

Abstract
Acetylcholine receptors (AChRs) mediate synaptic transmission at the neuromuscular junction, and structural and functional analysis has assigned distinct functions to the fetal (α2βγδ) and adult types of AChR (α2βɛδ). Mice lacking the ɛ-subunit gene die prematurely, showing that the adult type is essential for maintenance of neuromuscular synapses in adult muscle. It has been suggested that the fetally and neonatally expressed AChRs are crucial for muscle differentiation and for the formation of the neuromuscular synapses. Here, we show that substitution of the fetal-type AChR with an adult-type AChR preserves myoblast fusion, muscle and end-plate differentiation, whereas it substantially alters the innervation pattern of muscle by the motor nerve. Mutant mice form functional neuromuscular synapses outside the central, narrow end-plate band region in the diaphragm, with synapses scattered over a wider muscle territory. We suggest that one function of the fetal type of AChR is to ensure an orderly innervation pattern of skeletal muscle.
Keywords: AChR, mutant channel, end-plate distribution, innervation pattern, transgene
Introduction
The functional properties of end-plate channels in mammalian muscle change during postnatal development. With differentiation of the postsynaptic membrane, fetal acetylcholine receptors (AChRs) composed of α2βγδ subunits are gradually replaced by adult-type AChRs, where the ɛ-subunit replaces the fetal γ-subunit (Mishina et al, 1986). End-plate channel properties change to shorter mean open time and larger conductance with increased Ca2+ influx (Sakmann & Brenner, 1978; Villarroel & Sakmann, 1996); these attributes are essential to preserve functional neuromuscular synapses (Witzemann et al, 1996; Missias et al, 1997). Fetal-type AChRs, however, specified by the γsubunit, were shown to induce spontaneous contractile activity and thereby proposed to promote formation of the postsynaptic membrane specializations and muscle development. This raised the question of whether adult-type AChRs could replace fetal-type AChRs during development (Jaramillo et al, 1988). Furthermore, AChR activity was suggested to promote myoblast fusion to myotubes (Entwistle et al, 1988).
To address the issue of whether and how fetal-type AChRs might contribute to muscle innervation and muscle development, we have generated mutant mice in which the fetal γ-subunit is replaced by a chimaeric subunit (γɛ) with functional ɛ-subunit-like properties (Herlitze et al, 1996).
Results
The AChRγ subunit gene (Fig 1A) was modified by homologous recombination (Fig 1B) to replace the γsubunit with a subunit composed of the amino-terminal 113 amino acids (aa) of the γ-subunit and the carboxy-terminal 382 aa of the ɛ-subunit (Fig 1C). This chimaeric subunit is referred to as the γɛsubunit. Chimaeric mice were generated and mating between heterozygous males and females generated progeny with all three genotypes in a 129/sv/C57BL/6 mixed genetic background (Fig 1D). Homozygous mice (AChRγ(ɛ/ɛ)) seemed healthy and fertile, and behaved indistinguishably from wild-type littermates.
Figure 1.
