


Abstract
Phage DNA packaging is believed to be driven by a rotary device coupled to an ATPase ‘motor’. Recent evidence suggests that the phage DNA packaging motor is one of the strongest force-generating molecular motors reported to date. However, the ATPase center that is responsible for generating this force is unknown. In order to identify the DNA translocating ATPase, the sequences of the packaging/terminase genes of coliphages T4 and RB49 and vibriophages KVP40 and KVP20 have been analyzed. Alignment of the terminase polypeptide sequences revealed a number of functional signatures in the terminase genes 16 and 17. Most importantly, the data provide compelling evidence for an ATPase catalytic center in the N-terminal half of the large terminase subunit gp17. An analogous ATPase domain consisting of conserved functional signatures is also identified in the large terminase subunit of other bacteriophages and herpesviruses. Interestingly, the putative terminase ATPase domain exhibits some of the common features found in the ATPase domain of DEAD box helicases. Residues that would be critical for ATPase catalysis and its coupling to DNA packaging are identified. Com binatorial mutagenesis shows that the predicted threonine residues in the putative ATPase coupling motif are indeed critical for function.
INTRODUCTION
Double-stranded (ds)DNA packaging in icosahedral viruses is a fascinating biological problem. During packaging, a complex, metabolically active, concatemeric DNA is translocated into the confines of an empty capsid and condensed into a highly ordered, intact structure of near crystalline density. It is an ATP-driven process that incorporates a number of important fundamental biological mechanisms, including: reversible condensation and decondensation of DNA, high fidelity transport of genomes into daughter cells, DNA movement along protein complexes and transduction of ATP hydrolysis energy into mechanical work.
In the late stages of lytic infection, the head assembly and DNA replication pathways of T4 converge in preparation for DNA packaging (1). The head assembly pathway produces a mature empty prohead, whereas the DNA replication pathway results in a head-to-tail polymeric DNA, referred to as the concatemer. The terminase links these two pathways by recognizing the viral concatemer, making an endonucleolytic cut and joining it to the prohead through specific interactions with the unique portal vertex. Consequently, a DNA translocating pump is assembled, with the terminase as one of the key components. Following translocation of one headful-length DNA, terminase makes a second cut, dissociates from the packaged structure and repeats the DNA linkage to another prohead in a processive fashion (2–5).
Terminase is a hetero-multimeric complex consisting of one large and one small subunit, but the stoichiometry of the subunits in the holoenzyme is unknown. The small subunit has a weak DNA-binding activity and is thought to be involved in recognition of the viral genome substrate, whereas the large subunit is multifunctional and is required for cleavage of DNA to generate termini, linkage of cleaved DNA to the prohead connector and translocation of DNA into the empty prohead (4). In phage T4, the small terminase subunit is encoded by the 18 kDa gp16 and the large terminase subunit by the 70 kDa gp17 (6,7). gp17 exhibits an endonuclease activity, an in vitro DNA packaging activity and an ATPase activity, whereas gp16 lacks any of these activities but stimulates the latter two activities associated with gp17 (8,9).
Based on the data from the defined in vitro DNA packaging systems of T3 and φ29, it was estimated that 2 bp of dsDNA are packaged for every molecule of ATP hydrolyzed (10,11). This means that roughly 105 molecules of ATP are expended to package one molecule of T4 DNA at an estimated catalytic rate of ∼104 ATPs/min/packaging unit. Recent studies using optical tweezers have indicated that phage DNA is packaged against considerable internal pressure within the capsid and that the force generated by the packaging machine is the strongest measured for any biological motor (12). However, one key question remains unanswered in every phage system: which component of the packaging machine hydrolyzes ATP to generate this tremendous force?
There is good circumstantial evidence to suggest that the large terminase subunit is likely the DNA translocating ATPase. For instance, temperature-sensitive (ts) mutations in gp17 accumulate partially filled heads, suggesting a defect in DNA translocation (13), and conservative substitutions in the putative ATP-binding site I of gp17 result in complete loss of DNA packaging activity (14). Recent data from phages λ and T3 also provide evidence supporting the linkage of the large terminase protein to the DNA translocating ATPase (15,16).
In this study, we used a sequence alignment approach to analyze the coding sequences of terminase proteins. Considering the remarkable structural and functional conservation displayed by the components of the phage DNA packaging machines, we argued that their terminases must contain the signatures of a common ATPase catalytic center. This is a testable hypothesis, because the X-ray structures of a number of ATP-hydrolyzing biological machines show highly conserved functional motifs and catalytic residues in the ATPase center, despite their unrelated functions and lack of overall sequence similarity; indeed, preliminary evidence already exists to suggest that the terminases contain consensus Walker ATP-binding motifs (17,18).
We have analyzed four available T4 family terminase sequences and identified a number of putative functional motifs that are attributed to known functions of the terminase proteins. We have discovered an interesting, albeit limited, sequence similarity between the T4 terminase and the DEAD box helicases (19), which assisted in the identification of key elements of an ATPase catalytic center in the large terminase protein, gp17. These features are remarkably well conserved among the viral terminase families, including the herpesviruses. The functional significance of the predicted motifs was tested by combinatorial mutagenesis of the gp17 ATPase, which showed that substitutions at the signature residues are either not tolerated or are highly restricted. Thus, this study, for the first time, defines a common ATPase center in the terminases, supporting the hypothesis that the ATPase ‘motor’ function of the DNA packaging machine is localized in the large terminase protein.
MATERIALS AND METHODS
Terminase sequences
The amino acid sequences of T4 and RB49 gp16 and gp17 were obtained from GenBank. The KVP40 and KVP20 terminase genes were cloned using standard recombinant DNA procedures. Sequencing was done by the primer walk method and the reactions were analyzed on a PRISM 310 Genetic Analyzer (Applied Biosystems). The 5′-ends of the genes 16v and 17v were determined by the presence of a potential ribosome-binding site (AGGA) upstream of the open reading frame start codon, which corresponded to the analogous positions in the T4 and RB49 genomes. The completed 2993 bp (KVP40) and 2553 bp (KVP20) sequences consisting of the genes 16v and 17v were deposited in DDBJ (accession nos: KVP40, AB050094; KVP20, AB075036).
Sequence searches and alignments
Sequence analyses were performed using BLAST, PSI_ BLAST (20) and BLAST 2 (21) which are available on the National Center for Biotechnology Information (NCBI) server (http://www.ncbi.nlm.nih.gov/BLAST/). BLAST searches were conducted with default parameter values, except where noted.
Hydropathic profile of the small terminase subunit
The Kyte–Doolittle hydrophathy profile of the small terminase subunit multiple sequence alignment was done with the pepwindowall utility available on the Institut Pasteur bioweb (http://bioweb.pasteur.fr/seqanal/interfaces/pepwindowall.html) using a hydropathy averaging window size of 9.
Secondary structure predictions
For generating the secondary structure predictions, we used BLAST-retrieved terminase sequences (see legend to Fig. 5) to generate multiple sequence alignments. Multiple sequence alignments were prepared using CLUSTALW (22; http://barton.ebi.ac.uk/servers/jpred.html), which were then used to produce a secondary structure prediction by jpred2 (23; http://barton.ebi.ac.uk/servers/jpred.html). Both programs are located on the EMBL-European Bioinformatics Institute server. Other secondary structure predictions for single protein amino acid sequences were performed using the consensus secondary structure prediction program on the Pôle Bio-Informatique Lyonnais (PBIL) server (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_seccons.html).
Figure 5.
