

Abstract
Drosophila P-element somatic inhibitor protein (PSI) regulates splicing of the P-element transposase pre-mRNA by binding a pseudo-splice site upstream of the authentic splice site using four tandem KH-type RNA binding motifs. While the binding domains and specificity of PSI have been established, little is known about the contributions of each PSI KH domain to overall protein stability and RNA binding affinity. Using a construct containing only the RNA binding domain of PSI (PSI-KH03), we introduced a physiologically relevant point mutation into each KH domain of PSI individually and measured stability and RNA binding affinity of the resulting mutant proteins. Although secondary structure, as measured by circular dichroism spectroscopy, is only subtly changed for each mutant protein relative to wild type, RNA binding affinity is reduced in each case. Mutations in the second or third KH domains of the protein are significantly more deleterious to substrate recognition than mutation of the outer (first and fourth) domains. These results show that despite the ability of a single KH domain to bind RNA in some systems, PSI requires multiple tandem KH domains for specific and high-affinity recognition of substrate RNA.
Keywords: PSI, FMR1, KH domain, CD, alternative splicing
INTRODUCTION
Sequence- and structure-specific recognition of RNA targets is a hallmark of proteins involved in pre-messenger RNA splicing and other kinds of RNA processing. The hnRNP K-homology (KH) domain has emerged as one of the most prevalent protein motifs responsible for RNA binding in these systems (Siomi and Dreyfuss 1997; Adinolfi et al. 1999). Despite extensive research on the structure and RNA recognition properties of individual KH domains, it is not yet clear how the multiple KH domains found in many proteins contribute to recognition of specific RNA targets. A particularly well-characterized example is the fruit fly Drosophila melanogaster P-element somatic inhibitor (PSI), a 97-kDa, four-KH-domain protein that is responsible for alternative splicing of P-element transposase pre-mRNA in somatic tissues (Siebel and Rio 1990; Adams et al. 1997). P-element transposition is suppressed in non-germline cells because PSI binds to a pseudo-5′ splice site in the transposase pre-mRNA, leading to an alternatively spliced variant of the mRNA that encodes a shortened, repressor form of the transposase (Siebel et al. 1992; Fig. 1 ▶). Interestingly, PSI is required for Drosophila viability (Adams et al. 1997; Labourier et al. 2002), supporting an essential role of this protein as a splicing regulator.
FIGURE 1.

Mechanism of alternative splicing regulation of Drosophila P-element somatic inhibitor (PSI) protein. (A) Productive splicing of Drosophila P-element pre-mRNA in germ-line cells leads to the translation of an 87-kDa transposase protein. Alternative splicing in somatic cells leads to inclusion of intron 3. A stop codon (○) located in intron 3 prevents translation of the full-length transposase protein, resulting in a truncated 67-kDa protein that acts to repress transposition. (B) In germline cells, U1 snRNP (U1) binds to the actual 5′-ss (AS) or the F1 pseudo-splice site with approximately equal affinity. PSI, which binds to the F2 pseudo-splice site, recruits U1snRNP to the F1 pseudo-splice site in addition to preventing U1snRNP binding to the actual site, thus preventing spliceosome formation.
The N-terminal region of PSI contains four KH motifs that are required for specific binding to the pseudo-splice site sequence (Fig. 1B ▶). A C-terminal glutamine-rich AB motif interacts with the U1 snRNP, possibly to prevent its binding to the correct splice site in transposase pre-mRNA (Siebel et al. 1995; Labourier et al. 2001; Ignjatovic et al. 2005). Only the 4-KH region of PSI is required for Drosophila viability, though flies possessing a PSI version that lacks the AB motif display behavioral defects and male sterility (Labourier et al. 2002).
Like PSI, numerous other proteins possess multiple KH motifs that contribute to RNA and protein recognition. For example, the human PSI homolog KSRP contains both the four-KH domain region and the AB motif and is involved in alternative splicing of the neuron-specific c-src pre-mRNA among other functions (Gherzi et al. 2004; Hall et al. 2004; Linker et al. 2005). The protein vigilin, which is involved in a diverse array of functions including hetero-chromatin formation as well as RNA editing, contains 15 KH-domain repeats (Goolsby and Shapiro 2003; Wang et al. 2005). Additionally, some proteins, such as the fragile X related protein FMR1p, combine KH domains with other RNA binding motifs, such as the RGG box or RRM (Siomi and Dreyfuss 1997). The presence of multiple such binding motifs may enable recognition of a broad set of target sequences (Bomsztyk et al. 2004), as well as provide exquisite substrate specificity for a particular nucleic acid or protein target (Worbs et al. 2001; Gherzi et al. 2004).
Despite the importance of multiple KH domains to the biochemical function of many proteins, most studies on the structure and the function of KH domains have focused on proteins containing just one or two KH motifs. To gain insight into their roles in protein structural integrity and RNA recognition within the context of a larger protein, a single I-to-N mutation was introduced into each of the four KH domains of PSI individually, and the properties of the resulting proteins were investigated. This point mutation of a highly conserved isoleucine, first identified in the fragile X syndrome-associated protein FMR1p, disrupts the overall fold and RNA binding abilities of some KH domains (Musco et al. 1996, 1997; Lewis et al. 1999). However, in other proteins such as the splicing factor SF1, and when placed in proteins with two KH domains, the effect of this mutation seems to be attenuated (Rain et al. 1998; Liu et al. 2001; Pozdnyakova and Regan 2005). We show here that mutation of individual KH domains of PSI results in similar and relatively modest effects on protein secondary structure, as analyzed by circular dichroism (CD) spectroscopy, but differential defects in RNA substrate binding affinity and specificity. These findings indicate that the tandem KH domains of PSI interact to create high affinity RNA binding contacts to substrate RNAs.