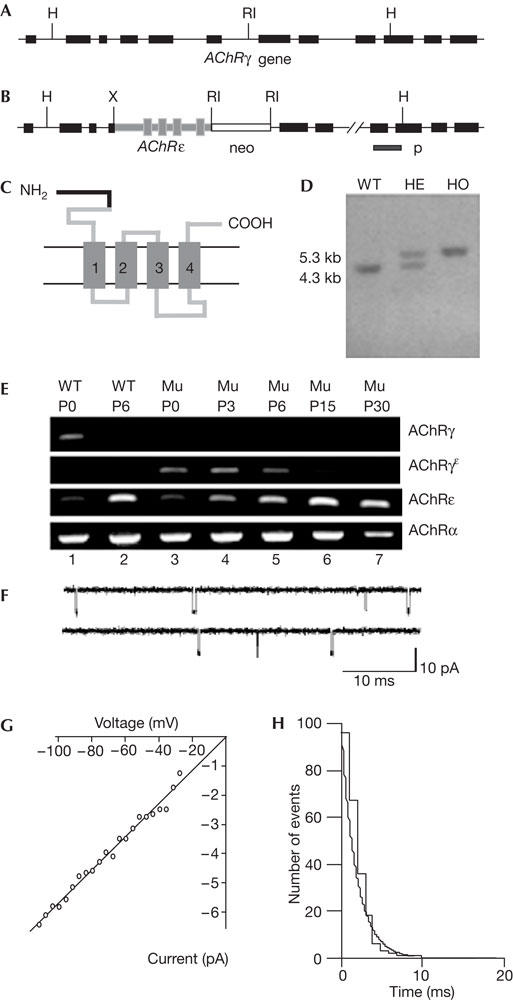
Substitution of the fetal-type acetylcholine receptors (AChRs) by adult-type AChRs. (A) Diagram of the murine AChR γ-subunit gene. Exons are indicated by black boxes. H, HindIII; RI, EcoRI. (B) Targeted allele. X, XbaI. (C) The recombinant γɛ-subunit is composed of the N-terminal 113 aa of the γ-subunit (black) and 382 aa of the ɛ-subunit (grey). Numbered boxes (1–4) indicate transmembrane segments. (D) HindIII digestion and hybridization with a probe (p) from outside the 3′ end of the targeting vector yielded a wild-type band of 4.3 kb and a mutant band of 5.3 kb. WT, wild type; HE, heterozygous; HO, homozygous. (E) RNA-dependent amplification of γ-, γɛ-, ɛ- and α-subunit transcripts of AChRγ(ɛ/ɛ) and WT mice. The γɛ-subunit transcripts were absent from WT. In mutants, only the γɛ-subunit, but not the γ-subunit, mRNA was detectable. Transcription of the γɛ-subunit is downregulated like γ-subunit transcription in WT. (F) Recordings of ACh-activated single-channel end-plate currents in AChRγ(ɛ/ɛ) mice at P2. Inward current is downwards. Mutant mice express only one class of Ach-activated currents. End-plate channel properties are indistinguishable from those in the adult muscle end-plate in conductance and mean open burst time. (G) Current–voltage relation of end-plate channels in AChRγ(ɛ/ɛ) mice measured in the cell-attached recording configuration. (H) Distribution of single-channel current durations fitted by a single exponential with decay time constant of τ=2.4 ms.
Expression profiling was made using AChR γ-, ɛ-, γɛ- and α-fragments generated by reverse transcription–PCR (RT–PCR) of total RNA from the skeletal muscle of neonatal mutant (AChRγ(ɛ/ɛ)) and control (C57BL/6) mice (Fig 1E). At birth, wild-type animals showed pronounced levels of γ- and αsubunit transcripts and low levels of ɛ-subunit transcripts, whereas γɛ-subunit transcripts were not detected (Fig 1E, lane 1). During the developmentally controlled replacement of fetal-type AChR by adult-type AChR (for a review, see Schuetze & Role, 1987), the levels of γ- and ɛsubunit-specific messenger RNAs changed in the wild type beginning at postnatal day 6 (P6), with an increase in ɛ-subunit mRNA and a decline in γ-subunit transcripts. The α-subunit transcript levels remained constant (Fig 1E, lane 2). AChRγ(ɛ/ɛ) mice lacked γsubunit transcripts, whereas α-, γɛ- and ɛ-subunit transcript levels seemed normal at birth (Fig 1E, lane 3). During postnatal development between P3 and P30 (Fig 1E, lanes 4–7), γɛsubunit transcript levels declined continuously and became undetectable at P30, demonstrating an AChR γ-subunit gene-directed regulation of the targeted allele (Witzemann et al, 1987). The α- and ɛsubunit transcript levels (Fig 1E, lanes 3–7) changed with development like the corresponding transcript levels in wild type. Thus, during fetal and neonatal development, mutant mice express exclusively AChR channels composed of α2-, β-, γɛ- and δsubunits. These are replaced postnatally by adult-type AChR composed of α2-, β-, ɛ- and δ-subunits by the same developmental time course as the fetal-type AChR in wild-type mice (for a review, see Schuetze & Role, 1987; Witzemann et al, 1987).
