

Abstract
Streptococcus pneumoniae (pneumococcus) is a major cause of morbidity and mortality world-wide. The initial event in invasive pneumococcal disease is the attachment of encapsulated pneumococci to epithelial cells in the upper respiratory tract. This work provides evidence that initial bacterial adhesion and subsequent ability to cause invasive disease is enhanced by pili, long organelles able to extend beyond the polysaccharide capsule, previously unknown to exist in pneumococci. These adhesive pili-like appendages are encoded by the pneumococcal rlrA islet, present in some, but not all, clinical isolates. Introduction of the rlrA islet into an encapsulated rlrA-negative isolate allowed pilus expression, enhanced adherence to lung epithelial cells, and provided a competitive advantage upon mixed intranasal challenge of mice. Furthermore, a pilus-expressing rlrA islet-positive clinical isolate was more virulent than a nonpiliated deletion mutant, and it out-competed the mutant in murine models of colonization, pneumonia, and bacteremia. Additionally, piliated pneumococci evoked a higher TNF response during systemic infection, compared with nonpiliated derivatives, suggesting that pneumococcal pili not only contribute to adherence and virulence but also stimulate the host inflammatory response.
Keywords: inflammation, Streptococcus pneumoniae, pili, adhesion, pathogenicity islet
Streptococcus pneumoniae (pneumococcus) is a major cause of morbidity and mortality world-wide and represents one of the four major infectious disease killers, together with HIV, malaria, and tuberculosis (1–5). It is a main cause of respiratory tract infections such as otitis media, sinusitis, and community-acquired pneumonia, but also an important pathogen in invasive diseases such as septicemia and meningitis. Even though pneumococcus is a devastating pathogen, it also harmlessly colonizes healthy children attending day-care centers to a high extent (6, 7). The major virulence factor in pneumococcal disease is the polysaccharide capsule, which groups pneumococci into at least 90 different serotypes (8). However, other genetic factors, such as CbpA (choline-binding protein A) and pneumolysin, have been described to be of importance for virulence (9–11).
Infection by S. pneumoniae leads to invasive disease triggered by initial colonization of the nasopharynx, but the mechanisms of adhesion are not well understood. In vitro adhesion of encapsulated pneumococci is much lower than for nonencapsulated nonvirulent derivatives (4), even though capsule expression is essential for successful colonization of the upper airways. These observations suggest that in vivo, pneumococci are adhesive despite the production of a thick capsule (5).
In Gram-positive bacteria, such as Corynebacterium diphtheriae (12, 13), Actinomyces spp. (14), and recently group A streptococci (GAS) and group B streptococci (GBS) (15, 16), pili-like surface structures have been identified by electron microscopy and characterized genetically as well as biochemically (12, 13, 15, 16). In Actinomyces spp. type 1 fimbrial genes mediate adhesion to dental and mucosal surfaces (17). However, among pathogenic Streptococcus spp., no functional data on their physiological role and function in infectious disease are available. Gram-positive pili are extended polymers formed by a transpeptidase reaction involving covalent cross-linking of LPXTG motifs containing subunit proteins assembled by specific sortases. Sortases are also responsible for covalent attachment of the pilus to the peptidoglycan cell wall.
Genetic comparative analysis with other pathogenic Streptococcus spp. allowed for the identification of a pilus-like operon encoding proteins belonging to the microbial surface cell recognition adhesion matrix molecule (MSCRAMM) family of adhesins. Here, we provide evidence that pneumococci have pilus-like structures on their surface encoded by the rlrA pathogenicity islet (18). This islet is present in some but not all clinical pneumococcal isolates. We show that this islet is important for pneumococcal adherence to lung epithelial cells as well as for colonization in a murine model of infection. Likewise, we find that the islet affects the development of pneumonia and bacteremia in mice. Furthermore, pilus-expressing pneumococci evoked a higher TNF response during systemic infections than nonpiliated isogenic mutants, suggesting a role in the host inflammatory response.
Results and Discussion
Evidence by Transmission Electron Microscopy for Pilus-Like Structures in Pneumococci.
By transmission electron microscopy and negative staining we found that pneumococci cultivated for up to 16 h on blood agar plates and in C+Y (5) or THY medium express pilus-like structures. These structures were found on strain T4 (TIGR4), belonging to the highly invasive serotype 4 clone of multilocus sequence type ST205, as well as on a clinical isolate of type 19F, with multilocus sequence type 162 (strain ST16219F) (Fig. 1A). This 19F clone is associated with both carriage and invasive disease in humans, and has been shown to be an efficient colonizer of the respiratory tract of C57BL/6 and BALB/c mice (5). Although a nonencapsulated mutant of T4 (T4R) was able to form pili, no pili were observed on the nonencapsulated laboratory strain R6 (data not shown).
Fig. 1.