RESULTS
Mutagenesis, expression, and purification of wild-type and mutant PSI-KH03 proteins
The minimal RNA binding domain of PSI, PSI-KH03, was identified by sequence homology to known KH-domain-containing proteins and by truncation of full-length PSI protein (Labourier et al. 2001; Fig. 2A ▶). This construct includes three “type I” KH motifs, KH1, −2, and −3, that are each expected to exhibit a βααββα secondary structure of ~70 residues followed by a ~40-residue linker region, and a fourth degenerate KH domain, KH0, near the N terminus (Siebel et al. 1995; Grishin 2001; Labourier et al. 2001; Fig. 2B ▶). The KH0 domain does not contain the conserved GXXG loop motif found in other KH domains, but sequence alignments (Altschul et al. 1990) reveal 31% identity to several KH domains, including those of KSRP (Min et al. 1997), and includes the conserved isoleucine residue corresponding to Ile304 in FMR1p. The 42-kDa PSI-KH03 protein is readily overexpressed and purified from bacterial cell cultures and was used as the basis for the mutagenesis and characterization presented here.
FIGURE 2.

PSI construct and sequence info. (A) A comparison of the full-length PSI protein and the construct used for binding experiments in this study. (B) Alignment of KH domains from several proteins showing conserved residues and secondary structure motifs. Arrow denotes location of conserved Ile residue mutated to Asn in individual PSI KH domains. Secondary structure was based on that observed by Lewis et al. (2000).
To determine the requirements for substrate recognition and binding of the PSI-KH03 protein, an isoleucine residue strictly conserved in the four KH domains of PSI and highly conserved throughout KH domains present in other proteins (Fig. 2B ▶) was mutated to asparagine. This Ile-to-Asn mutation corresponds to the mutation found in the second KH domain of the FMR1 protein of a patient with fragile X syndrome (De Boulle et al. 1993). Here, each domain of the PSI-KH03 RNA binding region was separately mutated to yield the following proteins: I145N, I234N, I338N, and I449N, corresponding to mutations in KH0, KH1, KH2, and KH3, respectively. Expression and purification of these mutant proteins followed the same basic procedure as for wild-type PSI-KH03 protein (see Materials and Methods); however, the mutant proteins were found to be less resistant to proteolysis than the wild-type counterpart, and the overall yields of these proteins were lower than wild-type PSI-KH03. In all cases, the proteins were purified to homogeneity as detected by Coomassie-stained SDS-PAGE gel electrophoresis.
KH03 mutant proteins maintain secondary structure
To determine if the mutations made in PSI-KH03 alter the secondary structure of the protein, CD spectroscopy was performed on the wild-type and mutant PSI-KH03 proteins. The far-UV wavelength scan of wild-type PSI-KH03 reveals a spectrum characteristic of a protein containing a mixed α/β structure, with a minimum at 222 nm, suggesting a high amount of helical character (Fig. 3A ▶). This agrees well with known structures of KH domains, as well as previously published spectra of single and tandem KH domains (Musco et al. 1996, 1997; Pozdnyakova and Regan 2005). As a control for denatured protein, a second data set was taken of the wild-type protein at 90°C, yielding a spectrum with a large degree of random coil. CD spectra of PSI-KH03 bound to RNA do not reveal any differences relative to wild-type protein alone (data not shown), suggesting there is no global change in secondary structure upon RNA binding. However, this does not rule out local RNA-induced conformational changes that are undetectable by CD. Each of the mutant PSI-KH03 proteins yields a spectrum with characteristics similar to that of the wild type at 25°C, although the magnitude of the spectrum is reduced for each of the mutant proteins (Fig. 3A ▶). Additionally, the two proteins with mutations in the N-and C-terminal KH domains, I145N and I449N, respectively, show subtle changes in the 208-nm region relative to wild type, which could be due to slight differences in the secondary structure of these domains. Repeated experiments show similar trends in the spectra, and the averages of the molar ellipticity at 222 nm reveal differences that are outside of one standard deviation of the average (Fig. 3B ▶). This suggests that the differences observed in the spectra between wild-type and mutant PSI-KH03 are real and not due to experimental variability.
FIGURE 3.

CD wavelength spectra of PSI-KH03 and mutant proteins (A) Far-UV CD wavelength scan of wild-type KH03 (native and denatured) and the four mutant proteins. (•) WT, (▴) I145N, (▪) I234N, (□) I338N, (▵); I449N, (○) WT denatured. (B) Absolute molar ellipticity at 222 nm.
PSI-KH03 mutant I234N displays altered thermal stability
To determine the effect of the Ile-to-Asn mutations on the stability of the PSI-KH03 construct, the thermal denaturation of each protein was measured by loss of CD signal (Porello et al. 1998). The change in ellipticity at 222 nm over a temperature range of 15–80°C was measured for the wild-type and each of the four point mutant protein variants. The raw data were converted to fractional change in ellipticity, and the melting temperature (Tm) was determined as the temperature at which 50% of the total change in ellipticity was observed (Fig. 4 ▶). Cooperative melting is observed for each protein, and three of the four mutant proteins yielded curves very similar to that of the wild-type protein. Indeed, the Tm values calculated from these experiments reveal that for all enzymes but I234N, the Tm is ~53°C (Table 1 ▶). In contrast, the I234N mutant has a Tm value of 46°C, 7°C lower than that observed for the other proteins. Taken together, these CD studies show that while three of the mutant proteins display overall fold and thermal stability similar to wild-type PSI-KH03, mutation of the second KH domain of the protein affects global stability to a greater degree than the other domains. This suggests the second KH domain of KH03 plays a more important role in maintaining PSI structural integrity than the other three domains.
