

Abstract
Human papillomaviruses (HPVs) cause a number of human tumors and malignancies, including cervical cancers. Epithelial differentiation is required for the complete HPV life cycle and can be achieved using the organotypic (raft) culture system. The CIN-612 9E cell line maintains episomal copies of HPV type 31b (HPV31b), an HPV type associated with cervical cancers. When grown in the raft system, CIN-612 9E cells form a differentiated epithelium such that infectious virions can be synthesized. Many aspects of the later stages of the HPV31b life cycle have been investigated in CIN-612 9E raft tissues. We used a biologically contained homogenization system for efficient virion extraction from raft epithelial tissues. Purified HPV31b virions were used to infect low-passage-number human foreskin keratinocytes and a variety of epithelial cell lines. Newly synthesized, spliced HPV31b transcripts were detected by reverse transcription and PCR (RT-PCR) following HPV31b infection. HPV31b infection was most efficient and reproducible in HaCaT cells. The onset of viral transcription following infection was also investigated using RT-PCR techniques. Spliced E1∗I,E2 RNAs were present as early as 4 h postinfection (p.i.), whereas the other major viral transcripts were detected by 8 to 10 h p.i. Furthermore, we characterized the structures and temporal expression of seven novel spliced early transcripts expressed following infection.
Papillomaviruses (PVs) are small DNA viruses that display remarkable species specificity and strong cellular tropism. These viruses produce benign and malignant tumors in their natural hosts (reviewed in reference 25). Humans are the sole hosts for human PVs (HPVs), and productive infections are restricted to cutaneous and mucosal epithelial tissues. Complete genomes have been cloned for over 85 types of HPVs, and an additional 130 types have been partially characterized by PCR techniques (8). Approximately one-third of HPV types can infect the anogenital tract. HPV type 16 (HPV16), HPV18, HPV31, HPV33, and HPV45 are known as high-risk anogenital viruses (reviewed in reference 26). These along with other less common high-risk types are involved in more than 99% of all cervical malignancies (57, 60). The capacity of HPVs to cause malignancies can be partially attributed to their ability to establish a persistent infection in host epithelial cells (18).
Complete HPV life cycles are dependent upon epithelial differentiation (4, 25, 32, 61). HPV infection of the mitotically active basal cells is generally thought to be necessary for the establishment of viral persistence. However, the late stages of viral DNA (vDNA) amplification and capsid gene expression require differentiation of infected epithelial cells (2, 14, 32). The organotypic (raft) tissue culture system supports epithelial differentiation and the HPV life cycle, allowing infectious virions to be purified from infected cells (31, 33, 35).
HPV31b is a prototype for high-risk HPVs. The study of the differentiation-dependent life cycle of HPV31b has been studied using latently infected CIN-612 9E cells in methylcellulose suspension and raft tissue culture systems (reviewed in references 38 and 50). A reverse-genetics system for HPVs has been recently developed and is adding to our understanding of these viruses (13, 33). These advances have permitted the characterization of many aspects of the latter phases of the differentiation-dependent HPV life cycle, including the spatial expression of viral proteins (14, 28, 32, 41), the regulation and structural characterization of viral transcripts (21-24, 36, 37, 39, 40, 48, 49, 51-54), and the replication of vDNA (2, 19, 20, 37, 55). We recently reported a biologically contained method for the isolation of large quantities of infectious HPV virions and showed that the immortalized human HaCaT keratinocyte cell line is susceptible to HPV31b infection (35). However, early events in HPV infection of host epithelial cells have yet to be characterized in detail.
In the present study, we used a stock of infectious HPV31b virions purified from the raft tissue culture system to test the susceptibility of various undifferentiated epithelial monolayer cells to HPV31b infection. We then performed a temporal analysis to determine the onset of HPV31b early transcription following epithelial cell infection. The permissiveness of infected cells for the HPV life cycle was investigated by differentiation of the infected cells in the raft tissue culture system and assay for viral late events.
MATERIALS AND METHODS
Cell and tissue culture.
The CIN-612 cell line was established from a biopsy specimen of a cervical intraepithelial neoplasm (CIN) (2), and the clonal derivative 9E maintains the HPV31b genome episomally at an average of 50 copies per cell (21). CIN-612 9E cells were maintained in monolayer culture with E medium containing 5% fetal bovine serum (FBS) (Summit Biotechnology) in the presence of mitomycin C-treated J2 3T3 mouse fibroblast feeder cells (29, 30). Epithelial organotypic (raft) tissue cultures were maintained as previously described (29, 30, 32). Raft tissues were treated with 10 μM 1,2-dioctanoyl-sn-glycerol (C8:0; Sigma Chemical Co., St. Louis, Mo.) in E medium containing 5% FBS every other day. Epithelial tissues were allowed to stratify and differentiate at the air-liquid interface for 14 days. HPV-negative human keratinocyte and other epithelial cell lines used for infections are summarized in Table 1. HaCaT, C-33A, and A431 cells were maintained in Dulbecco's modified Eagle medium-Ham's F-12 nutrient mixture (Sigma Chemicals) containing 10% FBS, 4× amino acids (Sigma Chemicals), 2 mM l-glutamine, 100 U of penicillin, and 1 μg of streptomycin per ml (Sigma Chemicals). N/TERT-1 cells (a generous gift of J. Rheinwald, Harvard University), NIKS cells (Stratatech, Madison, Wis.), and low-passage-number human foreskin keratinocytes (HFKs) (a gift of C. Wheeler, University of New Mexico School of Medicine) were grown either in keratinocyte serum-free medium (K-SFM; Gibco BRL) plus 30 μg of bovine pituitary extract per ml and 0.2 ng of epidermal growth factor per ml or in E medium containing 5% FBS plus 10 ng of epidermal growth factor per ml in the presence of mitomycin C-treated J2 3T3 feeder cells as described above.
TABLE 1.
HPV-negative cell lines used in infections
| Cell line | Characterization | Reference |
|---|---|---|
| HFK | Low-passage-number HFKs | None |
| HaCaT | Spontaneously immortalized human keratinocyte cell line from adult skin | 3 |
| NIKS | Spontaneously immortalized HFK cell line | 1 |
| N/TERT-1 | HFK cell line immortalized by telomerase catalytic subunit | 9 |
| C-33A | Cervical carcinoma cell line | 58 |
| A431 | Epidermoid carcinoma cell line | 15 |
| C127 | Murine mammary tumor cell line | 27 |
Virion purification and quantification.
CIN-612 9E raft tissues were extracted using a modified protocol of Favre et al. (12). Briefly, tissues were homogenized using a biologically contained BeadBeater device (BioSpec Products, Inc.), and the viral particles were purified using a series of low- and high-speed centrifugation steps as detailed previously (35). vDNA-containing particles were quantified by DNA hybridization as described previously (35).
HPV31b infections.
Cells were seeded at 3 × 105 cells per well in 4-cm2 wells or at 5 × 105 cells per well in 9-cm2 wells and allowed to attach overnight. The cells were 60 to 80% confluent at the time of infection. Virion stocks were thawed from −80°C at room temperature and were sonicated for 20 s at 0°C. For all cell lines, virus dilutions were added to each well in 0.25 ml for 4-cm2 wells or 0.5 ml for 9-cm2 wells of normal HaCaT medium (see above). The plates were rocked at 4°C for 1 h, then the viral inocula were removed, and the cells were washed with an excess of normal medium and then fed with their respective normal media and moved to 37°C. Media were changed every other day, and cells were expanded when they reached confluence.