Electron microscopic analysis of pneumococcal pili. (A) Negative staining of S. pneumoniae strain T4 showing abundant pili on the bacterial surface. (B) Negative staining of mutant strain T4Δ(rrgA-srtD) showing no pili. (C) Negative staining of the T4Δ(mgrA) mutant showing abundant pili. (D) Negative staining of the T4Δ(rrgA-srtD, mgrA) mutant showing no pili on the bacterial surface. (E) Immunogold labeling of T4 by using anti-RrgA. Anti-RrgA was shown to label the bacterial cell surface, suggesting that RrgA anchors the pilus structure to the cell wall. (F) Immunogold labeling of T4 with anti-RrgB (5 nm) and anti-RrgC (10 nm). Anti-RrgB was shown to decorate the entire pili. (Bar, 200 nm.) (G) High magnification of T4 pili double-labeled with anti-RrgB (5 nm) and anti-RrgC (10 nm). It shows specific labeling of a pilus tip by anti-RrgC as indicated by arrows. (Bar, 100 nm.) (H) Immunogold labeling of the deletion mutant S. pneumoniae T4Δ(rrgA-srtD) with no visible pili on the surface detectable by anti-RrgB and anti-RrgC. (Bar, 200 nm.)
The rlrA Islet in the Pneumococcal Genome Encodes Pili-Like Structures.
Comparison of the spaABC operon from C. diphtheriae (12) and adhesion islet 1 from group B streptococci (16) revealed a cluster of putative pilus genes within the T4 genome (Fig. 2). The pilus genes are located in the previously described S. pneumoniae rlrA pathogenicity islet (18, 19). The pneumococcal rlrA islet consists of seven genes of which rrgA, rrgB, and rrgC are predicted to encode LPXTG-containing microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) that bind to components of the extracellular matrix of the host (20). In addition, the rlrA islet also contains genes for three sortases, srtB, srtC, and srtD, as well as rlrA (rofA-like regulator) a positive regulator of the gene cluster (18) (Fig. 2). The genomic islet is flanked by IS1167 containing inverted repeats, characteristic of mobile genetic elements (Fig. 2). The sequenced strain R6 and its progenitor D39 are lacking the rlrA-pilus islet (Fig. 2). The transcriptional repressor mgrA is located external to the rlrA islet, and is involved in the regulation of the pilus genes (21). Sequence analysis after PCR amplification of the corresponding region in the clinical isolate ST16219F of serotype 19F revealed a homologous gene cluster with 98% identity to the T4 rlrA islet. A small ORF of unknown function in T4 was however absent in the ST16219F isolate (data not shown). Knockout mutants deleted for the mgrA gene of T4 and ST16219F were constructed by PCR ligation mutagenesis, thereby producing strains over-expressing the genes of the rlrA islet. In addition, we deleted the rrgA–srtD region in T4 (Fig. 1B) and ST16219F, as well as in their respective mgrA derivatives. Upon negative staining and electron microscopy the T4 mgrA and ST16219F mgrA mutants were found to produce abundant pili (Fig. 1C), whereas bacteria containing the rrgA–srtD deletion lacked pili altogether (Fig. 1D).
Fig. 2.

Genome organization of the rlrA islet in serotype 4 strain T4 (TIGR4) and comparison with the laboratory strain R6 from available sequences. The 19F strain, ST16219F, shares a similar organization with an overall 98% sequence identity, whereas the nonencapsulated strain R6 and its progenitor D39 are pilus-islet-negative strains. A couple of insertion sequences (IS1167) flank the locus in positive strains [one of the transposases is frame-shifted (fs)], whereas an RUP element (repeat unit in pneumococcus) is identified in the pilus-islet-negative strain. The size of the locus, as well as its relative G+C content, is shown. The position of the negative regulator mgrA is indicated. Also included is the genome organization of the islets encoding pilus structures in Streptococcus agalactiae (16) and C. diphtheriae (13).
Antisera were raised against His-tagged RrgA, RrgB, and RrgC proteins expressed in Escherichia coli, and used in immunogold labeling of the pilus expressed by T4. The RrgA antibodies localized to the bacterial cell surface, suggesting that this protein may associate the pilus to the cell wall. The RrgB antibodies decorated the entire pilus polymer, whereas the RrgC antibodies labeled the tip of the pilus (Fig. 1 E–G). FACS analysis, making use of RrgB-specific antibodies, revealed that 84% and 90% of the cells of T4 and ST16219F, respectively, expressed pili structures. In the mgrA mutant derivatives, almost all bacteria were piliated (Fig. 8, which is published as supporting information on the PNAS web site).
