

Abstract
Unknown mechanisms exist to ensure that exons are not skipped during biogenesis of mRNA. Studies have connected transcription elongation with regulated alternative exon inclusion. To determine whether the relative rates of transcription elongation and spliceosome assembly might play a general role in enforcing constitutive exon inclusion, we measured exon skipping for a natural two-intron gene in which the internal exon is constitutively included in the mRNA. Mutations in this gene that subtly reduce recognition of the intron 1 branchpoint cause exon skipping, indicating that rapid recognition of the first intron is important for enforcing exon inclusion. To test the role of transcription elongation, we treated cells to increase or decrease the rate of transcription elongation. Consistent with the “first come, first served” model, we found that exon skipping in vivo is inhibited when transcription is slowed by RNAP II mutants or when cells are treated with inhibitors of elongation. Expression of the elongation factor TFIIS stimulates exon skipping, and this effect is eliminated when lac repressor is targeted to DNA encoding the second intron. A mutation in U2 snRNA promotes exon skipping, presumably because a delay in recognition of the first intron allows elongating RNA polymerase to transcribe the downstream intron. This indicates that the relative rates of elongation and splicing are tuned so that the fidelity of exon inclusion is enhanced. These findings support a general role for kinetic coordination of transcription elongation and splicing during the transcription-dependent control of splicing.
Keywords: Yeast, 6-azauracil, RPB2, exon definition, intron definition
INTRODUCTION
In eukaryotes, the coding regions of genes are interrupted by noncoding introns that are removed from the precursor messenger RNA (pre-mRNA) by the splicing apparatus (for review, see Staley and Guthrie 1998; Brow 2002). Association between the U1 snRNP at the 5′ splice site and factors bound at the branchpoint near the 3′-end of the intron occurs in the early splicing complexes (i.e., the commitment complex) in vitro (Abovich et al. 1994; Abovich and Rosbash 1997). This critical step in splicing determines which splice sites will be paired and, thus, which exons will be included in the mature mRNA. Given that 5′ and 3′ splice sites seem universally compatible and that the nearest splice sites are not always paired (Smith and Valcarcel 2000), an average human gene with eight introns and seven internal exons could, in theory, generate as many as 128 different spliced RNAs, rather than the one to three bona fide mRNAs usually observed. Although many observations show that binding of splicing regulatory proteins can activate or repress the use of nearby splice sites (Lopez 1998), less is known about the general mechanisms that limit undesired pairing of the many possible combinations of authentic splice sites, so that all the exons are included.
The idea that some splice site pairing combinations are excluded by their order of transcription was originally expressed as the “first come, first served” model (Aebi and Weissman 1987). A prerequisite of this model is that the events leading to splice site pairing occur during transcription of the pre-mRNA. The model fell into disfavor for many years with the abundant demonstrations that accurate splicing can occur without transcription in vitro and, thus, that simple splicing events have no absolute requirement for ongoing transcription. However, natural genes are far more complex than the simple splicing substrates usually used. Since that time, numerous temporal and physical links between transcription and splicing in vivo have been established (for reviews, see Bentley 2002; Howe 2002; Neugebauer 2002; Proudfoot et al. 2002). It is not known how these connections influence the dynamics of splice site choice and exon inclusion.
The effects of promoter elements on alternative splicing have raised new interest in the first come, first served model. Promoter structure or the presence of pause sites in the template can influence the decision to skip or include certain regulated internal exons of the fibronectin and α tropomyosin mRNAs (Cramer et al. 1997; Roberts et al. 1998). Recent studies show that treatments designed to increase elongation stimulate skipping of the fibronectin EDI exon, whereas those that do not, favor exon inclusion (Kadener et al. 2001, 2002; Nogues et al. 2002). Thus, splice site pairing decisions can be influenced by states of ongoing transcription in the very direction predicted by the first come, first served model: delay elongation, favor exon inclusion; advance elongation, favor exon skipping. Exon skipping could be favored simply by allowing the advancing RNA polymerase to synthesize additional 3′ splice sites that compete for an upstream 5′ splice site.
To explore whether ongoing transcription generally influences the fidelity of exon inclusion, we used the simple yeast Saccharomyces cerevisiae. There are at least eight two-intron genes in S. cerevisiae (Blandin et al. 2000; Davis et al. 2000); thus, there are at least eight internal exons for which inclusion in the mature mRNA must be enforced. We previously found that exon inclusion in the two-intron YL8A (RPL7A) pre-mRNA in yeast is enforced in cis by intron self-complementarities that orchestrate correct pairing between each set of splice sites (Howe and Ares 1997). This mechanism is inadequate for genes such as DYN2 (Dick et al. 1996), which clearly lack intron self-complementarities. Here we show that subtle intron 1 branchpoint mutations have significant effects on DYN2 exon skipping. These effects can also be produced by alteration of the U2 snRNP in trans. We find that alterations of the second largest subunit of RNA polymerase II (RNAP II) or treatment of cells with elongation inhibitors partially rescue exon inclusion in dyn2 premRNAs in vivo. Furthermore, the elongation-promoting factor TFIIS enhances exon skipping. The results are consistent with the hypothesis that the relative local rates of splicing complex formation and RNAP II transit are generally coordinated to restrict splice site competition and ensure exon inclusion.
RESULTS
A reporter for studying exon inclusion during the splicing of Dyn2 pre-mRNA
To facilitate the genetic analysis of exon inclusion, we fused part of the DYN2 gene spanning both introns and the internal exon to the CUP1 reporter gene (Fig. 1A ▶, scheme i; Materials and Methods). Levels of Dyn2-Cup1 mRNA measured by reverse transcription (RT) by using a primer specific to CUP1 (Howe and Ares 1997) show that the internal exon is efficiently included in the mRNA (E2+; Fig. 1B ▶, lane 1). We also detect unspliced precursor (P), and singly spliced transcript retaining the second intron (I1−I2+); however, transcripts retaining the first intron (I1+I2−) are present only at very low levels. Similar relative amounts of spliced, partially spliced, and unspliced RNAs are found when the reporter is expressed from a low-copy plasmid (data not shown), indicating that high-copy expression does not greatly affect the splicing pattern. We also detect a weak signal corresponding to mRNAs that have skipped the internal exon (E2−), showing that the internal exon is rarely skipped. The E2− transcript is out of frame, and its biological role, if any, remains unknown. We conclude that the plasmid construct reasonably reflects accurate expression of the two-intron region of the DYN2 gene.
FIGURE 1.

Cloning and in vivo expression of dyn2-Cup1 constructs. (A) PCR amplification and cloning of DYN2 sequences into CUP1 expression vectors. The 5′ and 3′ PCR primers (L and R, respectively) introduce BamHI (b) and KpnI (k) sites for cloning. Lengths of the exons (filled boxes represent coding regions) and introns (lines) are indicated in the “pre-mRNA”. Variability in the DYN2 third exon length (see Materials and Methods) enabled us to use different constructs as Cup1 protein expression reporters for exon inclusion and skipping splicing. In schemes i–iii, full-length or truncated proteins are predicted, depending on the number of coding bases (indicated above) in the exon-included and exon-skipped mRNAs. For example, the pGSC1 plasmids allow Cup1p expression only from the exon included mRNA (scheme i), whereas exon skipped mRNA encodes a truncated polypeptide due to translation termination early in the CUP1 coding region. In contrast, the pGSC3 plasmids (scheme ii) permits Cup1p expression only from mRNAs lacking the internal exon, whereas exon-included mRNAs encode truncated polypeptide. Scheme iii was used for constructs derived from the low/moderate stringency screen described in the text and in Figure 2 ▶. (B) RT analysis of precursor and spliced mRNAs from wild type and mutant dyn2-Cup1 expression constructs (scheme ii translational reading frame). Sequences of the mutated splice sites are indicated (changes from the genomic sequence are shown in lowercase). WT indicates wild-type sequence. Expected products are as follows: P, pre-mRNA containing both introns; I1+I2−, intron 1 retained and intron 2 removed; I1−I2+, intron 1 removed and intron 2 retained; E2+, exon 2 included; E2−, both introns removed as a single intron and exon 2 skipped; and m, pUC13 Sau3AI DNA size markers. Reactions were normalized to cDNA derived from reverse transcription of SCR1, an RNAP III transcript.