FIGURE 4.

Tm measurements on KH03 and mutant proteins. (•) Wild-type protein, (□) I145N, (♦) I234N, (gray ▴) I338N, (○) I449N.
TABLE 1.
Tm calculations of wild-type and mutant PSI-KH03 proteins
| Enzyme | Tm (°C)a |
| Wild type | 52 |
| I145N | 52 |
| I234N | 46 |
| I338N | 53 |
| I449N | 52 |
aAll Tm measurements are the average of at least three separate experiments; errors are ±1°C.
KH03 substrate affinity is dependent on RNA length
Electrophoretic gel mobility shift assays were performed with the PSI-KH03 wild-type protein to determine the equilibrium dissociation constants (Kd) of PSI-KH03 with different lengths of substrate RNA (Fig. 5A ▶). The RNA substrate sequences were chosen based on the Δ11 69-nt RNA sequence originally used to probe the specificity of PSI (Siebel and Rio 1990). The PSI29 and PSI21 sequences represent truncations of the original Δ11 sequence that contain the F1 and F2 pseudo-splice sites required for PSI binding (Fig. 1B ▶). In addition, a 59-nt sequence (PL2) was used to test the non-specific binding affinity of PSI-KH03 to RNA. In previous studies, the PL2 oligonucleotide was unable to compete for binding with labeled Δ11 RNA and so should be an adequate measure of nonspecific RNA binding (Siebel and Rio 1990).
FIGURE 5.

Binding affinity (Kd) experiments on KH03 and RNA. (A) RNA sequences used. (Full length) Δ11 sequence, (italics) PSI29, (underlined italics) PSI21. The positions of the F1 and F2 pseudo-splice sites are shown. (B) Representative native gel of wild-type KH03 binding to PSI21 RNA substrate. The Kd calculated from this experiment was 229 nM. (C) Plot of binding isotherms comparing wild-type KH03 on three separate substrates. (□) Δ11, (▴) PSI29, (○) PSI21, (♦) PL2.
The results (Fig. 5B,C ▶) show similar affinities for the Δ11 and PSI29 sequences (Table 2 ▶). In addition, the Kd obtained for Δ11 RNA is similar to that reported previously (Amarasinghe et al. 2001). However, affinity of PSI-KH03 for the 21-nt sequence, PSI21, was reduced ninefold over PSI29. This translates to a −1.6 kcal/mol free energy difference between PSI21 and Δ11 RNA based on sequence length alone. The difference in binding affinity could be due to nonspecific binding of other PSI-KH03 proteins to the longer RNA sequences. However, this significant reduction in binding affinity could also imply that a sequence longer than that provided by the F1 and F2 pseudo-splice sites is required for high-affinity binding. The latter hypothesis is supported by the work of Siebel et al. (1992, 1994), who showed that PSI will bind to RNA sequences mutated in the F1 and F2 splice sites, indicating recognition of sequences outside of the pseudo-splice sites.
TABLE 2.
Binding (Kd) data of wild-type PSI-KH03 on RNA sequences of varying length and sequence context
| RNA | Kd (nM)a | ΔΔG (kcal/mol) |
| Δ11 | 11 ± 3 | n/ab |
| PSI29 | 22 ± 7 | −0.38 |
| PSI21 | 200 ± 30 | −1.6 |
| PL2 | 1300 ± 300 | −2.6 |
aAll binding determinations are averages of at least four separate experiments. Errors are reported as one standard deviation from the average.
bNot applicable.
All four KH domains are needed for PSI-KH03 high-affinity RNA substrate recognition
To determine the effect of the I-to-N mutation on substrate binding affinity of each KH domain of PSI-KH03, native gel mobility shift assays were performed on proteins mutated in KH0 (I145N), KH1 (I234N), KH2 (I338N), and KH3 (I449N). The data reveal that mutation of any one of the four KH domains leads to reduced binding affinity for the PSI29 substrate (Table 3 ▶). However, mutations of the internal domains (I234N and I338N) cause a much more pronounced reduction in binding affinity than mutation of the N- (I145N) or C-terminal (I449N) domains. While mutation of KH0 or KH3 leads to a five- or sevenfold decrease in RNA binding affinity, respectively, mutation of KH1 or KH2 causes a corresponding 59- or 23-fold reduction. The diminished binding affinity of I234N (in KH1), in particular, is consistent with its reduced Tm as determined from CD measurements. This suggests that the second KH domain of KH03 has a greater impact on overall protein stability and binding affinity than the other three domains.
TABLE 3.
Equilibrium dissociation constants (Kd) and free-energy differences (ΔΔG) for binding of wild-type and mutant PSI-KH03 proteins to PSI29
| Enzyme | Kd (nM)a | ΔΔG |
| Wild-type | 22 ± 7 | n/a |
| I145N | 100 ± 30 | −0.84 |
| I234N | 1300 ± 200 | −2.3 |
| I338N | 500 ± 100 | −1.7 |
| I449N | 160 ± 20 | −1.1 |
aAll Kd measurements are the average of at least three separate experiments, with errors reported as one standard deviation from the average.
bNot applicable.