Nucleic acid extraction and reverse transcription (RT)-PCR analyses.
Total RNAs were extracted from cells using TRIzol reagent (Invitrogen Life Technologies). RNA samples were treated with RNase-free DNase I (Promega Corp.) to remove copurifying viral and cellular DNA (36, 37). RNA concentrations were determined by optical density measurement; RNA concentrations and qualities were verified by electrophoresis through agarose gels containing ethidium bromide. RNAs were reverse transcribed by using random hexamer primers, and PCR was performed using a GeneAmp RNA PCR kit and AmpliTaq Gold DNA polymerase as instructed by the manufacturer (Applied Biosystems). All oligonucleotide primers (Table 2) were synthesized by Sigma Genosys and were used at 0.2 to 0.5 μM for PCR amplification. Individual primer pairs were optimized for annealing temperatures and MgCl2 concentration, and the relative sensitivities of primer pairs for a given template were previously determined (35). The PCR thermocycling profiles were as follows: 10-min time delay at 95°C; 30 to 45 cycles of 95°C for 45 or 60 s, 60°C for 30 or 60 s, and 72°C for 30 or 120 s; concluding with a 7-min extension at 72°C. For nested PCR, 4% of the first round of PCR was used for 30 cycles of PCR. PCR products were analyzed by electrophoresis through 2 or 3% agarose gels and ethidium bromide staining.
TABLE 2.
Oligonucleotide primers used to characterize HPV31 transcripts
| Primer name | Sequencea | Direction | Nucleotide position(s)a | ORF(s)b |
|---|---|---|---|---|
| E6A | 5′-CCT GCA GAA AGA CCT CGG-3′ | Sense | 120-137 | E6 |
| E6.2A | 5′-GCA TGA ACT AAG CTC GGC-3′ | Sense | 142-160 | E6 |
| E7.3A | 5′-GGA GAA GAC CTC GTA C-3′ | Sense | 526-541 | E7 |
| E7.4A | 5′-GGC TCA TTT GGA ATC GTG TGC-3′ | Sense | 812-832 | E7 |
| E7.2B | 5′-GCA CAC GAT TCC AAA TGA GCC-3′ | Antisense | 812-832 | E7 |
| 742A | 5′-CTA CAA TGG CTG ATC CAG-3′ | Sense | 857-874 | E7 |
| 742B | 5′-CTG GAT CAG CCA TTG TAC-3′ | Antisense | 857-874 | E7 |
| E1.4A | 5′-GCA GAA GCA GAG ACA GCA CAG GC-3′ | Sense | 1042-1064 | E1 |
| E1A | 5′-CAT GCA CAG GAA GCG GAG-3′ | Sense | 1072-1099 | E1 |
| E1B | 5′-TGT CCT CTT CCT CGT GC-3′ | Antisense | 2667-2683 | E1-E2 |
| E2.2B | 5′-GTT TCC AAT AGT CTA TAT GAT CAC-3′ | Antisense | 2772-2795 | E2 |
| E4.4B | 5′-GTT GGC TGT TGG TAG GTT TG-3′ | Antisense | 3327-3446 | E4 |
| E4B | 5′-CTT CAC TGG TGC CCA AGG-3′ | Antisense | 3395-3378 | E4 |
| E4.3B | 5′-GCT CTG TTC TTG GTC G-3′ | Antisense | 3425-3440 | E4 |
| L1.2B | 5′-C TAG CAC TGC CTG CGT G-3′ | Antisense | 5673-5657 | L1 |
| β-actin OA | 5′-GAT GAC CCA GAT CAT GTT TG-3′ | Sense | 1578-1587, 2029-2039 | β-Actin |
| β-actin IA | 5′-AAC ACC CCA GCC ATG TAC GTT G-3′ | Sense | 2046-2067 | β-Actin |
| β-actin IB | 5′-ACT CCA TGC CCA GGA AGG AAG G-3′ | Antisense | 2455-2467, 2563-2570 | β-Actin |
| β-actin OB | 5′-GGA GCA ATG ATC TTG ATC TTC-3′ | Antisense | 2735-2744, 2857-2867 | β-Actin |
Cloning and sequencing.
Molecular cloning was performed using standard techniques. All enzymes were purchased from Promega Corp. unless otherwise noted. PCR products were cloned using the TA Cloning kit (Invitrogen Life Technologies). Novel amplicons were cloned from multiple PCR assays. Double-stranded DNA sequencing was performed on multiple clones of each novel amplicon at the Center for Genetics in Medicine, Department of Biochemistry and Molecular Biology at the University of New Mexico School of Medicine. For the nomenclature of the amplicons, the open reading frames (ORFs) included are given with asterisks or carets signifying splicing, with subscripts indicating the HPV31 nucleotides at the junctions, which are shown in Fig. 1. For example, the structures of the major early spliced transcripts of HPV31b are described as follows: E6∗I210∧413 indicates that the splice site is in the E6 ORF and contains a junction from nucleotide (nt) 210 to 413; E1∗I begins at the E1 ORF in transcript E1∗I877∧ 2646,E2 and contains a splice joining nt 877 to 2646 and the E2 ORF follows; E1∧E4877∧ 3295 indicates the E1∧E4 fusion protein using the 877∧3295 splice junction; and E8∧E2C1296∧3295 represents the fusion of the E8 ORF with the C terminus of the E2 ORF using the splice joined at nt 1296 and 3295. A Roman numeral in the notations denotes that more than one splice variant exists for a given larger ORF.
FIG. 1.

Genomic organization of HPV31b and summary of the known early spliced polycistronic transcripts and L1-containing RNAs targeted by RT-PCR. The circular HPV31b genome of 7,912 bp is depicted linearized at the late polyadenylation (polyA) signal for the purpose of illustrating the ORFs and the characterized transcripts. The nucleotide numbering of the viral genome is given below the thin horizontal rule (17). The long control region (LCR) is indicated, and the open boxes show viral ORFs in all three reading frames of the genome. The viral promoters are shown by bent arrows with their initiation sites indicated; solid black arrows denote constitutively used promoters, light gray P7783 and P7850 are negatively regulated upon differentiation, and dark gray P742 is upregulated upon epithelial differentiation (21, 39, 40). Placement and orientation of the primers used in the first round of PCR to amplify specific spliced segments are illustrated with arrows below the ORFs. The basic RNA structures were characterized by sequencing cloned cDNAs obtained from CIN-612 9E cells and raft tissues (21, 37, 39, 49). Shaded boxes illustrate the ORFs contained within the polycistronic transcripts; thick lines represent noncoding sequences. Thin lines show regions spliced out of transcripts (introns); the nucleotide positions of splice donors and acceptors are indicated below the transcripts. The regions and ORFs contained in each mRNA are indicated to the right side of each.
RESULTS
HPV31b infection of monolayer epithelial cells from various sources.
