

Abstract
The mild phenotype associated with targeted disruption of the mouse OGG1 and NTH1 genes has been attributed to the existence of back-up activities and/or alternative pathways for the removal of oxidised DNA bases. We have characterised two new genes in human cells that encode DNA glycosylases, homologous to the bacterial Fpg (MutM)/Nei class of enzymes, capable of removing lesions that are substrates for both hOGG1 and hNTH1. One gene, designated HFPG1, showed ubiquitous expression in all tissues examined whereas the second gene, HFPG2, was only expressed at detectable levels in the thymus and testis. Transient transfections of HeLa cells with fusions of the cDNAs to EGFP revealed intracellular sorting to the nucleus with accumulation in the nucleoli for hFPG1, while hFPG2 co-localised with the 30 kDa subunit of RPA. hFPG1 was purified and shown to act on DNA substrates containing 8-oxoguanine, 5-hydroxycytosine and abasic sites. Removal of 8-oxoguanine, but not cleavage at abasic sites, was opposite base-dependent, with 8-oxoG:C being the preferred substrate and negligible activity towards 8-oxoG:A. It thus appears that hFPG1 has properties similar to mammalian OGG1 in preventing mutations arising from misincorporation of A across 8-oxoG and could function as a back-up repair activity for OGG1 in ogg1–/– mice.
INTRODUCTION
Reactive oxygen species (ROS) have been considered a major source of endogenous DNA damage in aerobic cells. ROS are formed as by-products of electron transport in the mitochondria and by various inflammatory responses, for instance the oxidative burst induced in macrophages upon bacterial and viral infections. In addition, ROS will be induced by various exogenous agents such as ionising irradiation and chemical carcinogens, like for instance 4-nitroquinoline-1-oxide. The main reaction products of ROS with DNA are base modifications, base loss and strand breaks (reviewed in 1). The major oxidised purine lesion is 8-oxo-7,8-dihydroguanine (8-oxoG), which is abundant and has strong promutagenic properties (2,3). During replication 8-oxoG in the template will pair with A as frequently as with C (4–6), yielding G:C→T:A transversions (7–9), a common mutation associated with human cancers (10). Other important purine lesions are formamidopyrimidines (faPy), which mostly have cytotoxic effects (11).
To withstand the deleterious effects of DNA oxidation damage different repair mechanisms have evolved, among which the base excision repair pathway (BER) is most important. BER is initiated by a DNA glycosylase that removes modified bases in a free form thus leaving an abasic site in the DNA. For bifunctional DNA glycosylases, the abasic site is acted upon by an AP lyase activity inherent in the glycosylase itself. For monofunctional DNA glycosylases the enzyme will protect the abasic site until acted upon by an AP endonuclease. In both cases a strand break is formed that needs further processing by other proteins (lyases and/or nucleases) in order to remove the sugar–phosphate residue remaining at the 3′- or the 5′-end, respectively. Repair is completed by the concerted action of a DNA polymerase to fill the gap and a DNA ligase to seal the strand (reviewed in 12). Several accessory factors such as X-ray repair cross- complementing gene I protein (XRCC1), replication protein A (RPA) and proliferating cell nuclear antigen (PCNA) are also involved in the repair process (13).
In mammalian cells, the gene functions responsible for the strand incision steps in repair of oxidised DNA include the glycosylases hNTH1 (14–17) and hOGG1 (18–20), as well as AP endonuclease 1 (HAP1/APE1) (21) and perhaps also a second minor AP endonuclease, APE2 (22). Human NTH1 is homologous to the bacterial Nth enzyme (endonuclease III) (14,15,17) and is mainly responsible for removal of oxidized pyrimidines such as thymine glycol, 5-hydroxycytosine (5-ohC), dihydrothymine and dihydrouracil (23,24). Human OGG1 is homologous to the OGG1 function in yeast (25), but sequence unrelated to the major enzyme for removal of oxidised purines in bacteria, formamidopyrimidine DNA glycosylase, Fpg (26,27). Both hNTH1 and hOGG1 belong to the helix–hairpin–helix (HhH) family of DNA glycosylases, whereas Fpg is part of another family of DNA glycosylases that also includes the bacterial endonuclease VIII (Nei) (28). Nei appears to serve as a back-up enzyme for Nth in bacteria and removes many of the same lesions as Nth, but also has a low but significant activity towards 8-oxoG (29).