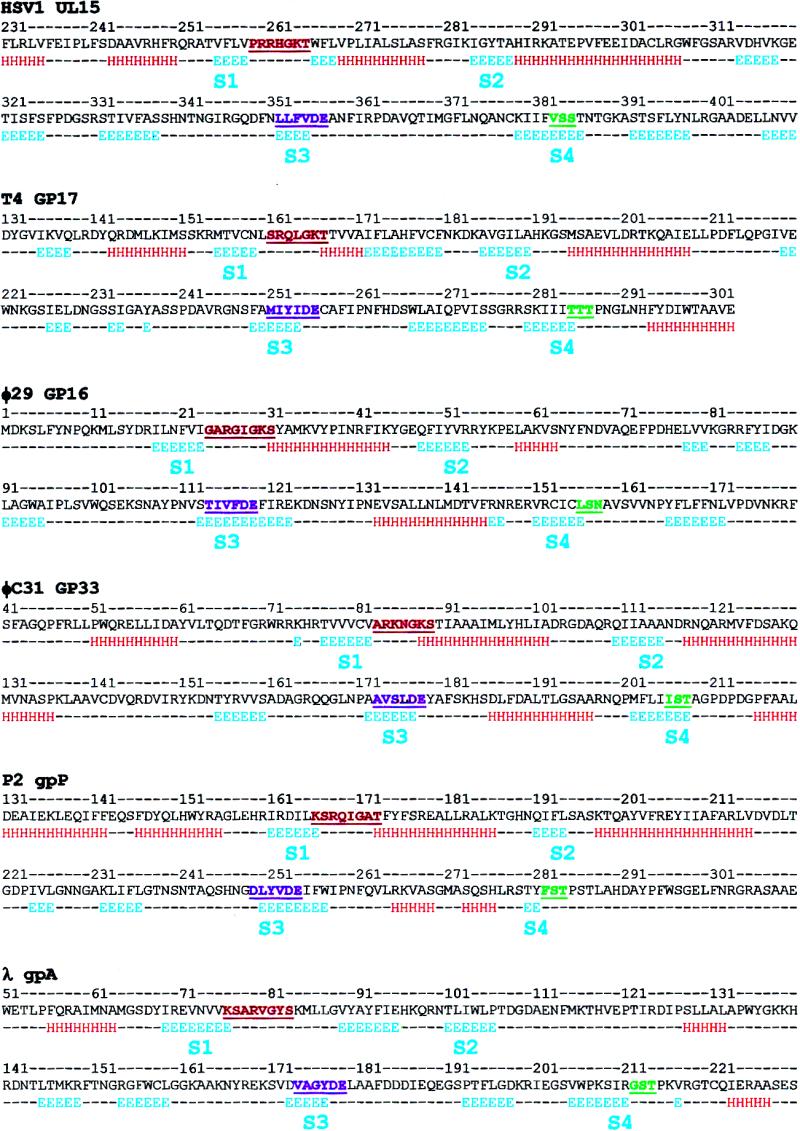
Secondary structure prediction for the ATPase domain of viral terminases. The putative ATPase domains of the represented terminase families were computed based on the input of multiple sequence alignments generated by using homologous terminase sequences available in the GenBank database (see Materials and Methods). H, α-helix; E, β-strand; dashes indicate no predicted structure. S1–S4, are the conserved β-strands (see Results). The Walker A motif (red), Walker B motif (purple) and motif III (green) are underlined and highlighted. The following viral terminase sequences were used (accession nos in parentheses). Herpes virus family: HSV-1 (P04295); EHV-1 (P28969); CHV-7 (NP_077457); VZV (P09294); GHV-2 (AAG14207); HCMV (P16732); THV (AAK00707); HSV-6B (NP_050241); HSV-6U (P24443); HSV-7J (P52462); AHV (NP-065527); EBV (P03219); SHV (Q01020); MHV-4 (AAF19294). T4 family: T4 (P17312); RB49 (AAF23757); KVP40 (BAB41133); KVP20 (BAB96804). φ29 family: φ29 (WMBP26); PZA (AAA88493); B103 (Q37897); GA-1 (CAC21537); Cp-1 (CAA87742); mycoplasma P1 (NP_064638). φC31 family: φC31 (CAA07103); HK97 (NP_037698); HK022 (AAF30354); A2 (CAB63682); φSLT (BAB21732); D3 (AAD38954); bIL286 (AAK08328); pi3 prophage (AAK05497); BK5-T (AAK56802); Sfi21 (AAD41029); φPVL (NP_058441); φPV83 prophage (BAA97845); φadh (NP_050148); Sfi19 (AAD44056); pi1 prophage (AAK04567); bIL309 (AAK08387); 7201 (AAF43515); bIL285 (AAK08266); sk1 (NP_044948); P008 (AAC63033); φ41 (AAB41467); BFK20 (CAB93906); bIL170 (AAC27181). P2 family: P2 (NP_542602); 186 (AAC34148); φCTX (BAA36228); HP1 (AAB09201); HP2 (AAK37798). λ family: λ (P03708); N15 (AAC19038); 21 (AAA32340); WO (BAA89621).
Construction of Thr285, Thr286 and Thr287 mutants
A site-specific, PCR-directed, splicing by overlap extension (SOE) strategy was used to construct combinatorial libraries at residues T285 and T286 and an amber mutant at T287. As described earlier (14), four oligonucleotides and three successive PCRs were used to incorporate the mutations and amplify the mutant. The common g17 end primers were: 5′-forward primer, 5′-CGCGGGATCCGATGGAACAACCGATTAATGTATTAAATG-3′ (nt 1–28); 3′-reverse primer, 5′-CGCGGGATCCTTATACCATTGACATACCATGAGATAC-3′ (nt 1836–1810) (italicized nucleotides represent the tag sequence added to the 5′-end for efficient cutting at the adjacent BamHI sequence, shown in bold; nucleotide numbers correspond to the coding sequence of gp17). The forward and reverse mutant primers were as follows: T285 combinatorial library, 5′-CGTTCGAAAATTATTATTNNNACGACTCCTAATGGATTAAATC-3′ and 5′-GATTTAATCCATTAGGAGTCGTNNNAATAATAATTTTCGAAC-3′; T286 com binatorial library, 5′-CGTTCGAAAATTATTATTACTNNNACTCCTAATGGATTAAATC-3′ and 5′-GATTTAATCCATTAGGAGTNNNAGTAATAATAATTTTCGAAC-3′; T287 amber mutant, 5′-CGTTCGAAAATTATTATTACTACGTAGCCTAATGGATTAAATC-3′ and 5′-GATTTAATCCATTAGGCTACGTAGTAATAATAATTTTCGAAC-3′ (the mutagenized nucleotides are shown in bold). The amplified mutant g17 DNA with BamHI ends was digested with BamHI and ligated with the BamHI-linearized and dephosphorylated pET15b vector. The recombinant DNA was then transformed into Escherichia coli BL21 and ampR colonies were selected.
The T4.17T287am phage mutant was constructed by transferring the T287am mutation from the plasmid into the T4 genome by recombinational exchange using T4.17K166am as the infecting phage and recovering the 17T287am mutant under the genetic background of E.coli-Ser suppressor (24,25).
To analyze the T285 and T286 combinatorial libraries, about 100 random ampR transformants were picked from each library and grown in LB-amp medium (1 ml). Each mutation was independently transferred into the T4 genome by a recombinational marker rescue strategy (14), using T4.17T287am as the infecting phage and T4.17K166am phage as a control. Each mutant was phenotypically scored as functional (lysis), small plaque (lysis with small plaque size) or null (no plaques/lysis). A set of 24–36 phenotypes were selected from each library, colony purified and the phenotype was confirmed. The g17 DNA encompassing the mutagenized site was PCR amplified and sequenced (Davis Sequencing, CA).
RESULTS
Similarity between T4 and viral terminases
BLAST searches of the NCBI database were performed using each of the four available T4 type terminase sequences. The results showed a close relationship between these proteins (Table 1), which is consistent with the recently established phylogenetic tree by Tétart et al. (26). RB49, a ‘pseudo T-even phage’, is a coliphage isolated from Long Island sewage (27) that is morphologically similar to the T-even phages, but has only a limited sequence similarity to the T4 genome. About 30% of the RB49 genome has no counterpart in T4, with the closest related region to date coding for the major capsid protein, which shows 70% identity to gp23*. KVP40 and KVP20, which are referred to as ‘schizo T-even phages’, are vibriophages isolated from highly polluted seawater in Urado Bay, Kochi, Japan. KVP40 is capable of infecting a broad range of species from the family Vibrionaceae (28), while KVP20 has a narrower host range (29). Electron micrographs of KVP40 and KVP20 show that they are large-tailed phages, morphologically similar to the T-even coliphages, with an elongated prolate head and contractile tail (30). Sequence analysis of the KVP40 and KVP20 major capsid proteins indicated that they are ∼96% identical to one another and ∼60% identical to T4 gp23*. In light of these data and the recent phylogenetic analysis of the gamma subdivision proteobacteria, which suggests placing the Vibrio genera and Enterobacteriaceae within the same monophyletic unit (31), the similarity observed between the small and large terminase subunits of T4, RB49, KVP40 and KVP20 (Table 1) is most likely due to divergent evolution from a common ancestor.
Table 1. Small and large terminase subunit protein sequences.
| No. | Phage | Protein | No. of amino acids | Identity percentage | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
|
|
|
1 |
2 |
3 |
4 |
5a |
6b |
7c |
| (A) Small terminase subunit protein sequences | ||||||||||
| 1 | T4 | gp16 | 164 | 100 | 55 | 43 | 44 | |||
| 2 | RB49 | gp16 | 158 | 100 | 39 | 39 | ||||
| 3 | KVP40 | gp16v | 182 | 100 | 87 | |||||
| 4 | KVP20 | gp16v | 182 | 100 | ||||||
| (B) Large terminase subunit protein sequences | ||||||||||
| 1 | T4 | gp17 | 610 | 100 | 69 | 54 | 53 | 25 | 26 | 21 |
| 2 | RB49 | gp17 | 607 | 100 | 51 | 51 | 25 | 22 | 19 | |
| 3 | KVP40 | gp17v | 600 | 100 | 96 | 27 | 25 | 22 | ||
| 4 | KVP20 | gp17v | 600 | 100 | 27 | 25 | 24 | |||
| 5 | HF2 | TerL | 563 | 100 | 20 | 16 | ||||
| 6 | 933W | L0113 | 568 | 100 | 15 | |||||
| 7 | HSV1 | UL15 | 735 | 100 | ||||||
aIn three PSI-BLAST iterations: in 160 (T4), 156 (RB49) and 157 amino acids (KVP40/20).
bIn three PSI-BLAST iterations: in 184 (T4), 508 (RB49), 112 (KVP40), 119 (KVP20) and 110 amino acids (HF2).
cIn three PSI-BLAST iterations: in 145 (T4), 147 (RB49), 143 (KVP40), 114 (KVP20), 309 (HF2) and 210 amino acids (933W).