Previously, we showed that the HaCaT spontaneously immortalized human keratinocyte cell line is susceptible to infection by HPV31b (35). As HaCaT cells are of cutaneous epithelial origin (3), we speculated that cell lines of genital epithelial origin might be infected more efficiently by HPV31b than HaCaT cells. Therefore, we surveyed the relative susceptibilities of several epithelial cell lines to HPV infection. The cell lines chosen to survey for efficient infection by HPV31b virions are summarized in Table 1. Cells were seeded in the absence of fibroblast feeder cells at equal numbers in their respective cell media for attachment overnight. HFK, NIKS, and N/TERT-1 cells were seeded in K-SFM. Each set of infection experiments included HaCaT cells to control for reproducibility and efficiency. To avoid the possibility that the components of the various cell media might influence HPV infection of the cells, all infections were performed with the media used to infect and culture HaCaT cells. All subconfluent monolayer cells were incubated with serial 10-fold dilutions of an HPV31b stock as described in Materials and Methods. The cells were harvested at 4 days postinfection (p.i.). Total RNAs were extracted, quantified, and subjected to RT-PCR as described above. Our previous work demonstrated that targeting of HPV31b E1∧E4 transcripts by RT-PCR was the most sensitive means of detecting infection at 4 days p.i (35). Therefore, HPV31b infection was assayed and compared among the cell lines via the detection of PCR amplimers arising from spliced E1∧E4 RNAs. Representative data from the cell lines are shown in Fig. 2.
FIG. 2.

RT-PCR analysis of HPV31b transcripts following infection of various cell lines. Subconfluent cell monolayers were inoculated with serial 10-fold dilutions of an HPV31b stock. Total RNAs were extracted and treated with DNase I. RT was preformed on 3 μg of RNA for each sample. Each cell line was mock-infected (lanes M), or infected with HPV31b doses corresponding to 0.1, 1.0, and 10 vDNA-containing particles per cell. CIN-612 9E monolayers (9E) and no RNA input (Ø) served as positive and negative amplification controls, respectively. (A) Primers E7.3A and E4.3B target a 498-bp amplimer arising from spliced HPV31b E1∧E4 RNA. The input RNA corresponded to 2.7 μg for each PCR. (B) Primers β-actin OA and β-actin OB detect a 641-bp amplimer derived from spliced cellular β-actin RNA. The input RNA corresponded to 0.3 μg. Molecular size standards (100-bp ladder, New England Biolabs) are shown at the edges of each panel.
The detection of spliced β-actin transcripts served as a positive control for the presence and integrity of RNA in samples, for RT, and for PCR amplification (Fig. 2B). RNA from CIN-612 9E monolayers was a positive control for HPV31b RNAs (Fig. 2A, lane 33), whereas no RNA input was a negative control (Fig. 2, lane 34). Samples from mock-infected cells (M) were negative for HPV31b RNAs (Fig. 2A). As previously reported, HPV31b infection in HaCaT cells was consistently detected at doses of vDNA containing particles equivalent to 10 and 1.0 genomes per cell in all experiments (35). Identical results were seen in six separate infections. Results from two separate HaCaT cell infections are shown in Fig. 2 (lanes 1 to 8).
We examined the ability of several cell lines derived from primary foreskin keratinocytes to be infected when grown in the presence or absence of fibroblast feeders. In some experiments, the low-passage-number HFK, NIKS, and N/TERT-1 cells were grown in the presence of mitomycin C-treated NIH J2 fibroblast feeders and the cultures were fed with E medium following infection. In experiments where mitomycin C-treated NIH J2 fibroblast feeder cells were not added, K-SFM was used to culture the HFK, NIKS, and N/TERT-1 cells. Out of two separate infections each where NIKS cells and N/TERT-1 cells were cultured in K-SFM, E1∧E4 transcripts were sporadically detected at a dose of 10 vDNA-containing particles per cell (data not shown). However, coculture of the NIKS cells with mitomycin C-treated NIH J2 fibroblast feeders following infection resulted in the reproducible detection of E1∧E4 RNAs from infections with doses of 10 and 1.0 vDNA-containing particles per cell (Fig. 2A, lanes 13 to 16). In infections of N/TERT-1 cells, E1∧E4 amplimers were detected reproducibly at a dose of 10 vDNA-containing particles per cell, and coculture with fibroblast feeders did not affect their HPV infection status (Fig. 2A, lanes 17 to 20).
In previous work using low-passage-number HFK cells grown in the presence of fibroblast feeder cells, we found it difficult to detect HPV18 infection by targeting spliced transcripts using RT-PCR (33). Furthermore, HFK cells derived from various donors show differing abilities to replicate HPV genomes following transfection (M. Ozbun, unpublished data; Laimonis A. Laimins, personal communication). With this in mind, we mixed low-passage-number HFKs from six separate donors for the present infections. We found that HPV31b infection could be reproducibly detected at a dose of 10 vDNA-containing particles per cell in this mixed-donor HFK population grown with mitomycin C-treated fibroblast feeders (Fig. 2A, lanes 9 to 12).
Three cell lines were found not to be able to support HPV31b infection. E1∧E4 RNAs were not detected following two separate infections each of the C-33A cervical cancer cell line or the A431 epidermoid carcinoma cell line (Fig. 2A, lanes 21 to 24, and 25 to 28, respectively). However, in a separate isolate of C-33A cells, we were able to detect infection (data not shown). Although PVs are considered to be species specific (reviewed in reference 25), bovine PV 1 (BPV1) can cross the species barrier and infect mouse C127 mouse mammary cells and NIH 3T3 mouse embryo fibroblasts, leading to morphological transformation of the cells (11). Roden and coworkers found that HPV16 VLPs were able to bind to C127 cells and inhibit their transformation by BPV1 (43). Furthermore, pseudotyped virions consisting of BPV1 genomes encapsidated by HPV16 late proteins could initiate transformation of C127 cells (42). Thus, we tested the possibility that C127 cells might be susceptible to HPV31b infection. However, we were not able to detect infection of C127 cells following three separate infections (Fig. 2A, lanes 29 to 32). For each infection experiment, the RT-PCRs were repeated under standardized reaction conditions to confirm the results.
HPV31b time course of infection of HaCaT monolayer cells.
A time course of infection was performed to determine the relative times of appearance of various spliced early viral transcripts. HaCaT cells were used in these experiments as we determined they could be efficiently and reproducibly infected. In addition, the growth and infectivity of HaCaT cells was not dependent upon coculture with fibroblast feeder cells as described above. Plates were either mock infected or inoculated with a dose of vDNA-containing virus particles equivalent to 20 genomes per cell. Cells were harvested for RNA at 2-h intervals beginning at 2 h p.i. and continuing to 16 h p.i. A second experiment was performed in which cells were harvested at 8-h intervals beginning at 8 h p.i. and continuing to 40 h p.i. Each RNA sample was subjected to one large RT reaction and then was divided into seven first-round PCR assays to detect the onset of viral transcription and to characterize the structures of the newly expressed viral RNAs. One set of PCR assays was used to detect β-actin RNAs as a control (Fig. 3A); the other six sets of PCR assays were specific for known HPV31b cDNAs (see Fig. 1, primer sets A to F). A fraction of the first round of PCR products was subjected to a second round of PCR using nested primers to obtain greater sensitivity. Upstream primers were selected to estimate the location of promoters used for HPV31b transcript initiation.
FIG. 3.