In order to elucidate the in vivo function of hOGG1, transgenic mice have been generated with a targeted deletion within the mouse OGG1 gene (30,31). Extracts from various organs of the null mutant mice appear to have a major defect in glycosylase activity for 8-oxoG removal when assayed under standard conditions. However, the mice are viable and without obvious phenotypic effects and there is only a modest increase in the steady-state level of 8-oxoG in DNA extracted from the mutant animals. Furthermore, when mouse embryonic fibroblasts are treated with agents that induce 8-oxoG there is only a small kinetic difference in 8-oxoG removal, implying that there must be some back-up mechanism operating in these animals (32). Recent evidence has indicated that transcription-coupled repair (TCR), first demonstrated for removal of bulky lesions by nucleotide excision repair, may also apply to BER lesions such as thymine glycol and 8-oxoG (33). Accordingly, analysis of mouse embryonic fibroblasts transfected with plasmids containing 8-oxoG in a transcribed or a non-transcribed sequence have suggested that ogg1–/– mice have a defect in global genome repair, but not in TCR (34).
In this study, we have looked for possible back-up enzymes for hOGG1 in human cells. Regular searches for expressed sequence tags (ESTs) with queries for known repair genes have recently revealed two new gene functions with sequence homology to Fpg/Nei in bacteria, potentially involved in 8-oxoG repair in human cells. Both proteins are shown to be nuclear and to possess DNA glycosylase activity for removal of faPy. However, different subcellular localisation and gene expression patterns suggest different roles in DNA repair for these two enzymes. One has been characterised with respect to enzymatic function and ability to remove 8-oxoG, and has properties making it a good candidate for a back-up enzyme for 8-oxoG removal in human cells.
MATERIALS AND METHODS
Sequence analysis
DNA and protein sequence database searches were performed using ParAlign (35). The multiple sequence alignment was created using T-Coffee v.1.37 (36) followed by manual editing and layout using GeneDoc (K.B.Nicholas and H.B.Nicholas, 1997, GeneDoc: a tool for editing and annotating multiple sequence alignments, distributed by the author). Single sequence entries were analysed with the Wisconsin Genetics Computer Group (GCG) Sequence Analysis Software Package (Wisconsin Package v.10.1; GCG, Madison, WI). The PSORTII program (37; http://psort.nibb.ac.jp/) was used to predict sequence motifs for intracellular sorting. The chromosomal location was determined using genomic BLAST (http://www.ncbi.nlm.nih.gov/genome).
Identification of human expressed sequence tags with homology to Escherichia coli fpg and nei
Two cDNA sequences (accession nos AK026055, termed hFPG1, and NM_018248, termed hFPG2) with sequence homology to E.coli Fpg and Nei were identified in the database of human ESTs. Plasmid clones (HRC08117, hFPG1, and 1089-f23, hFPG2) were obtained from the NEDO human cDNA sequencing project and from the IMAGE Consortium, respectively. The clones were sequenced. Clone HRC08117 contained an insert of ∼1.8 kb encompassing the entire hFPG1 open reading frame (ORF). This clone was used as a template for the generation of the hFPG1 expression clones described below. Clone 1089-f23 contained an insert of ∼1.0 kb and was lacking the 5′ 1290 bp of the hFPG2 cDNA.