The BLAST results also showed a surprising similarity between the putative ATPase domain of the large terminase subunits of the T4 family and the broad host range archaebacterial halophage HF2 (TerL, Table 1) (32), but it has thus far not been possible to identify the small terminase subunit by genome sequence analysis (M.Dyall-Smith, personal communication). This member of the Myoviridae family uses genome replication strategies that are similar to those of the dsDNA phages of the Bacteria, such as T3 or T7, which suggests either a distant evolutionary relationship that predates the divergence of Archaea and Bacteria or a lateral exchange of modules of phage genes (33).
Similarity is also evident between the T4 terminase and that of the lysogenic phage 933W, which was recently isolated from the enterohemorrhagic E.coli serotype O157:H7 strain EDL933 (34). 933W is an interesting phage, with a genome constituted of a mosaic of genes from various phages. Although much of the 933W genome and its organization resembles that of phage λ, the ends of the genome, like in phage T4, are apparently circularly permuted. Its large terminase subunit is more closely related to phage T4 than to any other phage; on the other hand, the small terminase subunit shows no similarity to T4 gp16 but does show a low level of similarity to PacA of phage P1. This mosaic genome suggests that the large terminase gene and probably many of the bacteriophage 933W genes were captured by horizontal gene transfer, rather than by divergent evolution.
Finally, the T4 large terminase shows a striking similarity to the putative terminase of herpesviruses in the region encompassing the Walker A and Walker B nucleotide-binding motifs (18). Although the small terminase has no clear sequence homolog in the herpesvirus genome, there is evidence to suggest that HSV-1 UL28 and its homologs are functionally equivalent to gp16 (35). Since no cellular counterpart to the terminase proteins has yet been discovered in eukaryotic or prokaryotic cells, this detected similarity suggests a distant relationship between these two viral families, as opposed to an independent acquisition of the protein from viral hosts (36).
Similarity between T4 terminases and DEAD box helicases
Helicases are a class of enzymes that use the free energy of ATP hydrolysis to translocate along the DNA or RNA backbone (19,37). Surprisingly, helicases are among the most frequently found genes in genomes; for example, they constitute the seventh largest group in the yeast genome (38). Walker A- and B-type NTP-binding motifs are universal features of the helicase polypeptide sequence, with additional motifs serving as identifiers for categorizing the established and putative helicases into distinct families, showing strong homology, or superfamilies (SF), showing a weaker homology (19). SF1 and SF2, to which the majority of helicases belong, have seven characteristic signature motifs, while members of SF3 and SF4 have three and five motifs, respectively. DEAD and DEXH box helicases, which have the DEAD and DEXH signatures in the Walker B motif, respectively, are among the families that form SF2.
We have detected similarities between gp17 and the DEXD/H box helicases through BLAST alignment of residues 138–291 of gp17 and the 166 amino acid long consensus sequence of the DEXD/H box helicases, which correspond to the respective ATPase domains (Fig. 1). This is a significant discovery for two reasons: first, it allowed the sequence analysis of terminases in the context of the available X-ray structural and biochemical information on DEAD box helicases, which led to the definition of an ATPase center in the N-terminal half of terminases; second, considering the analogous functions of helicases and terminases, i.e. ATP-driven translocation on the nucleic acid backbone, the similarities may have functional implications for the mechanisms of viral DNA packaging (see below).
Figure 1.

Alignment of the putative ATPase domains of the T4 family large terminase subunits and the DEXD/H helicases. Alignment of T4 terminases with DEXDc and P.abyssi Rad25 (accession no. NP_125889) was determined by BLAST with manual adjustment. DEXDc is a consensus sequence of DEAD box helicases from the Conserved Domain Database (CDD) of functional and/or structural domains; it represents the computed sequence (most frequently occurring residues at each position) from a collection of 703 DEAD-like helicase sequences (smart00487). Alignment of S.cerevisiae eIF-4a (accession no. NP_012985; pdb code 1QDEA), the probable ATP-dependent RNA helicase from M.jannaschii MJ0669 (accession no. Q58083; pdb code 1HV8), T.maritima RecG (accession no. 1GM5A; pdb code 1GM5) and A.fulgidus reverse gyrase (accession no. 1GL9B; pdb code 1GL9) was performed manually by aligning with the well-characterized helicase motifs. Helicase motifs I, II and III and the putative adenine-binding motif (YQ) are highlighted. The conserved residues are shown in bold.
Sequence identity among the terminases
As shown in Table 1, the gp16 and gp17 terminase proteins of T4 have a higher percentage of identity to their respective homologs in coliphage RB49 than to those of the two vibriophages KVP40 and KVP20. In addition, the terminase subunits of these KVP phages show less similarity to the pseudo T-even phage, RB49, than to T4, suggesting a more distant relationship between their respective proteins. Twenty-six percent of the small terminase subunit polypeptide sequence is identical among the three phages, while the overall sequence identity of the large terminase subunit is 42%. When the putative large terminase subunit of the prophage 933W is included in the alignment, the identity percentage drops to 14%, whereas each individual T-even large terminase subunit shares ∼25% identity with that of the 933W phage.
The T-even large terminase subunits are also ∼21% identical to the HSV-1 terminase, UL15, in the 140 amino acid region that encompasses the Walker A and Walker B motifs of the putative ATPase domain. However, similarity in other regions of the T4 and herpesvirus terminase proteins has also been noted (18). No significant similarity was detected between UL15 and the 933W terminase, even when the BLAST 2 search parameters were adjusted to improve the likelihood of an alignment.
Functional motifs in the small terminase subunit gp16
Oligomerization site
It is well documented that gp16 readily forms stable oligomeric rings that consist of an average of eight gp16 subunits per ring (39). gp16 oligomerization may play an important role in the initiation of DNA packaging and in the formation of a packaging-proficient holo-terminase complex. The central hydrophobic ‘domain’ of gp16 (Fig. 2A), as evident from the hydropathy profile (Fig. 2B), is a strong candidate for protein–protein interactions that lead to the oligomerization of the small subunit (39). Homologous peptides corresponding to this region were recently shown to interact with gp17 in a random peptide display strategy (L.Black, personal communication), which suggests that this region may also be involved in holoterminase assembly. An analogous hydrophobic region was identified in the phage λ small terminase subunit, gpNu1, the deletion of which resulted in a loss of oligomerization and holoenzyme formation (40).
Figure 2.

(A) Alignment of T4 family small terminase subunits. The polypeptide sequences were aligned by BLAST. The highlighted regions correspond to the following motifs: purple, helix–turn–helix; green, arginine switch; yellow, oligomerization. The clustered polar and charged residues in the C-terminus are shown in color [red, acidic (D and E); green, neutral hydrophilic (C, N, Q, S, T andY); blue, basic (H, K and R)]. The terminase small subunit polypeptide sequences are available from GenBank: T4 (accession no. P17311); RB49 (accession no. AAF18445); KVP40 (accession no. BAB41132); KVP20 (accession no. BAB96803). (B) Hydropathy profile of the small terminase multiple sequence alignment.
DNA-binding site
T4 gp16 exhibits a weak dsDNA-binding activity (39). This, combined with other genetic observations, led to the hypothesis that the small terminase protein is responsible for the recognition of the viral DNA substrate for packaging initiation. Analogous to the small terminase subunits of phages λ (41) and SPP1 (42), a helix–turn–helix (H-T-H) DNA-binding motif was predicted in the N-terminus of gp16 (39). These predictions were based on regional similarity with the H-T-H residues of other DNA-binding proteins and the presence of the characteristic ‘pivot residues’ S13, G17 and I23 (43; Fig. 2A). In the 434 Cro family H-T-H, glycine often initiates the connecting turn between the two helices, whereas the serine and isoleucine maintain the angle between them by Van der Waals interactions (43). The sequence alignments, however, show that glycine is the only residue of the putative H-T-H motif that is strictly conserved among all the small subunits, whereas substitutions are clearly evident with regard to the other two pivot residues (Fig. 2A). In the RB49 small terminase subunit, S13 is replaced by leucine, whereas I23 is replaced by aspartate or proline in the RB49 and vibriophage subunits, respectively. Secondary structure prediction programs do not predict this region to be strictly α-helical and oftentimes predict a β-strand instead (data not shown). Nevertheless, dsDNA-binding proteins with variations in the classical H-T-H DNA-binding domain have been identified, including some with a longer linker between the helices or with interrupting β-strands that either precede or follow the helices involved in DNA binding (44). Therefore, it is possible that this motif in the small terminase subunit represents a variant H-T-H motif, instead of the conserved H-T-H signature observed in many DNA-binding proteins. It may also reflect an apparently unique mode of DNA substrate recognition in the circularly permuted phages.
