

Abstract
Imd-mediated innate immunity is activated in response to infection by Gram-negative bacteria and leads to the activation of Jun amino-terminal kinase (JNK) and Relish, a nuclear factor-κB transcription factor responsible for the expression of antimicrobial peptides. Plenty of SH3s (POSH) has been shown to function as a scaffold protein for JNK activation, leading to apoptosis in mammals. Here, we report that POSH controls Imd-mediated immunity signalling in Drosophila. In POSH-deficient flies, JNK activation and Relish induction were delayed and sustained, which indicated that POSH is required for properly timed activation and termination of the cascade. The RING finger of POSH, possessing ubiquitin-ligase activity, was essential for termination of JNK activation. We show that POSH binds to and degrades TAK1, a crucial activator of both the JNK and the Relish signalling pathways. These results establish a novel role for POSH in the Drosophila immune system.
Keywords: JNK, POSH, Relish, TAK1, Imd
Introduction
Innate immunity is an evolutionarily conserved defence mechanism against infectious microorganisms (Hoffmann et al, 1999; Akira, 2001). Genetic approaches in Drosophila have uncovered two distinct pathways that are mediated by Toll and Imd (Hoffmann, 2003; Vodovar et al, 2004). The Toll pathway is primarily activated in response to infection by fungi and Gram-positive bacteria, whereas the Imd pathway is predominantly activated by Gram-negative bacteria, and in turn activates the Jun amino-terminal kinase (JNK) signalling cascade and a nuclear factor-κB (NF-κB) transcription factor, Relish, which induces the expression of a battery of genes encoding antimicrobial peptides (Lemaitre et al, 1995).
Among the components of the Imd pathway, TAK1 plays a crucial role as an activator of both the JNK and the Relish pathways (Vidal et al, 2001; Silverman et al, 2003; Park et al, 2004). Flies that are deficient in TAK1 do not produce antibacterial peptides and are therefore highly susceptible to Gram-negative bacterial infection (Vidal et al, 2001). In Drosophila S2 cells, TAK1 has been shown to be required for the peptidoglycan-induced activation of IκB kinase (IKK) and JNK (Leulier et al, 2003; Silverman et al, 2003; Kaneko et al, 2004). TAK1 is also important for limiting the duration of JNK activation, thereby resulting in a transient JNK-dependent response that precedes the sustained induction of the Relish-dependent genes.
Although it has been shown that proteasomal degradation of TAK1 leads to the rapid termination of JNK signalling (Park et al, 2004), the degradation mechanism has yet to be entirely clarified, and a ubiquitin ligase that promotes TAK1 degradation has not been reported. Here, we identify Plenty of SH3s (POSH) as a crucial component that controls the termination of immunity signalling in Drosophila. POSH, which was initially isolated as a Rac1-interacting protein, consists of a RING-finger domain and four SH3 domains, and has been shown to function as a scaffold protein for JNK signalling components leading to apoptosis in mammals (Tapon et al, 1998; Xu et al, 2003). In Drosophila, overexpression of POSH in imaginal discs activates JNK, which results in various defects in adult morphology (Seong et al, 2001). Using flies that are deficient in POSH, we provide evidence that POSH is required for the properly timed activation and termination of Imd-mediated immune responses, and that the RING finger of POSH, which shows ubiquitin-ligase activity, is essential for this regulation.
Results and Discussion
To assess the role of POSH in vivo, we created a null allele of POSH by the excision of a flanking P-element in EP(2)1026. We obtained a mutant bearing a deletion that included the start codon and extended 1,282 nucleotide bases into the coding region of POSH (supplementary Fig 1A,B online). Northern blot analysis indicated that POSH transcripts were not detectable in POSH74 homozygous animals (supplementary Fig 1C online). Most POSH74 homozygous individuals survived embryogenesis, and the JNK cascade operating during dorsal closure remained intact (data not shown; Harden, 2002). Furthermore, POSH74 showed no genetic interaction with the JNK pathway components in embryos (data not shown). Taken together, these results indicate that not every instance of JNK signalling requires POSH as a scaffold. Although mutants showed no obvious developmental defects, we observed that adult flies did not live as long as control flies, and that older cultures often contained dead larvae and pupae that were heavily melanized (Fig 1A). In addition, the viability of POSH-deficient flies after injection with Escherichia coli was significantly lower than that of wild type (Fig 1B). A possible cause for such phenotypes is defective immune function, and we therefore searched for a role for POSH in the immune response.
