, which leads to a slower (instead of faster) folding rate at lower T.
In a noncooperative folding process (Scenarios 3 and 4), the rollover can be caused by the formation of kinetic traps. In this case, as temperature is lowered, the detrapping rate kdetrap decreases, resulting in a decrease in the overall folding rate. In this case, rollover behavior corresponds to the transition from the cooperative to the noncooperative folding kinetics and Tr is determined by the condition for Scenario 3 to occur, which is kdetrap ∼ kf*. For the given sequence, we find Tr = 10°C.
As shown in Fig. 7 a, the two-state model cannot account for the rollover behavior. Only after we introduce the five-cluster model (C, In, Inn, Nn, Nnn) in Scenario 3 can we obtain the rollover. This clearly shows that the rollover for this sequence is caused by the second mechanism above. The strongly multi-state (and glassy) folding kinetics occurs at even lower temperatures.
Folding pathways
We discuss the folding pathways for three typical temperatures below. Fig. 8 shows the plot of the kinetic pathway partitioning probability 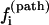 defined in Eq. 8 for all the 20 U → N pathways in Fig. 7 c. The dominant pathways have the largest
defined in Eq. 8 for all the 20 U → N pathways in Fig. 7 c. The dominant pathways have the largest 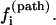
Cooperative folding at high temperature T = 90°C (T > Tm; Scenario 2). We find that the main folding pathways are U1 → N1 and U2 → N2, each contributing ∼14.5% and 51.6% to the total folding rate, respectively. This is because the populations of U2 and U1 overwhelmingly dominate the population in cluster U.
Cooperative folding at a lower temperature T = 30°C (Tr < T < Tm; Scenario 2). From Fig. 8 b we find the dominant folding pathway is U20 → N20. Approximately
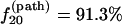 of the population in U folds along this pathway. This is because of U20 is the most stable fast-folding pathway conformation for T < Tm. As the temperature is increased, the population of the slow-folding (fully unfolded state) U1 increases and that of the fast-folding state U20 decreases, so the folding rate decreases.
of the population in U folds along this pathway. This is because of U20 is the most stable fast-folding pathway conformation for T < Tm. As the temperature is increased, the population of the slow-folding (fully unfolded state) U1 increases and that of the fast-folding state U20 decreases, so the folding rate decreases.Noncooperative folding at T = 10°C (T < Tr; Scenario 3). The folding kinetics can be described by the transitions between five clusters: C, In, Inn, Nn, Nnn. From the unfolded state C, there are a large number of pathways to form a nonnative state in Inn, so the chain would quickly fold to Inn from C and Inn becomes a kinetic trap. This is confirmed in the results of populational kinetics (data not shown). The later stage of the folding involves the detrapping from Inn and the formation of the rate-limiting stack s*. If detrapping is faster, the chain would first detrap, then form s*. As the temperature decreases, detrapping slows down and may eventually becomes slower than the formation of s*, which is T-independent in the model. So in such low T case, the chain would first form s*, then detrap to break the nonnative stacks.
SUMMARY
The kinetic-cluster approach allows us to study the kinetic rates, rate-limiting steps, and the pathways for biologically significant RNA hairpins. The overall hairpin-folding process can be rate-limited by the formation of the loop, the formation of the slow-forming native base stack, and the disruption of the slow-breaking nonnative base stack. The competition between these different processes leads to the great wealth of different RNA hairpin-folding kinetic scenarios. The detailed folding kinetics is sequence-specific. For many sequences, there exists a temperature Tr such that for T < Tr the folding rate decreases as the temperature is decreased because of the decreased rate for breaking a misfolded nonnative base stack or the increased stability of the (misfolded) nonpathway conformations. For T > Tr, however, depending on the competition between the stabilities of the fast and the slow pathway conformations and the nonpathway conformations, as the temperature is decreased, the folding rate can either increase if the fast pathway conformations are stabilized or decrease if otherwise.
As a caveat, we note several limitations of the model. For chain length longer than 30-nt, depending on the nucleotide sequence, the chain could possibly form complex branched secondary structures. Because the branched nonhairpin structures are not considered in the hairpin-folding theory, we cannot arbitrarily generalize the conclusions found in this study to the more complex, branched RNA structures of longer chains. Another limitation is that the present model does not consider the kinetic effect of the formation and breaking of single-strand stacking (23,61). Therefore, the model may not be valid for sequences (e.g., Adenine-rich sequences) that have a high tendency to form single-strand stacking in the unfolded state. For these sequences, to break the single-strand stacking in the folding process may involve a large kinetic barrier, which is neglected in the present model. A third limitation is that the model does not account for the temperature-dependence of the rate constant (prefactor) k0, which is dependent on the solvent viscosity and the hydration and electrostatic state of the bases. We assume that k0 has weaker temperature-dependence than the stability factor (e.g., 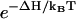 ). Moreover, the temperature-dependence of the (ΔH, ΔS) parameters (i.e., ΔCp) is neglected for the base stacks.