ATP-binding site
Although T4 gp16 is shown to have a weak ATP-binding activity (39), it does not exhibit detectable levels of ATPase activity (9). The presence of a Walker A or Walker B ATP-binding motif is not supported by the sequence alignment (Fig. 2A). The context of the sequence surrounding the two conserved lysine residues of T4 gp16 (K105 and K108) does not resemble the highly conserved Walker A P-loop motif (GKT/S) or any of its known variants. A Walker B-type motif was predicted in the central region of gp16 by Franklin and Mosig (45), presumably based on the predicted ATP reactive sites within the small terminase subunits of λ and SPP1 (46,47). However, neither of the aspartate residues located downstream of K105 and K108 are strictly conserved at the analogous positions of the RB49 and KVP40/KVP20 small terminase subunits and are, therefore, unlikely to be the metal-chelating aspartate of a Walker B motif. Furthermore, the mostly α-helical secondary structure prediction for the small subunit (data not shown) suggests that the protein does not contain the classic nucleotide-binding fold, which consists of β-strands at its core (48). The observed weak ATP-binding activity of gp16 must, therefore, be due to an anomalous ATP-binding site.
Stimulation of the gp17 ATPase activity
Perhaps the most interesting and well-documented feature of gp16 is its ∼50-fold stimulation of the ATPase and in vitro DNA packaging activities of gp17. Although the mechanism of this stimulation remains unknown, we have hypothesized that, analogous to the G protein GTPases, formation of the gp16–gp17 holoenzyme induces a conformational change in the large subunit which positions an arginine residue in the ATPase catalytic center (14). By stabilizing the transition state, this arginine stimulates nucleotide hydrolysis and the DNA translocation that is coupled to it. The conserved arginine residue could be provided either in cis by gp17 or in trans by gp16, analogous to the GTPase-activating proteins (GAPs) (49). In view of the latter possibility, the three strictly conserved arginine residues, R41, R53 and R81 (and possibly K105 and K108), should be considered as candidates for a role in ATPase stimulation. Mutagenesis experiments and the use of gp16 peptides for ATPase stimulation are currently underway to test this hypothesis.
Large terminase sequence analysis
ATPase catalytic center
Walker A motif. The consensus Walker A sequence, (G/A) XXXXGK(T/S), is present in a large number of enzymes capable of nucleotide binding and/or hydrolysis (50). Extensive X-ray structure analyses of numerous ATP-binding proteins reveal that the residues of this motif form a fairly rigid phosphate-binding loop (P-loop) that aligns the ε-amino group of lysine to interact with the β- and γ-phosphates of ATP and positions the hydroxyl group of the adjacent threonine (or serine) residue to directly bind to the Mg2+ of the MgATP complex.
Two putative Walker A motifs have been identified in gp17: the N-terminus proximal SRQLGKT161–167 (Walker AI) and a centrally located TAAVEGKS299–306 (Walker AII) (7,17). As shown in Figure 3, Walker AI is strictly conserved in all four T4 family terminases, suggesting its critical nature. Extensive combinatorial mutagenesis of this site revealed a striking conservation of its features, consistent with numerous studies of canonical Walker A P-loops. For example, substitutions in the highly conserved GKT signature sequence of the T4 gp17 Walker A motif, including the conservative substitutions G165A, K166R and T167A, resulted in a null phenotype (14). It is interesting, however, that the P-loops of KVP gp17v begin with proline, instead of the usual glycine or alanine. In T4 gp17, this residue is a rare serine, though mutagenesis studies showed that both glycine and alanine can substitute successfully. On the other hand, threonine substitution results in a cold-sensitive phenotype and an S161P substitution results in a null phenotype (14). These data and a shorter Walker A motif in gp17 (seven residues instead of eight) represent a slight departure from the classic Walker A P-loop and may reflect a greater structural flexibility in the region.
Figure 3.

Alignment of T4 family large terminase subunits. The terminase large subunits were aligned by BLAST. Putative functional motifs are highlighted as shown. Large terminase subunit polypeptide sequences are available from GenBank: T4 (accession no. P17312); RB49 (accession no. AAF23757); KVP40 (accession no. BAB41133); KVP20 (accession no. BAB96804).
We have also identified a well-conserved Walker A signature among the numerous terminase sequences available in the database (Fig. 4). Not only is this Walker A motif positionally conserved in the N-terminus of the large terminase protein, it also showed a conserved arginine in the ‘variant’ positions of the canonical Walker A signature (X of the G/AXXXXGKT/S sequence). This may represent a terminase Walker A signature, having functional significance to viral DNA packaging (see below). Interestingly, seven of the 24 terminases in Figure 4 show a variant Walker A motif in which the critical K residue in the GKT/S signature is present at the beginning of the Walker A motif. For example, the phage λ terminase has the sequence K76SARVGYS83 and the phage HF2, K74GRRIGVS81. Consistent with this attribution, while this manuscript was under review, Duffy and Feiss (15) reported that mutations at K76 of phage λ terminase resulted in a complete loss of DNA packaging, but not cos cleavage, which would be the predicted phenotype for Walker A lysine (14).
Figure 4.

Alignment of the ATPase domain of viral large terminase subunits. The putative functional motifs are highlighted as shown. The alignment was developed by PSI_BLAST searches using T4 gp17, limiting the Entrez query to the virus taxonomy. The φ29, T3 and λ terminases were inserted manually. BLAST determined alignments fit nicely with respect to the Walker A and B motifs, while, on a few occasions, the adenine-binding motif and motif III required manual adjustment when there was a significant deviation in the distance between the motifs. Information obtained from multiple alignment of terminase family sequences and data from secondary structure predictions guided the manual adjustments. Accession nos: T4, P17312; HK97, NP_037698; PBC5, NP_542306; Mu, Q9T1W6; φ-C31, NP_047924; TM4, NP_569740; HF2, NP_542602; HP2, NP_536821; φ-PVL, NP_058441; R1t, AAB18704; K139, NP_536648; P2, NP_046758; A2, CAB63682; φ-adh, NP_050148; φE125, NP_536358; P27, NP_543088; φKZ, AAL82926; Sk1, NP_044948; λ, P03708; T3, P10310; φ-29, WMBP26; P22, NP_059627; HSV-1, P04295; HCMV, P16732; CCV, NP_041153.
Adenine base-binding motif. A conserved RxYQ(K/R)(D/E) sequence is evident in all four T4 type terminases, starting 26 amino acids upstream of the Walker A lysine (Fig. 3). The YQ residues are also conserved in numerous other large terminase subunits at an analogous position (Fig. 4); in some cases, the tyrosine is replaced by another aromatic amino acid (e.g. tryptophan in φC31 gp33), whereas the glutamine is strictly conserved (Fig. 4). We suggest that the residue Y142 in T4 gp17 and other large terminase subunits interacts with the adenine base of the ATP substrate, while the glutamine residue is involved in hydrogen bonding with the N6 and N7 of the adenine nucleotide, as has been observed with Q49 of yeast eIF-4A (51). Recent experimental evidence with the phage λ terminase supports this hypothesis: Y46 of the gpA YQ motif crosslinks to 8-azido-ATP in the presence of UV light (52) and mutagenesis data further show that substitutions at this residue result in a loss of both the ATPase and in vitro DNA packaging activities but not the terminase activity. Our sequence analyses therefore lend support to the hypothesis that the YQ in λ gpA are part of a legitimate ATPase catalytic center.
Walker B motif. ATPase motors also contain a Walker B motif, with the consensus sequence ZZZZD (where Z represents a hydrophobic amino acid; 53) that is located 50–130 residues downstream of the Walker A lysine (54). The four hydrophobic amino acids of the Walker B motif form a β-strand that ends with the highly conserved aspartate. This well-characterized aspartate is responsible for chelating Mg2+ of the bound MgATP complex and for orienting the substrate for nucleophilic attack by an activated water molecule (55,56). Sequence analysis of the T4 large terminase shows that a Walker B motif, MIYID251–255, is located 89 amino acid residues downstream of the Walker A lysine. The critical elements of this Walker B motif not only appear to be strictly conserved in the four T4 family terminases, but also in HSV-1 UL15 and other terminases as well (Fig. 4). Recent mutagenesis and biochemical studies of this aspartate in gp17 (D255) have provided strong evidence in support of the assignment of this residue to the Walker B motif (M.S.Mitchell and V.B.Rao, unpublished data).