PCR amplification and Gateway cloning of hFPG1
PCR products of hFPG1 were inserted into plasmid vectors by in vitro recombination using the Gateway system as recommended by the manufacturer (Life Technologies Inc.). Primers with the following sequences were synthesized: P1 (forward), 5′-attB1-TAG AAG GAG ATA GAA CCA TGC CTG AGG GCC CCG AGC TGC A-3′; P2 (reverse), 5′-attB2-CCT AAG AGG CTG AGG TCC CCT-3′. The PCR products flanked by attB recombination sites were cloned into the donor vector pDONR201 using the Gateway BP reaction to generate the entry vector pEhFPG1. In the Gateway LR reaction the gene sequence was further subcloned from pEhFPG1 into the destination vectors pDEST14 (vector for native expression from a T7 promoter) and pDEST8 (vector for native expression from a polyhedrin promoter) to generate the plasmids pT7-hFPG1 and pBac-hFPG1, respectively. The presence of the correct hFPG1 cDNA sequence in pT7-hFPG1 and pBac-hFPG1 was confirmed by sequencing.
PCR amplification and protein expression constructs of hFPG2
A cDNA carrying the complete ORF was amplified with the Qiagen One Step RT–PCR kit (Qiagen) using mRNA extracted from HeLa S3 cells as the template. Oligodeoxy nucleotides based on the human cDNA sequence (P3, 5′-ATG GTG GAA GGA CCA GGC TGT ACT CTG AAT GGA G-3′, containing the putative start codon; P4, 5′-TTA GCA TCC AGG AAT AAT TTT TAT TCC TGG CCC ATT TTC-3′, containing the putative stop codon) were used for the reverse transcription and PCR amplification. The reverse transcription was carried out at 50°C for 30 min followed by inactivation of the reverse transcriptase and activation of the DNA polymerase at 95°C for 15 min. The cycling conditions were 94°C for 1 min, 64°C for 1 min and 72°C for 2 min, for 35 cycles, and finally 72°C for 5 min. After PCR, the amplification product was purified from an agarose gel and sequenced. This DNA fragment was used as DNA template for the PCR amplification of hFPG2. Primers P5 (5′-GGA ATT CGA ATG GTG GAA GGA CCA GGC TGT-3′) and P6 (5′-CGG GAT CCT TAG CAT CCA GGA ATA ATT TT-3′) were used to introduce EcoRI and BamHI restriction sites at the start and stop of the coding region. The amplified fragment was ligated into pT7SCII (vector for native expression from a T7 promoter) and pFastBac1 (Life Technologies Inc; vector for native expression from a polyhedrin promoter). The corresponding plasmids pT7-hFPG2 (T7 promoter) and pBac-hFPG2 (polyhedrin promoter) were sequenced.
Northern blot hybridisation
Northern blots containing multiple human adult samples purchased from Clontech (catalog nos 7759-1 and 7760-1) were probed for hFPG1 and hFPG2 expression. Northern blot hybridisation was carried out using ExpressHyb solution (Clontech) as recommended by the manufacturer. Probes were labelled using the Rediprime DNA labelling system (Amersham Corp.)
Cellular localisation of hFPG1 and hFPG2
hFPG1 and hFPG2 were amplified by PCR and fused to the ORF of EGFP in the directions hFPG1–EGFP/hFPG2–EGFP (cloned into pEGFPN1) and EGFP–hFPG1/EGFP–hFPG2 (cloned into pEGFPC1). Evaluation of the subcellular localisation was accomplished using the HeLa S3 cell line. Cells were grown in Dulbecco’s modified Eagle’s medium with 4.5 g/l glucose, 10% foetal calf serum, 3 mg/ml glutamine, 0.1 mg/ml penicillin and 0.1 mg/ml streptomycin, and plated out to a density of 15 000 cells/cm2. Twenty-four hours after plating, the cells were transfected with pEGFPN1-hFPG1/pEGFPN1-hFPG2, pEGFPC1-hFPG1/pEGFPC1-hFPG2 or pEGFPN1/pEGFPC1 vector alone using the FuGENE™ Transfection Reagent (Roche Diagnostics). The concentration ratio between DNA (plasmid) and FuGENE™ was 3:2. Cells were fixed with 96% ethanol for 10 min at room temperature. Cells were washed with PBS, rinsed in distilled water and air dried before incubation with primary and secondary antibodies diluted in PBS with 1% BSA. As primary antibody monoclonal anti-nucleolin (clone 3G4B2; Upstate Biotechnology, Lake Placid, NY) or monoclonal anti-RPA/p32 (9H8; Neo Markers) were used and incubated overnight at room temperature. Incubation with secondary antibody (Alexa 594, 590/617; Molecular Probes) was carried out for 30 min at room temperature. DNA was stained with 4′,6′-diamino-2-phenylindole (DAPI) for 10 min at room temperature. Slides were mounted using the Dako fluorescence mounting medium and examined with a Zeiss Axioplan 2 fluorescence microscope. Images were obtained using CoolSNAP and then processed for publication using Jasc Pain Shop Pro.