Dyn2 pre-mRNA is not spliced through the exon definition pathway
Molecular phenotypes of splicing caused by mutations at the conserved branchpoint sequence and 5′ splice site immediately flanking an internal exon have been used as diagnostic predictors of the so-called exon definition or intron definition pathways (Robberson et al. 1990; Talerico and Berget 1990, Talerico and Berget 1994). We investigated the effects of the diagnostic mutations on DYN2 in the context of the reporter construct. The 5′ splice site flanking the internal exon was altered (Fig. 1B, ▶top panel), and splicing of the mutant pre-mRNA was evaluated. If Dyn2 pre-mRNA is spliced by exon definition, the mutation should activate exon skipping or activate the use of cryptic splice sites within the exon (Berget 1995). If classical intron definition is occurring, the mutation should cause retention of the affected intron or activation of intronic cryptic splice sites (Robberson et al. 1990; Talerico and Berget 1990, Talerico and Berget 1994). Destroying the 5′ splice site downstream of the internal exon (I2Δ5′ss) results in retention of intron 2 but does not activate exon skipping (Fig. 1B ▶, lane 3). Therefore, Dyn2 pre-mRNA is not spliced by the exon definition pathway.
Consistent with an intron definition mechanism, destroying the branchpoint sequence in the first intron (I1ΔBPS) causes retention of the affected intron (Fig. 1B ▶, lane 2; note that levels of the partially spliced I1+I2− RNA are higher than for the wild type pre-mRNA). However, this mutation also greatly activates skipping of the internal exon (E2−). In fact, exon skipping appears to be more efficient than the mutually exclusive removal of the downstream intron (E2− comprises 73.6% of the mature dyn2 RNA versus 26.4% for the I1+I2− RNA). The phenotype of the branchpoint mutation does not fall neatly into the classification used to identify intron versus exon definition (Berget 1995). Exon skipping, rather than simple intron retention or activation of intronic cryptic splice sites, suggests that the distinction between exon and intron definition may not be useful in this case. Destroying any of the remaining conserved splicing signals or increasing the pyrimidine content of the downstream intron failed to activate exon skipping (data not shown). We conclude that the branchpoint region is key to enforcing exon inclusion, and appears necessary to prevent competition by the 3′ splice site of intron 2 that leads to exon skipping.
A broad search for cis-acting exon skipping mutants
In a comprehensive search for other cis-acting elements that enforce exon inclusion, we introduced mutations throughout the DYN2 sequence in the context of the CUP1 fusion plasmid. We selected for the increase in copper resistance (CuR) expected to occur when the internal exon of Dyn2 is skipped during splicing (Fig. 1A ▶, schemes ii, iii). This yielded 26 different plasmids able to grow on media containing up to 500 μM CuSO4, and three different plasmids that confer moderate CuR (growth up to 325 μM, data not shown). Sequencing shows that each plasmid carries between one and 10 mutations, and a total of 140 well-distributed base changes were identified, altering nearly one third of the positions in the 237-bp region tested (Fig. 2A ▶). Surprisingly 28 of the 29 plasmids have a change within or adjacent to the first intron branchpoint (Fig. 2A ▶). The single exception was a deletion in the internal exon that restored the reading frame and allowed expression of the reporter without exon skipping. Because most plasmids shared at least one alteration in the intron 1 branchpoint region and because no other region was consistently altered, we focused on this element.
FIGURE 2.

Cis-acting exon skipping mutations. (A) Schematic of positions altered by mutation. Each bar represents a base position changed within a unique mutant sequence, and the height of the bar indicates the frequency with which a change at that position was identified in the library (many of the sequences carried multiple mutations). Dark bars indicate mutations within the conserved splicing signals. Shown in detail is the branchpoint region in intron 1 (numbering relative to the conserved branchpoint sequence). Changes are indicated beneath the wild type sequence. X indicates XhoI site used for subcloning. (B) Primer extension analysis of exon skipping mutants. Single base mutants derived from the high CuR selection (left gel, lanes 2–10) and low/moderate CuR selection (right gel, lanes 12,13) are shown. Products were normalized to SCR1 cDNA and are labeled as described in Figure 1B ▶.
Single branchpoint region mutations cause widely varying levels of exon skipping
To verify that changes in the intron 1 branchpoint sequence are responsible for activating exon skipping, we resolved multiple mutations into their component single mutations. Nearly every construct carrying a single base change within the branchpoint sequence of the first intron exhibited levels of exon skipping comparable to the multiple mutant from which it was derived (Fig. 2B ▶; data not shown). Mutations at different positions in the branchpoint sequence cause varying levels of skipping (Fig. 2B ▶). Different substitutions at the same position within the branchpoint sequence have different effects on exon skipping (e.g., Fig. 2B ▶, cf. lanes 2 and 3, cf. lanes 7 and 8). The strength of the exon skipping phenotype correlates approximately with the strength of the splicing inhibition observed in single intron substrates with similar mutations (Newman et al. 1985; Fouser and Friesen 1986; Vijayraghavan et al. 1986; Parker et al. 1987; Rain and Legrain 1997). Surprisingly, even mutations in the less well conserved positions of the branchpoint sequence give rise to significant exon skipping. For example, U[1] is conserved in only 87% of natural yeast introns (Rain and Legrain 1997; Grate and Ares 2002), and its mutation has a weak effect in single intron reporters (Rain and Legrain 1997), yet it causes a substantial increase in the amount of exon-skipped RNA (Fig. 2B ▶, lanes 2,3). Mutations near the consensus sequence also cause skipping (lanes 12,13), suggesting that subtle elements of branchpoint sequence context are critical for exon inclusion. These results indicate that exon skipping is a consequence of inefficient recognition of the branchpoint of intron 1.
A mutation in U2 snRNA enhances exon skipping
Given the above results, we tested whether reducing the activity of the U2 snRNP, a generic splicing factor that recognizes the branchpoint, could increase exon skipping. In strains expressing the U2 RNA C41G mutation, slight growth defects are observed only at 37°C (Yan and Ares 1996). This part of U2 is not known to interact directly with the pre-mRNA but is thought to be important for the association of conserved splicing factors (Yan and Ares 1996). When we test the wild-type DYN2-CUP1 reporter in a strain expressing the U2 mutation at 26°C, we observe increased exon skipping (Fig. 3 ▶, lanes 1–3). This exon skipping could be enhanced by sensitizing the reporter plasmid for skipping with the U-to-C mutation at position −2 upstream of the intron 1 branchpoint consensus (U[−2]C). In the presence of wild-type U2, the effect of the sensitizing mutation on the reporter is small (cf. lanes 2 and 4). However, in the presence of only mutant U2, exon skipping increased significantly (Fig. 3B ▶, cf. lanes 3 and 5, lanes 4 and 5). There is no evidence for base pairing between this region of the pre-mRNA and the altered region of U2 snRNA despite the pairing nearby. We conclude that modestly reduced function of the U2 snRNA can lead to exon skipping and reduced fidelity of splicing.
FIGURE 3.

The U2 C41G mutation promotes exon skipping. The U2 snRNA/branchpoint interaction sequence with the bulged adenosine branch residue is shown. Wild-type U2 snRNA is present in lanes 1, 2, and 4 and is absent in lanes 3 and 5. Figure labels are as described in Figure 1B ▶.
RNAP II mutants suppress exon skipping
The above results suggest that slowing the recognition of the intron 1 branchpoint sequence through either cis-acting branchpoint region mutations or trans-acting mutations in the U2 snRNP leads to exon skipping. If this reflects increased competition by the downstream 3′ splice site due to additional time for elongating polymerase to complete the downstream intron, then slowing of RNA polymerase should have the opposite effect and should suppress exon skipping (enhance exon inclusion). To test this, we used two mutations in the gene encoding the second largest subunit of RNAP II, RPB2, because multiple lines of evidence implicate this subunit in the process of transcript elongation (Treich et al. 1992; Powell and Reines 1996; Lennon et al. 1998). One mutant, rpb2-10, has been shown to exhibit a reduced rate of transcript elongation in vitro (Powell and Reines 1996). Both mutants also display sensitivity to the nucleotide-depleting drug 6-azauracil (6AU) in vivo (Powell and Reines 1996), a phenotype associated with defects in transcript elongation (Uptain et al. 1997). Total cellular RNAs expressed from various dyn2 reporters in wild-type and mutant rpb2 strains were analyzed by RT (Fig. 4A,B ▶ ; data not shown). Figure 4 ▶ shows that a modest shift from exon skipping to inclusion occurs in both rpb2 mutant strains. For two different sensitized dyn2 reporters, the fraction of the total dyn2 mRNA as E2+ increases, whereas the E2− fraction decreases. Despite their small magnitudes, these shifts are reproducible and statistically significant (Fig. 4C ▶). Both of rpb2 mutants appear to suppress exon skipping in vivo. Thus, we conclude that reducing the activity of the Rpb2p subunit of RNAP II increases the tendency to include the internal DYN2 exon.