Catalytic carboxylate. Biochemical, structural and sequence alignment studies of many ATPases show that a ‘catalytic carboxylate’ residue, a glutamate (or aspartate), is required to activate a water molecule for an in-line attack on the ATP γ-phosphate (56–58). Although originally proposed to be located between the Walker A and B motifs (about 24 ± 2 residues downstream of the Walker A lysine), it was shown that in some ATPases the glutamate immediately following the Walker B aspartate is spatially and functionally equivalent to the catalytic carboxylate (57).
In the T4 family terminases, three of the four glutamate residues located between the Walker A and B motifs are strictly conserved (E198, E208 and E220). However, we assign the role of catalytic carboxylate to the glutamate residue E256 that is immediately adjacent to the Walker B motif (Fig. 3), because this glutamate is also strictly conserved in λ gpA, HSV-1 UL15 and numerous other terminases (Fig. 4). Recent molecular genetic analysis with the T4 gp17 provides strong evidence for this hypothesis (K.R.Goetzinger and V.B.Rao, in preparation). We suggest that this carboxylate in terminases is spatially equivalent to the catalytic carboxylate of RecA, F1-ATPase and other ATPase motors. This feature makes the terminase ATPase domain further resemble the catalytic center of the SF2 DEXD/H box helicases, in which the conserved glutamate residue following the Walker B aspartate has been well established as the catalytic carboxylate (Fig. 1; 58).
Catalytic arginine(s). As discussed above, an attractive hypothesis is the linkage of an appropriately oriented arginine finger to the DNA translocating state of the packaging machine, thereby stimulating ATP hydrolysis and triggering DNA movement. The conserved arginines of gp16 (trans) and gp17 (cis) are being considered as candidates for arginine fingers, although recent analysis warrants a focus on those located either within the Walker A motif or about 100 residues downstream of the Walker B aspartate (49). Therefore, mutational and biochemical studies are currently underway on the strictly conserved R162, R245, R321 and R406 residues of gp17. Of particular interest is R162, which is conserved in numerous terminase sequences (Fig. 4; 14); substitutions are not tolerated at this residue, potentially making it analogous to the P-loop arginine finger of yeast thymidylate kinase (59).
ATP-binding site II
A second Walker A P-loop has previously been proposed in T4 gp17 (TAAVEGKS299–306) and a number of terminases, with its apparent Walker B motif close to the C-terminus in gp17 (GVSVAKSLYMD468–478) (17,60). Unlike the striking conservation of Walker AI, the multiple sequence alignments (Fig. 3) show that the sequence conservation in Walker AII and its surrounding region is quite poor. Moreover, (i) substitution of the critical lysine (K305 in T4 gp17) with isoleucine in KVP40 and valine in KVP20, (ii) the predicted α-helical nature of this sequence with a bulky tryptophan preceding the proposed P-loop and (iii) the recent mutagenesis data (14) rule out this site as a critical functional site.
Terminase cutting site
A potential metal-binding motif was identified in the C-terminal half of gp17 and other terminases (17,25), with suggested roles in DNA binding and endonucleolytic activity. Site-directed mutagenesis of H436 showed that none of the 12 substitutions made were tolerated, suggesting that this is a critical residue (25). In addition, certain aspartate residues in this region (e.g. D401 and D409) are strictly conserved among the T4 family terminases as well as the 933W terminase (Fig. 3). Recent mutagenesis studies provide evidence that these residues are critical for terminase cutting (F.J.Rentas and V.B.Rao, in preparation).
Prohead-binding site
The terminase–DNA complex, following DNA cleavage and generation of one of the termini, docks onto the unique portal vertex of the prohead, thus linking the ‘primed’ DNA substrate to the empty prohead container. The C-terminal end of the large terminase subunit has been implicated in portal binding in phages T3 and λ (61,62). The portal–terminase interactions appear to be mediated by the negatively charged and hydrophobic residues at the C-terminus. In T4 gp17, the analogous negatively charged sequence ELQDMSDD YAP578–588 at the C-terminus is considered a good candidate for the portal-binding site. However, recent data indicate that this sequence is not critical for function, since a recombinant gp17 that is truncated at K577 is fully functional for in vitro DNA packaging (V.B.Rao, unpublished data). Genetic data, however, suggest that the other negatively charged clusters, LYNDEDIFDD322–331 and IDYADKDD560–567, are likely candidates for the portal-binding site. Second site suppressors of g20 mutants [17tsR1 (I364F) and 17tsL51 (S583N); 63] map close to these sites, which are well preserved among the T4 family terminases (Fig. 3). It is likely that additional residues in the flanking sequence are also involved in the interactions, which may provide specificity for docking the terminase with the corresponding portal protein.
Secondary structure analysis of the ATPase domain
Considering the evidence that ATPase motors (e.g. helicases, F1-ATPases and ABC transporters) share a RecA-like topology among their ATPase domains, even though they lack overall sequence similarity (64,65), we generated a preliminary secondary structure model for the ATPase domain of T4 gp17 (residues 131–301 in gp17) based on the limited sequence similarity between the T4 family large terminase proteins and the DEAD box helicases (Fig. 1). Since it is likely that the ATPase domains of various terminases are architecturally conserved, the secondary structure predictions for HSV-1 UL15, φ29 gp16, φC31 gp33, P2 gpP and λ gpA were generated in parallel, using their homologous sequences to generate multiple sequence alignments, which were then used as input to generate secondary structure predictions for each terminase family (Fig. 5; multiple sequence alignment data of various terminase families are not shown). The data predict a RecA-like ATPase domain consisting of six β-strands for gp17 and other terminases. As in helicases, the core β-strands S1 (preceding the Walker A motif), S2, S3 (in the Walker B motif) and S4 (preceding motif III, see below) that form the nucleotide-binding pocket appear to be strongly conserved among the terminase families (Fig. 5).
Discovery of an ATPase coupling ‘helicase motif III’ in terminases
It is known that in SF2 helicases, the loop following the β-strand S4 contains the helicase motif III (S/TAT) and is usually located about 30 residues downstream of the Walker B aspartate (S3) (see Fig. 1). Recent structural data suggest that the threonine residue(s) of motif III makes direct contact with the nucleic acid phosphate backbone, while the last arginine of the helicase motif VI (Q/HxxGRxxR) serves as a γ-phosphate sensor that is involved in triggering conformational changes in response to ATP hydrolysis (37). Together, these apparently constitute the ATPase coupling switch that is part of the energy transduction mechanism.
At the C-terminal end of the putative terminase β-strand S4, we have discovered the presence of a well-conserved motif III consensus sequence (Fig. 5). It is most apparent in the T4 terminases, which have a conserved TT(S/T) sequence located 29 residues downstream of the Walker B motif. In HSV-1 UL15 terminase, the corresponding sequence is VSS383–385, which is located 28 residues from the Walker B motif. Similar sequences appear 27–29 residues downstream of the putative Walker B aspartate in the large terminase of φC31 (IST206–208), P2 (FST282–284), φ29 (LSN156–158), λ (GST212–214) and numerous other terminases (Figs 4 and 5).
The presence of a conserved motif III threonine(s) in terminases implies a functional role. Although the terminases do not appear to have the helicase motif VI equivalent, it is possible that these residues may be connected to a spatially equivalent γ-phosphate sensor(s), such as the arginine finger residues discussed earlier.
Experimental evidence
A combinatorial mutagenesis approach was applied to test the importance of motif III residues (Fig. 5). This represents a rigorous test, because, unlike the Walker A and Walker B motifs, which were predicted based on straightforward sequence analysis, motif III was revealed following our discovery of a sequence relationship between the helicases and terminases, and the extensive multiple sequence alignments and secondary structure analyses of the various terminase families. Furthermore, its well-characterized role in helicases in the coupling of DNA binding, translocation and ATP hydrolysis makes it an important target for evaluating its functional significance in the terminase ATPase center.
We first constructed an amber mutant phage at the T287 residue (T4.17T287am), which is viable in the genetic background of the serine suppressor E.coli-Ser (no E.coli-Thr suppressor is available); clearly, the conservative serine substitution at T287 did not affect function. Three independent T4.17T287am plaques were selected and the mutant sequence was confirmed by DNA sequencing. The mutant phages were then tested for their ability to form plaques on a battery of 12 E.coli suppressors (G, A, L, K, C, E, Q, R, P, H, F and Y), each substituting the corresponding amino acid at the T287 codon (24). No plaque formation was evident on any of the suppressor strains, suggesting that none of the 12 substitutions, including the small side chain-containing alanine, cysteine or glycine, were tolerated at the T287 position (Fig. 6).
Figure 6.