Figure 1.
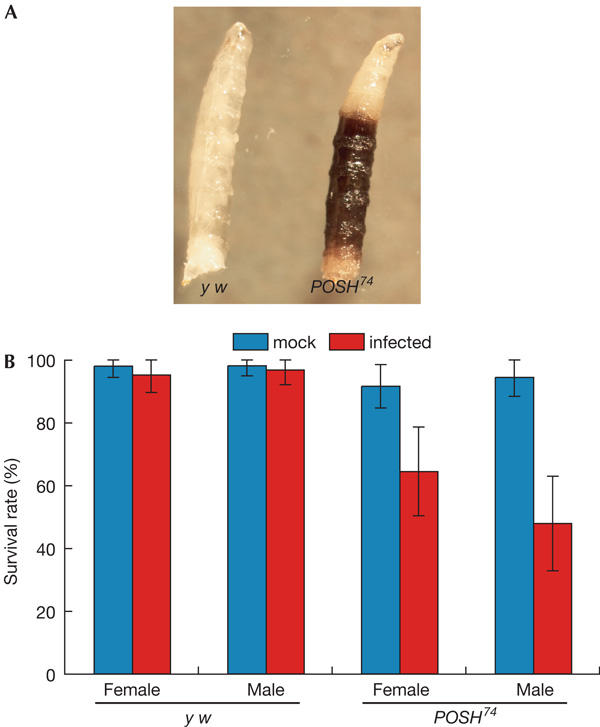
Plenty of SH3s-deficient flies show defects in the innate immune system. (A) Melanized larvae observed in POSH74 stocks. (B) Viability of flies infected with Gram-negative bacteria. Wild-type and POSH mutant adult flies were injected with a needle dipped in Escherichia coli culture. Viability was determined at 3 days after infection.
We tested whether the TAK1-mediated immune response was compromised by the POSH74 mutation. Infection with E. coli activates TAK1, which consequently phosphorylates JNK and induces the expression of Relish/NF-κB, resulting in the subsequent induction of a battery of genes encoding antimicrobial peptides, such as Diptericin (Park et al, 2004). Northern blot analysis indicated that the induction of relish and diptericin (dpt) messenger RNAs after E. coli infection was significantly delayed in POSH74 flies compared with that in controls (Fig 2A). Interestingly, we also observed that expression of relish was sustained for a longer period after infection in POSH74 flies.
Figure 2.
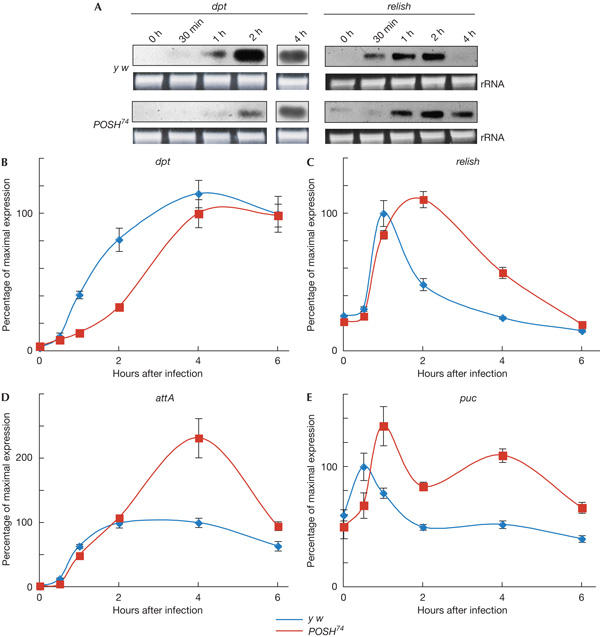
The immune response induced by bacterial infection is compromised in POSH74 mutants. (A) Northern blot analysis of immune response genes, diptericin (dpt) and relish. Total RNA was isolated at an indicated time point after Escherichia coli infection and subjected to northern blot analysis. The inductions of both dpt and relish are significantly delayed compared with those in control flies. Expression of relish is sustained for a longer period in POSH74. rRNA, ribosomal RNA. (B–E) Real-time PCR analysis of immune response genes, dpt, relish, attacin A (attA) and puckered (puc). In POSH74, induction of dpt is delayed (B), relish expression is sustained for a longer period (C) and induction of attA, the expression of which depends on Relish, is slightly delayed, but its expression level at 4 h after infection is higher than that of the control (D). Expression of puc, a Jun amino-terminal kinase target gene, is delayed and sustained in POSH74 (E).