Single-channel currents recorded in the presence of 0.1–0.5 μM ACh from P2 foot muscle end-plates showed that mutant mice express only a single class of end-plate channels (Fig 1F) with a mean open duration of 2.5±0.5 ms (n=5), resembling adult-type end-plate channels (Witzemann et al, 1996). During the postnatal second week, mutant mice continue to express a single class of adult muscle-like channels. Single-channel current–voltage relationships indicated that the conductance of mutant end-plate channels in the neonatal muscle was 61±9 pS (Fig 1G), identical to that of the adult-type AChR channel (62±3 pS; Witzemann et al, 1987). Wild-type mice during this time express both a fetal (mean open duration 7.9±1.2 ms; n=10) and an adult muscle class of end-plate channels (n=5; Mishina et al, 1986; Witzemann et al, 1987). Hence, during their entire lifespan, the AChRγ(ɛ/ɛ) mice exclusively express AChR with the functional properties of the adult-type end-plate channel.
The presence of the fetal-type AChR could be a key signal for the formation of end-plates in embryonic muscle. The replacement of the fetal-type AChR by the adult-type AChR could, in this case, affect the distribution of end-plates within a characteristic ‘band' of end-plates, which is most pronounced in the diaphragm. Indeed, in AChRγ(ɛ/ɛ) mice, the innervation pattern of the diaphragm is markedly altered. The pattern seemed disordered and end-plates are scattered over a much wider territory in comparison to the wild type, where the distribution of end-plates fluctuates only a little along the dorsoventral axis of the diaphragm. To quantify the differences in end-plate distribution, we analysed the corresponding segments of diaphragms stained for acetylcholinesterase (Koelle & Friedenwald, 1949) in mutant and wild-type animals at P6 and P118 (segment 4; Fig 2A). The lateral distance of end-plates from the central tendinous segment of the muscle fibre was measured in each muscle fibre and then averaged over segments of 100 μm width. In mutant animals at P6 (Fig 2B), synapses were more scattered than in the wild type (Fig 2C). When fitted with a single gaussian, the means of half-maximum width of the distribution measured in mutant muscle at P6 (mean±s.d. of three muscles (1,566 synapses): 605.37±83.16 μm) showed an ≈2.2-fold increase compared with wild-type muscle at P6 (mean of three muscles (1,509 synapses): 267.74±13.73 μm). In adult mutant diaphragm muscle (Fig 2D) examined at the age of P118, the scatter of end-plates was also larger than in wild-type mice (Fig 2E). The much broader territory containing mutant end-plates (mean of three muscles (P118–P120) (1,778 synapses): 1,189.77±93.6 μm) was reflected by the same ≈2.2-fold increase of the half-maximum width of a gaussian fitted to their distribution and compared with wild-type muscle (mean of three muscles (P118–P120) (1,517 synapses): 530.37±17.33 μm). This observation indicated that the large changes in synaptic patterning present shortly after birth (P6) persist in the adult muscle. The broadening of the end-plate zone is observed throughout the entire diaphragm muscle and is not restricted to selected segments (see also Fig 3). In one muscle at P6, the distribution of end-plates in segment 4 was fitted better by the sum of two gaussians, an observation that might reflect the increased splitting of the phrenic nerve (supplementary Fig 1A online).
Figure 2.