Reduction in substrate binding affinity is not due to a loss of protein activity
To test whether the observed loss of RNA binding affinity in the PSI mutant proteins results from lower fractions of functional molecules in these samples, titration experiments to determine the quantity of protein active for RNA binding were performed with PSI-KH03 and PSI29 RNA (Fig. 6 ▶). Using concentrations of protein and RNA at least 25-fold above the measured Kd values, wild-type PSI-KH03 binds completely to RNA at a protein:RNA ratio of 0.4:1 (Table 4 ▶) as calculated from the stoichiometric equivalence point of the data fit. In contrast, both PSI-KH03 variants with a point mutation in an internal KH domain (I234N or I338N) require a protein:RNA ratio of 1:1 for complete complex formation. The PSI-KH03 variants containing a point mutation in the Nor C-terminal KH domain (I145N or I449N) require a protein:RNA ratio between 0.5:1 and 1:1 for full complex formation. These data show that all of the protein samples tested contain fractions of active molecules within twofold of each other. While reduced activity of the mutant proteins may explain the differences observed in the CD spectra (Fig. 3 ▶), the 5- to 59-fold reductions in binding affinity of these mutant enzymes must be due to something other than a lower fraction of active molecules in the experiment. Interestingly, the titration data advocate that each wild-type PSI-KH03 molecule binds on average to two RNA molecules, and the decreased activity seen with the mutant proteins could reflect a loss of ability to bind multiple RNA molecules rather than a lower active fraction.
FIGURE 6.

Titration experiments to determine active protein fraction. Wild-type or mutant PSI-KH03 was added to RNA in the ratios described. (•) Wild-type PSI-KH03, (▪) I145N, (□) I234N, (⋄) I338N, (▴) I449N. Each experiment was repeated two to four times, with one data set represented in the plot.
TABLE 4.
Stoichiometric activity titrations of wild-type and mutant PSI-KH03
| Enzyme | Eq. Pt.a |
| Wild type | 0.37 ± 0.03 |
| I145N | 0.71± 0.05 |
| I234N | 0.8 ± 0.2 |
| I338N | 0.8 ± 0.2 |
| I449N | 0.54 ± 0.05 |
aAll measurements of the stoichiometric equivalence point are the average of two to four separate experiments, with errors reported as one standard deviation from the average.
DISCUSSION
In D. melanogaster, PSI is a vitally important splicing regulator. The protein uses four KH domains to distinguish and bind target RNA sequences in somatic cells, but contributions of individual KH domains in PSI to substrate affinity and discrimination remain unknown. Using a construct containing just the four KH domains of PSI (PSI-KH03), we have addressed this issue by individually mutating each domain and measuring the differences in secondary structure and binding affinity relative to wild-type PSI-KH03. The results of this study provide a plausible explanation for why PSI, and possibly other proteins, use multiple RNA binding motifs for high affinity substrate recognition.
Mutation of a highly conserved isoleucine residue located in the second KH domain of FMR1p (Ile 304) (Fig. 2B ▶) was originally linked to fragile X syndrome (De Boulle et al. 1993). In many studies on single KH domains, mutation of this residue to asparagine disrupts the global fold of the domain in vitro (Musco et al. 1996, 1997; Lewis et al. 1999). However, in the context of an extended domain, such as the KH-QUA2 domain of splicing factor SF1 or in the 2-KH domain-containing protein dFXRP (the Drosophila homolog to FMR1p), the effect of this mutation on secondary structure is minimal (Liu et al. 2001; Pozdnyakova and Regan 2005). These data argue that elements outside the KH motif in native proteins help maintain overall protein structure. Nonetheless, in dFXRP, the Ile-to-Asn mutation leads to significant defects in protein function both in vivo and in vitro (Wan et al. 2000; Pozdnyakova and Regan 2005). The data obtained on PSI-KH03 suggest this holds true for larger constructs as well. Mutation of the individual KH domains leads to relatively minor disruptions of protein secondary structure, as detected by CD spectroscopy, and thermal stability is maintained at wild-type levels for mutations in all domains except KH1 (I234N). However, RNA binding affinity is reduced in PSI-KH03 for all of the mutant proteins tested, with the most severe effects occurring in the internal KH1 (I234N) and KH2 (I338N) domains.
Recent structures of KH domains bound to nucleic acid have shown that the conserved isoleucine residue makes specific contacts to substrate nucleic acid (Lewis et al. 2000; Liu et al. 2001; Braddock et al. 2002a,b). It follows that mutation would lead to altered binding affinity, even when secondary structure is not affected. Indeed this is the case for SF1, where the Ile-to-Asn mutation causes a 50% decrease in binding affinity relative to wild type (Rain et al. 1998). In the Xenopus protein Vg1RBP, which contains four KH motifs localized to two didomains, mutations in any of the individual KH domains reduce RNA binding by a factor of two or less (Git and Standart 2002). The effect is more pronounced in PSI, where the magnitude of the effect depends on the domain that is mutated. Binding effects of mutations in the flanking KH domains, KH0 and KH3, are not as severe (five- to sevenfold) as the effect of mutating the inner domains, KH1 and KH2 (59- and 23-fold, respectively), suggesting the internal domains are more important for substrate recognition than the outer domains. As with Vg1RBP, highest affinity binding is achieved with all four PSI-KH03 domains intact.