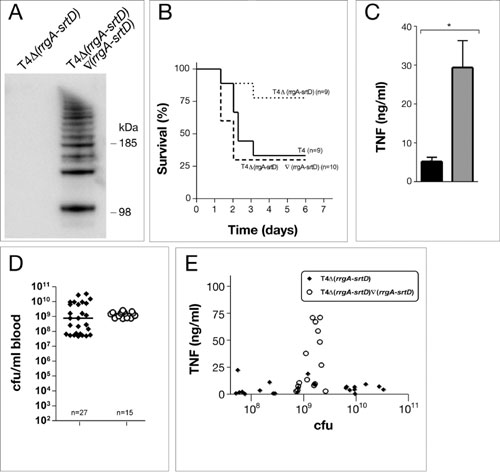
To verify the polymeric nature of the pili structures observed in T4 and ST16219F, total extracts of these strains and their respective rrgA–srtD deletion derivatives were treated with mutanolysin, separated on 4–12% (Fig. 3) polyacrylamide gradient gels, and immunoblotted with antisera specific for RrgB. We observed a ladder of high molecular weight (HMW) polymers ranging from <100 kDa to > 1,000 kDa, similar to those previously described in C. diphtheriae (12, 13). Even though equal amounts of protein extract were loaded onto the gel, the bands stained by the RrgB antibodies were more intense for the mgrA mutants than for their respective wild-type strains, supporting the data from transmission electron microscopy and FACS analysis that a greater percentage of pneumococci expressed pilus structures in the mgrA mutant background. As expected, the deletion mutants of rrgA–srtD, in T4 and ST16219F, respectively, showed no RrgB-reactive bands (Fig. 3A). However, when the pilus islet was reintroduced into the deletion mutant T4Δ(rrgA–srtD), Western blot analysis with the RrgB antiserum detected HMW polymers similar to those for the wild type T4 strain (Fig. 9, which is published as supporting information on the PNAS web site). Interestingly, we found by using Western blotting that pili were present in pneumococcal strains cultivated both in liquid media and on plates, even though we could not always find them by using transmission electron microscopy, suggesting why pili have never been found previously.
Fig. 3.

Detection of pili polymers by Western blot analysis. (A) Western blot using a 4–12% polyacrylamide gradient gel with the RrgB antiserum detects a ladder of HMW polymers in strains expressing pili (T4, T4Δ(mgrA), ST16219F, and ST16219FΔ(mgrA)), whereas the mutant strains lacking pili (T4Δ(rrgA–srtD, T4Δ(rrgA–srtD, mgrA)) and ST16219FΔ(rrgA–srtD)) have no HMW polymers. The mutant mgrA shows an increased intensity when compared with the respective wild type. (B) Western blot with the RrgB antiserum using a 4–12% gradient gel for D39 lacking the islet, the mutant D39 with the rlrA islet introduced (D39∇(rlrA–srtD), and its rlrA deletion derivative (D39∇(rlrA–srtD)∇(rlrA)).
The rlrA Islet Is Important for Pneumococcal Adherence to Lung Epithelial Cells.
The serotype 2 strain D39, like its nonencapsulated derivative R6, lacks the rlrA islet (Fig. 2). The complete rlrA islet from T4 was introduced into D39 (D39∇(rlrA–srtD)). This islet insertion mutant of D39 expressed pili as evidenced by a ladder of HMW polymers based on Western blotting with anti-RrgB (Fig. 3B). Pilus expression in D39∇(rlrA–srtD) was dependent on the positive regulator rlrA, because no HMW polymers were detected in an rlrA mutant derivative of D39∇(rlrA–srtD) (Fig. 3B). D39, D39∇(rlrA–srtD), and D39∇(rlrA–srtD)Δ(rlrA) were used to study adherence to A549 lung epithelial cells (Fig. 4). Only pilus-expressing D39∇(rlrA–srtD) bound to these cells (Fig. 4). This binding was similar to that of pilus-expressing T4, whereas an rlrA mutant of T4 showed no detectable binding to A549 cells. (Fig. 10 A–C, which is published as supporting information on the PNAS web site).
Fig. 4.

Adherence of piliated S. pneumoniae strain D39. (A) Adherence of D39 and D39∇(rlrA–srtD), as well as D39∇(rlrA–srtD)Δ(rlrA) to monolayers of A549 lung epithelial cells. (B–D) Immunofluorescence microscopy of D39 (B), D39∇(rlrA–srtD) (C), and D39∇(rlrA–srtD)Δ(rlrA) (D) adhering to A549 lung epithelial cells. Shown are labeling of pneumococci with anti-capsular antibody (green) and visualization of epithelial F-actin with rhodamine (red).
The rlrA Islet Affects Virulence in Mouse Models.
To investigate the role of the pilus in pneumococcal colonization and in invasive disease, strains T4 and T4Δ(rrgA–srtD) were used in murine infection models. To mimic the natural route of infection, 6- to 8-week-old C57BL/6 mice were inoculated intranasally with high [5 × 106 colony-forming units (cfu)], and medium (5 × 105 cfu) doses of pneumococci. Colonization was estimated by performing nasopharyngeal–tracheal lavages in animals postmortem. The nonpiliated mutant was less virulent than the wild-type strain as revealed by a higher survival rate of mice infected by the mutant (Fig. 5A and B). This defect in virulence could be restored by reintroducing the rlrA islet (Fig. 9B).
Fig. 5.