FIGURE 4.

Mutations in RNAP II subunit RPB2 rescue exon inclusion. (A) Splicing of dyn2 U[−2]C. (B) Splicing of dyn2 A[2]G. All reactions were normalized to the E2+ signal before loading on the gel, so that levels of skipping can be evaluated by observing changes in the amount of the E2− signal. (C) Quantitation of dyn2 cDNA signals and calculation of alternative splicing ratios (±SEM; n = 3 for U[−2]C and n = 4 for A[2]G). Each dyn2 cDNA was measured and is expressed as the percentage of the total dyn2 cDNA signal in each lane. Measures of the statistical confidences of E2+, E2−, and E2−/E2+ values relative to the wild-type RPB2 strain were calculated. Student’s t test was applied to compare E2+, E2−, or E2−/E2+ quantities in mutant with the wild-type strain. P values <0.05 indicate that the differences between the compared values are unlikely to be due to sampling error.
Inhibitors of nucleotide biosynthesis suppress exon skipping
Sensitivity to mycophenolic acid (MPA) and the base analog 6AU are hallmarks of defects in transcript elongation in yeast (Nakanishi et al. 1992, 1995; Powell and Reines 1996). Both drugs cause depletion of cellular nucleotide substrate pools required by RNA polymerases (Franklin and Cook 1969; Exinger and Lacroute 1992). In vitro, RNAP II complexes pause and arrest more frequently in the presence of limiting amounts of nucleotides (for review, see Uptain et al. 1997), suggesting the in vivo hypersensitivity to these compounds is due to increased dependence on factors that promote elongation by RNAP II. If exon inclusion is enforced by correct relative rates of transcription elongation and splicing, we would expect these drugs, like the rpb2 mutants, to suppress exon skipping. At drug concentrations that slow but do not arrest growth, both 6AU and MPA shift the splicing pathway back toward exon inclusion (Fig. 5 ▶).
FIGURE 5.

Rescue of exon skipping by treatment with inhibitors of nucleotide biosynthesis. (A) Primer extension reactions of RNAs isolated from untreated (lanes 1,2) cells and from cultures treated with 6AU (lanes 3,4) or MPA (lanes 5,6). Reactions were normalized to the signal for SCR1. (B) Statistical analysis of dyn2 cDNA signals and ratios of exon skipping-to-inclusion. The 1-h or 2-h E2+, E2−, or E2−/E2+; quantities were compared with the 1-h or 2-h MOCK treatment, respectively. The 2-h MOCK was compared with the 1-h measurement. P value indicates the likelihood that the differences between the compared values are due to sampling error.
In the 6AU experiment, the fraction of U[1]A pre-mRNA spliced by the exon inclusion pathway (E2+ relative to all spliced species) increased from ~56% before treatment to 60% within the first hour (P < 0.01; Fig. 5B ▶). This level remains nearly constant into the second hour of treatment. A similar increase in exon inclusion is observed for the MPA treatment after 1 h (67.9%), but increases to 78% at the second hour of drug exposure (P < 0.01). Although these differences are relatively modest, they are statistically significant (Fig. 5B ▶). Importantly, the total dyn2 mRNA levels (percentage total relative to MOCK) increase between the first and second hours of treatment for either drug (data not shown), indicating that differential decay is not an explanation for this effect. Consequently, the faster rate of appearance of E2+ mRNAs relative to E2− mRNAs in the second hour of drug treatment argues that a shift in the choice of splicing pathways from exon skipping to exon inclusion has occurred, and in fact, this change is greater than that observed due to the persistence of RNA made prior to drug addition. The simplest explanation for our results is that reduction in transcript elongation relative to splicing allows more time for intron 1 branchpoint recognition to occur before intron 2 is transcribed, resulting in suppression of exon skipping.
TFIIS-dependent exon skipping can be suppressed by targeting lacI DNA binding to the second intron
Transcript elongation factor TFIIS is encoded by the PPR2 gene in yeast and is not essential for viability (Nakanishi et al. 1992). In vitro, TFIIS activates an intrinsic transcript cleavage activity in arrested polymerases (Izban and Luse 1992). This activity is believed to assist RNAP II in overcoming blocks to elongation by repositioning the 3′ end of the nascent mRNA in the active site of arrested transcription complexes (Reines et al. 1992, 1993; Christie et al. 1994). The lack of TFIIS renders cells hypersensitive to 6AU and is lethal in combination with mutations in RPB2 (Archambault et al. 1992). We tested several different dyn2 reporters in strains expressing different levels of TFIIS. These levels were manipulated with low- and high-copy PPR2-containing plasmids in comparison to a strain expressing no TFIIS. Although no changes in exon skipping are observed for some of the reporters, detectable differences are observed in others (Fig. 6A ▶; data not shown). For example, the U[−2]C reporter displays a modest but reproducible and statistically significant enhancement in exon skipping when TFIIS is present (Fig. 6A ▶). Thus, the presence of TFIIS can stimulate exon skipping, most likely by increasing elongation through the dyn2 template.
FIGURE 6.

A transcriptional block to elongation abrogates TFIIS-dependent exon skipping. (A, upper panel) Presence of TFIIS enhances exon skipping in dyn2 U[−2]C. Strains JAY698 (ppr2Δ) and JAY699 (PPR2) were transformed with pU[−2]C and grown at 30°C. (A, lower panel) Analysis of signals, as described in previous figure legends (n = 3). E2+, E2−, or E2−/E2+ values were compared between TFIIS+ and TFIIS− strains; P values indicate the likelihood that the differences in these values are due to sampling error. (B) Schematic of the pGSlacIAt template with the lacI operator sequence (laco). Yeast strain CMKY21 (ppr2Δ) carrying plasmids pGSlacIAt and a methionine-repressible lacI expression construct were transformed with a high-copy (H) or low-copy (L) PPR2 expression construct or appropriate high-copy or low-copy empty vector (−). (C) Primer extension (upper panel) and graph of exon skipping ratios (lower panel; E2−/E2+ ±SEM, n = 3).
To test whether we could reduce the ability of TFIIS to stimulate dyn2 exon skipping, we engineered a construct with a template that contains an obstacle to elongation through intron 2 (Fig. 6B ▶). A region between the 5′ splice site and branchpoint sequence of the U[−1]G dyn2 reporter on a low-copy plasmid was altered to create the lac operator sequence, and introduced into yeast. The lac repressor (lacI) was conditionally expressed in yeast under the control of a methionine-repressible promoter (Luukkonen and Seraphin 1998). In the absence of expression of the lacI protein, TFIIS produced from a low-copy plasmid detectably stimulates exon skipping (Fig. 6C ▶, cf. lanes 1 and 2). Increased expression of TFIIS on a high-copy plasmid has no further effect (Fig. 6C ▶, cf. lanes 2 and 4). When expression of lacI protein is allowed, TFIIS is no longer capable of stimulating exon skipping (Fig. 6C ▶, lanes 5–8). Expression of the lacI protein has no effect on splicing of transcripts from templates lacking the operator sequence, regardless of whether TFIIS is absent or present (data not shown). Thus, although TFIIS can stimulate exon skipping in dyn2, its ability to do so is relatively weak and can be abolished by expression of lacI, provided the second intron contains a lac operator sequence. In the absence of TFIIS, levels of dyn2 mRNA are not significantly reduced in the presence of lacI protein. In addition, the expression of lacI alone is insufficient to stimulate exon skipping. These observations suggest that the obstacle presented by lacI does not greatly impede transcription even in the absence of TFIIS. Perhaps binding of lacI alters the chromatin structure in a way that does not increase pausing but prevents TFIIS from acting efficiently on transcriptionally arrested complexes. As above, the predicted effects of TFIIS and the lac repressor are consistent with the idea that the relative rates of elongation and definition of intron 1 influence the choice between exon skipping and exon inclusion.