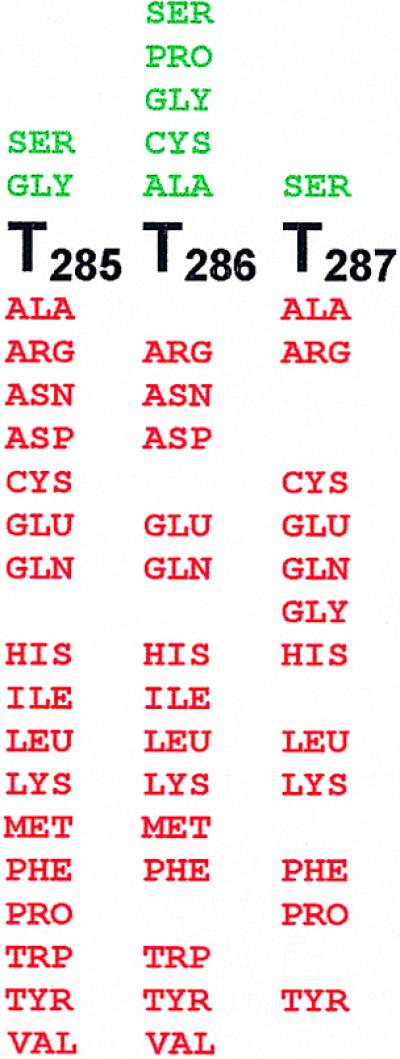
Phenotypes of amino acid substitutions in gp17 motif III. Amino acid substitutions below the native sequence (black) resulted in a null phenotype (red) and the ones above resulted in a functional phenotype (green). Some of the null phenotypes shown were inferred from the marker rescue data. Each substitution shown corresponds to a single amino acid substitution (see Materials and Methods).
Single amino acid libraries were constructed at the T285 and T286 residues of motif III and random mutants were tested for null or functional phenotypes by marker rescue using the T4.17T287am mutant constructed as described above. In order to identify all the substitutions that can give rise to a functional phenotype, more than 20 mutants that showed positive marker rescue were selected from each library and sequenced. The data, as shown in Figure 6, clearly showed that substitutions at both positions are highly restricted, more so at the T285 residue. Only a glycine substitution was tolerated at residue T285, which, in fact, showed a small plaque phenotype. At T286, only glycine, alanine, cysteine and proline substitutions were tolerated. Not surprisingly, serine substitution at either of the positions preserved gp17 function.
These data provide strong evidence to suggest that terminase motif III is a critical functional motif. Sub stitutions are either not tolerated (T287) or highly restricted (T285 and T286) at the threonine residues that constitute this motif. These data are also consistent with the sequence alignment in Figure 4, which shows a virtual conservation of the third threonine in numerous terminases. It would be interesting to see whether the motif III mutants exhibit a biochemical phenotype showing an uncoupling of the ATPase activity from DNA packaging. These, and mutagenesis experiments to construct two amino acid libraries at the threonine residues, are currently underway.
DISCUSSION
Terminases are multifunctional molecular machines that apparently drive the ATP-powered DNA packaging in dsDNA viruses. Among the multiple functions they carry out, the terminases recognize the viral DNA substrate, cut and attach it to the empty prohead through interactions with the portal ring, translocate one headful of DNA and make a second cut to terminate DNA packaging (2–5). These functions are carefully orchestrated to generate a highly ordered, metabolically inactive DNA condensate. While strong genetic and biochemical evidence exists for most of these functions, the role of terminase in the energetics of DNA translocation is poorly understood and the evidence for a causal role is meagre.
Using a sequence alignment approach, we identified a number of motifs in the T4 terminase proteins, which are assigned to specific terminase functions. Perhaps the most important of these is the definition of an ATPase catalytic center in the N-terminal half of the large terminase subunit gp17. Critical motifs for ATP binding (the adenine-binding, Walker A and Walker B motifs), ATP hydrolysis (the catalytic carboxylate) and ATPase coupling (motif III) are predicted. Equally important, this center is identified in the N-terminal half of all the large terminase proteins we have analyzed. The functional signatures and the structural framework are remarkably well conserved among the terminases, despite the lack of significant overall similarity among the peptide sequences. Interestingly, the T4 ATPase domain showed a much closer resemblance to its counterpart from the herpesviruses than to the ATPase domain of other phages (18). This is surprising, but consistent with some commonalities found in the head assembly pathways of T4 and herpesviruses; for example, proteolytic cleavage of the scaffolding core components preceding DNA packaging (66).
A very interesting similarity is discovered between the ATPase domains of terminases and SF2 DEXD/H box helicases (19). Two important signatures of the helicases, the adjacent aspartate and catalytic glutamate residues in the Walker B motif (DE in the DEXD/H box helicases) and the threonine(s) of motif III, are strictly conserved in the terminases. In view of the established DNA/RNA unwinding and translocating functions associated with the helicases, the question of whether the viral terminases have descended from the more ‘ancient’ helicases by acquiring a novel dsDNA translocating ATPase function is an interesting one and is being explored.
Extensive testing of our predictions by a combinatorial mutagenesis paradigm showed that the proposed ATPase center in T4 gp17 is not only a legitimate catalytic center but is, indeed, critical for DNA packaging. No amino acid substitutions, including conservative substitutions such as G165A, K166R and T167A, are tolerated at the highly conserved GKT sequence in the Walker A motif (14). Biochemical data further showed that the gp17-associated ATPase and the in vitro DNA packaging activities are lost in the mutants, while their DNA cutting function remained undisturbed (14). A similar biochemical phenotype was revealed in the mutants of the predicted Walker B motif and the catalytic carboxylate residues (M.S.Mitchell, K.R.Goetzinger and V.B.Rao, in preparation). Compelling evidence has also been obtained recently using Walker A motif mutants of the λ, T3 and HSV-1 large terminase proteins (15,16,67).
Combinatorial mutagenesis of the putative ATPase coupling motif further substantiated the predicted ATPase center in terminases. X-ray structure and biochemical analyses suggest that the threonine(s) of DEXD/H box helicase motif III is one of a network of residues that couple DNA binding to ATP hydrolysis (37,68). In one model, the threonine interaction with the nucleic acid backbone positions a γ-phosphate sensor, such as a glutamine and/or an arginine side chain (motif VI in DEXD/H helicases), to interact with the bound ATP, triggering ATP hydrolysis (68). This motif may also interface with the bound ATP via interactions with the Walker B DEXD/H signature residues, as was proposed for the putative DEXD helicase from Methanocaldococcus jannaschii (37). The motif III we have discovered in the T4 terminase family (TT/ST) is well conserved in all the viral terminase families we have analyzed. The last threonine is especially invariant in virtually all of the terminases. Although we have not been able to identify the spatial equivalent of the γ-phosphate sensor(s) in terminases, the presence of the highly conserved motif III suggested an analogous ATPase coupling function for this motif. Consistent with this prediction, mutational analyses clearly showed that the strictly conserved third threonine is a critical residue, since no substitutions, including alanine, glycine or cysteine substitution, were tolerated at this residue; only the highly conservative serine substitution was acceptable for function.
The residues that contact DNA during the active translocation process are unknown for any terminase. These interactions, unlike the intitial terminase–DNA interactions involved in substrate recognition, should necessarily be weak, sequence non-specific and transient. In addition, these interactions must be regulated in conjunction with their communication with the ‘force-generating’ ATPase catalytic site. In the context of what is known about the helicase motif III residues, which have well-established roles in DNA binding, and interfacing and coupling to the ATPase motif (69), it is tempting to speculate that this motif may directly participate in the dynamics of DNA movement. This issue has not been formally considered in any packaging models and biochemical analyses of the conservative motif III mutants (e.g. T287A), which are currently underway, should shed light on this critical piece of the DNA packaging mechanism.
Current packaging models propose that DNA packaging is driven by an ATP-powered DNA translocating pump assembled at the unique portal vertex of the prohead. This pump is presumed to consist of the dodecameric portal ring, at least a portion of the capsid protein subunits (that are in contact with the portal) and the two non-structural terminase proteins. An explicit model proposed by Simpson et al. (70) suggests that the DNA is translocated translationally through the portal channel by rotation of the portal ring, which is energetically coupled to an ATPase motor. Although the ATPase motor has not been identified in any viral system, biophysical measurements suggest that this is one of the strongest motors measured to date (12). The evidence presented here for a conserved common ATPase catalytic center in the N-terminal half of the large terminase proteins is significant in this regard. Considering the fact that the key features of the packaging mechanism appear to be conserved among the dsDNA viruses (e.g. the conserved dodecameric portal ring structure with no significant amino acid homology), we believe that the evidence points to this being the elusive translocating ATPase. The biochemical evidence supports this assertion because ‘subtle’ changes in the predicted catalytic residues resulted in a loss of ATPase and DNA packaging activities (14). However, more direct evidence is needed in order to assign a causal role, because it is possible that this ATPase may be required for a critical packaging function other than DNA translocation; for example, assembly of the packaging pump by linking the terminase–DNA complex to the portal protein. In this scenario, DNA translocation may be catalyzed by a second ‘cryptic’ ATPase, which is activated upon assembly of the complete packaging machine. However far fetched, this possibility cannot be ruled out by the available evidence. We are currently analyzing the biochemical parameters of specific mutants, including their ability to assemble on the prohead, in order to test whether a direct linkage exists between this site and DNA translocation.