RT-nested-PCR analysis of β-actin and spliced HPV31b E6 transcripts expressed following infection of HaCaT cells. Subconfluent HaCaT monolayers were infected with an HPV31b stock at 20 vDNA-containing particles per cell. The cells were harvested for total RNAs at 2-h intervals beginning at 2 h p.i. and continuing to 16 p.i. Infected cells were also harvested at 8-h intervals up to 40 h p.i. For each sample an RT reaction was performed using 12.0 μg of DNase I-treated total RNA. Samples included mock-infected HaCaT cells (M); HaCaT cells harvested at 2, 4, 6, 8, 10, 12, 14, 16, 24, 36, 48 h p.i; and CIN-612 9E monolayers (9E). No RNA input (Ø) served as a negative amplification control. RT assays were divided into seven different first round PCR amplifications of 45 cycles using primer sets A, B, C, D, E, and F from Fig. 1 and also targeting spliced cellular β-actin. Following the first round of PCR, 4% of each of these assays was used in nested PCR of 30 cycles. (A) Primers β-actin OA and β-actin OB were used in the first round, and primers β-actin IA and β-actin IB were used in nested PCR to detect a 429-bp amplimer derived from spliced β-actin RNA. Input RNA in the first round of PCR corresponded to 380 ng. (B) Primers E6A and 742B were used in the first round, primers E6.2A and E7.2B were used in nested PCR. For each reaction, the input RNA in the first round of PCR corresponded to 1.9 μg. Molecular size standards (100-bp ladder; New England Biolabs) are shown at the edges of each panel. (C) Early region of the HPV31b genome as described in the legend to Fig. 1. Below the genome diagram, the structures of the observed amplicons derived from viral RNAs or DNAs are shown with lines pointing to the respective amplicons in panels A and B. Boldface letters denote previously characterized ORFs, with their splice junctions indicated in parentheses. The newly identified RNA structure containing the 210∧739 splice junction is indicated. Both spliced structures were verified by sequencing from at least one of the samples shown. Placement and orientation of HPV31b PCR primers are given with primer pairs used for the first round of PCR indicated first. Primer pairs used in nested PCR are given in parentheses.
Representative data from RT and nested PCR are shown in Fig. 3 to 6. As expected, all the RNA samples were strongly positive for the amplification of the cellular β-actin control (Fig. 3A, lanes 1 to 13). Mock-infected controls showed no amplification of spliced HPV transcripts (Fig. 3 to 6, panels A and B, lanes 1), whereas the latently infected 9E cell monolayers yielded products of expected sizes as well as novel products (Fig. 3 to 6, panels A and B, lanes 13). The early infection-specific RT-PCR products were similar in size to those observed from the latently infected 9E cell monolayers; no amplicons were present in newly infected cells that were not also present in the latently infected 9E monolayer cells.
FIG. 6.

RT-nested-PCR analysis of HPV31b spliced E8∧E2C transcripts expressed following infection of HaCaT cells. Details of the experiment are given in the legend to Fig. 3. (A) First round of RT-PCR using primers E1.4A and E4B to illustrate the two amplicons present in CIN-612 9E cells. (B) Nested PCR using primers E1A and E4.4B. For each reaction, the input RNA in the first round of PCR corresponded to 1.9 μg. (C) Early region of the HPV31b genome and explanation of observed RNA structures as described in the legends to Fig. 1 and 3C. Boldface letters denote previously characterized ORFs, with their splice junctions indicated in parentheses. The novel RNA structure containing the 1124∧3295 splice junction is shown. The E8∧E2C structure was verified by sequencing from two positive samples in panel B, and the novel 1124∧3295 splice junction was characterized by sequencing of products from 9E cells.
Targeting the E6 region (Fig. 3C), the upstream primers E6A and E6.2A were used to identify transcripts initiating near the major early promoter, P99, upstream of the E6 ORF. We detected a major amplimer of 489 bp derived from the E6∗I spliced transcript, and this product was consistently present at 8 h p.i. (Fig. 3B, lane 5). Also evident in samples inoculated with virus were amplimers of 755 bp arising from vDNA or unspliced RNA. An intermediately sized product was also present in many samples positive for E6∗I. This amplimer has been refractory to cloning and further characterization (35) and could result from heteroduplex formation between E6 and E6∗I. A novel amplimer of 163 bp was also detected beginning at 12 h p.i. and in the latently infected 9E monolayer cells (Fig. 3B, lanes 7 to 13, respectively). Cloning and sequencing revealed this product to be a result of splicing between the donor at nt 210 and a novel splice acceptor at nt 739.
Targeting spliced E2 transcripts (Fig. 4C), upstream primers positioned near the major early promoter P99 plus downstream primers in the E2 ORF yielded the expected amplimers of 572 bp containing E6∗I,E7,E1∗I,E2 beginning at 10 h p.i. (Fig. 4A, lanes 6 to 12). Also present at this time were amplimers of 705 bp consisting of the splice donor at nt 877 joined to a novel acceptor at nt 2513 (Fig. 4A, lanes 6 to 12). Two additional novel amplimers were also detected at 16 h p.i. and in 9E cells (Fig. 4A, lanes 9, 13, respectively). The amplimer of 240 bp results from the joining of the donor at nt 210 to the acceptor at nt 2513. The product of 107 bp uses the splice donor at nt 210 in the E6 ORF joined to the acceptor at nt 2646 also used in E1∗I,E2 transcripts. Upstream primers positioned in the E7 ORF paired with downstream primers in the E2 ORF produced amplicons of 59 bp from spliced E1∗I,E2 transcripts as early as 4 h p.i. (Fig. 4B, lanes 3 to 12). These amplimers were also found in 9E cells (Fig. 4B, lane13). Amplicons of 192 bp containing the 877∧2513 splice were also slightly visible by 4 to 6 h p.i. and were more obvious by 14 h p.i. and in latently infected 9E cells (Fig. 4B, lanes 3 and 4 and 8 to 13).
FIG. 4.

RT-nested-PCR analysis of HPV31b spliced E2 transcripts expressed following infection of HaCaT cells. Details of the experiment are given in the legend to Fig. 3. (A) Primers E6A and E2.2B were used in the first round, and primers E6.2A and E1B were used in nested PCR. (B) Primers E7.4A and E2.2B were used in the first round, and primers 742A and E1B were used in nested PCR. For each reaction, the input RNA in the first round of PCR corresponded to 1.9 μg. (C) Early region of the HPV31b genome and explanation of observed RNA structures as described in the legend to Fig. 1 and 3C. Boldface letters denote previously characterized ORFs, with their splice junctions indicated in parentheses. The novel RNA structures containing the 877∧2513 splice junction, the 210∧2513 splice junction, and the 210∧2646 splice junction are indicated. Each of the novel spliced structures was verified by sequencing from at least one of the samples as shown.
Spliced E1∧E4 transcripts were targeted using upstream primers positioned near the major early promoter P99 paired with downstream primers in the E4 ORF (Fig. 5C). This yielded expected amplimers of 635 bp arising from the E6∗I,E7,E1∧E4 transcripts and of 170 bp derived from the E6∧E4 (E6∗III) RNA as early as 8 h p.i. (Fig. 5A, lanes 5 to 13). A novel amplimer of 133 bp was also detected at the same times. This product contains the donor at nt 210 joined to a novel acceptor at nt 3332 (Fig. 5A, lanes 5 to 13). Similar to what was observed using upstream primers near P99, upstream primers positioned in the E7 ORF paired with downstream E4 primers (Fig. 5C) yielded E1∧E4 amplimers of 122 bp beginning at 8 h p.i. (Fig. 5B, lanes 5 to 13). Also observed were novel amplimers of 85 bp and ∼150 bp (Fig. 5B, lanes 5 to 13). In the 85-bp amplimer, the splice donor at nt 877 is joined to an acceptor at nt 3332; this product can best be seen in Fig. 5B, lane 5. The structure of the amplimer of ∼150 bp is under investigation.
FIG. 5.