). Moreover, the temperature-dependence of the (ΔH, ΔS) parameters (i.e., ΔCp) is neglected for the base stacks.
Acknowledgments
We are grateful to Drs. Anjum Ansari and Herve Isambert for useful discussions.
This research was supported by the National Institutes of Health (NIH/NIGMS) through grant No. GM063732 (to S.-J.C.).
References
- 1.Bartley, L. E., X. Zhuang, R. Das, S. Chu, and D. Herschlag. 2003. Exploration of the transition state for tertiary structure formation between an RNA helix and a large structure RNA. J. Mol. Biol. 328:1011–1026. [DOI] [PubMed] [Google Scholar]
- 2.Treiber, D. K., and J. R. Williamson. 2001. Beyond kinetic traps in RNA folding. Curr. Opin. Struct. Biol. 11:309–314. [DOI] [PubMed] [Google Scholar]
- 3.Woodson, S. A. 2000. Recent insights on RNA folding mechanisms from catalytic RNA. Cell. Mol. Life Sci. 57:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brenowitz, M., M. R. Chance, G. Dhavan, and K. Takamoto. 2002. Probing the structural dynamics of nucleic acids by quantitative time-resolved and equilibrium hydroxyl radical “footprinting”. Curr. Opin. Struct. Biol. 12:648–653. [DOI] [PubMed] [Google Scholar]
- 5.Thirumalai, D., N. Lee, S. A. Woodson, and D. K. Klimov. 2001. Early events in RNA folding. Annu. Rev. Phys. Chem. 52:751–762. [DOI] [PubMed] [Google Scholar]
- 6.Sosnick, T. R., and T. Pan. 2003. RNA folding: models and perspectives. Curr. Opin. Struct. Biol. 13:309–316. [DOI] [PubMed] [Google Scholar]
- 7.Uhlenbeck, O. C. 1990. Nucleic-acid structure—tetraloops and RNA folding. Nature. 346:613–614. [DOI] [PubMed] [Google Scholar]
- 8.Herschlag, D., and T. R. Cech. 1990. Catalysis of RNA cleavage by the Tetrahymena thermophila ribozyme. I. Kinetic description of the reaction of an RNA substrate complementary to the active site. Biochemistry. 29:10159–10171. [DOI] [PubMed] [Google Scholar]
- 9.Herschlag, D., and T. R. Cech. 1990. Catalysis of RNA cleavage by the Tetrahymena thermophila ribozyme. II. Kinetic description of the reaction of an RNA substrate that forms a mismatch at the active site. Biochemistry. 29:10172–10180. [DOI] [PubMed] [Google Scholar]
- 10.van Duin, J. 1988. Single-stranded RNA bacteriophages. In The Bacteriophages, Vol. 1. R. Calendar, editor. Plenum Press, New York. 117–167.
- 11.Porschke, D. 1974. Thermodynamic and kinetic parameters of an oligonucleotide hairpin helix. Biophys. Chem. 1:381–386. [DOI] [PubMed] [Google Scholar]
- 12.Hall, K. B., and J. Williams. 2004. Dynamics of the IRE RNA hairpin loop probed by 2-aminopurine fluorescence and stochastic dynamics simulations. RNA. 10:34–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jean, J. M., and K. B. Hall. 2001. 2-Aminopurine fluorescence quenching and lifetimes: role of base stacking. Proc. Natl. Acad. Sci. USA. 98:37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall, K. B., and C. G. Tang. 1998. C-13 relaxation and dynamics of the purine bases in the iron responsive element RNA hairpin. Biochemistry. 37:9323–9332. [DOI] [PubMed] [Google Scholar]
- 15.Proctor, D. J., H. Ma, E. Kierzek, R. Kierzek, M. Gruebele, and P. C. Bevilacqua. 2004. Folding thermodynamics and kinetics of YNMG RNA hairpins: specific incorporation of 8-bromoguanosine leads to stabilization by enhancement of the folding rate. Biochemistry. 43:14004–14014. [DOI] [PubMed] [Google Scholar]
- 16.Liphardt, J., B. Onoa, S. B. Smith, I. J. Tinoco, and C. Bustamante. 2001. Reversible unfolding of single RNA molecules by mechanical force. Science. 292:733–737. [DOI] [PubMed] [Google Scholar]
- 17.Munoz, V., P. A. Thompson, J. Hofrichter, and W. A. Eaton. 1997. Folding dynamics and mechanism of β-hairpin formation. Nature. 390:196–199. [DOI] [PubMed] [Google Scholar]
- 18.Bonnet, G., O. Krichevsky, and A. Libchaber. 1998. Kinetics of conformational fluctuations in DNA hairpin-loops. Proc. Natl. Acad. Sci. USA. 95:8602–8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ansari, A., S. V. Kunznetsov, and Y. Shen. 2001. Configurational diffusion down a folding funnel describes the dynamics of DNA hairpins. Proc. Natl. Acad. Sci. USA. 98:7771–7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuznetsov, S. V., Y. Shen, A. S. Benight, and A. Ansari. 2001. A semiflexible polymer model applied to loop formation in DNA hairpins. Biophys. J. 81:2864–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallace, M. I., L. Ying, S. Balasubramanian, and D. Klenerman. 2001. Non-Arrhenius kinetics for the loop closure of a DNA hairpin. Proc. Natl. Acad. Sci. USA. 98:5584–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallace, M. I., L. Ying, S. Balasubramanian, and D. Klenerman. 2000. FRET fluctuation spectroscopy: exploring the conformational dynamics of DNA hairpin loop. J. Phys. Chem. B. 104:11551–11555. [Google Scholar]