The exon-skipped and exon-included dyn2 RNAs have similar decay rates
Because the E2+ and E2− mRNAs differ only in the presence or absence of the 22-nt internal exon, respectively, only one of these forms is capable of encoding full-length fusion protein (Fig. 1A ▶). In the dyn2-CUP1 constructs used in our cis selection (Fig. 1A ▶, schemes ii, iii), only the E2− mRNA encodes Dyn2-Cup1 protein. The E2+ mRNA is out-of-frame for Cup1p expression and translation terminates prematurely. This can impact the stability of the mRNA through the nonsense-mediated decay (NMD) pathway (He et al. 1993). To determine whether the dyn2 E2+ and E2− mRNAs have significantly different relative stabilities, we used strains that possess or are deficient in NMD. The dyn2 mutant U[−1]A was used because it yields high levels of both (E2+ and E2−) mRNAs.
Reporters expressing mRNA in either of the two translational reading frames consistent with splicing were constructed carrying the sensitizing U[−1]A mutation (Fig. 1 ▶, scheme ii). Plasmid pU[−1]A allows Cup1 protein expression from the E2− mRNAs, whereas E2+ mRNAs have a premature stop codon in the Cup1 coding region. Conversely, plasmid pU[−1]A* allows Cup1 protein expression from the E2+ mRNAs, whereas E2− mRNAs have a premature stop codon. Plasmids pU[−1]A and pU[−1]A* were expressed separately in both UPF1 (NMD+) and upf1− (NMD-deficient) strains. Results summarized in Figure 7A ▶ show that the ratios of E2+ and E2− mRNAs do not significantly differ whether NMD is active or not. This demonstrates that the differences in the steadystate levels of the RNAs are not substantially affected by the presence or absence of the internal exon itself, inframe stop codons in the third exon, or an intact NMD pathway.
FIGURE 7.

Differences in levels of exon skipped and included mRNAs are not caused by differences in their relative steady-state stabilities or decay rates. (A) Strains HFY114 (UPF1) and HFY870 (upf1Δ) (Table 1 ▶) carry dyn2-CUP1 plasmids pU[−1]A and pU[−1]A*, which are identical except that they possess translational opposite reading frames (RFs). E2+ and E2− levels (percentage of all dyn2 RNAs) and alternative splicing ratios (E2−/E2+) were calculated (±SEM, n = 3). Probability values (P) comparing E2+ or E2−, or E2−/E2+ from both reading frames were determined using Student’s t test. P values >0.05 signify the compared values are not significantly different. (B) 1,10-Phenanthroline treatment of strain KH51B4 expressing pU[−2]C. Because the drug blocks nascent transcription by all DNA-dependent RNA polymerases, absolute decay rates cannot be measured (equivalent amounts of SCR1 cDNA were loaded in each lane except lane 12). Rather, relative decay rates of the E2+ and E2− mRNAs were measured by taking the E2−/E2+ ±SEM ratio (n = 4). P values were determined by comparing E2−/E2+ values in each lane to lane 1. Time points for lanes 1–12, respectively, are (in minutes): 0, 3, 6, 10, 15, 20, 30, 40, 60, 90, 120, 180.
To measure the relative decay rates of the exon included versus skipped mRNAs, yeast were treated for increasing amounts of time with the DNA intercalating drug 1,10-phenanthroline sufficient to arrest DNA-dependent RNA transcription (Santiago et al. 1986). Transcripts from four different dyn2 reporters were assayed by this method, and all yielded equivalent results (data not shown). Shown in Figure 7B ▶ is a representative gel from a strain expressing the U[−2]C mutant and measurements of mRNA levels representing four independent assays. In this case, there is no pattern of change in relative levels consistent with differential decay, and only the 15- and 180-min time points showed a significant deviation from the initial time point. However, when all of the data are considered, it is clear that the two alternatively spliced RNAs do not decay at substantially different rates following arrest of transcription. We conclude that the different relative amounts of exon-skipped and exon-included RNAs in our experiments are primarily due to changes in splicing and are not due to differences in the relative decay rates of the two spliced products. Note that because 1,10-phenanthroline also blocks RNA polymerase III transcription of the SCR1 internal control we used for normalization, no conclusion can be made about the absolute decay rates of any of the RNA species.
DISCUSSION
Multiple mechanisms enforce exon inclusion
We have investigated the mechanisms that enforce exon inclusion in multi-intron transcripts, using a natural two-intron yeast gene. Under normal conditions, nearly all wild-type Dyn2 transcripts produce mRNAs that contain the internal exon (Dick et al. 1996), (Fig. 1 ▶; data not shown). The distance between the 5′ splice site of intron 1 and the 3′ splice site of intron 2 is 199 nucleotides, a distance shorter than the length of the majority of yeast introns (Davis et al. 2000). How does the splicing machinery preferentially select against competing splice sites so that the internal exon is included in virtually every spliced mRNA? We previously identified intron self-complementarities that direct appropriate splice site pairing in pre-mRNAs transcribed from the yeast RPL7A and RPL7B genes (Howe and Ares 1997); however, the DYN2 pre-mRNA lacks such sequence elements. We excluded the possibility that the internal exon is included by a mechanism related to the vertebrate “exon definition” mechanism (Robberson et al. 1990), because weakening the 5′ splicing signals flanking an internal exon does not lead to exon skipping (Fig. 1B ▶). Instead, our search for cis-acting exon skipping mutations exposes the crucial role of the intron 1 branchpoint sequence in ensuring exon inclusion (Fig. 2 ▶). We did not identify any accessory sequences that act as splicing enhancers to enforce recognition of the exon (Carlo et al. 1996, 2000).
The first come, first served model
The efficiency of recognition of the intron 1 branchpoint appears to be key, because subtle mutations not observed to have great effect on the splicing of single intron pre-mRNAs greatly stimulate exon skipping. A slight weakening of the intron 1 branchpoint could allow the intron 2 branchpoint to compete more effectively for pairing to the intron 1 5′ splice site, leading to exon skipping (Fig. 8 ▶). This competition could be manifested even without coupled transcription if the efficiency of pairing of the intron 1 5′ splice site with the branchpoint of intron 2 is favored over the intron 1 branchpoint. Because DYN2 substrates are inefficiently spliced in standard in vitro splicing reactions, we were unable to rigorously test the effect of weakening the first intron branchpoint on exon skipping in a context uncoupled from transcription (data not shown). Despite this, the results of our in vivo manipulations are fully consistent with the predictions of the first come, first served model. A manipulation designed to slow branchpoint recognition relative to the rate of transcription elongation (mutation of U2 snRNA), increases skipping (Fig. 3 ▶). Several manipulations designed to lower the rate of transcription elongation relative to branchpoint recognition (RNA polymerase mutants, addition of 6AU or MPA, removal of TFIIS, blocking of TFIIS-stimulated elongation) reduced exon skipping (Figs. 4– ▶6 ▶; see also Cramer et al. 1997; Roberts et al. 1998; Kadener et al. 2001, 2002; Nogues et al. 2002). Thus, the weight of the evidence points to a generic role for the first come, first served mechanism in enforcing exon inclusion, and implies that in genes that contain multiple introns, the local rates of intron recognition and transcription elongation may be tuned so that constitutive exons are included with high fidelity. In addition, the rate of either process could be influenced in order to regulate exon inclusion (Cramer et al. 1997; Roberts et al. 1998; Kadener et al. 2001, 2002; Nogues et al. 2002), because it is the relative rates that would matter (Fig. 8 ▶).
FIGURE 8.

A model showing how relative rates of elongation and intron recognition can lead to exon skipping or inclusion. Factors that affect the relative rates of spliceosome assembly and transcript elongation will influence the amount of time available for establishment of splice site pairing as in (A), which will lead to exon inclusion. Slow recognition of the first intron or rapid transit of polymerase through the second intron provides a greater opportunity for exon skipping, as in (B).
Accurate splicing in vitro does not require transcription, and “recursive” splicing can occur in vivo (Lopez and Seraphin 2000). Thus, the effects we observe may be operating on a fraction of all transcripts, or the rates of intron recognition and transcript elongation may be so different and our ability to affect them so limited that the extent of the effect we can observe is not large. For example, a mutation that slows the global rate of splicing may be generally more tolerable than one that slows the global rate of elongation. Thus mutations in RPB2 or PPR2 (TFIIS) may have weaker effects on exon inclusion. In our attempt to address this, we tried to position a specific transcriptional block comprised of the lac operator in intron 2 of the reporter. Because the overall reporter mRNA levels were not severely reduced (Fig. 6C ▶), this block is not especially strong, even in the absence of TFIIS. Nonetheless, we were able to block TFIIS-stimulated exon skipping in a lacI-dependent fashion (Fig. 6C ▶). Although other mechanisms for this effect are possible, the simplest interpretation is that binding of lacI to the DNA of intron 2 interferes with the ability of TFIIS to promote elongation through this region of the gene, providing additional time for recognition of the intron 1 branchpoint and subsequent exon inclusion.