Piliated pneumococci are more virulent and outcompete nonpiliated mutants. (A–E) Intranasal challenge of C57BL/6 mice with piliated T4 and its isogenic nonpiliated deletion mutant T4Δ(rrgA-srtD). (A and B) Survival of mice after inoculation with 5 × 106 cfu (high dose, A) or 5 × 105 cfu (medium dose, B). Survival was analyzed by using the Kaplan–Meier log rank test. (C–E) In vivo competition infection experiments where T4 and its isogenic mutant T4Δ(rrgA–srtD) were mixed in a ratio of 1:1 before intranasal infection. The competitive index (CI) was calculated as described in Methods; each circle represents the CI for one individual mouse in each set of competition experiments. A CI below 1 indicates a competitive disadvantage of the mutant in relation to the wild-type strain. CI values <10−4 were set to 10−4. All mice were colonized. (C) CI in colonization, pneumonia, and bacteremia after high-dose challenge (n = 20). Of 20 mice, only 14 presented pneumonia (defined as bacteria recovered from the lungs), and 14 were bacteremic. We have shown previously that TIGR4 evoke an inflammatory response in murine lungs (26). (D) CI in colonization after medium dose challenge (n = 10). Of 10 mice, only 5 presented pneumonia and only 1 was bacteremic. (E) CI in colonization after low dose challenge (n = 10). Of 10 mice, only 4 presented pneumonia and none developed bacteremia. (F) CI in colonization and pneumonia after with mixed infection with wild-type D39 and its isogenic pilus islet insertion derivative D39∇(rlrA–srtD), or D39∇(rlrA–srtD)Δ(rlrA) with the rlrA gene inactivated. A CI above 1 indicates a virulence gain by the presence of the rlrA islet in D39∇(rlrA–srtD).
Both wild-type and mutant pneumococci administered separately were able to colonize mice to a similar degree (nonsignficant by Mann–Whitney U test, P > 0.05) (Fig. 10D). However, when equal numbers of wild-type and mutant T4 bacteria were given together intranasally, the pilus-deficient mutant was out-competed by the wild type in the upper airways, lungs, and blood, in the majority of cases (Fig. 5 C–E). The type 2 strain D39, the islet insertion derivative D39∇(rlrA–srtD), and the rlrA mutant D39∇(rlrA–srtD)Δ(rlrA), were also used in competition experiments for nasopharyngeal carriage and pneumonia. The nonpiliated wild-type D39 was out-competed by the piliated islet insertion mutant D39∇(rlrA–srtD), whereas the mutant lacking rlrA was not (Fig. 5F). The present data suggest that pneumococcal pili play a role in colonization, pneumonia, and invasive disease.
The rlrA Islet Plays a Role in Host Inflammatory Responses.
The outcome of a pneumococcal infection is affected by the host inflammatory response, which can promote bacterial clearance as well as contribute to local damage (pneumonia) or systemic damage (of which the most severe form is septic shock). We have recently shown that diverse pneumococcal clones evoke distinct proinflammatory cytokine responses when given i.p. to mice. A serotype 6B strain and the T4 and ST16219F strains, shown here to produce pili, all evoked a high TNF response after i.p. challenge (5). In contrast, a serotype 19F strain of a different clonal type, ST42519F, was not as efficient in colonizing the upper airways of mice and evoked a low TNF response (5). This was also true for a serotype 1 and a serotype 7F isolate (5, 22), which belong to invasive clonal types associated with relatively mild invasive disease and no mortality in humans (22). These clones were analyzed for the presence of the rlrA pilus islet by PCR, sequencing, and Southern blot hybridization (data not shown). Results demonstrated that rlrA islet-positive pneumococcal strains (ST2054 and ST16219F of type 4 and 19F, respectively) elicited a high cytokine response, whereas rlrA islet-negative strains (ST1917F, ST2281, and ST3061 of type 7F and 1, respectively) induced a low TNF response (ref. 5 and data not shown). Presence or absence of the pneumococcal pilus islet might therefore explain the difference in TNF response. To test this possibility directly, the inflammatory response was measured during invasive pneumococcal infection after challenging mice i.p. with piliated wild-type and rrgA–srtD deletion mutants lacking pili. Infections with the two deletion mutants were also performed with higher infection doses to ensure that the low TNF responses was not due to lower numbers of bacteria in the blood stream. The pilus deletion mutants in T4 as well as ST16219F backgrounds showed a significantly lower TNF response (Fig. 6) and IL-6 response (Fig. 7) compared with their respective wild-type strains. By plotting TNF values against bacterial numbers it was evident that the TNF response to piliated pneumococci was significantly higher than to the equivalent number of nonpiliated pneumococci (Fig. 6 C and D). Furthermore, reintroduction of the rlrA islet into T4Δ(rrgA-srtD) restored the high TNF response of piliated T4 (Fig. 9 C, D, and E).
Fig. 6.