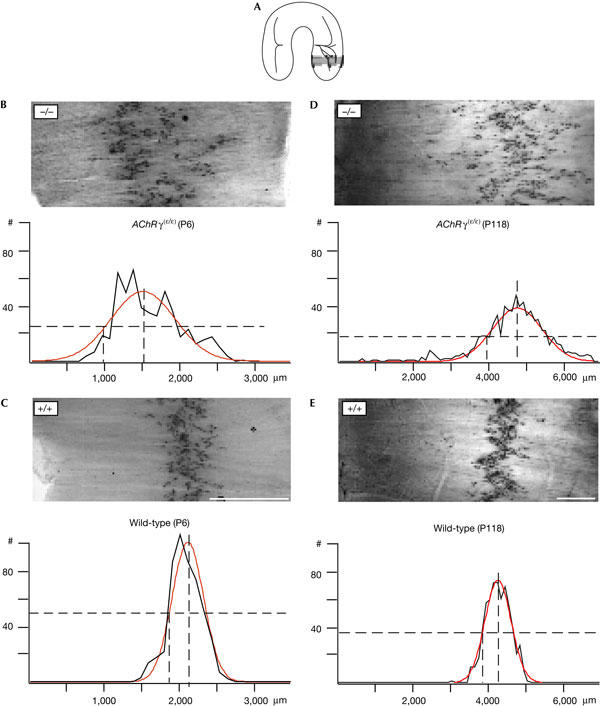
Scattering of end-plate locations in diaphragm muscle. (A) Segments of diaphragm that were compared at P6 and in adult animals. The locations of end-plates in the boxed portion of the diaphragm were measured in AChE-stained muscle as the distance from the tendinous muscle insertion using IP lab (Scanalytics). Data were copied to Excel and the distribution was calculated using Igor (WaveMetrics). (B) AChRγ(ɛ/ɛ) mice at P6 (n=537 fibres, one muscle). Upper panel: end-plates are spread over a large muscle territory. Lower panel: the fitted gaussian distribution is superimposed on a histogram of distances (black; peak height=51.86±4.65; x0=1,523.7±68.8 μm; half-maximal width=578.73±68.8 μm). (C) Wild-type mice at P6 (n=510 fibres, one muscle). Upper panel: end-plates form a narrow band. The lower panel shows a gaussian distribution superimposed on the histogram of distances (peak=102.66±3.95; x0=2,059.6±8.5 μm; half-maximal width=276.83±12.9 μm). (D) AChRγ(ɛ/ɛ) mice at P118 (n=755 fibres, one muscle): distribution indicates laterally displaced and scattered locations of end-plates. Gaussian fit is in red (peak=39.72±1.1; x0=4,839.4±20.7 μm; half-maximal width=937.8±33.7 μm). (E) Wild-type mice at P118 (n=630 fibres, one muscle): end-plates are concentrated in a narrow band (peak=74.94±1.43; x0=4,316.5±7.45 μm; half-maximal width=486.42±11.1 μm). #, number of synapses/100 μm. Dashed lines indicate peak and half-maximum. Scale bar, 1 mm. Distributions are shown in black. Fitted Gaussians are shown in red. Values±1 s.d.
Figure 3.
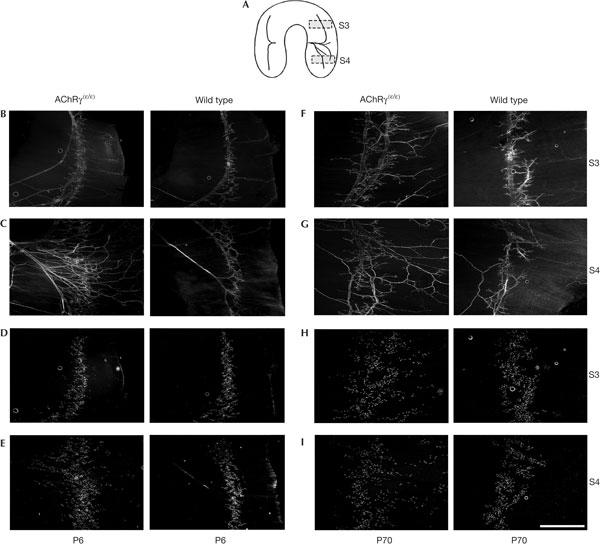
Pattern of nerve branching and distribution of end-plates in diaphragm muscle of mutant and wild type at P6 and P70. (A) Scheme of the innervation of the diaphragm indicating the approximate location of segments 3 (B,D,F,H) and 4 (C,E,G,I) that were analysed. (B,C) Neurofilament staining at P6 shows a progressive rate in branching of the phrenic nerve as compared with wild type. (D,E) r-bgt-stained end-plates in the same muscle segments 3 (D) and 4 (E) show larger scatter of end-plate locations in mutants. (F,G) Neurofilament staining in segments 3 (F) and 4 (G) at P70 shows massively increased branching in mutant compared with wild type. (H,I) r-bgt-stained AChRs in the same segments 3 (H) and 4 (I). Scale bar, 1 mm for all panels.