RT-nested-PCR analysis of HPV31b spliced E4 transcripts expressed following infection of HaCaT cells. Details of the experiment are given in the legend to Fig. 3. (A) Primers E6A and E4.3B were used in the first round, primers E6.2A and E4B were used in nested PCR. (B) Primers E7.4A and E4.3B were used in the first round, primers 742A and E4B were used in nested PCR. For each reaction, the input RNA in the first round of PCR corresponded to 1.9 μg. (C) Early region of the HPV31b genome and explanation of observed RNA structures as described in the legend to Fig. 1 and 3C. Boldface letters denote previously characterized ORFs, with their splice junctions indicated in parentheses. The novel RNA structures containing the 210∧3332 and the 877∧3332 splice junctions are indicated. The novel 210∧3332 spliced structure was verified by sequencing from at least one of the samples in panel A and the 877∧3332 splice junction was determined from multiple positive samples as shown in panel B.
In previous work, we were unable to detect spliced E8∧E2C transcripts using RT and a single round of PCR following infection of HaCaT cells at various doses of virus inoculum (35). As the promoter(s) for transcripts containing the E8∧E2C ORF has not been identified, the first round of assays using primers E6A and E4.3B spanning E6 to E4 (Fig. 1, primer set D) or primers E7.4A and E4.3B spanning E7 to E4 (Fig. 1, primer set E) were subsequently subjected to nested PCR to test for the presence of E8∧E2C transcripts. Primers E1A and E4.4B were used in the nested PCR experiments (Fig. 6C); however, we were unable to detect products corresponding to spliced E8∧E2C RNA using this approach (data not shown). Next we used first-round primers E1.4A and E4B to target the E8∧E2C region (Fig. 6C). Spliced E8∧E2C transcripts were readily detected in the 9E cells along with a distinct amplicon found to use a novel splice donor at nt 1124 joined to the acceptor at nt 3295 (Fig. 6A, lane 13). Nested PCR permitted the detection of E8∧E2C RNAs at 4, 10, and 14 h p.i. and in 9E monolayer cells (Fig. 6B, lanes 3, 6, 8, 13). The novel 1124∧3295 spliced RNA was found at 16 h p.i. and in 9E cells (Fig. 6B, lanes 9, 13). RT-PCR and nested PCR experiments were performed on the time course infections at least four times to verify the results.
Persistence of HPV31b infection in HaCaT cells.
Experiments were performed to assess the persistence of HPV31b infection in HaCaT cells and to determine whether infected HaCaT cells could support a productive HPV infection. HaCaT cells infected with a dose of vDNA-containing particles equivalent to 100 genomes per cell were passaged for approximately 5.5 population doublings. This corresponds to each cell in the original population giving rise to nearly 50 daughter cells. Infected cells were either harvested for RNA or were seeded for growth in the raft system to allow for differentiation-dependent late viral gene expression. Raft tissues were harvested at 14 days postlifting to the air-liquid interface, and then total RNAs were extracted and DNase I treated. RT-PCR was performed targeting the spliced E6 transcripts and the spliced late transcripts as we have characterized previously (36). All samples were positive for spliced β-actin amplicons (Fig. 7A). Samples from all infected cells were positive for E6 amplicons, including the unspliced RNA or vDNA, the spliced E6∗I, and the novel 210∧739 spliced products (Fig. 7B, lanes 1 to 3). Furthermore, the samples from 9E rafts and from HPV31b-infected HaCaT raft tissues were positive for spliced L1 RNAs (Fig. 7C, lanes 1 and 3) indicating late stage viral events. Samples from uninfected HaCaT cells showed no amplification of HPV31b-specific targets (Fig. 7B and C, lanes 4 to 5). These data suggest that HaCaT cells can support a productive HPV31b infection and confirms the validity of using these cells to study the early events of viral infection.
FIG. 7.
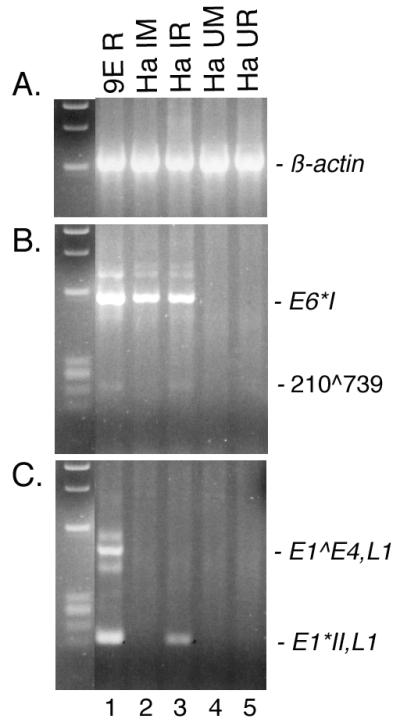
RT-PCR for HPV31b transcripts in infected HaCaT cells and raft tissues. HaCaT cells were infected with a dose of HPV31b corresponding to 100 genomes per cell. The cells were passaged through approximately 5.5 population doublings (Ha IM), and infected cells were seeded on collagen-fibroblast matrices for raft epithelial growth (Ha IR). 9E raft tissues (9E R), uninfected HaCaT monolayers (Ha UM), and uninfected HaCaT rafts (Ha UR) were used as controls. DNase I-treated, total RNAs (5 μg) were subjected to RT and divided for PCR amplifications of 40 cycles. PCR was performed using primers specific for the spliced products of β-actin (β-actin OA and β-actin OB), spliced HPV31b E6 RNAs (E6A and 742B), or spliced HPV31b L1 RNAs (E7.4B and L1.2B). As size standards, ΦX174 DNA digested with HaeIII is shown to the left of each panel.
DISCUSSION
Infectious stocks of the high-risk prototype HPV, HPV31b, cultivated in the organotypic (raft) tissue culture system were used to investigate the early stages of infection of human keratinocytes, the host cell type for HPVs. Newly synthesized, spliced viral RNAs were detected by RT-PCR and used as an indication of infection. In recent work, we found that HPV31b infection could be detected at a dose as low as 1.0 vDNA-containing particle per cell in HaCaT cells (35) and in this study we demonstrate that it is highly sensitive to HPV infection in vitro. HPV infections tend to be specific to either mucosal or cutaneous epithelium (reviewed in reference 25). Because HaCaT cells are of cutaneous origin, we hypothesized that HPV-negative cell lines of genital epithelial origin would be more susceptible to HPV31b infection. Low-passage-number HFKs were shown to be susceptible to HPV infection using stocks of HPV types 11, 40, and LVX82/MM7 produced in the mouse xenograft system (6, 10, 45, 47) and HPV18 that we synthesized in the raft system (33). Using a mixture of low-passage-number HFKs from six donors, we were able to detect HPV31b infection at a dose as low as 10 vDNA-containing particles per cell in this population of cells. The neonatal HFK line, N/TERT-1, immortalized by the catalytic subunit of the telomerase gene (9), was infected with an efficiency similar to that of low-passage-number HFKs. The spontaneously immortalized NIKS neonatal HFK cell line (1) was infected at a higher efficiency, similar to that of the HaCaT cells (at a dose as low as 1.0 vDNA-containing particle per cell) but only when the NIKS cells were grown in the presence of fibroblast feeder cells after infection. Previous observations indicate that HPV infection and genome replication varies among low-passage-number HFKs from different donors (33) and (M. A. Ozbun, unpublished data; L. A. Laimins, personal communication). The finding that the N/TERT-1 HFK cells appear less susceptible than the NIKS HFK cells to infection also may be explained by the differences in HFK donors. These data from HFK infections suggest that host genetics may play a role in susceptibility to HPV infection and genome replication. The C-33A cell line is the only known HPV-negative cervical carcinoma line (C. Wheeler and E.-M. de Villiers, personal communications). It was unexpected that HPV31b infection was not detected in one isolate of the C-33A cervical cancer cell line. Perhaps during passage of the cells in culture, critical receptors or intracellular factors were mutated or down regulated. A431 epidermoid carcinoma cells were exposed to HPV31b virus with no preconception of whether they would be susceptible to infection. We were consistently unable to detect spliced HPV31b RNAs in the A431 cells following viral exposure. Although C127 cells are susceptible to infection by BPV1 and pseudotyped HPV16 particles carrying BPV1 genomes, and HPV16 VLPs can bind to C127 cells (11, 42, 43), we were unable to detect spliced E1∧E4 RNAs following three separate HPV31b infections. These data suggest that HPV capsids can initiate infection in C127 cells to the extent of delivering BPV1 vDNA to the nucleus, but that HPV-specific transcription or replication factors are lacking in these mouse cells. This supports the concept that all PVs could use the same cellular receptor(s) to attach to and penetrate cells but that HPV host range and cell type specificity (i.e., aspects of bona fide infection) may be determined at point downstream of viral attachment (34, 43, 46, 56).