Can intron 1 be recognized before RNAP II gets to the end of intron 2?
The changes in exon skipping and inclusion we observe are relatively small but are not due to sampling error. Importantly, their directionality is consistently in agreement with that expected for cotranscriptional splice site pairing decisions. The small magnitude of the changes could be due to several factors. It may be that the tendency for correct pairing of the intron 1 splice sites is intrinsically efficient even in the absence of cotranscriptional enforcement of exon inclusion, simply due to the proximity of the splice sites. The distance separating the branchpoint sequences of intron 1 and 2 is <100 nt. The in vivo rate of transcription by RNAP II is estimated at ~30 nt/sec, indicating that the time advantage enjoyed by the intron 1 branchpoint sequence over its counterpart in intron 2 is ~3 sec. The kinetics of spliceosome assembly in vivo are unknown, but it has been estimated based on the number of introns that must be spliced per hour by the 500 molecules of U2 snRNA that a complete cycle of intron removal and U2 snRNP regeneration must take <3 min (Ares et al. 1999). Depending on the fraction of the average cycle time devoted to branchpoint recognition, the 3 sec that the intron 1 branchpoint spends in the nucleoplasm before the intron 2 branchpoint is made could provide a significant advantage in the competition to pair with the 5′ splice site.
Recent application of chromatin-immunoprecipitation experiments to questions about cotranscriptional events reveal that proteins known to be involved in splicing and mRNA export remain associated at least indirectly with the DNA template from which their RNA substrates are derived (Lei et al. 2001; Lei and Silver 2002; Neugebauer 2002; K. Neugebauer, pers. commun.). The simplest interpretation of the observed physical association is that the events carried out by these proteins on the RNA are initiated during the process of transcription. Recently, mutations in general elongation factors have been shown to inhibit splicing (Lindstrom et al. 2003). Furthermore, work from several laboratories indicates that the presence of introns, in particular the association of the U1 snRNP with the nascent transcript, has direct stimulatory effects on transcription (Fong and Zhou 2001; Tian 2001; Furger et al. 2002; Kwek et al. 2002). Abundant evidence now indicates that during transcription, splicing takes place (Bentley 2002; Howe 2002; Neugebauer 2002; Proudfoot et al. 2002). The work presented here provides evidence that the two processes are dynamically coupled in a way that can influence the structure of the final mRNA and thus the qualitative nature of the information expressed from the gene.
MATERIALS AND METHODS
S. cerevisiae strains and media
The S. cerevisiae strains used are listed in Table 1 ▶. Except when specifically described, media used was either synthetic complete with dextrose (SCD) lacking the appropriate nutrients to ensure stable maintenance of plasmids, or rich media (YEPD) as previously described (Sherman 1991). When required to test for CuR, copper is added to the growth medium as CuSO4, following autoclaving (Hamer et al. 1985). Yeast cell transformation was performed essentially as described (Hill et al. 1991), except that temperature-sensitive strains were incubated with plasmid and carrier DNA at 22°C–25°C instead of at 30°C.
TABLE 1.
S. cerevisiae strains
| Name | Genotype | Source |
| KH51B4 | MAT a ura3-52 leu2-3, 112 trp1 lys2 his3 ade3 cup1::ura3-52 | This work |
| KH46H3 | MATα ura3-52 leu2-3, 112 trp1 lys2 his3 ade3 cup1::ura3-52 | This work |
| IH1097 × IH930 | MAT a/α trp1/trp1 his1/HIS1 leu2/LEU2 ura3-52/URA3 mal1/MAL1 gal2/GAL2 prb1-1122/prb1-1122 pep4-3/pep4-3 prc1-407/prc1-407 | This work |
| BJ81 | MAT a leu2-3, 112 ura3-52 trp1 URA3::GALI:SNR20 pep4-3 prb1 prc1 [pU2-C41G LEU2] | Miraglia et al. 1991 |
| CMKy21 | MAT a ade2 ade3 ura3 leu2 lys2 ppr2Δ::hisG trp1::hisG | Davie and Kane 2000 |
| JAY697 | MAT a can1-100 his3-11,15 leu2-3, 112 trp1-1 ura3-1 ade2-1 PPR2 | Archambault et al. 1992 |
| JAY698 | MAT a can1-100 his3-11,15 leu2-3, 112 trp1-1 ura3-1 ade2-1 ppr2 | Archambault et al. 1992 |
| HFY114 | MAT a ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 UPFI | A. Jacobson, University Massachusetts Medical School, Worcester |
| HFY870 | MAT a ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 upf1::HIS3 | A. Jacobson, University Massachusetts Medical School, Worcester |
| GHY283 (RPB2) | MAT a his3-200 lys2-128 ura3-52 leu2-1 rpb2-297:HIS3 trp1-63 [pRP212] | G. Hartzog, University of California at Santa Cruz |
| GHY486 (rpb2-7) | MAT a his3-200 lys2-128 ura3-52 leu2-1 rpb2-297:HIS3 trp1-63 [pRP2-7(U)] | G. Hartzog, University of California at Santa Cruz |
| GHY492 (rpb2-10) | MAT a his3-200 lys2-128 ura3-52 leu2-1 rpb2-297:HIS3 trp1-63 [pRP2-10(U)] | G. Hartzog, University of California at Santa Cruz |
Plasmids and splicing reporter construction
Plasmids used are listed in Table 2 ▶. Plasmid pGEMSLC1r was constructed by amplifying a 281-bp BamHI-KpnI PCR fragment of DYN2/SLC1 (Dick et al. 1996; GenBank accession no. L13282) from diploid yeast strain IH1097 × IH930 and inserting it into the bacterial plasmid pGEM7zf+. PCR amplification was performed by using primers SLC1-5′BamHI (L in Fig. 1A ▶; 5′p-dAATTggAtC CAAAATGAGCGATA-3′) and SLC1-3′KpnI (R in Fig. 1A ▶; 5′p-dCAGAGCATCggTACcGATcGTTAAA-3′; lowercase letters represent changes from the sequence that create restriction sites) and Taq DNA polymerase (Promega). Sequencing confirmed that the cloned BamHI-KpnI fragment is identical to the GenBank sequence except at the positions altered by the oligonucleotide primers. The DYN2-CUP1 yeast reporter pGSC1t was made by replacing the ACT1 BamHI-KpnI fragment of pGAC14 (Lesser and Guthrie 1993) with the BamHI-KpnI fragment from pGEMSLC1r. This construct fuses DYN2 to CUP1, such that CuR is dependent on splicing of the DYN2 RNA. Plasmid pGSC1u construction was identical to pGSC1t except that the parental plasmid (pGAC14Uf; Table 2 ▶) contains the URA3 instead of TRP1 nutritional marker. CuR due to expression of Dyn2p-Cup1p fusion protein from pGSC1t and pGSC1u occurs when the internal (second) exon of DYN2 is included in the mRNA. The mRNA lacking the internal exon is out of frame with respect to Cup1p. To generate reporter constructs that express a fusion protein when the internal DYN2 exon is skipped, plasmids pGSC1t and pGSC1u were digested with KpnI, blunt ended with Klenow enzyme, and religated to generate plasmids pGSC3t and pGSC3u, respectively. This process removes four base pairs from the junction between DYN2 and CUP1, and changes the reading frame of the reporter. In the KH51B4 strain, the wild-type pGSC1 plasmids provide resistance to >2mM CuSO4, whereas the pGSC3 plasmids are resistant to no >250 μM CuSO4. All recombinant DNA manipulations were performed following established procedures (Sambrook et al. 1989).
TABLE 2.