Sf9 cell culture and protein extracts
pBac-hFPG1 and pBac-hFPG2 were transposed into the baculovirus genome according to the manufacturer’s instructions for overexpression of the proteins in Sf9 insect cells (Life Technologies, Inc.). Sf9 cells in suspension were infected with pBac-hFPG1 and pBac-hFPG2 at a multiplicity of infection (MOI) of 1 p.f.u./ml and harvested after 72 h. Protein extracts were prepared according to the manufacturer’s instructions (Life Technologies).
hFPG1 expression and purification
Escherichia coli BK3004 (fpg::kan) was transformed with pT7-hFPG1 and grown in 10 l of LB medium at 37°C to an optical density of 0.9. IPTG (1 mM) was added and incubation continued for 2 h. The cell pellet was resuspended in 100 ml of buffer A (50 mM Tris, pH 7.0, 100 mM NaCl, 10 mM β-mercaptoethanol) and cell-free extract was prepared by sonication. An assay for faPy activity was used to monitor hFPG1 purification. The cell extract was applied to a HiTrapQ column (5 ml × 2) (Amersham) equilibrated with buffer A and eluted with a linear gradient of 0.1–2 M NaCl in buffer A. Active fractions (eluting at 0.4 M NaCl) were pooled, diluted with 2 vol of buffer B (50 mM Tris, pH 7.0, 10 mM β-mercaptoethanol) and applied to a ResourceQ column (Amersham) equilibrated with buffer A. The ResourceQ column was eluted with a linear gradient of 0.1–2 M NaCl in buffer A and active fractions (eluting at 0.3 M NaCl) were pooled, diluted with 2 vol of buffer C (50 mM Tris, pH 8.0, 10 mM β-mercaptoethanol) and applied to a MonoQ column equilibrated with buffer D (50 mM Tris, pH 8.0, 50 mM NaCl, 10 mM β-mercaptoethanol). The MonoQ column was eluted with a linear gradient of 0.1–2 M NaCl in buffer D and active fractions (eluting at 0.2 M NaCl) were collected.
Assay for faPy DNA glycosylase activity
FaPy DNA glycosylase activity was assayed in a reaction buffer containing 50 mM 3-(N-morpholino)propane sulfonic acid, pH 7.5, 1 mM dithiothreitol, 1 mM EDTA and 5% glycerol for 30 min at 37°C. N-[3H]methyl-N′-nitrosourea (18 Ci/mmol) was used to prepare poly(dG·dC) DNA containing faPy residues (5000 d.p.m./µg DNA) as described (38). faPy DNA glycosylase activity was measured in a total volume of 50 µl containing 0.4 µg faPy DNA substrate as described (39).
Assay for enzyme cleavage of 8-oxoG-, AP site- and 5-ohC-containing DNA fragments
Duplex DNA substrates containing either a single 8-oxoG residue or an AP site at position 14 were generated by 32P-end-labelling the 5′-end of the 24mer oligonucleotides [5′-GGCGGCATGACCC(8-oxoG or uracil)GAGGCCCATC-3′] by T4 polynucleotide kinase (MBI Fermentas) and [γ-32P]ATP (3000 Ci/mmol; Amersham). The labelled oligonucleotides were annealed to complementary strands with adenine, cytosine, guanine or thymidine opposite to 8-oxoG or uracil. AP DNA substrate was generated by incubation of the uracil-containing oligonucleotide with uracil DNA glycosylase (New England Biolabs) for 30 min at 37°C prior to use. Similarly, a 40 bp long duplex DNA substrate containing a single 5-ohC at position 22 [5′-AATTGCGATCTAGCTCGCCAG(5-ohC) AGCGACCTTATCTGATGA-3′] was 5′-32P-end-labelled as described above and hybridised to a complementary strand with guanine opposite to 5-ohC. The enzyme activities were assayed in a reaction buffer containing 25 mM 3-(N-morpholino)propane sulfonic acid, pH 7.5, 0.5 mM dithiothreitol, 0.5 mM EDTA and 2.5% glycerol for 30 min at 37°C. Reaction mixtures contained 100–500 fmol substrate and enzymes as indicated in a total volume of 10 µl. The products of the reactions were analysed by 20% denaturing DNA sequencing gels and phosphorimaging.