To examine how the branching pattern of the diaphragm's motor nerve (nervus phrenicus) and the structure of individual nerve terminals of motoneurons were affected, segments (Fig 3A) of mutant and wild-type diaphragm from P6 and P70 mice were labelled with antibodies directed against neurofilament 150 kDa that specifically labels nerve fibres, and simultaneously with rhodamine-labelled α-Bungarotoxin (r-bgt) that specifically stains end-plates. In AChRγ(ɛ/ɛ) mice at P6, the main nerve trunk of the phrenic nerve splits after its entry into the diaphragm into many more branches than in wild-type mice (Fig 3B,C). Corresponding to the changes observed with AChE staining, the AChR distribution apparent in r-bgt-labelled muscles at P6 showed a pattern with end-plates scattered over a much wider territory compared with the wild type (Fig 3D,E). At P70, the territory innervated by the highly branched phrenic nerve was still enlarged (Fig 3F,G) and the distribution of synapses in the end-plate band remained wide (Fig 3H,I). These results suggest that, in AChRγ(ɛ/ɛ) mice, a specific developmental mechanism is lacking or compromised that is required to establish neuromuscular synapses in the central portion of myofibres early during development. Further, the disordered innervation pattern is not rescued later in the adult muscle. In this context, AChR-type-dependent voltage changes at the nascent end-plate might be a factor that regulates convergence of postsynaptic AChR clusters and ingrowing motor nerve. Quantitative fluorescence measurements of r-bgt-labelled end-plate AChR showed no significant changes of mutant and wild-type AChR densities (supplementary Fig 1B–D online).
To assess whether the cellular and subcellular architectures of nerve terminals and muscle end-plates were altered in AChRγ(ɛ/ɛ) animals, diaphragm muscle was doubly labelled with r-bgt and antibodies directed against neurofilament 150 kDa. Confocal microscopy showed that, independent of their localization, nerve terminals and end-plates appear similar in AChRγ(ɛ/ɛ) mutant and wild-type mice (Fig 4A,B). Dual stain with r-bgt and antisynaptophysin antibodies perfectly superimposed and AChRs are apposed to nerve terminals in mutant as in wild-type mice (Fig 4C,D). Consistently, the distribution of agrin (McMahan, 1990) is unaltered (supplementary Fig 1E online) and the postsynaptic aggregating protein MuSK (Gautam et al, 1995; DeChiara et al, 1996; Sanes & Lichtman, 2001) seemed comparable in crosssections through soleus muscle of mutant and wild-type mice (Fig 4E,F). These results indicate that mutant neuromuscular synapses, although distributed differently in the innervation zone, are the same as wild type.
Figure 4.