Role of the rlrA pilus islet in systemic host inflammatory response. Mice were challenged i.p. with high challenge dose (5 × 106 to 2 × 107 cfu) of T4, ST16219F, and their isogenic mutants T4Δ(rrgA–srtD), and ST16219FΔ(rrgA–srtD) and killed at 6 h after infection. (A) Bacterial outgrowth in blood after high-dose i.p. challenge. Results from individual mice are shown. Horizontal lines represent the medians, and analysis by Mann–Whitney U test gives no significant differences (P > 0.05). (B) Serum TNF response. Data are presented as means and SEMs. Statistical significance was established by Mann–Whitney U test (∗∗, P < 0.0001; ∗, P < 0.001). (C and D) TNF response for individual mice correlated to the bacteremic levels after inoculation with T4 and T4Δ(rrgA–srtD) (C) or ST16219F and ST16219FΔ(rrgA–srtD) (D).
Fig. 7.

Analysis of the IL-6 response for the same i.p. challenges as shown in Fig. 6. Bacterial growth in blood is shown in Fig. 6A. (A) Serum IL-6 response at 6 h after infection. Data are presented as means and SEMs (Mann–Whitney U test; ∗, P < 0.0001). (B) IL-6 response for individual mice correlated to the bacteremia levels after inoculation with T4 and T4Δ(rrgA–srtD).
Concluding Remarks.
We show here that S. pneumoniae produces pilus-like structures, which project from the bacterial cell surface. The pneumococcal pilus is encoded by the rlrA pilus islet, found in some but not all pneumococcal strains. In encapsulated S. pneumoniae, pili contribute to adhesion to cultured epithelial cells, and to colonization and invasive disease in murine models of infection. Pili expression also augments the host inflammatory response. Pneumococci use a variety of mechanisms to interact with their host at different stages of infection. Expression of pili may facilitate the initial bacterial adherence, promoting colonization of the nasopharynx. Simultaneously, bacteria expressing these structures may be more prone to trigger mucosal inflammation that could promote clearance, but potentially could also lead to invasion of pneumococci into the tissue, if inflammation leads to damage of the mucosal barrier. Pneumococcal strains lacking pili, belonging to clones of serotype 1 and 7F, are rarely found among healthy carriers, but may cause invasive disease of a relatively mild character. Hence, presence or absence of the pneumococcal pilus may be an important factor in colonization of the nasopharynx, and once bacteria have gained entrance into the blood stream, it may have a profound effect on the severity of the disease.
Methods
Construction of Pneumococcal Mutants.
Pneumococcal strains, and deletion mutants created in these backgrounds are described in Table 1. To create knockout mutants of T4 and ST16219F we used PCR ligation mutagenesis (23). Fragments upstream and downstream of the target genes were amplified with specific primer pairs. The upstream fragments were constructed with ApaI sites and the downstream fragments with BamHI sites. Primers used for construction and screening of deletion alleles are listed in Table 2, which is published as supporting information on the PNAS web site. The PCR products (1,000 bp) were digested with corresponding restriction enzymes, purified, and ligated with the erm cassette (1,306 bp) (GenBank accession no. AB057644) or the Km–rpsL cassette, Janus (24) (1,368 bp) containing ApaI and a BamHI sites. The ligation mix was then transformed as described in ref. 25 into the recipient pneumococcal strain and plated on blood agar plates containing either erythromycin (1 μg/ml) of kanamycin (400 μg/ml). The correct insertion was confirmed by PCR and sequencing.
Table 1.
S. pneumoniae strains used
| Strain | Relevant characteristics | Source |
|---|---|---|
| T4 | Type 4 strain TIGR4 | www.tigr.org |
| T4Δ(rlrA) | rlrA::erm (EmR) | This study |
| T4Δ(rrgA–srtD) | rrgABC–srtBCD::erm (EmR) | This study |
| T4Δ(mgrA) | mgrA::erm (EmR) | This study |
| T4Δ(rrgA–srtD, mgrA) | (rrgABC–srtBCD::erm)::(mgrA::km–rpsL) (Emr, KmR) | This study |
| T4Δ(rrgA–srtD)∇(rrgA–srtD) | T4Δ(rrgA–srtD) where (rrgABC–srtBCD)::erm (EmR) was replaced by (rrgABC–srtBCD–km) (KmR) | This study, ref. 24 |
| T4R | CmR inactivation of cps4A in T4 | (27, 28) |
| T4RΔ(rrgA–srtD) | rrgABC–srtBCD::erm (EmR) in T4R | This study |
| ST16219F | Clinical isolate of type 19F, excellent colonizer in mice | (5) |
| ST16219FΔ(rrgA–srtD) | rrgABC–srtBCD::erm (EmR) | This study |
| ST16219FΔ(mgrA) | mgrA::erm (EmR) | This study |
| ST16219FΔ(rrgA–srtD, mgrA) | rrgABC–srtBCD::erm (EmR), mgrA::km-rpsL (KmR) | This study |
| D39 | Type 2 strain lacking the rlrA islet | (29) |
| D39∇(rlrA–srtD) | rlrA islet IS167::magellan5 (SpcR, SmR) | This study |
| D39∇(rlrA–srtD)Δ(rlrA) | rlrA islet IS167::magellan5rlrA::magellan2 (SpcR, CmR, SmR) | This study |
EmR, erythromycin-resistant; KmR, kanamycin-resistant; SpcR, spectinomycin-resistant; SmR, streptomycin-resistant; CmR, chloramphenicol-resistant.