NaCNBH3-mediated trapping of enzyme–substrate intermediates
Aliquots of 100 fmol of 5′-32P-end-labelled AP- or 5-ohC-containing duplex DNA was incubated with 30 ng of enzyme as indicated in the presence of 32 mM sodium cyanoborohydride (NaCNBH3), 25 mM 3-(N-morpholino)propane sulfonic acid, pH 7.5, 0.5 mM dithiothreitol, 0.5 mM EDTA and 2.5% glycerol in a total volume of 12.5 µl. After incubation for 30 min at 37°C samples were separated on 10% Tricine–SDS gels and analysed by phosphorimaging.
RESULTS
Human proteins with homology to E.coli Fpg and Nei
We have identified and characterised two new entries in the human expressed sequence database that translate into sequences with significant homology to E.coli Fpg and Nei, termed hFPG1 and hFPG2. The ORF of hFPG1 encodes a polypeptide of 390 amino acids with a predicted molecular mass of 43.7 kDa, whereas the cDNA sequence of hFPG2 encodes a predicted protein of 605 amino acids with a Mr of 67.9. Alignment of hFPG1 and hFPG2 with E.coli Fpg and Nei reveals significant similarity to characteristic motifs and conserved residues of the bacterial Fpg/Nei family, including the catalytic residues at the N-terminus and the DNA-binding helix–two-turns–helix (H2TH) motif (Fig. 1A). The zinc finger DNA-binding motif found in all bacterial Fpg/Nei enzymes characterised so far is conserved in hFPG2 but absent from hFPG1. As a possible substitute, hFPG1 has a large number of positively charged residues at the C-terminus, which may serve as an alternative DNA-binding domain. hFPG2 also contains a RANbp-like zinc finger motif, which might be involved in nuclear transport (see below). Furthermore, hFPG2 contains a repeated putative zinc ribbon domain (CHCC). This domain is also present in topoisomerase IIIα, the base excision repair enzyme APE2, an RNA polymerase II transcription termination factor as well as two other uncharacterised proteins of the human database (Fig. 1B). The zinc ribbon domain is also duplicated in topoisomerase IIIα and for this protein has been reported to promote DNA binding with preference for single-stranded DNA (40).
Figure 1.

(A) Aligned sequences of hFPG1 (accession no. BAB15337), hFPG2 (NP_060718), E.coli Fpg (FPG_ECOLI) and E.coli Nei (END8_ECOLI). The conserved N-terminal domain and the helix–two-turns–helix motif are boxed in red. The conserved zinc finger motifs of E.coli Fpg, Nei and hFPG2 are boxed in green. Putative nuclear localisation signals of hFPG1 and hFPG2 are boxed in blue. The RANbp-like zinc finger motif and the duplicated zinc ribbon domains of hFPG2 are boxed in purple. The alignment was made using T-Coffee v.1.37 (36) followed by manual editing and layout using GeneDoc (K.B.Nicholas and H.B.Nicholas, GeneDoc: a tool for editing and annotating multiple sequence alignments, distributed by the authors). (B) Alignment of the duplicated zinc ribbon domain of hFPG2 with those of related sequences in the protein database.