In conclusion, this study has, for the first time, defined an ATPase catalytic center in the large terminase proteins of dsDNA phages and herpesviruses. It has established a common structural framework with predicted catalytic signatures, which should allow testing of specific hypotheses of the DNA translocating ATPase question in a number of viral packaging systems. In the phage T4 system at least, as shown here and elsewhere, the validity of the key predicted motifs have been quite rigorously tested using a combinatorial mutagenesis paradigm, which has paved the way to analyzing more defined questions about the causal relationship between this center and DNA translocation. Finally, the discovered ‘helicase connection’ may generate new insights into the mechanisms of viral dsDNA packaging and help elucidate the modalities of DNA/RNA translocation mechanisms.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Drs Jörg Benz, Alan Senior, Dale Wigley, Andrew Davison, Nigel Stow, Joel Baines and Michael Dyall-Smith for their prompt and helpful correspondence and Drs Henri Krisch and Lindsay Black for providing the sequence of RB49 terminase. Research in V.B.R.’s laboratory is supported by the National Science Foundation (MCB-0110574).
REFERENCES
- 1.Black L.W., Showe,M.K. and Steven,A.C. (1994) Morphogenesis of the T4 head. In Karam,J.D. (ed.), Molecular Biology of Bacteriophage T4. ASM Press, Washington, DC, pp. 218–258.
- 2.Earnshaw W.C. and Casjens,S.R. (1980) DNA packaging by the double-stranded DNA bacteriophages. Cell, 21, 319–331. [DOI] [PubMed] [Google Scholar]
- 3.Feiss M. (1986) Terminase and the recognition, cutting, and packaging of lambda chromosomes. Trends Genet., 2, 100–104. [Google Scholar]
- 4.Black L.W. (1989) DNA packaging in dsDNA bacteriophages. Annu. Rev. Microbiol., 43, 267–292. [DOI] [PubMed] [Google Scholar]
- 5.Catalano C.E., Cue,D. and Feiss,M. (1995) Virus DNA packaging: the strategy used by phage lambda. Mol. Microbiol., 16, 1075–1086. [DOI] [PubMed] [Google Scholar]
- 6.Rao V.B. and Black,L.W. (1988) Cloning, overexpression and purification of the terminase proteins gp16 and gp17 of bacteriophage T4. Construction of a defined in-vitro DNA packaging system using purified terminase proteins. J. Mol. Biol., 200, 475–488. [DOI] [PubMed] [Google Scholar]
- 7.Powell D., Franklin,J., Arisaka,F. and Mosig,G. (1990) Bacteriophage T4 DNA packaging genes 16 and 17. Nucleic Acids Res., 18, 4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattacharyya S.P. and Rao,V.B. (1993) A novel terminase activity associated with the DNA packaging protein gp17 of bacteriophage T4. Virology, 196, 34–44. [DOI] [PubMed] [Google Scholar]
- 9.Leffers G. and Rao,V.B. (2000) Biochemical characterization of an ATPase activity associated with the large packaging subunit gp17 from bacteriophage T4. J. Biol. Chem., 275, 37127–37136. [DOI] [PubMed] [Google Scholar]
- 10.Morita M., Tasaka,M. and Fujisawa,H. (1993) DNA packaging ATPase of bacteriophage T3. Virology, 193, 748–752. [DOI] [PubMed] [Google Scholar]
- 11.Guo P., Peterson,C. and Anderson,D. (1987) Initiation events in in-vitro packaging of bacteriophage phi 29 DNA-gp3. J. Mol. Biol., 197, 219–228. [DOI] [PubMed] [Google Scholar]
- 12.Smith D.E., Tans,S.J., Smith,S.B., Grimes,S., Anderson,D.L. and Bustamante,C. (2001) The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature, 413, 748–752. [DOI] [PubMed] [Google Scholar]
- 13.Black L.W. (1995) DNA packaging and cutting by phage terminases: control in phage T4 by a synaptic mechanism. Bioessays, 17, 1025–1030. [DOI] [PubMed] [Google Scholar]
- 14.Rao V.B. and Mitchell,M.S. (2001) The N-terminal ATPase site in the large terminase protein gp17 is critically required for DNA packaging in bacteriophage T4. J. Mol. Biol., 314, 401–411. [DOI] [PubMed] [Google Scholar]
- 15.Duffy C. and Feiss,M. (2002) The large subunit of bacteriophage lambda’s terminase plays a role in DNA translocation and packaging termination. J. Mol. Biol., 316, 547–561. [DOI] [PubMed] [Google Scholar]
- 16.Morita M., Tasaka,M. and Fujisawa,H. (1994) Analysis of functional domains of the packaging proteins of bacteriophage T3 by site-directed mutagenesis. J. Mol. Biol., 235, 248–259. [DOI] [PubMed] [Google Scholar]
- 17.Guo P., Peterson,C. and Anderson,D. (1987) Prohead and DNA-gp3-dependent ATPase activity of the DNA-packaging protein gp16 of bacteriophage phi 29. J. Mol. Biol., 197, 229–236. [DOI] [PubMed] [Google Scholar]
- 18.Davison A.J. (1992) Channel catfish virus: a new type of herpesvirus. Virology, 186, 9–14. [DOI] [PubMed] [Google Scholar]
- 19.Gorbalenya A.E. and Koonin,E.V. (1993) Helicases: amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol., 3, 419–429. [Google Scholar]
- 20.Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tatusova T.T. and Madden,T.L. (1999) BLAST 2 sequences—a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett., 174, 247–250. [DOI] [PubMed] [Google Scholar]
- 22.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cuff J.A. and Barton,G.J. (2000) Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins, 40, 502–511. [DOI] [PubMed] [Google Scholar]
- 24.Kleina L.G., Masson,J.M., Normanly,J., Abelson,J. and Miller,J.H. (1990) Construction of Escherichia coli amber suppressor tRNA genes. II. Synthesis of additional tRNA genes and improvement of suppressor efficiency. J. Mol. Biol., 213, 705–717. [DOI] [PubMed] [Google Scholar]
- 25.Kuebler D. and Rao,V.B. (1998) Functional analysis of the DNA-packaging/terminase protein gp17 from bacteriophage T4. J. Mol. Biol., 281, 803–814. [DOI] [PubMed] [Google Scholar]
- 26.Tétart F., Desplats,C., Kutateladze,M., Monod,C., Ackermann,H.W. and Krisch,H.M. (2001) Phylogeny of the major head and tail genes of the wide-ranging T4-type bacteriophages. J. Bacteriol., 183, 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monod C., Repoila,F., Kutateladze,M., Tétart,F. and Krisch,H.M. (1997) The genome of the pseudo T-even bacteriophages, a diverse group that resembles T4. J. Mol. Biol., 267, 237–249. [DOI] [PubMed] [Google Scholar]
- 28.Matsuzaki S., Tanaka,S., Koga,T. and Kawata,T. (1992) A broad-host-range vibriophage, KVP40, isolated from sea water. Microbiol. Immunol., 36, 93–97. [DOI] [PubMed] [Google Scholar]
- 29.Matsuzaki S., Inoue,T., Kuroda,M., Kimura,S. and Tanaka,S. (1998) Cloning and sequencing of major capsid protein (mcp) gene of a vibriophage, KVP20, possibly related to T-even coliphages. Gene, 222, 25–30. [DOI] [PubMed] [Google Scholar]
- 30.Matsuzaki S., Kuroda,M., Kimura,S. and Tanaka,S. (1999) Vibriophage KVP40 and coliphage T4 genomes share a homologous 7-kbp region immediately upstream of the gene encoding the major capsid protein. Arch. Virol., 144, 2007–2012. [DOI] [PubMed] [Google Scholar]
- 31.Ruimy R., Breittmayer,V., Elbaze,P., Lafay,B., Boussemart,O., Gauthier,M. and Christen,R. (1994) Phylogenetic analysis and assessment of the genera Vibrio, Photobacterium, Aeromonas, and Plesiomonas deduced from small-subunit rRNA sequences. Int. J. Syst. Bacteriol., 44, 416–426. [DOI] [PubMed] [Google Scholar]
- 32.Nuttall S.D. and Dyall-Smith,M.L. (1993) HF1 and HF2: novel bacteriophages of halophilic Archaea. Virology, 197, 678–684. [DOI] [PubMed] [Google Scholar]