To create an insertion mutant of D39 (serotype 2 strain) containing the rlrA islet, competent D39 cells were transformed with genomic DNA from CH155, a serotype 4 S. pneumoniae strain with a magellan5 transposon insertion in one of the IS1167 elements flanking the rlrA islet. The double recombination event was selected for by plating on spectinomycin, and islet presence was confirmed by PCR. To generate an rlrA mutant derivative of D39∇(rlrA–srtD), PCR amplification of the mutated region in the mutant serotype 4 strain was performed with primer pairs RLRAFR/RLRARX and the purified amplicon transformed into required serotype 2 background. The recombination event was selected for by plating on chloramphenicol for rlrA and confirmed by PCR.
Cloning, Expression, and Purification of RrgA, RrgB, and RrgC.
Standard recombinant DNA techniques were used to construct all expression plasmids. pET 21b+ was purchased from Invitrogen. PCRs were performed with Pfu Turbo Taq (Roche) during 25 cycles of amplification with genomic DNA. PCR products were purified, digested, ligated into a His6 expression vector, transformed into E. coli TOPO10, and subsequently subcloned into E. coli BLR(DE3). Recombinant proteins were expressed and purified from transformed bacteria according to the instructions of the manufacturer.
Animal Sera.
Purified recombinant His6-tagged RrgA, RrgB, and RrgC were concentrated with the Centricon YM-30 spin column (Millipore) and subsequently used to immunize BALB/c mice (20 μg) and New Zealand White rabbits (100 μg) (Charles River Laboratory).
Negative Staining.
For negative staining, bacteria were grown on blood agar for up to 16 h and colonies were resuspended in PBS. An aliquot of 4 μl was added to a grid coated with a Formvar supporting film for 5 min. The excess solution was soaked off by a filter paper and the grid was stained with 0.5% uranyl acetate in water for 5 sec and air-dried. The samples were examined in an FEI Tecnai 10 electron microscope (Phillips, FEI, Eindhoven, The Netherlands) at 80 kV.
Immunoelectron Microscopy.
S. pneumoniae was grown overnight in liquid THY medium. One milliliter of bacterial suspension O with an OD600 of 0.5 was centrifuged at 3,000 rpm in an Eppendorf MiniSpin plus at 4°C and resuspended in 500 μl of sterile filtered PBS. Twenty microliters of sample was added to Formvar-coated nickel grids and let stand for 5 min. The grids were subsequently fixed in 1% paraformaldehyde/PBS and incubated with 1:10 polyclonal mouse antibodies to RrgA, RrgB, or RrgC in blocking buffer (1% normal rabbit serum, 1% BSA, 1× PBS). Samples were washed five times for 5 min in blocking buffer and incubated with secondary gold-conjugated antibodies at 1:20 (goat anti-mouse IgG, 5-nm gold particles; goat anti-rabbit IgG, 10 nm). Samples were washed five times in blocking buffer for 5 min, and subsequently fixed for 30 min in 1% paraformaldehyde/PBS. Samples were washed in distilled water five times for 5 min and let dry. Grids were stained for 15 sec with aqueous uranyl acetate and processed in a Tecnai high-field transmission electron microscope.
Western Blotting.
Bacteria were grown on blood agar plates for up to 16 h. Bacteria (30 mg wet weight) were resuspended in 1 ml of 50 mM Tris·HCl, pH 6.8, containing 400 units of Mutanolysin (Sigma) and incubated 2 h at 37°C. After three cycles of freezing and thawing, cellular debris was removed by centrifugation at 13,000 rpm for 15 min. Fifty microliters of the supernatant was treated with NuPage sample buffer and mercaptoethanol for 10 min at 70°C, and 10 μl was loaded on a 4–12% or 3–8% NuPage Novex Bis-Tris Gel (Invitrogen). The electroblotting and detection with RrgB antibody (mouse immune sera) diluted 1:500 was performed according to Invitrogen’s instructions.
A549 Adherence Assays.
S. pneumoniae cells grown to midexponential phase (OD600 = 0.3–0.4) were incubated with A549 cells for 30–40 min at 37°C in a 5% CO2/95% air atmosphere, and washed three times with PBS (pH 7.4) to remove nonadherent bacteria. For enumeration of adherent and/or internalized bacteria, epithelial cells were detached from the wells by treatment with 200 μl of 0.25% trypsin/1 mM EDTA and lysed by the addition of 800 μl of ice-cold 0.025% Triton X-100. Appropriate dilutions were plated on blood agar plates to count the number of bacteria adherent to the eukaryotic cells. The titer of adherent bacteria for each strain was compared to the input titer, and the percentage of adherent bacteria was determined.