Plasmids
| Name | Features/construction | Source |
| pGEM7zf+ | Standard cloning vector | Promega |
| pGEMSLC1r | BamHI-KpnI PCR fragment of DYN2 inserted into pGEM7Zf+ | This work |
| pGAC14 | ACT1-CUP1 splicing reporter, GPD promoter; TRP1; 2u | Lesser and Guthrie 1993 |
| pGAC14Uf | URA3 gene insertion into Bg1II site of TRP1 gene of pGAC14 | This work |
| pGSC1t | pGAC14 ACT1 replaced by DYN2 frag.; CUP1 in E2+ frame | This work |
| pGSC1u | Same as pGSC1t except pGAC14Uf is parental plasmid | This work |
| pGSC3t | pGSC1t KpnI (blunted), religated; CUP1 in E2− frame | This work |
| pGSC3u | pGSC1u KpnI (blunted), religated; CUP1 in E2− frame | This work |
| pGSC621 | pGSC1t with the I1ΔBP mutation in DYN2 | This work |
| pGSC431 | pGSC1t with the I2Δ5′ss mutation in DYN2 | This work |
| pU[−2]C | DYN2 intron 1 branchpoint sequence U[−2]C; E2− frame | This work |
| pU[−1]A | DYN2 intron 1 branchpoint sequence U[−1]A; E2− frame | This work |
| pU[−1]A* | Same as pU[−1]A except in E2+ frame | This work |
| pU[−1]G | DYN2 intron 1 branchpoint sequence U[−1]G; E2− frame | This work |
| pU[1]A | DYN2 intron 1 branchpoint sequence U[1]A; E2− frame | This work |
| pA[2]G | DYN2 intron 1 branchpoint sequence A[2]G; E2− frame | This work |
| pGSlacIAt | pU[−1]G carrying the lac operator in intron 2 | This work |
| pRS316 | Empty vector control; URA3; CEN | Sikorski and Hieter 1989 |
| pJD4 | PPR2; URA3; CEN | Davie and Kane 2000 |
| pFL44D | Empty vector control; URA3, 2u | Archambault et al. 1992 |
| pPPR2 | PPR2 gene inserted into pFL44D | Archambault et al. 1992 |
Site-specific mutagenesis of DYN2
Mutations were introduced by the two-step PCR method (Good and Nazar 1992). The following oligodeoxynucleotides were used (lowercase letters denote base changes; restriction sites are underlined): SLC1-I1ØBP (5′p-cctcgagctcgtaaagaaaaattg-3′), SLC1-I2Ø5′ss (5′p-caattgtattctatgtcagac-3′), SLC1-I2bpA[2]G (5′p-dCT TCCTCGAGTTAGcACAGAAAAATTG-3′), SLC1-lacIA (5′p-dAAATATATATATTCAatTGTgagCgCtCAcAAttTGTATATCAT GGC-3′). Flanking primers used were SLC1-5′BamHI and SLC1-3′KpnI (see above).
Construction and screening of dyn2 mutant libraries
Two libraries were constructed by degenerate PCR according to the protocol previously described (Cadwell and Joyce 1992). For the first library, a 100-μL PCR contained the following: 250 ng pGEMSLC1r, 200 pmole each SLC1-5′BamHI and SLC1-3′KpnI primers, 50 mM KCl, 10 mM Tris-Cl (pH 8.3), 0.01% gelatin, 7 mM MgCl2, 0.5 mM MnCl2, 200 μM each dATP and dGTP, 1 μM each dCTP and dTTP, and 5 U TAQ polymerase (Promega). After an initial 5 min at 94°C denaturation step, amplification was carried out for 30 cycles of 94°C (60 sec), 51°C (20 sec), 31°C (20 sec), and 72°C (60 sec) and was finished with a single 72°C (7 min) extension step. The BamHI-KpnI fragment was gel-purified and cloned into pGAC14 as described above. Sequencing of 12 randomly selected clones from a pool of ~1850 indicated that an average of 2.6 mutations were present per sequence (~1.1 percent misincorporation rate). Accordingly, the probability that all of the bases have been mutated at least once to each of the other three bases within this library is >0.99 (based on Clarke and Carbon 1976). The pooled transformants were grown and plasmid DNA isolated and, then subjected to KpnI digestion and blunt-ending using Klenow fragment. The resultant pool of linearized/blunted plasmid DNA was religated and then transformed into bacterial strain XL-1 Blue (Stratagene). Based on restriction analysis of plasmids from randomly chosen bacterial transformants, <1% of the plasmids still retained the KpnI site following these steps. The library was transformed into yeast strain KH51B4 (which is cup1−) and plated onto SCD media lacking both tryptophan and copper. Roughly 3900 yeast transformants were tested for CuR by replica plating to SCD plates containing 500 μM CuSO4. A total of 262 colonies (6.7%) were resistant to 500 μM CuSO4. Of these, ~75% retained an intact KpnI site from the original library and were discarded. The plasmids lacking KpnI sites were examined further. Plasmids rescued from 29 independent CuR yeast transformants were sequenced. Twenty-four mutants were considered unique because they did not share overlapping sets of mutations. For mutants with multiple base changes, specific mutations were tested by swapping restriction fragments between wild-type and mutant dyn2 sequences by using the unique XhoI site located just downstream of the intron 1 branchpoint sequence.
A second library with a lower target misincorporation rate was constructed by using a similar approach, except that PCR reactions contained 60 pmole each of the primers SLC1-5′BamHI and SLC1-KpnI-3 (R2 in Fig. 1A ▶; 5′p-dGCATCTggtaccATGGTTAA AATATCCTCTTTC-3′) and 1U TAQ polymerase. Amplification was carried out for only 15 cycles. Primer SLC1-KpnI-3 was substituted for SLC1-3′KpnI so that clones containing BamHI-KpnI PCR fragments could be directly screened without the extra KpnI digestion/Klenow blunting step. After amplification in XL-1 Blue, this library was transformed into yeast strain KH51B4. Colonies were selected for CuR in the low to moderate range (325 to 350 μM CuSO4) and tested to identify those that were CuS >350 μM CuSO4 (CuS, copper sensitive). Plasmids from five transformants were isolated and sequenced.
RNA preparation and RT analysis
Liquid yeast cultures were harvested in mid-log growth at an optical density (A600) of between 0.4 and 0.8. In some strains, the exon skipping-to-inclusion ratios varied in a culture density-dependent manner. Therefore, cultures for any set of data were grown to within ±0.1 O.D. of each other. Cells were pelleted at 4000 rpm for 5 min at RT, washed with one culture volume of ice cold sterile water, and either were used to prepare total cellular RNA immediately or were flash frozen and stored at −20°C. Total RNA was isolated as previously described (Ares and Igel 1990) and stored at −20°C in diethylpyrocarbonate (DEPC)-treated water at a concentration of 0.5 to 2 mg/mL. For splicing analysis, labeled dyn2-CUP1 cDNAs were synthesized by RT using primer CUP1 (Howe and Ares 1997). Primer scr1-108, which is complementary to the pol III transcript encoded by SCR1, was included as an internal standard (Howe and Ares 1997). Between 7.5 and 15 fmole of each 5′-32P–labeled primer was annealed to 5 to 11 μg total RNA (5 min at 65°C followed by 25 min at 42°C). Extension reactions were performed at 42°C as previously described (Ares and Igel 1990) using either AMV RT (Life Sciences) or M-MLV RT (GIBCO; Fig. 6B ▶) in the buffer suggested by the manufacturers. Products were separated on 6% acrylamide gels (19:1 mono:bis) containing 7 M urea, and the dried gels were exposed to phosphorimager screens. Radioactivity was measured using the Molecular Dynamics ImageQuant software (version 1.2).
Mycophenolate and 6AU treatment
For liquid cultures, 5mL of a saturated culture of KH51B4 transformed with plasmid pU[1]A was used to inoculate 150 mL SCD media lacking uracil. The culture was grown at 30°C with shaking to A600 = 0.5 (approximately three doublings), at which point the culture was split equally into three 125-mL flasks of 50 mL each. To one flask, 1 mL 6AU (4 mg/mL in water; Fisher Scientific) was added; to the second, 0.5 mL MPA (10 mg/mL in ethanol; Fisher Scientific) was added. The third culture was treated with 0.5 mL ethanol only. At 60 and 120 min of treatment, 20 mL aliquots were removed from each flask, added separately to 30 mL loose crushed ice, which rapidly and completely melted. The mixture was centrifuged for RNA extraction as described above.
Transcription arrest with 1,10-phenanthroline
Yeast strains derived from KH51B4, carrying different dyn2-CUP1 reporters were grown at 30°C in 150 mL SCD media lacking uracil. When the cultures had reached A600 = 0.5, 0.75 mL of 1,10-phenanthroline (100 mg/mL in ethanol; Fisher Scientific; kindly provided by H. Noller, University of California at Santa Cruz) was added (Santiago et al. 1986). At specified times after addition, 10 mL aliquots of the cultures were removed, added to 15 mL loose crushed ice, and pelleted for RNA isolation as described above.