To investigate the effects of the POSH mutation more quantitatively, we carried out real-time PCR analysis of immune response genes (Fig 2B,C). Consistent with northern blot analysis, the induction of dpt was delayed, and relish induction was sustained, with a maximum expression at 2 h after infection. We also analysed the expression pattern of attacin A (attA), which is dependent on Relish (Fig 2D). Induction of attA was almost normal, but unlike in the control flies, its expression level continued to increase up to 4 h after infection. The maximum level of expression was twice that of control flies, consistent with Relish activity being elevated in POSH mutant flies. We also analysed the expression profile of puckered (puc), a target gene of the JNK pathway (Fig 2E). Induction of puc was slightly delayed, with the highest level of expression at 1 h after infection, and expression was sustained for a longer period in POSH74 flies compared with that in controls. These results indicate that POSH is required for properly timed activation and rapid termination of both JNK- and Relish-mediated immune response pathways.
TAK1 has been shown to be required for the maintenance of relish mRNA expression, and its proteasomal degradation is responsible for the rapid termination of Relish and JNK signalling (Park et al, 2004). POSH contains a CH4C4-type RING-finger domain near its N terminus. It has been shown that the RING-finger domain of mammalian POSH possesses E3 ubiquitin-ligase activity and regulates the level of POSH protein through the proteasomal pathway (Xu et al, 2003). We confirmed that Drosophila POSH also had E3 ubiquitin-ligase activity, by demonstrating its auto-ubiquitination (Fig 3A). To assess the functional importance of the RING domain, we constructed POSHmR bearing point mutations in the RING domain, and POSHΔR lacking the RING-finger domain altogether. Neither of these proteins was able to auto-ubiquitinate after expression in cultured Drosophila cells (S2). It is conceivable, therefore, that POSH is responsible for the feedback regulation of the immune response through TAK1 ubiquitination and its subsequent degradation.
Figure 3.