Several reasons have been proposed to explain the presence of multiple KH domains in proteins. In the X-ray crystal structures of NusA transcription factor protein from Mycobacterium tuberculosis (Gopal et al. 2001) and Thermotoga maritima (Worbs et al. 2001), three RNA binding motifs (one S1 and two KH motifs) form a rigid surface for binding of an extended sequence of RNA. Additionally, multiple domains can be used to bind multiple substrates. The three KH domains of hnRNP K bind both ssDNA and RNA substrates, providing a very diverse functionality for this protein (Bomsztyk et al. 2004). The KH domains of Nova-1 have been proposed to provide a surface for inter-or intramolecular self-association (Ramos et al. 2002), and the KH3 domain of KSRP is responsible for interactions with the exosome (Gherzi et al. 2004), implicating a role in protein–protein interactions for these domains as well.
For PSI, two previous studies support a role for multiple KH domains in targeting a longer and more specific RNA sequence than could be distinguished by a single KH domain. Microarray analysis of several proteins involved in pre-mRNA splicing shows that PSI affects the smallest number of splicing events of all the proteins tested in the study, suggesting a narrow target specificity for PSI relative to other splicing factors (Blanchette et al. 2005). Additionally, work by Siebel et al. (1994) shows that RNA substrate specificity for PSI extends beyond the pseudo-splice sites located in exon 3 of Drosophila P-element transposase pre-mRNA. This is supported in the current work by the ninefold increase in Kd for substrates that include sequence elements 5′ of the pseudo-splice sites (PSI21 vs. PSI29) (Fig. 4 ▶; Table 2 ▶). Taken together, these studies implicate the multiple KH domains of PSI in increasing binding affinity for a relatively small number of target substrates.
Mutation of the N-terminal KH domain of PSI, KH0, shows the smallest overall effect on substrate binding. KH0 is degenerate in that it retains 31% identity to other KH domain sequences but lacks the conserved GXXG motif involved in substrate binding (Fig. 2B ▶). Several other proteins have been found to contain degenerate KH domains, such as human vigilin protein (Musco et al. 1996), though the purpose of these domains is unknown. Musco et al. (1996) suggested these domains could help modulate binding affinity. This would be very important for PSI in choosing between the authentic and the pseudo-splice sites present in P-element transposase pre-mRNA. Additionally, KH0 could be involved in nonspecific RNA recognition; while it lacks the GXXG motif, identity to other KH domains reveals that it most likely shares the same global fold and could contact the phosphate backbone of RNA. This does not eliminate the possibility that KH0 is involved in interactions with other proteins recruited to the pseudo-splice site. For instance, PSI could directly bind to hrp48, a splicing factor that associates with the pseudo-splice sites in the P-element transposase pre-mRNA (Siebel et al. 1992).
The introduction of a missense mutation into the individual KH domains of Drosophila PSI has yielded insight into the role multiple KH domains play in stability and substrate affinity of this protein. The presence of this mutation reveals a decrease in RNA binding affinity for PSI, the severity dependent on the domain mutated. Previous work on the role of this mutation in structure and activity of KH domain-containing proteins has mainly focused on systems containing only one or two domains. This work shows that this mutation can have varied effects on a protein with four KH domains, yet reveals a necessity for all domains for highest affinity substrate recognition.
MATERIALS AND METHODS
Enzyme cloning
The pSV272 plasmid containing the wild-type KH03 region of PSI places the sequence for KH03 downstream of a (His)6 affinity tag, a maltose binding protein, and a Tobacco etch virus (TEV) pro-tease site. Point mutations in the KH03 construct were made using QuikChange site-directed mutagenesis (Stratagene) and the following primers:
I145N, 5′-ccagtaacgacaccaacacccacatccagg-3′,5′-cctggatgtgg gtgttggtgtcgttactgg-3′;
I234N, 5′-ggcggcgataccaataaacagctg caggag-3′,5′-ctcctgcagct gtttattggtatcgccgcc-3′;
I338N, 5′-caaaggcggcgacatgaaccgtaaaatacaaactgag-3′,5′-ctca gtttgtattttacggttcatgtcgccgc ctttg-3′;
I449N, 5′-cgcggcggtgagaccaacaagctgatcaaccag-3′,5′-ctggttga tcagcttgttggtctcaccgccgcg-3′.
All clones were verified by gene sequencing.
RNA preparation
The PSI21 sequence was ordered from Dharmacon and purified by ion exchange HPLC on a Nucleopac PA-100 column (Dionex). The RNA was dissolved in HPLC buffer A (20 mM NaOAc, 20 mM LiClO4, and 10% AcCN) and eluted using a gradient with HPLC buffer B (20 mM NaOAc, 600 mM LiClO4, and 10% AcCN). The fractions containing RNA were precipitated with N-butanol and stored at −20°C. PSI29 was isolated by in vitro transcription from a linearized plasmid encoding the PSI29 sequence flanked by a 5′-hammerhead ribozyme and a 3′-hepatitis delta virus ribozyme (Ferre-D’Amare and Doudna 1996).
T7 transcription (Δ11) and SP6 transcription (PL2) of plasmids containing the sequence for Δ11 and PL2 RNA were performed as previously described (Siebel and Rio 1990). All transcribed RNAs were CIP treated as per the manufacturer’s protocol (NEB). All RNA sequences were 5′-32P end-labeled using T4 polynucleotide kinase (NEB) as per the manufacturer’s protocol and gel-purified on a 12%–15% denaturing polyacrylamide gel. Labeled RNA was extracted and ethanol precipitated prior to storage at −20°C. The nucleotide sequences for PL2, PSI21, PSI29, and Δ11 RNA are listed in Figure 5A ▶.