- 33.Nuttall S.D. and Dyall-Smith,M.L. (1995) Halophage HF2: genome organization and replication strategy. J. Virol., 69, 2322–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plunkett G. III, Rose,D.J., Durfee,T.J. and Blattner,F.R. (1999) Sequence of the Shiga toxin 2 phage 933W from Escherichia coli O157:H7: Shiga toxin as a phage late-gene product. J. Bacteriol., 181, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adelman K., Salmon,B. and Baines,J.D. (2001) Herpes simplex virus DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc. Natl Acad. Sci. USA, 98, 3086–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davison A.J. (1998) The genome of salmonid herpesvirus 1. J. Virol., 72, 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Story R.M., Li,H. and Abelson,J.N. (2001) Crystal structure of a DEAD box protein from the hyperthermophile Methanococcus jannaschii. Proc. Natl Acad. Sci. USA, 98, 1465–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubin G.M., Yandell,M.D., Wortman,J.R., Miklos,G.L.G., Nelson,C.R., Hariharan,I.K., Fortini,M.E., Li,P.I., Apweiler,R., Fleischmann,W. et al. (2000) Comparative genomics of the eukaryotes. Science, 287, 2204–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin H., Simon,M.N. and Black,L.W. (1997) Purification and characterization of the small subunit of phage T4 terminase, gp16, required for DNA packaging. J. Biol. Chem., 272, 3495–3501. [DOI] [PubMed] [Google Scholar]
- 40.Yang Q., Berton,N., Manning,M.C. and Catalano,C.E. (1999) Domain structure of gpNu1, a phage lambda DNA packaging protein. Biochemistry, 38, 14238–14247. [DOI] [PubMed] [Google Scholar]
- 41.Kypr J. and Mrazek,J. (1986) Lambda phage protein Nu 1 contains the conserved DNA binding fold of repressors. J. Mol. Biol., 191, 139–140. [DOI] [PubMed] [Google Scholar]
- 42.Chai S., Bravo,A., Luder,G., Nedlin,A., Trautner,T.A. and Alonso,J.C. (1992) Molecular analysis of the Bacillus subtilis bacteriophage SPP1 region encompassing genes 1 to 6. The products of gene 1 and gene 2 are required for pac cleavage. J. Mol. Biol., 224, 87–102. [DOI] [PubMed] [Google Scholar]
- 43.Pabo C.O. and Sauer,R.T. (1984) Protein-DNA recognition. Annu. Rev. Biochem., 53, 293–321. [DOI] [PubMed] [Google Scholar]
- 44.Wintjens R. and Rooman,M. (1996) Structural classification of HTH DNA-binding domains and protein-DNA interaction modes. J. Mol. Biol., 262, 294–313. [DOI] [PubMed] [Google Scholar]
- 45.Franklin J. and Mosig,G. (1996) Expression of the bacteriophage T4 DNA terminase genes 16 and 17 yields multiple proteins. Gene, 177, 179–189. [DOI] [PubMed] [Google Scholar]
- 46.Becker A. and Gold,M. (1988) Prediction of an ATP reactive center in the small subunit, gpNu1, of the phage lambda terminase enzyme. J. Mol. Biol., 199, 219–222. [DOI] [PubMed] [Google Scholar]
- 47.Chai S., Kruft,V. and Alonso,J.C. (1994) Analysis of the Bacillus subtilis bacteriophages SPP1 and SF6 gene product 1: a protein involved in the initiation of headful DNA packaging. Virology, 202, 930–939. [DOI] [PubMed] [Google Scholar]
- 48.Rossmann M.G., Moras,D. and Olsen,K.W. (1974) Chemical and biological evolution of nucleotide-binding protein. Nature, 250, 194–199. [DOI] [PubMed] [Google Scholar]
- 49.Via A., Ferrè,F., Brannetti,B., Valencia,A. and Helmer-Citterich,M. (2000) Three-dimensional view of the surface motif associated with the P-loop structure: cis and trans cases of convergent evolution. J. Mol. Biol., 303, 455–465. [DOI] [PubMed] [Google Scholar]
- 50.Walker J., Saraste,M., Runswick,M. and Gay,N. (1982) Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benz J., Trachsel,H. and Baumann,U. (1999) Crystal structure of the ATPase domain of translation initiation factor 4A from Saccharomyces cerevisiae—the prototype of the DEAD box protein family. Structure, 6, 89–100. [DOI] [PubMed] [Google Scholar]
- 52.Hang J.Q., Tack,B.F. and Feiss,M. (2000) ATPase center of bacteriophage lambda terminase involved in post-cleavage stages of DNA packaging: identification of ATP-interactive amino acids. J. Mol. Biol., 302, 777–795. [DOI] [PubMed] [Google Scholar]
- 53.Myles G.M., Hearst,J.E. and Sancar,A. (1991) Site-specific mutagenesis of conserved residues within Walker A and B sequences of Escherichia coli UvrA protein. Biochemistry, 30, 3824–3834. [DOI] [PubMed] [Google Scholar]
- 54.Yoshida M. and Amano,T. (1995) A common topology of proteins catalyzing ATP-triggered reactions. FEBS Lett., 359, 1–5. [DOI] [PubMed] [Google Scholar]
- 55.Pai E.F., Krengel,U., Petsko,G.A., Goody,R.S., Kabsch,W. and Wittinghofer,A. (1990) Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 Å resolution: implications for the mechanism of GTP hydrolysis. EMBO J., 9, 2351–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Story R.M. and Steitz,T.A. (1992) Structure of the recA protein-ADP complex. Nature., 355, 318–325. [DOI] [PubMed] [Google Scholar]
- 57.Subramanya H.S., Bird,L.E., Brannigan,J.A. and Wigley,D.B. (1996) Crystal structure of a DExx box DNA helicase. Nature, 384, 379–383. [DOI] [PubMed] [Google Scholar]
- 58.Pause A. and Sonenberg,M. (1992) Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J., 11, 2643–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lavie A., Konrad,M., Brundiers,R., Goody,R.S., Schlichting,I. and Reinstein,J. (1998) Crystal structure of yeast thymidylate kinase complexed with the bisubstrate inhibitor P1-(5′-adenosyl) P5-(5′-thymidyl) pentaphosphate (TP5A) at 2.0 Å resolution: implications for catalysis and AZT activation. Biochemistry, 37, 3677–3686. [DOI] [PubMed] [Google Scholar]
- 60.Franklin J., Haseltine,D., Davenport,L. and Mosig,G. (1998) The largest (70 kDa) product of the bacteriophage T4 DNA terminase gene 17 binds to single stranded DNA segments and digests them towards junctions with double-stranded DNA. J. Mol. Biol., 277, 541–557. [DOI] [PubMed] [Google Scholar]
- 61.Morita M., Tasaka,M. and Fujisawa,H. (1995) Structural and functional domains of the large subunit of the bacteriophage T3 DNA packaging enzyme: importance of the C-terminal region in prohead binding. J. Mol. Biol., 245, 635–644. [DOI] [PubMed] [Google Scholar]
- 62.Yeo A. and Feiss,M. (1995) Specific interaction of terminase, the DNA packaging enzyme of bacteriophage λ, with the portal protein of the prohead. J. Mol. Biol., 245, 141–150. [DOI] [PubMed] [Google Scholar]
- 63.Lin H., Rao,V.B. and Black,L.W. (1999) Analysis of a capsid portal protein and terminase functional domains: interaction sites required for DNA packaging in bacteriophage T4. J. Mol. Biol., 289, 249–260. [DOI] [PubMed] [Google Scholar]
- 64.Milner-White E.J., Coggins,J.R. and Anton,I.A. (1991) Evidence for an ancestral core structure in nucleotide-binding proteins with the type A motif. J. Mol. Biol., 221, 751–754. [DOI] [PubMed] [Google Scholar]
- 65.Smith C.A. and Rayment,I. (1996) Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys. J., 70, 1590–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Homa F.L. and Brown,J.C. (1997) Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol., 7, 107–122. [DOI] [PubMed] [Google Scholar]
- 67.Yu D. and Weller,S.K. (1998) Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology, 243, 32–44. [DOI] [PubMed] [Google Scholar]
- 68.Kim J.L., Morgenstern,K.A., Griffith,J.P., Dwyer,M.D., Thomson,J.A., Murcko,M.A., Lin,C. and Caron,P.R. (1998) Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure, 6, 89–100. [DOI] [PubMed] [Google Scholar]
- 69.Schmid S.R. and Linder,P. (1991) Translation initiation factor 4A from Saccharomyces cerevisiae: analysis of residues conserved in the D-E-A-D family of RNA helicases. Mol. Cell Biol., 11, 3463–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Simpson A.A., Tao,Y., Leiman,P.G., Badasso,M.O., He,Y., Jardine,P.J., Olson,N.H., Morais,M.C., Grimes,S., Anderson,D.L., Baker,T.S. and Rossmann,M.G. (2000) Structure of the phi29 DNA packaging motor. Nature, 408, 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]