For fluorescence microscopy, A549 monolayers were grown on coverslips in 24-well tissue culture plates. Infected cell layers on coverslips were fixed in 3% paraformaldehyde and labeled with antibodies after the 30- to 40-min incubation and washing with PBS. Bacteria were labeled with anti-capsular antibody and epithelial cells were visualized after permeabilization by staining F-actin with rhodamine-conjugated phalloidin. All experiments were performed in quadruplicate and each experiment was replicated three times on different days.
Mouse Challenge.
T4 and ST16219F and their respective isogenic mutants were grown for 16 h on blood agar plates at 37°C under 5% CO2. Colonies were taken directly from plates and resuspended gently in PBS to OD620 = 0.5 or inoculated into semisynthetic C+Y (Casamino acids + yeast extract) medium and grown to midlogarithmic phase (OD620 = 0.5) for intranasal inoculation, and OD620 = 0.2 for i.p. inoculation. Appropriate dilutions were made to obtain the desired concentration. Six- to 8-week-old C57BL/6 mice were used for intranasal and i.p. bacterial challenge of T4, and ST16219F and their mutants as described in ref. 5. D39 and its isogenic mutants were grown in THY (Todd–Hewitt + 0.05% yeast extract) broth supplemented with appropriate antibiotics. Six- to 10-week-old female CD1 (UK) mice (Charles River Laboratories) were used for intranasal challenge with 1 × 107 bacteria.
For competition experiments, mutant and wild-type bacteria were mixed in a 1:1 ratio. The output of mutant cfu compared to the wild-type cfu was determined by selection on erythromycin, streptomycin, and/or chloramphenicol blood agar plates. In vivo competition indices (CI) were calculated as the ratio of mutant to wild-type output cfu divided by the mutant to wild-type input cfu.
Determination of TNF and IL-6 after i.p. challenge in serum was performed by using commercial ELISA kits (BD Biosciences).
Statistical Analysis.
Data were analyzed for statistical significance by using GraphPad prism Version 4. Continuous variables were compared by using the t test or the nonparametric Mann–Whitney test. Statistical significance was defined as P < 0.05.
FACS Analysis and Creation of Revertant in T4Δ(rrgA–srtD).
These techniques are described in Supporting Methods, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
We thank Ingrid Andersson, Gunnel Möllerberg, and Christina Johansson for excellent technical assistance. We also thank Marco Tortoli for animal care and Markus Hilleringmann for the RrgA antibody, Ilaria Ferelinghi and Fabiola Giusti for electron microscopy work, Sandra Nuti and Simona Tavarini for FACS analysis, and Giorgio Corsi for the artwork. This work was supported by grants from the Swedish Research Council, Svenska Läkaresällskapet, the European Union Project PREVIS within the 6th Framework Programme Torsten and Ragnar Söderbergs Foundation, the Foundation for Strategic Research in Sweden (SSF), and the Wellcome Trust (U.K.) through the award of a clinical training fellowship (to C.H.).
Abbreviations
- HMW
high molecular weight
- cfu
colony-forming units
Footnotes
Conflict of interest statement: M.A.B., M.M., V.M., and R.R. are employees of Chiron Corporation.
References
- 1.Bruyn G. A. W., van Furth R. Eur. J. Clin. Microbiol. Infect. Dis. 1991;10:897–910. doi: 10.1007/BF02005442. [DOI] [PubMed] [Google Scholar]
- 2.Ryan M. W., Antonelli P. J. Laryngoscope. 2000;110:961–964. doi: 10.1097/00005537-200006000-00014. [DOI] [PubMed] [Google Scholar]
- 3.Cutts F. T., Zaman S. M., Enwere G., Jaffar S., Levine O. S., Okoko C., Oluwalana A., Vaughan S., Obaro A., Leach A., et al. Lancet. 2005;365:1139–1146. doi: 10.1016/S0140-6736(05)71876-6. [DOI] [PubMed] [Google Scholar]
- 4.Swiatlo E., Champlin F. R., Holman S. C., Wilson W. W., Watt J. M. Infect. Immun. 2002;70:412–415. doi: 10.1128/IAI.70.1.412-415.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandgren A., Albiger B., Orihuela C., Tuomanen E., Normark S., Henriques-Normark B. J. Infect. Dis. 2005;192:791–800. doi: 10.1086/432513. [DOI] [PubMed] [Google Scholar]