Mutant PSI-KH03 overexpression and purification
Plasmids containing the genes for wild-type PSI-KH03 or the PSI-KH03 mutant proteins were transformed into BL21(DE3)pLysS cells. Approximately 6 L of cells were grown in LB media at 37°C to an absorbance at 600 nm of 0.6. IPTG was added to a final concentration of 1 mM, and the cells were incubated at 30°C for an additional 3 h. Cells were harvested and resuspended in Buffer A (20 mM HEPES at pH 7.5, 1 M NaCl, 10% glycerol, 0.1% Triton X-100) to a final volume of 5 mL/g of cells. Cells were frozen at −80°C.
Cells were thawed, sonicated, and centrifuged (25,000g for 15 min). The supernatant was removed and filtered through a 0.45 μm membrane filter before loading onto a 5-mL HiTrap Chelating column (Amersham Biosciences) charged with 100 mM NiSO4 and equilibrated in Buffer A. A gradient (Buffer A containing 5–250 mM imidazole) was run, and the (His)6-MBP-TEV-KH03 construct eluted around 75 mM imidazole. The fractions containing protein were combined, concentrated slightly, and TEV protease was added to cleave the (His)6-MBP-TEV construct from KH03 protein. The protein construct including TEV protease was dialyzed against Buffer A overnight.
To remove the (His)6-MBP-TEV sequence, the dialyzed protein was reapplied to the nickel-charged HiTrap column in 10 mM imidazole, and the eluant containing cleaved PSI-KH03 was collected. The eluant was then dialyzed against Buffer B (25 mM MES at pH 6.5, 50 mM KCl, 1 mM EDTA, 1 mM DTT, 1 mM TCEP, 5 mM methionine, and 10% glycerol). The dialyzed protein was loaded onto a column containing Poros HiS resin (2.3 mL; Applied Biosystems, Inc.) equilibrated in Buffer B, and eluted in Buffer B containing 1 M KCl. The protein eluted at ~240 mM KCl. Fractions containing protein were combined and concentrated. Glycerol was added to 20% total volume, and the protein was subsequently stored in small aliquots at ~80°C. Homogeneous purification was verified by SDS-PAGE.
Wild-type KH03 overexpression and purification
Wild-type KH03 was expressed and purified as above except after purification through Poros-HS; the fractions were combined, dialyzed into Buffer C (40 mM HEPES at pH 8.0, 200 mM KCl, 1 mM EDTA, 10% glycerol, 1 mM DTT, 1 mM TCEP, 5 mM methionine), and purified over a Superdex 200 gel filtration column (Amersham Biosciences). The fractions containing protein were combined and concentrated. Glycerol was added to 20% final volume, and protein aliquots were stored at −80 °C.
CD wavelength scans and Tm measurements
CD spectroscopy was performed on wild-type and mutant KH03 proteins using an Aviv 62 spectrophotometer. All wavelength spectra were measured in a 1-cm quartz cuvette, scanning a range of 290–200 nm. Proteins were diluted into CD buffer (12.5 mM potassium phosphate at pH 7.5, 50 mM KCl, 0.5 mM EDTA, 2.5% glycerol) to a final concentration of 1 μM as determined by the Bradford method (Bradford 1976). The ellipticity values for each protein were subsequently converted to mean residue molar ellipticity, and the spectra were plotted in Microsoft Excel. Melting temperatures (Tm values) were obtained by measuring the CD signal at a fixed wavelength of 222 nm. The temperature was increased from 15°C to 80°C in steps of 1° or 2° with an equilibration time of 1 min between steps. The data were converted to fractional ellipticity and fit to Equation 1
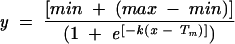 |
(1) |
in the program GraFit (Erithacus software). The temperature at which 50% of the total ellipticity was lost was defined as the Tm (Porello et al. 1998).
Equilibrium dissociation constant (Kd) measurements
Electromobility gel shift assays to determine equilibrium binding dissociation constants (Kd) for wild-type and mutant KH03 enzyme were performed as described (Amarasinghe et al. 2001). Briefly, the protein was serially diluted into binding buffer (25 mM HEPES at pH 7.6, 100 mM KCl, 10 mg/mL heparin, 0.05% NP-40, and 6% glycerol) and added to RNA kept at a concentration well below the observed Kd (100 pM–1 nM, depending on the enzyme and RNA sequence used). The samples were left to equilibrate at 4°C for 30 min, and subsequently loaded onto a 4.2% acrylamide gel (4.2% 29:1 bis:acrylamide, 50 mM Tris, 50 mM glycine, 5% glycerol). The gel was run at 10 W for 45 min, dried, and exposed overnight to a Molecular Dynamics storage phosphor screen. The autoradiogram was quantitated in ImageQuant v.5.2 (Molecular Dynamics) and the data were fit to the equation for single-site binding (Equation 2) using GraFit
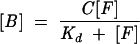 |
(2) |
where [B] and [F] are the concentrations of protein-bound and free RNA, respectively; C is the total binding capacity; and Kd is the apparent dissociation constant. The Kd values reported are the averages of at least three separate experiments.