- 23.Goddard, N. L., G. Bonnet, O. Krichevsky, and A. Libchaber. 2000. Sequence-dependent rigidity of single-stranded DNA. Phys. Rev. Lett. 85:2400–2403. [DOI] [PubMed] [Google Scholar]
- 24.Shen, Y., S. V. Kunznetsov, and A. Ansari. 2001. Loop dependence of the dynamics of DNA hairpins. J. Phys. Chem. B. 105:12202–12211. [Google Scholar]
- 25.Onuchic, J. N., Z. Luthey-Schulten, and P. G. Wolynes. 1997. Theory of protein folding: the energy landscape perspective. Ann. Rev. Phys. Chem. 48:545–600. [DOI] [PubMed] [Google Scholar]
- 26.Dill, K. A., and H. S. Chan. 1997. From Levinthal to pathways to funnels. Nat. Struct. Biol. 4:10–19. [DOI] [PubMed] [Google Scholar]
- 27.Zhang, W. B., and S. J. Chen. 2002. RNA hairpin folding kinetics. Proc. Natl. Acad. Sci. USA. 99:1931–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorin, E. J., B. J. Nakatani, Y. M. Rhee, G. Jayachandran, V. Vishal, and V. S. Pande. 2004. Does native state topology determine the RNA folding mechanism? J. Mol. Biol. 337:789–797. [DOI] [PubMed] [Google Scholar]
- 29.Sorin, E. J., Y. M. Rhee, B. J. Nakatani, and V. S. Pande. 2003. Insights into nucleic acid conformational dynamics from massively parallel stochastic simulations. Biophys. J. 85:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorin, E. J., M. A. Engelhardt, D. Herschlag, and V. S. Pande. 2002. RNA simulations: probing hairpin unfolding and the dynamics of a GNRA tetraloop. J. Mol. Biol. 317:493–506. [DOI] [PubMed] [Google Scholar]
- 31.Flamm, C., W. Fontana, I. L. Hofacker, and P. Schuster. 2000. RNA folding at elementary step resolution. RNA. 6:325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isambert, H., and E. D. Siggia. 2000. Modeling RNA folding paths with pseudoknots: application to hepatitis δ-virus ribozyme. Proc. Natl. Acad. Sci. USA. 97:6515–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gultyaev, A. P., F. H. van Batenburg, and C. W. Pleij. 1995. The computer-simulation of RNA folding pathways using a genetic algorithm. J. Mol. Biol. 250:37–51. [DOI] [PubMed] [Google Scholar]
- 34.Galzitskaya, O., and A. V. Finkelstein. 1998. Folding rate dependence on the chain length for RNA-like heteropolymers. Fold. Des. 3:69–78. [DOI] [PubMed] [Google Scholar]
- 35.Cocco, S., J. F. Marko, and R. Monasson. 2003. Slow nucleic acid unzipping kinetics from sequence-defined barriers. Eur. Phys. J. E. 10:153–161. [DOI] [PubMed] [Google Scholar]
- 36.Zhang, W. B., and S. J. Chen. 2003. Master equation approach to finding the rate-limiting steps in biopolymer folding. J. Chem. Phys. 118:3413–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang, W. B., and S. J. Chen. 2003. Analyzing the biopolymer folding rates and pathways using kinetic cluster method. J. Chem. Phys. 119:8716–8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner, D. H., N. Sugimoto, and S. M. Freier. 1988. RNA structure prediction. Annu. Rev. Biophys. Biophys. Chem. 17:167–192. [DOI] [PubMed] [Google Scholar]
- 39.Borer, P. N., B. Dengler, I. Tinoco, Jr., and O. C. Uhlenbeck. 1974. Stability of Ribonucleic-acid double-stranded helices. J. Mol. Biol. 86:843–853. [DOI] [PubMed] [Google Scholar]
- 40.Turner, D. H., N. Sugimoto, R. Kierzek, and D. D. Dreiker. 1987. Free energy increments for hydrogen-bonds in nucleic-acid basepairs. J. Am. Chem. Soc. 109:3783–3785. [Google Scholar]
- 41.Chen, S. J., and K. A. Dill. 2000. RNA folding energy landscapes. Proc. Natl. Acad. Sci. USA. 97:646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marky, L. A., and D. W. Kupke. 2000. Enthalpy-entropy compensation in nucleic acids: contribution of electrostriction and structure hydration. Methods Enzymol. 323:419–441. [DOI] [PubMed] [Google Scholar]
- 43.van Kampen, N. G. 1981. Stochastic Processes in Physics and Chemistry. North-Holland, Amsterdam, The Netherlands.