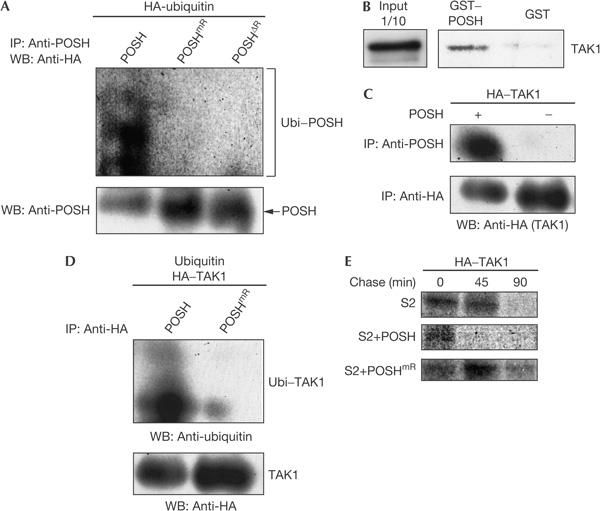
Plenty of SH3s binds to and affects TAK1 stability by proteasomal degradation. (A) Auto-ubiquitination occurs with wild-type Plenty of SH3s (POSH), but not with POSHmR harbouring point mutations in the RING domain or with POSHΔR lacking the RING-finger domain altogether. HA, haemagglutinin; IP, immunoprecipitation; WB, western blotting, Ubi, ubiquitin (B) Glutathione S-transferase (GST) pull-down assay showing that POSH binds to in vitro transcribed/translated TAK1. (C) Interaction between POSH and TAK1 in S2 cells. TAK1 and POSH were transfected into S2 cells, and co-immunoprecipitated with an anti-POSH antibody. (D) Ubiquitination of TAK1 by POSH, but not by POSHmR. S2 cells were transfected with ubiquitin, HA–TAK1 and either POSH or POSHmR. Lysates were prepared and incubated for 1 h at 4°C with an anti-HA antibody. Immunoprecipitates were subjected to western blot analysis with either an anti-ubiquitin or an anti-HA antibody. (E) Pulse–chase experiments showing POSH-dependent degradation of TAK1. Cells were pulse labelled for 1 h in 500 μl of PBS containing 100 μCi [35S]methionine and then chased with Schneider's medium supplemented with 10% fetal bovine serum. The cells were then lysed, and HA–TAK1 protein was immunoprecipitated with an anti-HA antibody and analysed by SDS–polyacrylamide gel electrophoresis, followed by fluorography. HA–TAK1 is rapidly degraded in cells transfected with POSH, but not with POSHmR.
To test this hypothesis, we carried out a glutathione S-transferase (GST) pull-down assay to investigate whether POSH was able to form a complex with TAK1. A bacterially expressed GST–POSH fusion protein pulled down TAK1 protein generated by in vitro transcription/translation (Fig 3B). We also tested the interaction between POSH and TAK1 in S2 cells by immunoprecipitation assay (Fig 3C). Lysate from cells co-transfected with POSH and haemagglutinin (HA)-tagged TAK1 (HA–TAK1) was immunoprecipitated with an anti-POSH antibody and then subjected to immunoblot analysis with an anti-HA antibody. HA–TAK1 was precipitated with an anti-POSH antibody, but not from the lysate of cells lacking the POSH expression construct (Fig 3C). These results indicate that POSH is able to bind to TAK1. To determine whether POSH could ubiquitinate TAK1, we transfected S2 cells with HA–TAK1, ubiquitin and either POSH or RING-defective POSH. HA–TAK1 was immunoprecipitated with an anti-HA antibody and then subjected to immunoblot analysis with an anti-ubiquitin antibody. This assay showed that the RING finger of POSH was able to ubiquitinate TAK1 (Fig 3D). Finally, we examined the effect of POSH overexpression on TAK1 stability using a pulse–chase experiment (Fig 3E). HA-tagged TAK1 was pulse labelled with [35S]methionine for 1 h and then chased under various conditions. Pulse-labelled HA–TAK1 was detected in S2 cells after 45 min of chasing, but not in POSH-overexpressing S2 cells at the same time point, which suggests that POSH promotes TAK1 degradation. In contrast, HA–TAK1 was exceptionally stable in S2 cells expressing POSHmR, a mutant form lacking ubiquitin-ligase activity. Overexpression of POSHmR might exert a dominant-negative effect on interactions between any endogenous POSH and TAK1, and thereby inhibit the degradation of TAK1. These results indicate that the RING-finger function of POSH is essential for the degradation of TAK1.