Protein activity titration experiments
Experiments were performed in a manner similar to that of Rambo and Doudna (2004). Briefly, 2.5 pmol of 32P-5′-end-labeled PSI29 was added to unlabeled PSI29 for a total of 100 pmol RNA (wild-type PSI-KH03, I145N, I449N), 125 pmol RNA (I338N), or 300 pmol RNA (I234N). PSI-KH03 wild-type or mutant protein was added to the RNA in protein:RNA molar ratios of 0.25:1, 0.5:1, 1:1, 1.25:1, 1.5:1, 1.75:1, and 2.0:1. The protein and RNA were diluted in PSI binding buffer to a total volume of 10 μL. The samples were incubated at 4°C for 30 min and loaded onto a 4.2% native gel as described above. Dried gels were exposed to a storage phosphor screen overnight, and the autoradiogram was quantitated in ImageQuant 5.2. The data were normalized for the amount of RNA bound and fit to Equation 3
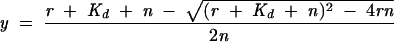 |
(3) |
where y, the fraction of RNA bound, is plotted against r, the molar ratio of [KH03]:[RNA]. N is the stoichiometric equivalence point, and Kd is the apparent dissociation constant.
Acknowledgments
The authors acknowledge K. Karbstein, W. Gilbert, and R. Spanggord for critical reading of the manuscript, and thank members of the S. Marqusee lab for help and advice on CD spectroscopy techniques. This work was supported in part by the NIH.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2175706.
REFERENCES
- Adams, M.D., Tarng, R.S., and Rio, D.C. 1997. The alternative splicing factor PSI regulates P-element third intron splicing in vivo. Genes & Dev. 11: 129–138. [DOI] [PubMed] [Google Scholar]
- Adinolfi, S., Bagni, C., Morelli, M.A.C., Fraternali, F., Musco, G., and Pastore, A. 1999. Novel RNA-binding motif: The KH module. Biopolymers 51: 153–164. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Amarasinghe, A.K., MacDiarmid, R., Adams, M.D., and Rio, D.C. 2001. An in vitro-selected RNA-binding site for the KH domain protein PSI acts as a splicing inhibitor element. RNA 7: 1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette, M., Green, R.E., Brenner, S.E., and Rio, D.C. 2005. Global analysis of positive and negative pre-mRNA splicing regulators in Drosophila. Genes & Dev. 19: 1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsztyk, K., Denisenko, O., and Ostrowski, J. 2004. hnRNP K: One protein, multiple processes. Bioessays 26: 629–638. [DOI] [PubMed] [Google Scholar]
- Braddock, D.T., Baber, J.L., Levens, D., and Clore, G.M. 2002a. Molecular basis of sequence-specific single-stranded DNA recognition by KH domains: Solution structure of a complex between hnRNP K KH3 and single-stranded DNA. EMBO J. 21: 3476–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braddock, D.T., Louis, J.M., Baber, J.L., Levens, D., and Clore, G.M. 2002b. Structure and dynamics of KH domains from FBP bound to single-stranded DNA. Nature 415: 1051–1056. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- De Boulle, K., Verkerk, A.J.M.H., Reyniers, E., Vits, L., Hendrickx, J., Van Roy, B., Van Den Bos, F., de Graaff, E., Oostra, B.A., and Willems, P.J. 1993. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet. 3: 31–35. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare, A.R. and Doudna, J.A. 1996. Use of cis- and trans-ribozymes to remove 5′ and 3′ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic Acids Res. 24: 977–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherzi, R., Lee, K.Y., Briata, P., Wegmuller, D., Moroni, C., Karin, M., and Chen, C.Y. 2004. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol. Cell 14: 571–583. [DOI] [PubMed] [Google Scholar]
- Git, A. and Standart, N. 2002. The KH domains of Xenopus Vg1RBP mediate RNA binding and self-association. RNA 8: 1319–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goolsby, K.M. and Shapiro, D.J. 2003. RNAi-mediated depletion of the 15 KH domain protein, vigilin, induces death of dividing and non-dividing human cells but does not initially inhibit protein synthesis. Nucleic Acids Res. 31: 5644–5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal, B., Haire, L.F., Gamblin, S.J., Dodson, E.J., Lane, A.N., Papa-vinasasundaram, K.G., Colston, M.J., and Dodson, G. 2001. Crystal structure of the transcription elongation/anti-termination factor NusA from Mycobacterium tuberculosis at 1.7 Å resolution. J. Mol. Biol. 314: 1087–1095. [DOI] [PubMed] [Google Scholar]
- Grishin, N.V. 2001. KH domain: One motif, two folds. Nucleic Acids Res. 29: 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, M.P., Huang, S., and Black, D.L. 2004. Differentiation-induced colocalization of the KH-type splicing regulatory protein with polypyrimidine tract binding protein and the c-src pre-mRNA. Mol. Biol. Cell 15: 774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignjatovic, T., Yang, J.C., Butler, J., Neuhaus, D., and Nagai, K. 2005. Structural basis of the interaction between P-element somatic inhibitor and U1–70k essential for the alternative splicing of P-element transposase. J. Mol. Biol. 351: 52–65. [DOI] [PubMed] [Google Scholar]