- 6.Henriques-Normark B., Christensson B., Sandgren A., Noreen B., Sylvan S., Burman L. G., Olsson-Liljequist B. Microb. Drug Resist. 2003;9:337–344. doi: 10.1089/107662903322762761. [DOI] [PubMed] [Google Scholar]
- 7.Nunes S., Sá-Leão R., Carriço J., Alves C. R., Mato R., Avô A. B., Saldanha J., Almeida J. S., Sanches I. S., de Lencastre H. J. Clin. Microbiol. 2005;43:1285–1293. doi: 10.1128/JCM.43.3.1285-1293.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henrichsen J. J. Clin. Microbiol. 1995;33:2759–2762. doi: 10.1128/jcm.33.10.2759-2762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau G. W., Haataja S., Lonetto M., Kensit S. E., Marra A., Bryant A. P., McDevitt D., Morrison D. A., Holden D. W. Mol. Microbiol. 2001;40:555–571. doi: 10.1046/j.1365-2958.2001.02335.x. [DOI] [PubMed] [Google Scholar]
- 10.Rosenow C., Ryan P., Weiser J. N., Johnson S., Fontan P., Ortqvist A., Masure H. R. Mol. Microbiol. 1997;25:819–829. doi: 10.1111/j.1365-2958.1997.mmi494.x. [DOI] [PubMed] [Google Scholar]
- 11.Tuomanen E. Current Opin. Biol. 1999;2:35–39. doi: 10.1016/s1369-5274(99)80006-x. [DOI] [PubMed] [Google Scholar]
- 12.Ton-That H., Marraffini L. A., Schneewind O. Mol. Microbiol. 2004;53:251–261. doi: 10.1111/j.1365-2958.2004.04117.x. [DOI] [PubMed] [Google Scholar]
- 13.Ton-That H., Schneewind O. Mol. Microbiol. 2003;50:1429–1438. doi: 10.1046/j.1365-2958.2003.03782.x. [DOI] [PubMed] [Google Scholar]
- 14.Kelstrup J., Theilade J., Fejerskov O. Scand. J. Dent. Res. 1979;87:415–423. doi: 10.1111/j.1600-0722.1979.tb00702.x. [DOI] [PubMed] [Google Scholar]
- 15.Mora M., Bensi G., Capo S., Falugi F., Zingaretti C., Manetti A. G. O., Maggi T., Taddei A. R., Grandi G., Telford J. L. Proc. Natl. Acad. Sci. USA. 2005;102:15641–15646. doi: 10.1073/pnas.0507808102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lauer P., Rinaudo C. D., Soriani M., Margarit I., Maione D., Rosini R., Taddei A. R., Mora M., Rappuoli R., Grandi G., Telford J. L. Science. 2005;309:105. doi: 10.1126/science.1111563. [DOI] [PubMed] [Google Scholar]
- 17.Li T., Khah M. K., Slavnic S., Johansson I., Strömberg N. Infect Immun. 2001;69:7224–7233. doi: 10.1128/IAI.69.12.7224-7233.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hava D. L., Hemsley C. J., Camilli A. J. Bacteriol. 2003;185:413–421. doi: 10.1128/JB.185.2.413-421.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hava D. L., Camilli A. Mol. Microbiol. 2002;45:1389–1406. [PMC free article] [PubMed] [Google Scholar]
- 20.Schwarz-Linek U., Hook M., Potts J. R. Mol. Microbiol. 2004;52:631–641. doi: 10.1111/j.1365-2958.2004.04027.x. [DOI] [PubMed] [Google Scholar]
- 21.Hemsley C., Joyce E., Hava D. L., Kawale A., Camilli A. J. Bacteriol. 2003;185:6640–6647. doi: 10.1128/JB.185.22.6640-6647.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sjöström K., Spindler K., Örtqvist Å., Kalin M., Sandgren A., Henriques Normark B. Clin. Infect. Dis. 2006 doi: 10.1086/499242. in press. [DOI] [PubMed] [Google Scholar]
- 23.Lau P. C. Y., Sung C. K., Lee J. H., Morrison D. A., Cvitkovitch G. D. J. Microbiol. Methods. 2002;49:193–205. doi: 10.1016/s0167-7012(01)00369-4. [DOI] [PubMed] [Google Scholar]
- 24.Sung C. K., Li H., Claverys J. P., Morrison D. A. Appl. Environ. Microbiol. 2001;67:5190–5196. doi: 10.1128/AEM.67.11.5190-5196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bricker A. L., Camilli A. FEMS Microbiol. Lett. 1999;172:131–135. doi: 10.1111/j.1574-6968.1999.tb13460.x. [DOI] [PubMed] [Google Scholar]
- 27.Albiger B., Sandgren A., Katsuragi H., Meyer-Hoffert U., Beiter K., Wartha F., Hornef M., Normark S., Henriques Normark B. Cell. Microbiol. 2005;7:1603–1615. doi: 10.1111/j.1462-5822.2005.00578.x. [DOI] [PubMed] [Google Scholar]
- 27.Fernebro J., Andersson I., Sublett J., Morfeldt E., Novak R., Tuomanen E., Normark S., Henriques Normark B. J. Infect. Dis. 2004;189:328–338. doi: 10.1086/380564. [DOI] [PubMed] [Google Scholar]
- 28.Gosink K. K., Mann E. R., Guglielmo C., Tuomanen E. I., Masure H. R. Infect. Immun. 2000;68:5690–5695. doi: 10.1128/iai.68.10.5690-5695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iannelli F., Pearce B. J., Pozzi G. J. Bacteriol. 1999;181:2652–2654. doi: 10.1128/jb.181.8.2652-2654.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}