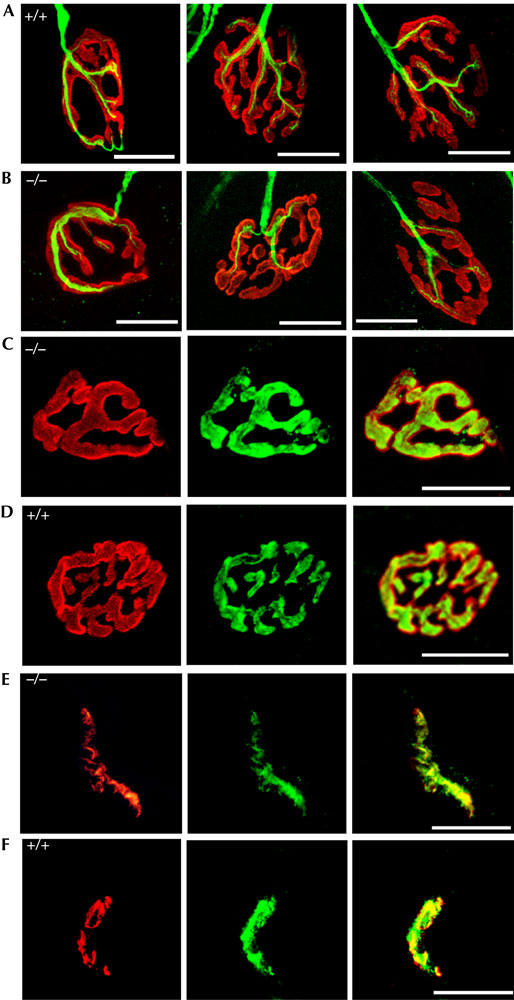
Pre- and postsynaptic molecular architecture of neuromuscular junctions at P70. (A,B) Innervation at the level of individual synapses in mutant (A) and control (B) diaphragm shows no differences. Neurofilament is in green and AChR in red. (C,D) Immune staining of presynaptic synaptophysin (green) and r-bgt-stained postsynaptic AChR (red) superpose perfectly in end-plates of diaphragm muscle of AChRγ(ɛ/ɛ) (C) and control mice (D). (E,F) MuSK staining (green) in cross-sections of mutant (E) and control soleus muscle (F) counter-labelled with r-bgt (red).
Muscle morphology of mutant mice, analysed by comparing haematoxylin/eosin-stained cross-sections through soleus muscle from mutant and wild-type mice at P70, showed no differences in size, density and number of fibres (supplementary Fig 1F online), indicating that localization of end-plates in the central innervation zone is not essential for the development of contractile muscle. Mutant end-plates act properly outside the narrow and central end-plate band present in wild type, and did not show differences in secondary postsynaptic folds (supplementary Fig 1G online). Mutant animals have the same ability to maintain balance and locomotor coordination as their wild-type littermates (data not shown).
Thus, in AChRγ(ɛ/ɛ) mice, myoblasts differentiate into muscle fibres as in wild-type mice, demonstrating that the AChR γ-subunit and, consequently, fetal-type AChRs are not required for terminal differentiation into contractile muscle (Takahashi et al, 2002). The molecular architecture of the junctions is indistinguishable between wild-type and mutant mice, and the main difference is their altered distribution and the disordered pattern of innervation by the motor nerve.
Discussion
It has been shown that pre-patterning of AChRs is a process that is intrinsic to muscle and independent of agrin, neuregulin, and even independent of innervation (Arber et al, 1999; Yang et al, 2000, 2001; Lin et al, 2001). Furthermore, the phenotype of choline acetyltransferase (ChAT)-deficient mouse embryos suggested that ACh release from the motor nerve is required to restrict axon growth, to generate functional neuromuscular synapses and to establish the characteristic central end-plate band (Misgeld et al, 2002; Brandon et al, 2003). However, whether presynaptically released ACh or postsynaptic induced AChR-dependent current is the signal that determines the site at which a synapse is formed remains unknown. The broadening of the central end-plate band in aneural muscle (Lin et al, 2001; Yang et al, 2001) and in the muscle of ChAT-null mice (Misgeld et al, 2002; Brandon et al, 2003) suggests that transmitter release is a signal that may determine the size of a muscle territory susceptible to innervation. Alternatively, postsynaptic AChR channel activity induced by ACh released from growing nerve terminals (Young & Poo, 1983) might promote precise target recognition and control the size of the region where the end-plate band is formed. In the latter case, genetic manipulations that alter AChR properties should modify the fraction of the muscle surface covered by end-plates.