A time course of HPV31b infection was performed in HaCaT cells to analyze the onset of viral transcription. RNAs containing the E1 ORF were not targeted as the majority are unspliced (23, 37). Thus, it would be impossible to distinguish between unspliced transcripts and vDNA, which might not be indicative of a genuine infection. The major spliced viral RNAs, including E6∗I210∧ 413, E1∗I877∧ 2646,E2 and E1∧E4877∧ 3295, were reproducibly present by 8 to 10 h p.i. However, a subset of the spliced E1∗I,E2 RNAs that appear to initiate at a promoter other than the P99 major early promoter were present as early as 4 h p.i. (Fig. 4B). We do not believe that detection of the E1∗I,E2 RNA at 4 h p.i. is a simple reflection of a more-efficient RT or PCR amplification for the shorter transcript, as one set of cDNA templates was used for all the PCR assays. Furthermore, we previously found the primer sets used to amplify spliced E1∗I,E2 RNAs (i.e., E6A and E2.2B or E7.4A and E2.2B and their nested counterparts E6.2A and E1B or 742A and E1B) have similar sensitivities of detection as low as 102 copies of template (35). However, the possibilities that the RT efficiency is template dependent or that the shorter amplicons are preferentially detected cannot be discounted completely. Transcripts containing the E1∗I,E2 splice are believed to be important for the translation of the E2 ORF (37). The E2 protein is required for vDNA replication and for transcriptional modulation (reviewed in reference 50); thus, it is not surprising that the E1∗I,E2 RNAs would be among the first detected following infection. Klumpp and Laimins showed that upon differentiation, initiation of HPV31 transcripts containing E1 and E2 ORFs switches from the major early promoter P99 to the late P742 promoter (23). Our data suggest that the P742 promoter, or some promoter other than the major early promoter, might also be used for E2 transcripts upon initial infection, which occurred in the absence of differentiation. We attempted to perform primer extension and nuclease protection assays on the RNA samples from our infections to determine promoter usage. However, the levels of viral transcripts present were below the level of detection of these assays. We are currently evaluating the utility of 5′ rapid amplification of cDNA ends techniques for determining the start sites of these early RNAs. The E8∧E2C transcript was detected as early as 4 h p.i. The E8∧E2C protein is negative regulator of both vDNA replication and transcription (49, 52). The early presence of an E8∧E2C RNA along with the E1∗I,E2 RNA makes it tempting to speculate that the E8∧E2C protein might be important to restrain the initial burst in vDNA replication following E2 expression upon infection. The majority of transcripts that appear to be expressed continuously following their onset were also detected by a single round of PCR at the same time points determined by nested PCR (data not shown). Other cDNAs were discontinuously detected by nested PCR, such as the 210∧2513 and 210∧2646 species in Fig. 4A, 877∧3332 in Fig. 5B, and E8∧E2C and 1124∧3295 in Fig. 6B. This latter phenomenon could reflect regulated expression; however, only a fraction of cells are infected and it is also likely that the levels of each of these RNA are low in infected cells as they were not detected without nested PCR. A low number of these RNA species in a given aliquot could give rise to stochastic sampling by PCR. Due to the number of splice-variants (twelve total), we found it unfeasible to target each cDNA for quantitative or semiquantitative analysis in this initial qualitative characterization of HPV31b early transcripts.
The amplicon corresponding to the full-length E6 target in samples harvested at 2, 4, and 6 h p.i. most likely arises from vDNA. PCR analyses on the samples in the absence of RT also yielded full-length E6 amplimers, indicating vDNA was present (data not shown). Detection of an amplicon from vDNA is important to illustrate two points about infection. First, the vDNA could be present in virions that were simply attached to cells, or the vDNA could reflect virions present in the cytoplasm of the cells. In either case mere detection of vDNA in this PCR assay would not be indicative of true infection per se. Thus, targeting of newly synthesized, spliced viral RNAs is a more appropriate assay for initiation of infection. Second, the only HPV-specific product detected at 2 h p.i. was that of the full-length E6 target (see lane 2 in Fig. 3 to 6, panels A and B). This result demonstrates that HPV31b virions do not carry spliced viral RNAs or processed genomes that might mimic newly synthesized spliced viral transcripts in this assay. These data further support the targeting of newly synthesized, spliced viral RNAs rather than vDNA as an appropriate assay for HPV infection.
We detected HPV31b infection of human keratinocytes by 4 h p.i. using RT-PCR techniques to assay newly synthesized, spliced viral RNAs. Thus, viral penetration, nuclear transport, and uncoating can be a relatively quick process. In agreement with our data, Zhou and coworkers used electron microscopy to show that most BPV1 particles were cytoplasmic by 30 min p.i. and were in a perinuclear zone by 60 min p.i. (59). They also detected capsid proteins, but not particles, in the nuclei by 90 min p.i., suggesting that uncoating of the vDNA had occurred (59). Volpers and colleagues also found that the bulk of HPV33 VLPs were internalized by 1 h at 37°C (56). Day et al. recently showed the internalization of PVs via a clathrin dependent pathway (7). That clathrin is typically internalized with a half-life of 5 to 15 min (16, 44) is in accordance with our detection of spliced viral RNAs by 4 h p.i. However, other data from Day and coworkers and from Christensen et al. suggest a slower kinetics on the order of 8 to 10 h for BPV1 and cottontail rabbit PV binding and internalization (5, 7). These disparities could be due to differences in some of the assay systems, differences in the sensitivities of the techniques, or differences in the viruses themselves. The design and implementation of standardized techniques for studying PVs will be necessary to resolve these disparities.