- Labourier, E., Adams, M.D., and Rio, D.C. 2001. Modulation of P-element pre-mRNA splicing by a direct interaction between PSI and U1 snRNP 70K protein. Mol. Cell 8: 363–373. [DOI] [PubMed] [Google Scholar]
- Labourier, E., Blanchette, M., Feiger, J.W., Adams, M.D., and Rio, D.C. 2002. The KH-type RNA-binding protein PSI is required for Drosophila viability, male fertility, and cellular mRNA processing. Genes & Dev. 16: 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, H.A., Chen, H., Edo, C., Buckanovich, R.J., Yang, Y.Y., Musunuru, K., Zhong, R., Darnell, R.B., and Burley, S.K. 1999. Crystal structures of Nova-1 and Nova-2 K-homology RNA-binding domains. Structure 7: 191–203. [DOI] [PubMed] [Google Scholar]
- Lewis, H.A., Musunuru, K., Jensen, K.B., Edo, C., Chen, H., Darnell, R.B., and Burley, S.K. 2000. Sequence-specific RNA binding by a Nova KH domain: Implications for paraneoplastic disease and the fragile X syndrome. Cell 100: 323–332. [DOI] [PubMed] [Google Scholar]
- Linker, K., Pautz, A., Fechir, M., Hubrich, T., Greeve, J., and Kleinert, H. 2005. Involvement of KSRP in the post-transcriptional regulation of human iNOS expression-complex interplay of KSRP with TTP and HuR. Nucleic Acids Res. 33: 4813–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z., Luyten, I., Bottomley, M.J., Messias, A.C., Houngninou-Molango, S., Sprangers, R., Zanier, K., Kramer, A., and Sattler, M. 2001. Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science 294: 1098–1102. [DOI] [PubMed] [Google Scholar]
- Min, H., Turck, C.W., Nikolic, J.M., and Black, D.L. 1997. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes & Dev. 11: 1023–1036. [DOI] [PubMed] [Google Scholar]
- Musco, G., Stier, G., Joseph, C., Morelli, M.A.C., Nilges, M., Gibson, T.J., and Pastore, A. 1996. Three-dimensional structure and stability of the KH domain: Molecular insights into the fragile X syndrome. Cell 85: 237–245. [DOI] [PubMed] [Google Scholar]
- Musco, G., Kharrat, K., Stier, G., Fraternali, F., Gibson, T.J., Nilges, M., and Pastore, A. 1997. The solution structure of the first KH domain of FMR1, the protein responsible for the fragile X syndrome. Nat. Struct. Biol. 4: 712–716. [DOI] [PubMed] [Google Scholar]
- Porello, S.L., Cannon, M.J., and David, S.S. 1998. A substrate recognition role for the [4Fe-4S]2+ cluster of the DNA repair glycosylase MutY. Biochemistry 37: 6465–6475. [DOI] [PubMed] [Google Scholar]
- Pozdnyakova, I. and Regan, L. 2005. New insights into fragile X syndrome. Relating genotype to phenotype at the molecular level. FEBS J. 272: 872–878. [DOI] [PubMed] [Google Scholar]
- Rain, J.C., Rafi, Z., Rhani, Z., Legrain, P., and Kramer, A. 1998. Conservation of functional domains involved in RNA binding and protein–protein interactions in human and Saccharomyces cerevisiae pre-mRNA splicing factor SF1. RNA 4: 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo, R.P. and Doudna, J.A. 2004. Assembly of an active group II intron-maturase complex by protein dimerization. Biochemistry 43: 6486–6497. [DOI] [PubMed] [Google Scholar]
- Ramos, A., Hollingworth, D., Major, S.A., Adinolfi, S., Kelly, G., Muskett, F.W., and Pastore, A. 2002. Role of dimerization in KH/RNA complexes: The example of Nova KH3. Biochemistry 41: 4193–4201. [DOI] [PubMed] [Google Scholar]
- Siebel, C.W. and Rio, D.C. 1990. Regulated splicing of the Drosophila P transposable element third intron in vitro: Somatic repression. Science 248: 1200–1208. [DOI] [PubMed] [Google Scholar]
- Siebel, C.W., Fresco, L.D., and Rio, D.C. 1992. The mechanism of somatic inhibition of Drosophila P-element pre-mRNA splicing: Multiprotein complexes at an exon pseudo-5′ splice site control U1 snRNP binding. Genes & Dev. 6: 1386–1401. [DOI] [PubMed] [Google Scholar]
- Siebel, C.W., Kanaar, R., and Rio, D.C. 1994. Regulation of tissue-specific P-element pre-mRNA splicing requires the RNA-binding protein PSI. Genes & Dev. 8: 1713–1725. [DOI] [PubMed] [Google Scholar]
- Siebel, C.W., Admon, A., and Rio, D.C. 1995. Soma-specific expression and cloning of PSI, a negative regulator of P element pre-mRNA splicing. Genes & Dev. 9: 269–283. [DOI] [PubMed] [Google Scholar]
- Siomi, H. and Dreyfuss, G. 1997. RNA-binding proteins as regulators of gene expression. Curr. Opin. Genes Dev. 7: 345–353. [DOI] [PubMed] [Google Scholar]
- Wan, L., Dockendorff, T.C., Jongens, T.A., and Dreyfuss, G. 2000. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell. Biol. 20: 8536–8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q., Zhang, Z., Blackwell, K., and Carmichael, G.G. 2005. Vigilins bind to promiscuously A-to-I-edited RNAs and are involved in the formation of heterochromatin. Curr. Biol. 15: 384–391. [DOI] [PubMed] [Google Scholar]
- Worbs, M., Bourenkov, G.P., Bartunik, H.D., Huber, R., and Wahl, M.C. 2001. An extended RNA binding surface through arrayed S1 and KH domains in transcription factor NusA. Mol. Cell 7: 1177–1189. [DOI] [PubMed] [Google Scholar]