Our results clearly show that patterning of end-plates in a central ‘innervation band' of the diaphragm is dependent on the expression of fetal-type AChR channels during embryonic development. Substitution of the normally expressed γ-subunit by the γɛ-subunit caused expression of adult-type AChR channels (α2βγɛδ) with much shorter (three- to fivefold) mean open time, however, with larger (1.5-fold) conductance and increased Ca2+ permeability (Sakmann & Brenner, 1978; Villarroel & Sakmann, 1996). The differences in conductance and gating between fetal- and adult-type end-plate channels are expected to decrease the charge transfer during the end-plate potential, at very early stages of innervation. It has been suggested that the long durations of currents mediated by fetal-type AChR are essential for spontaneous muscle activity (Jaramillo et al, 1988), which may be required for AChR cluster stabilization at a particular site. How substitution of fetal-type AChR by adult-type AChR activity is converted into an altered distribution of neuromuscular synapses remains to be determined. We show that synapse distribution is coupled to the size and duration of currents and determined by the subunit composition of postsynaptic AChRs. Due to the size and duration of end-plate currents evoked at nascent synapses, fetal-type AChR may induce Ca2+ influx through voltage-dependent Ca2+ channels and thereby promote the structural organization of the central and narrow end-plate band. In the case of adult-type AChRs, however, specified by their much shorter mean open time, this Ca2+ influx might be reduced to a lower level interfering with an orderly establishment and the stabilization of initial nerve–muscle contacts. Thus, AChR subtype-controlled submembranous Ca2+ levels could be a determinant for the branching pattern of motor nerves in the available muscle territory and determine the size of the muscle surface covered by end-plates.
In the developing postnatal central nervous system (CNS), changes in subunit composition of the NMDAR and GABAR channels are thought to be essential for their accumulation and localization (Monyer et al, 1994; Essrich et al, 1998). Possibly, in an analogy to the muscle innervation, the subunit composition of these receptors may contribute to specify the detailed innervation pattern in CNS circuits.
Methods
Reverse transcription–PCR analysis. AChR γ-, mutant γɛ-, ɛ- and α-subunit expression were assayed by amplification of reverse-transcribed total RNA isolated from normal and mutant skeletal muscle using Hot Star Taq Polymerase (Qiagen, Hilden, Germany). RT–PCR products were obtained after 15 min at 94°C, followed by 30 cycles of a ‘touchdown' PCR with 45 s at 94°C, 45 s annealing at 57°C (ɛ,γɛ) or 67°C (α,γ), and a 130 s synthesis step at 72°C. The annealing temperature decreased by 0.25°C every cycle to the ‘touchdown' annealing temperature of 49.5°C (ɛγɛ) or 59.5°C (αγ amplification was finished by a 10 min step at 72°C). Primers in 5′ to 3′ orientation are as follows: γ: forward GATGCGAAACTACGACCCC; reverse AGGAGGAGCGGAAGATGG; γɛ: forward GGTGCGACTATCGCCTGCGC; reverse AGATGAACTCCACCTCCT; ɛ: forward GATTGGCATTGACTGGCACG; reverse CCACTCCAAACTGCCCATC; α: forward ATGACGGCTCTGTGGTGG; reverse CGGTGGTGTGTGTTGATGAC.
Anti-neurofilament staining of adult diaphragm. Diaphragm muscles were dissected, rinsed in phosphate-buffered saline (PBS) and incubated in collagenase (Type IA, Sigma, Munich, Germany, C-9891)/PBS (1 mg/ml) for 15 min at 22°C, rinsed in PBS and fixed in 1% formaldehyde (60 min). Subsequently, the tissue was rinsed in PBS and incubated in 0.1 M glycine/PBS at 4°C overnight. Connective tissue was removed and tissue was permeabilized in 1% Triton X-100/PBS at 22°C for 8 h. Antibody staining was performed using a 1:500 dilution of an anti-neurofilament 150 kDa antibody (AB1981, Chemicon, Hofheim, Germany) in 2% bovine serum albumin (BSA)/PBS at 4°C for 36 h. Samples were washed for 3 × 20 min in PBS and incubated at 4°C overnight using a 1:100 dilution of a fluorescently labelled secondary antibody (B-2766, Molecular Probes) in 2% BSA/PBS. Muscles were washed in 2% BSA/PBS (3 × 20 min), rinsed in PBS and mounted in Citifluor AF2 (glycerol solution/Plano).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
We thank Dr S. Petrou and Dr J. Waters for critically reading the manuscript, Dr R. Kühn and Dr K. Rajewsky for providing the ES cell line E14-1 and Dr M. Rüegg for providing the anti-MuSK antibody. We acknowledge the excellent technical assistance of U. Mersdorf, U. Warnke and K. Barenhoff. The lab of A.V. was supported by the Natural Sciences and Engineering Research Council of Canada.