In this study, seven unique, spliced viral RNA structures were detected. One novel splice donor at nt 1124 was found, and three new splice acceptors were discovered at nt 739, 2513, and 3332. The ORFs created by these newly identified splicing patterns have the ability to yield viral peptides as small as 2.9 kDa. Larger ORFs created by the splices are predicted to be 9.9 kDa for a fusion of the N and C termini of the E1 ORF using the 877∧2513 splice, 13.5 kDa for a fusion of the E6 N terminus with the E1 C terminus using the 210∧2513 splice, and 28.4 kDa for a fusion of the E1 N terminus with the E4 ORF using the 1124∧3295 splice. Small viral regulatory proteins are well known for other viruses, including the HIV Vpr and Nef proteins, the human polyomavirus agno proteins, and the hepatitis B X protein. The use of the HPV reverse genetics system will permit testing the functions of these potential proteins in the viral life cycle. Finally, that HPV31b infection of HaCaT cells in monolayers can become productive for late gene synthesis when the infected cells are grown in the raft system leads us to conclude that our data reflect initial viral activities that lead to a persistent and productive infection of HaCaT cells. Recently, we have purified infectious virions from HaCaT raft tissues derived from cells transfected with HPV31 genomes (B. K. Steele and M. A. Ozbun, unpublished results). This further substantiates the idea that HaCaT cells are permissive for the HPV31 life cycle.
Acknowledgments
This work was supported by Public Health Service grants CA-85747 and AI-52049 from the NIH and American Cancer Society grants IRG#192 and RPG-00-276.
Rosa L. Otero and Catherine A. Monserrat provided outstanding technical assistance. I am indebted to Laimonis A. Laimins, Cosette M. Wheeler, Jesse Summers, and Stephanie L. Lowe for helpful discussions and for critical comments on the manuscript. I am grateful to Norbert Fusenig for providing the HaCaT cell line, to James Rheinwald for the N/TERT-1 cell line, and to Cosette Wheeler for the gift of low-passage-number HFKs. I thank Patricia Day and John Schiller for sharing data on BPV1 infections prior to publication.
REFERENCES
- 1.Allen-Hoffmann, B. L., S. J. Schlosser, C. A. R. Ivarie, C. A. Sattler, L. F. Meisner, and S. L. O'Connor. 2000. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J. Investig. Dermatol. 114:444-455. [DOI] [PubMed] [Google Scholar]
- 2.Bedell, M. A., J. B. Hudson, T. R. Golub, M. E. Turyk, M. Hosken, G. D. Wilbanks, and L. A. Laimins. 1991. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 65:2254-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boukamp, P., R. T. Petrussevska, D. Breitkreutz, J. Hornung, A. Markham, and N. E. Fusenig. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106:761-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broker, T. R., and M. Botchan. 1986. Papillomaviruses: retrospectives and prospectives. Cancer Cells 4:17-36. [Google Scholar]
- 5.Christensen, N. D., N. M. Cladel, and C. A. Reed. 1995. Postattachment neutralization of papillomaviruses by monoclonal and polyclonal antibodies. Virology 207:136-142. [DOI] [PubMed] [Google Scholar]
- 6.Christensen, N. D., W. A. Koltun, N. M. Cladel, L. R. Budgeon, C. A. Reed, J. W. Kreider, P. A. Welsh, S. D. Patrick, and H. Yang. 1997. Coinfection of human foreskin fragments with multiple human papillomavirus types (HPV-11, -40, and -LVX82/MM7) produces regionally separate HPV infections within the same athymic mouse xenograft. J. Virol. 71:7337-7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Day, P. M., D. R. Lowy, and J. T. Schiller. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology, in press. [DOI] [PubMed]
- 8.de Villiers, E.-M. 1999. Human papillomavirus. Introduction. Semin. Cancer Biol. 9:337. [DOI] [PubMed] [Google Scholar]
- 9.Dickson, M. A., W. C. Hahn, Y. Ino, V. Ronfard, J. Y. Wu, R. A. Weinberg, D. N. Louis, F. P. Li, and J. G. Rheinwald. 2000. Human keratinocytes that express hTERT and also bypass a p16INK4a-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20:1436-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dollard, S. C., L. T. Chow, J. W. Kreider, T. R. Broker, N. L. Lill, and M. K. Howett. 1989. Characterization of an HPV type 11 isolate propagated in human foreskin implants in nude mice. Virology 171:294-297. [DOI] [PubMed] [Google Scholar]
- 11.Dvoretzky, I., R. Shober, S. K. Chattopadhyay, and D. R. Lowy. 1980. A quantitative in vitro focus assay for bovine papilloma virus. Virology 103:369-375. [DOI] [PubMed] [Google Scholar]
- 12.Favre, M., F. Breitburd, O. Croissant, and G. Orth. 1975. Structural polypeptides of rabbit, bovine, and human papillomaviruses. J. Virol. 15:1239-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frattini, M. G., H. B. Lim, J. Doorbar, and L. A. Laimins. 1997. Induction of human papillomavirus type 18 late gene expression and genomic amplification in organotypic cultures from transfected DNA templates. J. Virol. 71:7068-7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frattini, M. G., H. B. Lim, and L. A. Laimins. 1996. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc. Natl. Acad. Sci. USA 93:3062-3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giard, D. J., S. A. Aaronson, G. J. Todaro, P. Arnstein, J. H. Kersey, H. Dosik, and P. W. P. 1973. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 51:1417-1423. [DOI] [PubMed] [Google Scholar]
- 16.Goldenthal, K. L., K. Hedman, J. W. Chen, J. T. August, P. Vihko, I. Pastan, and M. C. Willingham. 1988. Pre-lysosomal divergence of alpha 2-macroglobulin and transferring: a kinetic study using a monoclonal antibody against a lysosomal membrane glycoprotein (LAMP-1). J. Histochem. Cytochem. 36:391-400. [DOI] [PubMed] [Google Scholar]
- 17.Goldsborough, M. D., D. DiSilvestre, G. F. Temple, and A. T. Lorincz. 1989. Nucleotide sequence of human papillomavirus type 31: a cervical neoplasia-associated virus. Virology 171:306-311. [DOI] [PubMed] [Google Scholar]
- 18.Ho, G. Y. F., R. D. Burk, S. Klein, A. S. Kadish, C. J. Chang, P. Palan, J. Basu, R. Tachezy, R. Lewis, and S. Romney. 1995. Persistent genital human papillomavirus infection as a risk factor for persistent cervical dysplasia. J. Natl. Cancer Inst. 87:1365-1371. [DOI] [PubMed] [Google Scholar]
- 19.Hubert, W. G., T. Kanaya, and L. A. Laimins. 1999. DNA replication of human papillomavirus type 31 is modulated by elements of the upstream regulatory region that lie 5′ of the minimal origin. J. Virol. 73:1835-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hubert, W. G., and L. A. Laimins. 2002. Human papillomavirus type 31 replication modes during the early phases of the viral life cycle depend on transcriptional and post transcriptional regulation of E1 and E2 expression. J. Virol. 76:2263-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hummel, M., J. B. Hudson, and L. A. Laimins. 1992. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 66:6070-6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hummel, M., H. B. Lim, and L. A. Laimins. 1995. Human papillomavirus type 31b late gene expression is regulated through protein kinase C-mediated changes in RNA processing. J. Virol. 69:3381-3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klumpp, D. J., and L. A. Laimins. 1999. Differentiation-induced changes in promoter usage for transcripts encoding the human papillomavirus type 31 replication protein E1. Virology 257:239-246. [DOI] [PubMed] [Google Scholar]