To characterize further the role of POSH in the regulation of JNK signalling, we used immunocompetent S2 cells in an in vitro model system used to study the immune response triggered by peptidoglycan in a commercial lipopolysaccharide preparation (Leulier et al, 2003; Silverman et al, 2003; Kaneko et al, 2004). Consistent with puc expression results in POSH mutant flies, the reduction of POSH by RNA interference (RNAi) significantly decreased the level of JNK activation, but it was sustained for a longer period than in control cells (Fig 4A). In contrast, overexpression of POSH induced JNK activation at a high level, but then inactivated it rapidly. When POSHmR was transfected into S2 cells, phosphorylated JNK persisted for a longer period than in control cells. We also assessed the effects of POSH overexpression and the role of the RING-finger function in vivo using armadillo-GAL4 (arm-GAL4) as a ubiquitous expression driver (Fig 4B). Following bacterial infection, the expression level of puc, a target of JNK signalling, rapidly declined in flies overexpressing POSH (arm>POSH). In contrast, puc expression was sustained in flies overexpressing POSHmR (arm>POSHmR), which suggests that POSHmR has a dominant-negative effect on JNK signalling. Together, these results indicate that POSH affects both activation and inactivation of JNK signalling induced by microbial infection, and that the RING-finger domain of POSH is essential for the negative feedback regulation of JNK signalling. The profile of puc expression in POSH74 flies is remarkably similar to that seen in relish flies (Park et al, 2004), which supports the idea that POSH mediates Relish-dependent inactivation of JNK in the immune system.
Figure 4.
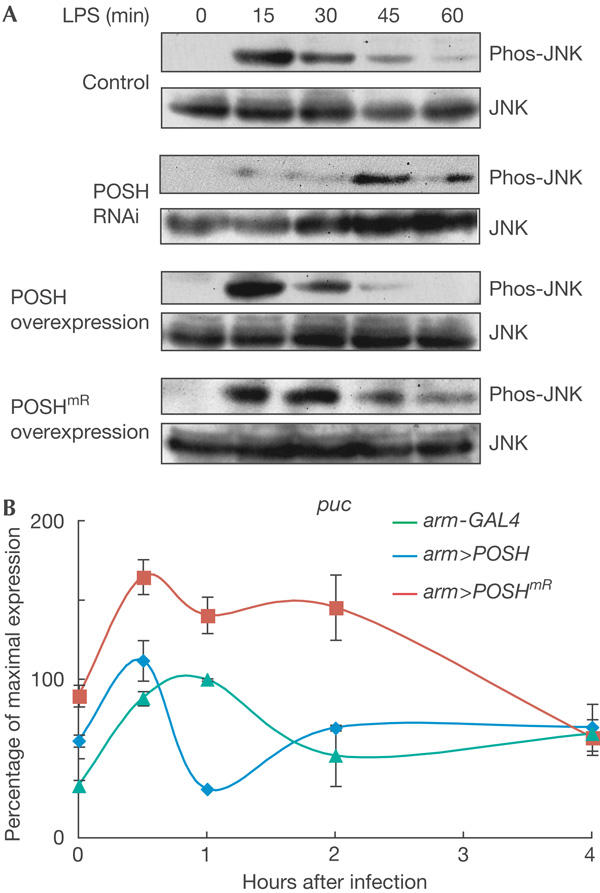
Regulation of Jun amino-terminal kinase signalling by Plenty of SH3s. (A) Time-course analysis of Jun amino-terminal kinase (JNK) phosphorylation after peptidoglycan stimulation of S2 cells transfected with Plenty of SH3s doublestranded RNA (POSH dsRNA), POSH or POSHmR DNA. Peptidoglycan-induced JNK activation is delayed and sustained in cells treated with POSH dsRNA. In contrast, JNK activation is rapidly terminated when wild-type POSH is overexpressed, whereas the activated JNK persists for a longer time period when POSHmR is overexpressed. LPS, lipopolysaccharide; phos-JNK, phorphorylated JNK. (B) Real-time PCR analysis showing the time-course analysis of puckered (puc) expression. Expression level of puc, a target of the JNK pathway, is rapidly reduced in flies overexpressing POSH, but is sustained in those overexpressing POSHmR, which suggests that RING-defective POSH has a dominant-negative effect on JNK signalling activated by infection.