- 44.Flory, P. J. 1969. Statistical Mechanics of Chain Molecules. Wiley, New York.
- 45.Czerminski, R., and R. Elber. 1990. Reaction-path study of conformational transitions in flexible systems—applications to peptides. J. Chem. Phys. 92:5580–5601. [Google Scholar]
- 46.Becker, O. M., and M. Karplus. 1997. The topology of multidimensional potential energy surfaces: theory and application to peptide structure and kinetics. J. Chem. Phys. 106:1495–1517. [Google Scholar]
- 47.Krivov, S. V., and M. Karplus. 2002. Free energy disconnectivity graphs: application to peptide models. J. Chem. Phys. 117:10894–10903. [Google Scholar]
- 48.Evans, D. A., and D. J. Wales. 2003. The free energy landscape and dynamics of Met-encephalin. J. Chem. Phys. 119:9947–9955. [Google Scholar]
- 49.Evans, D. A., and D. J. Wales. 2004. Folding of the GB1 hairpin peptide from discrete path sampling. J. Chem. Phys. 121:1080–1090. [DOI] [PubMed] [Google Scholar]
- 50.Konishi, Y., T. Ooi, and H. A. Scheraga. 1982. Regeneration of ribonuclease A from the reduced protein rate-limiting steps. Biochemistry. 21:4734–4740. [DOI] [PubMed] [Google Scholar]
- 51.Cieplak, M., M. Henkel, J. Karbowski, and J. R. Banavar. 1998. Master-equation approach to protein folding and kinetic traps. Phys. Rev. Lett. 80:3654–3657. [Google Scholar]
- 52.Ozkan, S. B., I. Bahar, and K. A. Dill. 2001. Transition states and the meaning of Φ-values in protein folding kinetics. Nat. Struct. Biol. 8:765–769. [DOI] [PubMed] [Google Scholar]
- 53.Chan, H. S., and K. A. Dill. 1994. Transition states and folding dynamics of proteins and heteropolymers. J. Chem. Phys. 100:9238–9257. [Google Scholar]
- 54.Makarov, D. E., P. K. Hansma, and H. Metiu. 2001. Kinetic Monte Carlo simulation off titin unfolding. J. Chem. Phys. 114:9663–9673. [Google Scholar]
- 55.Ulitsky, A., and D. Shalloway. 1998. Variational calculation of macrostate transition rates. J. Chem. Phys. 109:1670–1686. [Google Scholar]
- 56.Serra, M. J., and D. H. Turner. 1995. Predicting thermodynamic properties of RNA. Methods Enzymol. 259:242–261. [DOI] [PubMed] [Google Scholar]
- 57.Craig, M. E., D. M. Crothers, and P. Doty. 1971. Relaxation kinetics of dimer formation by self-complementary oligonucleotides. J. Mol. Biol. 62:383–401. [DOI] [PubMed] [Google Scholar]
- 58.Porschke, D., O. C. Uhlenbeck, and F. H. Martin. 1973. Thermodynamics and kinetics of the helix-coil transitions of oligomers containing GC pairs. Biopolymers. 12:1313–1335. [Google Scholar]
- 59.SantaLucia, J. J. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA. 95:1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hilbers, C. W., C. A. G. Hassnoot, S. H. de Bruin, J. J. M. Joordens, G. A. Van Der Marel, and J. H. Van Boom. 1985. Hairpin formation in synthetic oligonucleotide. Biochimie. 67:685–695. [DOI] [PubMed] [Google Scholar]
- 61.Aalberts, D. P., J. M. Parman, and N. L. Goddard. 2003. Single-strand stacking free energy from DNA beacon kinetics. Biophys. J. 84:3212–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]