- 24.Klumpp, D. J., F. Stubenrauch, and L. A. Laimins. 1997. Differential effects of the splice acceptor at nucleotide 3295 of human papillomavirus type 31 on stable and transient viral replication. J. Virol. 71:8186-8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowy, D. R., and P. M. Howley. 2001. Papillomaviruses, p. 2231-2264. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 26.Lowy, D. R., R. Kirnbauer, and J. T. Schiller. 1994. Genital human papillomavirus infection. Proc. Natl. Acad. Sci. USA 91:2436-2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lowy, D. R., E. Rands, and E. M. Scolnick. 1978. Helper-independent transformation by unintegrated Harvey sarcoma virus DNA. J. Virol. 26:291-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayer, T. J., and C. Meyers. 1998. Temporal and spatial expression of the E5a protein during the differentiation-dependent life cycle of human papillomavirus type 31b. Virology 248:208-217. [DOI] [PubMed] [Google Scholar]
- 29.McCance, D. J., R. Kopan, E. Fuchs, and L. A. Laimins. 1988. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc. Natl. Acad. Sci. USA 85:7169-7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyers, C. 1996. Organotypic (raft) epithelial tissues culture system for the differentiation-dependent replication of papillomavirus. Methods Cell Sci. 18:201-210. [Google Scholar]
- 31.Meyers, C., J. L. Bromberg-White, J. Zhang, M. E. Kaupas, J. T. Bryan, R. S. Lowe, and K. U. Jansen. 2002. Infectious virions produced from a human papillomavirus type 18/16 genomic DNA chimera. J. Virol. 76:4723-4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyers, C., M. G. Frattini, J. B. Hudson, and L. A. Laimins. 1992. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 257:971-973. [DOI] [PubMed] [Google Scholar]
- 33.Meyers, C., T. J. Mayer, and M. A. Ozbun. 1997. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J. Virol. 71:7381-7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Müller, M., L. Gissman, R. J. Cristiano, X.-Y. Sun, I. H. Frazer, A. B. Jenson, A. Alonso, H. Zentgraf, and J. Zhou. 1995. Papillomavirus capsid binding and uptake by cells from different tissues and species. J. Virol. 69:948-954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozbun, M. A. Infectious human papillomavirus type 31b: purification and infection of an immortalized human keratinocyte cell line. J. Gen. Virol., in press. [DOI] [PubMed]
- 36.Ozbun, M. A., and C. Meyers. 1997. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J. Virol. 71:5161-5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ozbun, M. A., and C. Meyers. 1998. Human papillomavirus type 31b E1 and E2 transcript expression correlates with vegetative viral genome amplification. Virology 248:218-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozbun, M. A., and C. Meyers. 1999. Human papillomavirus type 31b transcription during the differentiation-dependent viral life cycle. Curr. Top. Virol. 1:203-217. [Google Scholar]
- 39.Ozbun, M. A., and C. Meyers. 1998. Temporal usage of multiple promoters during the life cycle of human papillomavirus type 31b. J. Virol. 72:2715-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozbun, M. A., and C. Meyers. 1999. Two novel promoters in the upstream regulatory region of human papillomavirus type 31b are negatively regulated by epithelial differentiation. J. Virol. 73:3505-3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pray, T. R., and L. A. Laimins. 1995. Differentiation-dependent expression of E1∧E4 proteins in cell lines maintaining episomes of human papillomavirus type 31b. Virology 206:679-685. [DOI] [PubMed] [Google Scholar]
- 42.Roden, R. B. S., H. L. Greenstone, R. Kirnbauer, F. P. Booy, J. Jessie, D. R. Lowy, and J. T. Schiller. 1996. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J. Virol. 70:5875-5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roden, R. B. S., R. Kirnbauer, A. B. Jenson, D. R. Lowy, and J. T. Schiller. 1994. Interaction of papillomavirus with the cell surface. J. Virol. 68:7260-7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmid, S. L., R. Fuchs, P. Male, and I. Mellman. 1988. Two distinct subpopulations of endosomes involved in membrane recycling and transport to lysosomes. Cell 52:73-83. [DOI] [PubMed] [Google Scholar]
- 45.Smith, L. H., C. Foster, M. E. Hitchcock, and R. Isseroff. 1993. In vitro HPV-11 infection of human foreskin. J. Investig. Dermatol. 101:292-295. [DOI] [PubMed] [Google Scholar]
- 46.Steinberg, B. M., K. J. Auborn, J. L. Brandsma, and L. B. Taichman. 1989. Tissue site-specific enhancer function of the upstream regulatory region of human papillomavirus type 11 in cultured keratinocytes. J. Virol. 63:957-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stoler, M. H., A. Whitbeck, S. M. Wolinsky, T. R. Broker, L. T. Chow, M. K. Howett, and J. W. Kreider. 1990. Infectious cycle of human papillomavirus type 11 in human foreskin xenografts in nude mice. J. Virol. 64:3310-3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stubenrauch, F., A. M. E. Colbert, and L. A. Laimins. 1998. Transactivation by the E2 protein of oncogenic human papillomavirus type 31 is not essential for early and late viral functions. J. Virol. 72:8115-8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stubenrauch, F., M. Hummel, T. Iftner, and L. A. Laimins. 2000. The E8∧E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J. Virol. 74:1178-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stubenrauch, F., and L. A. Laimins. 1999. Human papillomavirus life cycle: active and latent phases. Cancer Biol. 9:379-386. [DOI] [PubMed] [Google Scholar]
- 51.Stubenrauch, F., H. B. Lim, and L. A. Laimins. 1998. Differential requirement for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J. Virol. 72:1071-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stubenrauch, F., T. Zobel, and T. Iftner. 2001. The E8 domain confers a novel long-distance transcriptional repression activity on the E8∧E2C protein of high-risk human papillomavirus type 31. J. Virol. 75:4139-4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terhune, S. S., W. G. Hubert, J. T. Thomas, and L. A. Laimins. 2001. Early polyadenylation signals of human papillomavirus type 31 negatively regulate capsid gene expression. J. Virol. 75:8147-8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Terhune, S. S., C. Milcarek, and L. A. Laimins. 1999. Regulation of human papillomavirus type 31 polyadenylation during the differentiation-dependent life cycle. J. Virol. 73:7185-7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas, J. T., W. G. Hubert, M. N. Ruesch, and L. A. Laimins. 1999. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal keratinocytes. Proc. Natl. Acad. Sci. USA 96:8449-8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Volpers, C., F. Unckell, P. Schirmacher, R. E. Streeck, and M. Sapp. 1995. Binding and internalization of human papillomavirus type 33 virus-like particles by eukaryotic cells. J. Virol. 69:3258-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walboomers, J. M. M., M. V. Jacobs, M. M. Manos, F. X. Bosch, J. A. Kummer, K. V. Shah, P. J. F. Snijders, J. Peto, C. J. L. M. Meijer, and N. Muñoz. 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 189:12-19. [DOI] [PubMed] [Google Scholar]
- 58.Yee, C., I. Krishnan-Hewlett, C. C. Baker, R. Schlegel, and P. M. Howley. 1985. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am. J. Pathol. 119:361-366. [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou, J., L. Gissmann, H. Zentgraf, H. Müller, M. Picken, and M. Müller. 1995. Early phase in the infection of cultured cells with papillomavirus virions. Virology 214:167-176. [DOI] [PubMed] [Google Scholar]
- 60.zur Hausen, H. 1989. Papillomaviruses in anogenital cancer as a model to understand the role of viruses in human cancers. Cancer Res. 49:4677-4681. [PubMed] [Google Scholar]
- 61.zur Hausen, H., and A. Schneider. 1987. The role of papillomaviruses in human anogenital cancer, p. 245-263. In N. P. Salzman and P. M. Howley (ed.), The Papovaviridae, vol. 2. The papillomaviruses. Plenum Press, New York, N.Y. [Google Scholar]