Error determination and statistical analysis
Quantification of cDNA signals was performed using ImageQuant (version 1.2). Background corrections were performed by subtracting an area of density of identical size from a region of the gel immediately adjacent to and within the same gel lane (i.e., above or below) as the sampled signal. The total dyn2 signal was calculated for each lane as the sum of all dyn2 RNA species, and the proportion of each RNA species relative to the total for each lane was determined. The exon skipping ratios (E2−/E2+) were calculated directly from the absolute signal densities. Each experiment was performed at least three times (n ≥ 3), and the average of these values (X) and the standard error of the means (SEM) are reported in the figures. SEM was calculated by dividing the standard deviation of the mean (σ) by the square root of n. Propagated error (σC) was calculated using Formula 1:
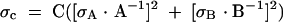 |
(1) |
where value C is the quotient derived from values A and B.
Student’s t test of compared values was performed by first determining the pooled variance (σP) using Formula 2,
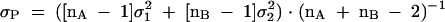 |
(2) |
which was used to calculate the t value using Formula 3:
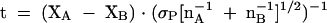 |
(3) |
The t values were used to determine the probability (P, where 0 ≤ P ≥ 1) that the differences between the compared values measured for the dependent variable are due to chance. Where P ≤ 0.05, the differences between the compared values are considered unlikely to be due to sampling error, and are therefore likely due to the experimental manipulation of the independent variable.
Acknowledgments
We thank the following people for generously providing strains and reagents: Harry Noller, Doug Kellogg, and Grant Hartzog (University of California at Santa Cruz); Jim Friesen (University of Toronto); and Allan Jacobson (University of Massachusetts). Also, thanks to Karla Neugebauer (Max Plank Institute, Germany) and to Elissa Lei and Pam Silver (Harvard) for sharing results and ideas prior to publication. Thanks to Rhonda Perriman and Grant Hartzog for critical reading of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.5390803.
REFERENCES
- Abovich, N. and Rosbash, M. 1997. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89: 403–412. [DOI] [PubMed] [Google Scholar]
- Abovich, N., Liao, X.C., and Rosbash, M. 1994. The yeast MUD2 protein: An interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes & Dev. 8: 843–854. [DOI] [PubMed] [Google Scholar]
- Aebi, M. and Weissman, S.M. 1987. Precision and orderliness in splicing. Trends Genet. 3: 102–107. [Google Scholar]
- Archambault, J., Lacroute, F., Ruet, A., and Friesen, J.D. 1992. Genetic interaction between transcription elongation factor TFIIS and RNA polymerase II. Mol. Cell. Biol. 12: 4142–4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ares Jr., M. and Igel, A.H. 1990. Lethal and temperature-sensitive mutations and their suppressors identify an essential structural element in U2 small nuclear RNA. Genes & Dev. 4: 2132–2145. [DOI] [PubMed] [Google Scholar]
- Ares Jr., M., Grate, L., and Pauling, M.H. 1999. A handful of intron-containing genes produces the lion’s share of yeast mRNA. RNA 5: 1138–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley, D. 2002. The mRNA assembly line: Transcription and processing machines in the same factory. Curr. Opin. Cell. Biol. 14: 336–342. [DOI] [PubMed] [Google Scholar]
- Berget, S.M. 1995. Exon recognition in vertebrate splicing. J. Biol. Chem. 270: 2411–2414. [DOI] [PubMed] [Google Scholar]
- Blandin, G., Durrens, P, Tekaia, F., Aigle, M., Bolotin-Fukuhara, M., Bon, E., Casaregola, S., de Montigny, J., Gaillardin, C., Lepingle, A., et al. 2000. Genomic exploration of the hemiascomycetous yeasts, 4: The genome of Saccharomyces cerevisiae revisited. FEBS Lett. 487: 31–36. [DOI] [PubMed] [Google Scholar]
- Brow, D.A. 2002. Allosteric cascade of spliceosome activation. Annu. Rev. Genet. 36: 333–360. [DOI] [PubMed] [Google Scholar]
- Cadwell, R.C. and Joyce, G.F. 1992. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 2: 28–33. [DOI] [PubMed] [Google Scholar]
- Carlo, T., Sterner, D.A., and Berget, S.M. 1996. An intron splicing enhancer containing a G-rich repeat facilitates inclusion of a vertebrate micro-exon. RNA 2: 342–353. [PMC free article] [PubMed] [Google Scholar]
- Carlo, T., Sierra, R., and Berget, S.M. 2000. A 5′ splice site-proximal enhancer binds SF1 and activates exon bridging of a microexon. Mol. Cell. Biol. 20: 3988–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie, K.R., Awrey, D.E., Edwards, A.M., and Kane, C.M. 1994. Purified yeast RNA polymerase II reads through intrinsic blocks to elongation in response to the yeast TFIIS analogue, P37. J. Biol. Chem. 269: 936–943. [PubMed] [Google Scholar]
- Clarke, L. and Carbon, J. 1976. A colony bank containing synthetic Col El hybrid plasmids representative of the entire E. coli genome. Cell 9: 91–99. [DOI] [PubMed] [Google Scholar]
- Cramer, P., Pesce, C.G., Baralle, F.E., and Kornblihtt, A.R. 1997. Functional association between promoter structure and transcript alternative splicing. Proc. Natl. Acad. Sci. 94: 11456–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie, J.K. and Kane, C.M. 2000. Genetic interactions between TFIIS and the Swi-Snf chromatin-remodeling complex. Mol. Cell. Biol. 20: 5960–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, C.A., Grate, L., Spingola, M., and Ares Jr., M. 2000. Test of intron predictions reveals novel splice sites, alternatively spliced mRNAs and new introns in meiotically regulated genes of yeast. Nucleic Acids Res. 28: 1700–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick, T., Surana, U., and Chia, W. 1996. Molecular and genetic characterization of SLC1, a putative Saccharomyces cerevisiae homolog of the metazoan cytoplasmic dynein light chain 1. Mol. Gen. Genet. 251: 38–43. [DOI] [PubMed] [Google Scholar]
- Exinger, F. and Lacroute, F. 1992. 6-Azauracil inhibition of GTP biosynthesis in Saccharomyces cerevisiae. Curr. Genet. 22: 9–11. [DOI] [PubMed] [Google Scholar]
- Fong, Y.W. and Zhou, Q. 2001. Stimulatory effect of splicing factors on transcriptional elongation. Nature 414: 929–933. [DOI] [PubMed] [Google Scholar]
- Fouser, L.A. and Friesen, J.D. 1986. Mutations in a yeast intron demonstrate the importance of specific conserved nucleotides for the two stages of nuclear mRNA splicing. Cell 45: 81–93. [DOI] [PubMed] [Google Scholar]
- Franklin, T.J. and Cook, J.M. 1969. The inhibition of nucleic acid synthesis by mycophenolic acid. Biochem. J. 113: 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furger, A., O’Sullivan, J.M., Binnie, A., Lee, B.A., and Proudfoot, N.J. 2002. Promoter proximal splice sites enhance transcription. Genes & Dev. 16: 2792–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good, L. and Nazar, R.N. 1992. An improved thermal cycle for two-step PCR-based targeted mutagenesis. Nucleic Acids Res. 20: 4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grate, L. and Ares Jr., M. 2002. Searching yeast intron data at Ares lab Web site. Methods Enzymol. 350: 380–392. [DOI] [PubMed] [Google Scholar]
- Hamer, D.H., Thiele, D.J., and Lemontt, J.E. 1985. Function and autoregulation of yeast copperthionein. Science 228: 685–690. [DOI] [PubMed] [Google Scholar]
- He, F., Peltz, S.W., Donahue, J.L., Rosbash, M., and Jacobson, A. 1993. Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1− mutant. Proc. Natl. Acad. Sci. 90: 7034–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, J., Ian, K.A., Donald, G., and Griffiths, D.E. 1991. DMSO-enhanced whole cell yeast transformation. Nucleic Acids Res. 19: 5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe, K.J. 2002. RNA polymerase II conducts a symphony of pre-mRNA processing activities. Biochim. Biophys. Acta 1577: 308–324. [DOI] [PubMed] [Google Scholar]