Our results show that POSH modulates the TAK1-mediated innate immune response. Following microbial infection, POSH facilitates rapid JNK activation and the induction of relish/NF-κB expression, both of which are mediated by TAK1 (Silverman et al, 2003). Conversely, POSH is also required for the rapid termination of both JNK activation and relish transcription through the E3 ubiquitin-ligase activity of the RING-finger domain. Our results indicate that POSH is involved in a mechanism of negative feedback regulation of JNK and NF-κB signalling. The Drosophila TAK1-mediated innate immune response is similar to the tumour necrosis factor (TNF) signalling cascade in mammals (Leulier et al, 2002). Imd protein contains a death domain with homology to that of mammalian TNF-receptor-interacting protein (RIP; Georgel et al, 2001). Most of the pathway components—such as FADD, TAK1—and Drosophila homologues of IKKγ and IKKβ are conserved in mammalian TNF signalling. The activation of Relish involves its phosphorylation and cleavage by DREDD, the Drosophila homologue of caspase 8 (Silverman et al, 2000; Stoven et al, 2000). Eiger, a Drosophila homologue of the TNF-α superfamily ligand, has recently been identified (Igaki et al, 2002; Moreno et al, 2002; Kauppila et al, 2003). Overexpression of Eiger induces cell death by activating the JNK signalling pathway, as in mammals. Although the Eiger signalling pathway has not been fully elucidated in Drosophila, it has been clearly shown that TAK1 is essential to transduce the cell-death signal. It is possible that POSH is involved in Eiger signalling and modulates TAK-mediated JNK signalling as in the immune system. It will be of interest to determine whether POSH functions in mammalian TNF signalling.
Methods
Fly stocks. y w (y w67c23Df(1)w67c23) was used as a control strain. EP(2)1206 (Rorth et al, 1998) and arm-GAL4 were obtained from Szeged and the Kyoto Stock Center, respectively.
DNA constructs. Mutant POSH complementary DNAs were constructed using PCR mutagenesis and standard molecular biology techniques. POSHmR has the point mutations C28S, C33S and C36S, and POSHΔR lacks the N-terminal 107 residues containing the entire RING domain.
Immune response assays. An overnight culture of E. coli was resuspended in PBS and injected into adult flies using a microinjection needle. Injected flies were collected at an appropriate time and were subjected to northern blot analysis. Total RNA was isolated from adult flies using an RNeasy Total RNA kit (QIAgen, Tokyo, Japan). Northern blot analysis was carried out with digoxigenin-UTP-labelled RNA probes (Roche, Tokyo, Japan) prepared using dpt or relish cDNA clone, and signals were visualized by the CDP-star system (Roche). For real-time PCR, cDNAs were synthesized using the Superscript II reverse transcriptase system (Invitrogen, Carlsbad, CA, USA). Real-time PCR was carried out using SYBER Premix Ex Taq (Takara, Shiga, Japan) and a Chromo 4 Detector (MJ Research, Hercules, CA, USA). Results were normalized to the rp49 mRNA level. All the experiments were repeated at least three times. Primer sequences are available on request. The peptidoglycan-induced immune response was examined using S2 cells, as described previously (Park et al, 2004). Cells were collected at an appropriate time after induction and were subjected to western blot analysis with an anti-phospho-JNK antibody G9 (Cell Signaling Technology, Beverley, MA, USA). Primary and secondary antibodies were removed by treating with stripping buffer (10 mM Tris–HCl (pH 6.7), 100 mM 2-β-mercaptoethanol and 20% SDS) at 50°C for 30 min and were subjected to western blot analysis with an anti-JNK antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Ubiquitination analysis. To analyse the self-ubiquitination of POSH, S2 cells were transfected with pUAS–HA–ubiquitin (a gift from S. Hatakeyama, Kyusyu University), pWAGAL4 (a gift from Y. Hiromi, National Institute of Genetics), and pUAS–POSH, pUAS–POSHmR or pUAS–POSHΔR. At 48 h after transfection, cells were treated with 5 μM lactacystin for 3 h. Cell lysates were incubated for 1 h at 4°C with a rabbit anti-POSH antibody (raised against a GST fusion protein of amino acids 479–781 of POSH) immobilized using an ImmunoPure Plus IgG Orientation Kit (Pierce, Rockford, IL, USA). The immunoprecipitates were analysed by western blot with either an anti-HA antibody 12CA5 (Sigma, St Louis, MO, USA) or an anti-POSH antibody. To detect the ubiquitination of TAK1, cells were transfected with pWAGAL4, pUAS–ubiquitin, pUAS–HA–TAK1 and either pUAS-POSH or pUAS-POSHmR. Cells were treated with lactacystin and lysed. The lysates were incubated for 1 h at 4°C with an anti-HA antibody Y-11 (Santa Cruz Biotech.). Protein G beads were added to solution for 1 h at 4°C, followed by centrifugation. The immunoprecipitates were washed five times with lysis buffer and subjected to western blot analysis with either an anti-ubiquitin antibody 6C1 (Sigma) or an anti-HA antibody 12CA5.