References
- Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, Sockanathan S (1999) Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron 23: 659–674 [DOI] [PubMed] [Google Scholar]
- Brandon EP et al. (2003) Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci 23: 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara TM et al. (1996) The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 85: 501–512 [DOI] [PubMed] [Google Scholar]
- Entwistle A, Zalin RJ, Warner AE, Bevan S (1988) A role for acetylcholine receptors in the fusion of chick myoblasts. J Cell Biol 106: 1703–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Lüscher B (1998) Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nat Neurosci 1: 563–571 [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP (1995) Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 377: 232–236 [DOI] [PubMed] [Google Scholar]
- Herlitze S, Villarroel A, Witzemann V, Koenen M, Sakmann B (1996) Structural determinants of channel conductance in fetal and adult rat muscle acetylcholine receptors. J Physiol (Lond) 492: 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo F, Vicini S, Schuetze SM (1988) Embryonic accetylcholine receptors guarantee spontaneous contractions in rat developing muscle. Nature 335: 66–68 [DOI] [PubMed] [Google Scholar]
- Koelle GB, Friedenwald JS (1949) A histochemical method for localizing cholinesterase activity. Proc Soc Exp Biol Med 70: 617–622 [DOI] [PubMed] [Google Scholar]
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF (2001) Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 410: 1057–1064 [DOI] [PubMed] [Google Scholar]
- McMahan UJ (1990) The agrin hypothesis. Cold Spring Harb Symp Quant Biol 55: 407–418 [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR (2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36: 635–648 [DOI] [PubMed] [Google Scholar]
- Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, Methfessel C, Sakmann B (1986) Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature 321: 406–411 [DOI] [PubMed] [Google Scholar]
- Missias AC, Mudd J, Cunningham JM, Steinbach JH, Merlie JP, Sanes JR (1997) Deficient development and maintenance of postsynaptic specializations in mutant mice lacking an ‘adult' acetylcholine receptor subunit. Development 124: 5075–5086 [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12: 529–540 [DOI] [PubMed] [Google Scholar]
- Sakmann B, Brenner HR (1978) Change in synaptic channel gating during neuromuscular development. Nature 276: 401–402 [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (2001) Induction, assembly, maturation and maintenance of postsynaptic apparatus. Nat Rev Neurosci 2: 791–805 [DOI] [PubMed] [Google Scholar]
- Schuetze SM, Role LW (1987) Developmental regulation of nicotinic acetylcholine receptors. Annu Rev Neurosci 10: 403–457 [DOI] [PubMed] [Google Scholar]
- Takahashi M, Kubo T, Mizoguchi A, Carlson CG, Endo K, Ohnishi K (2002) Spontaneous muscle action potentials fail to develop without fetal-type acetylcholine receptors. EMBO Rep 3: 674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroel A, Sakmann B (1996) Calcium permeability increase of end-plate channels in rat muscle during postnatal development. J Physiol (London) 496: 331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzemann V, Barg B, Nishikawa Y, Sakmann B, Numa S (1987) Differential regulation of muscle acetylcholine receptor γ- and ɛsubunit mRNAs. FEBS Lett 223: 104–112 [DOI] [PubMed] [Google Scholar]
- Witzemann V, Schwarz H, Koenen M, Berberich C, Villarroel A, Wernig A, Brenner HR, Sakmann B (1996) Acetylcholine receptor ɛsubunit deletion causes muscle weakness and atrophy in juvenile and adult mice. Proc Natl Acad Sci USA 93: 13286–13291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Li W, Prescott ED, Burden SJ, Wang JC (2000) DNA topoisomerase IIβ and neural development. Science 287: 131–134 [DOI] [PubMed] [Google Scholar]
- Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, Birchmeier C, Burden SJ (2001) Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 30: 399–410 [DOI] [PubMed] [Google Scholar]
- Young SH, Poo MM (1983) Spontaneous release of transmitter from growth cones of embryonic neurons. Nature 305: 634–637 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information