- Howe, K.J. and Ares Jr., M. 1997. Intron self-complementarity enforces exon inclusion in a yeast pre-mRNA. Proc. Natl. Acad. Sci. 94: 12467–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izban, M.G. and Luse, D.S. 1992. The RNA polymerase II ternary complex cleaves the nascent transcript in a 3′-5′ direction in the presence of elongation factor SII. Genes & Dev. 6: 1342–1356. [DOI] [PubMed] [Google Scholar]
- Kadener, S., Cramer, P., Nogues, G., Cazalla, D., de la Mata, M., Fededa, J.P., Werbajh, S.E., Srebrow, A., and Kornblihtt, A.R. 2001. Antagonistic effects of T-Ag and VP16 reveal a role for RNA pol II elongation on alternative splicing. EMBO J. 20: 5759–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadener, S., Fededa, J.P., Rosbash, M., and Kornblihtt, A.R. 2002. Regulation of alternative splicing by a transcriptional enhancer through RNA pol II elongation. Proc. Natl. Acad. Sci. 99: 8185–8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwek, K.Y., Murphy, S., Furger, A., Thomas, B., O’Gorman, W., Kimura, H., Proudfoot, N.J., and Akoulitchev, A. 2002. U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat. Struct. Biol. 9: 800–805. [DOI] [PubMed] [Google Scholar]
- Lei, E.P. and Silver, P.A. 2002. Intron status and 3′-end formation control cotranscriptional export of mRNA. Genes & Dev. 16: 2761–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, E.P., Krebber, H., and Silver, P.A. 2001. Messenger RNAs are recruited for nuclear export during transcription. Genes & Dev. 15: 1771–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon III, J.C., Wind, M., Saunders, L., Hock, M.B., and Reines, D. 1998. Mutations in RNA polymerase II and elongation factor SII severely reduce mRNA levels in Saccharomyces cerevisiae. Mol. Cell. Biol. 18: 5771–5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser, C.F. and Guthrie, C. 1993. Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics 133: 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom, D.L., Squazzo, S.L., Muster, N., Burckin, T.A., Wachter, K.C., Emigh, C.A., McCleery, J.A., Yates III, J.R., and Hartzog, G.A. 2003. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol. Cell. Biol. 23: 1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez, A.J. 1998. Alternative splicing of pre-mRNA: Developmental consequences and mechanisms of regulation. Annu. Rev. Genet. 32: 279–305. [DOI] [PubMed] [Google Scholar]
- Lopez, P.J. and Seraphin, B. 2000. Uncoupling yeast intron recognition from transcription with recursive splicing. EMBO Rep. 1: 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen, B.G. and Seraphin, B. 1998. Construction of an in vivo–regulated U6 snRNA transcription unit as a tool to study U6 function. RNA 4: 231–238. [PMC free article] [PubMed] [Google Scholar]
- Miraglia, L., Seiwert, S., Igel, A.H., and Ares Jr., M. 1991. Limited functional equivalence of phylogenetic variation in small nuclear RNA: Yeast U2 RNA with altered branchpoint complementarity inhibits splicing and produces a dominant lethal phenotype. Proc. Natl. Acad. Sci. 88: 7061–7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi, T., Nakano, A., Nomura, K., Sekimizu, K., and Natori, S. 1992. Purification, gene cloning, and gene disruption of the transcription elongation factor S-II in Saccharomyces cerevisiae. J. Biol. Chem. 267: 13200–13204. [PubMed] [Google Scholar]
- Nakanishi, T., Shimoaraiso, M., Kubo, T., and Natori, S. 1995. Structure-function relationship of yeast S-II in terms of stimulation of RNA polymerase II, arrest relief, and suppression of 6-azauracil sensitivity. J. Biol. Chem. 270: 8991–8995. [DOI] [PubMed] [Google Scholar]
- Neugebauer, K.M. 2002. On the importance of being co-transcriptional. J. Cell. Sci. 115: 3865–3871. [DOI] [PubMed] [Google Scholar]
- Newman, A.J., Lin, R.J., Cheng, S.C., and Abelson, J. 1985. Molecular consequences of specific intron mutations on yeast mRNA splicing in vivo and in vitro. Cell 42: 335–344. [DOI] [PubMed] [Google Scholar]
- Nogues, G., Kadener, S., Cramer, P., Bentley, D., and Kornblihtt, A.R. 2002. Transcriptional activators differ in their abilities to control alternative splicing. J. Biol. Chem. 277: 43110–43114. [DOI] [PubMed] [Google Scholar]
- Parker, R., Siliciano, P.G., and Guthrie, C. 1987. Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell 49: 229–239. [DOI] [PubMed] [Google Scholar]
- Powell, W. and Reines, D. 1996. Mutations in the second largest subunit of RNA polymerase II cause 6-azauracil sensitivity in yeast and increased transcriptional arrest in vitro. J. Biol. Chem. 271: 6866–6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot, N.J., Furger, A., and Dye, M.J. 2002. Integrating mRNA processing with transcription. Cell 108: 501–512. [DOI] [PubMed] [Google Scholar]
- Rain, J.C. and Legrain, P. 1997. In vivo commitment to splicing in yeast involves the nucleotide upstream from the branch site conserved sequence and the Mud2 protein. EMBO J. 16: 1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines, D., Ghanouni, P., Li, Q.Q., and Mote Jr., J. 1992. The RNA polymerase II elongation complex: Factor-dependent transcription elongation involves nascent RNA cleavage. J. Biol. Chem. 267: 15516–15522. [PMC free article] [PubMed] [Google Scholar]
- Reines, D., Ghanouni, P., Gu, W., Mote Jr., J., and Powell, W. 1993. Transcription elongation by RNA polymerase II: Mechanism of SII activation. Cell. Mol. Biol. Res. 39: 331–338. [PubMed] [Google Scholar]
- Robberson, B.L., Cote, G.J., and Berget, S.M. 1990. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol. 10: 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, G.C., Gooding, C., Mak, H.Y., Proudfoot, N.J., and Smith, C.W. 1998. Co-transcriptional commitment to alternative splice site selection. Nucleic Acids Res. 26: 5568–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Santiago, T.C., Purvis, I.J., Bettany, A.J., and Brown, A.J. 1986. The relationship between mRNA stability and length in Saccharomyces cerevisiae. Nucleic Acids Res. 14: 8347–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 194: 3–21. [DOI] [PubMed]
- Sikorski, R.S. and Hieter, P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C.W. and Valcarcel, J. 2000. Alternative pre-mRNA splicing: The logic of combinatorial control. Trends Biochem. Sci. 25: 381–388. [DOI] [PubMed] [Google Scholar]
- Staley, J.P. and Guthrie, C. 1998. Mechanical devices of the spliceosome: Motors, clocks, springs, and things. Cell 92: 315–326. [DOI] [PubMed] [Google Scholar]
- Talerico, M. and Berget, S.M. 1990. Effect of 5′ splice site mutations on splicing of the preceding intron. Mol. Cell. Biol. 10: 6299–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1994. Intron definition in splicing of small Drosophila introns. Mol. Cell. Biol. 14: 3434–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, H. 2001. RNA ligands generated against complex nuclear targets indicate a role for U1 snRNP in co-ordinating transcription and RNA splicing. FEBS Lett. 509: 282–286. [DOI] [PubMed] [Google Scholar]
- Treich, I., Carles, C., Sentenac, A., and Riva, M. 1992. Determination of lysine residues affinity labeled in the active site of yeast RNA polymerase IIB by mutagenesis. Nucleic Acids Res. 20: 4721–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uptain, S.M., Kane, C.M., and Chamberlin, M.J. 1997. Basic mechanisms of transcript elongation and its regulation. Annu. Rev. Biochem. 66: 117–172. [DOI] [PubMed] [Google Scholar]
- Vijayraghavan, U., Parker, R., Tamm, J., Iimura, Y., Rossi, J., Abelson, J., and Guthrie, C. 1986. Mutations in conserved intron sequences affect multiple steps in the yeast splicing pathway, particularly assembly of the spliceosome. EMBO J. 5: 1683–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, D. and Ares Jr., M. 1996. Invariant U2 RNA sequences bordering the branchpoint recognition region are essential for interaction with yeast SF3a and SF3b subunits. Mol. Cell. Biol. 16: 818–828. [DOI] [PMC free article] [PubMed] [Google Scholar]