Glutathione S-transferase pull-down assay and immunoprecipitation assay. To make GST fusion proteins, the protein-coding regions of POSH cDNAs were cloned into pGEX-41 (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Bacterially expressed fusion proteins were immobilized onto glutathione–Sepharose 4B beads according to the manufacturer's instructions (Amersham Pharmacia Biotech). Proteins labelled with [35S]methionine were produced by the in vitro transcription and translation system (Promega, Madison, WI, USA). A GST–POSH fusion protein was incubated with labelled TAK1 protein, and then analysed by SDS–polyacrylamide gel electrophoresis, followed by fluorography. For immunoprecipitation assay, cells were transfected with pWAGAL4 and pUAS–HA–TAK1 with or without pUAS–POSH. At 48 h after transfection, cell lysates were incubated for 1 h at 4°C with an immobilized anti-POSH antibody. The immunoprecipitates were subjected to western blot analysis with either an anti-HA antibody 12CA5 (Sigma) or an anti-POSH antibody.
RNA interference experiments. A 500 bp fragment of POSH cDNA flanked by a T7 promoter at both ends was generated by PCR using appropriate primers. Double-stranded RNA (dsRNA) was produced using a MEGAscript RNAi Kit (Ambion, Austin, TX, USA). S2 cells were incubated with serum-free medium containing 25 μg/ml dsRNA for 4 h. The RNAi-treated cells were grown in serum-containing medium for 72 h before immune response experiments.
Pulse–chase experiment. Cells were pulse labelled for 1 h in 500 μl of PBS containing 100 μCi [35S]methionine and then chased with Schneider's medium supplemented with 10% fetal bovine serum. The cells were then lysed with 250 μl of lysis buffer (100 mM potassium phosphate buffer (pH 7.8), 1% Triton X-100, 1 mM dithiothreitol and protease inhibitor cocktail tablets (Roche)). HA-tagged TAK1 protein was immunoprecipitated from the lysates with an anti-HA antibody 12CA5 (Sigma) and analysed by SDS–polyacrylamide gel electrophoresis, followed by fluorography.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Fig 1
Acknowledgments
We thank S.A. Wasserman, S. Hatakeyama and Y. Hiromi for plasmids, and the Kyoto Stock Center for fly stocks. This study was supported by grants from the Ministry of Education, Culture, Sports, Science & Technology of Japan (T.A.), the National Cancer Institute of Canada (N.H.) and the Japan Society for the Promotion of Science (M.T.).
References
- Akira S (2001) Toll-like receptors and innate immunity. Adv Immunol 78: 1–56 [DOI] [PubMed] [Google Scholar]
- Georgel P, Naitza S, Kappler C, Ferrandon D, Zachary D, Swimmer C, Kopczynski C, Duyk G, Reichhart JM, Hoffmann JA (2001) Drosophila immune deficiency (IMD) is a death domain protein that activates antibacterial defense and can promote apoptosis. Dev Cell 1: 503–514 [DOI] [PubMed] [Google Scholar]
- Harden N (2002) Signaling pathways directing the movement and fusion of epithelial sheets: lessons from dorsal closure in Drosophila. Differentiation 70: 181–203 [DOI] [PubMed] [Google Scholar]
- Hoffmann JA (2003) The immune response of Drosophila. Nature 426: 33–38 [DOI] [PubMed] [Google Scholar]
- Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA (1999) Phylogenetic perspectives in innate immunity. Science 284: 1313–1318 [DOI] [PubMed] [Google Scholar]
- Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, Miura M (2002) Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J 21: 3009–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T, Goldman WE, Mellroth P, Steiner H, Fukase K, Kusumoto S, Harley FW, Fox A, Golenbock D, Silverman N (2004) Monomeric and polymeric gram-negative peptidoglycan but not purified LPS stimulate the Drosophila IMD pathway. Immunity 20: 637–649 [DOI] [PubMed] [Google Scholar]
- Kauppila S et al. (2003) Eiger and its receptor, Wengen, comprise a TNF-like system in Drosophila. Oncogene 22: 4860–4867 [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Kromer-Metzger E, Michaut L, Nicolas E, Meister M, Georgel P, Reichhart JM, Hoffmann JA (1995) A recessive mutation, immune deficiency (imd), defines two distinct control pathways in the Drosophila host defense. Proc Natl Acad Sci USA 92: 9465–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulier F, Vidal S, Saigo K, Ueda R, Lemaitre B (2002) Inducible expression of doublestranded RNA reveals a role for dFADD in the regulation of the antibacterial response in Drosophila adults. Curr Biol 12: 996–1000 [DOI] [PubMed] [Google Scholar]
- Leulier F, Parquet C, Pili-Floury S, Ryu JH, Caroff M, Lee WJ, Mengin-Lecreulx D, Lemaitre B (2003) The Drosophila immune system detects bacteria through specific peptidoglycan recognition. Nat Immunol 4: 478–484 [DOI] [PubMed] [Google Scholar]
- Moreno E, Yan M, Basler K (2002) Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 12: 1263–1268 [DOI] [PubMed] [Google Scholar]
- Park JM et al. (2004) Targeting of TAK1 by the NF-κB protein Relish regulates the JNK-mediated immune response in Drosophila. Genes Dev 18: 584–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorth P et al. (1998) Systematic gain-of-function genetics in Drosophila. Development 125: 1049–1057 [DOI] [PubMed] [Google Scholar]
- Seong KH, Matsuo T, Fuyama Y, Aigaki T (2001) Neuralspecific overexpression of Drosophila Plenty of SH3s (DPOSH) extends the longevity of adult flies. Biogerontology 2: 271–281 [DOI] [PubMed] [Google Scholar]
- Silverman N, Zhou R, Stoven S, Pandey N, Hultmark D, Maniatis T (2000) A Drosophila IκB kinase complex required for Relish cleavage and antibacterial immunity. Genes Dev 14: 2461–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman N, Zhou R, Erlich RL, Hunter M, Bernstein E, Schneider D, Maniatis T (2003) Immune activation of NF-κB and JNK requires Drosophila TAK1. J Biol Chem 278: 48928–48934 [DOI] [PubMed] [Google Scholar]
- Stoven S, Ando I, Kadalayil L, Engstrom Y, Hultmark D (2000) Activation of the Drosophila NF-κB factor Relish by rapid endoproteolytic cleavage. EMBO Rep 1: 347–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Nagata K, Lamarche N, Hall A (1998) A new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-κB signalling pathways. EMBO J 17: 1395–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal S, Khush RS, Leulier F, Tzou P, Nakamura M, Lemaitre B (2001) Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-κB-dependent innate immune responses. Genes Dev 15: 1900–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodovar N, Acosta C, Lemaitre B, Boccard F (2004) Drosophila: a polyvalent model to decipher host–pathogen interactions. Trends Microbiol 12: 235–242 [DOI] [PubMed] [Google Scholar]
- Xu Z, Kukekov NV, Greene LA (2003) POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J 22: 252–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig 1