Different expression patterns of HFPG1 and HFPG2 in human tissues
Expression patterns of HFPG1 and HFPG2 were investigated in several human adult tissues by northern blot hybridisation to full-length cDNA probes (Fig. 2). Three different bands (1.8, 4.0 and 7.5 kb) were consistently detected with the hFPG1 cDNA. The expected 1.8 kb mature mRNA was only faintly detectable but could be observed in almost all tissues examined, with the highest levels in skeletal muscle, spleen, prostate, testis and ovary. The larger transcripts (7.5 and 4.0 kb) were readily observed in all tissues examined. The nature of these transcripts is presently unknown but could possibly reflect the presence of larger transcripts that are only partially processed. Expression of HFPG2 was only detectable in testis and thymus as mature transcripts of 2.4 kb.
Figure 2.

Northern blot analysis of HFPG1 and HFPG2 expression. Poly(A)+ mRNA (2 µg/lane) extracted from various normal human tissues as indicated were hybridised with full-length cDNAs of hFPG1 and hFPG2.
hFPG1 and hFPG2 are nuclear proteins
The intracellular localisation of hFPG1 and hFPG2 was investigated by transient transfections of HeLa S3 cells with four constructs containing hFPG1 or hFPG2 fused either C- or N-terminal to the EGFP protein. The green fluorescence was confined to the nuclei of cells transfected with all the four expression vectors (Fig. 3A, C, D and F). In contrast, EGFP alone was distributed rather evenly throughout the cells (Fig. 3G and I). Both proteins contain putative nuclear localisation signals at the C-terminus (residues 359–378 of hFPG1 and residues 462–469 of hFPG2) as indicated by the PSORTII algorithm. However, the subnuclear localisation of hFPG1 and hFPG2 was different. EGFP-tagged hFPG1 showed an accumulation in the nucleoli as revealed by co-localisation with the major nucleolar protein, nucleolin (Fig. 3A and B). It thus appears that hFPG1 has a subcellular localisation that resembles hOGG1 with accumulation in the nucleoli (41). Conversely, hFPG2 appeared to be excluded from the nucleoli, as shown by the anti-nucleolin staining (Fig. 3D and E). Staining with antibodies against the 30 kDa subunit of RPA showed co-localisation with hFPG2; however, there is also an even background staining of hFPG2 fusions elsewhere in the nucleus (Fig. 3J and K).
Figure 3.

Nuclear localisation of the hFPG1–EGFP and hFPG2–EGFP fusion proteins. Exponentially growing asynchronous HeLa S3 cells were transiently transfected with constructs expressing hFPG1–EGFP (A–C), EGFP–hFPG2 (D–F and J–L) and pEGFP–N1 (G–I). The cells were imaged directly by fluorescence microscopy for EGFP detection (green, A, D, G and J). Nucleolin (red, B, E and H) and RPA (red, K) were both visualised by immunofluorescence with specific monoclonal antibodies followed by Alexa-conjugated secondary antibodies. DNA was stained by DAPI (blue, C, F, I and L).
Excision of faPy by hFPG1 and hFPG2 expressed in Sf9 insect cells
DNA glycosylase assays were used to determine whether HFPG1 and HFPG2 encode enzymes that possessed the predicted DNA repair function for removal of oxidized base residues. The cDNAs were expressed in the baculovirus system and crude extracts of Sf9 insect cells assayed for the ability to release faPy (Fig. 4A). A significant increase relative to background was observed in the removal of faPy by extracts of both hFPG1- and hFPG2-expressing cells but not in a glycosylase-negative control expressing APE2. These findings confirm that hFPG1 and hFPG2 are indeed novel DNA glycosylases involved in the removal of oxidative DNA damage.
Figure 4.

FaPy DNA glycosylase activity of hFPG1 and hFPG2. (A) Different amounts of cell extracts from uninfected and baculovirus-infected insect cells expressing APE2, hFPG2 or hFPG1 from appropriate cDNA constructs were assayed for removal of faPy from [3H]-methyl-faPy-poly(dG·dC) (0.4 µg). Diamonds, hFPG1; small squares, hFPG2; triangles, APE2; large squares, uninfected. (B) Removal of faPy from [3H]-methyl-faPy-poly(dG·dC) DNA by increasing amounts of purified E.coli Fpg (triangles), hOGG1 (squares) and hFPG1 (diamonds).
Expression and purification of hFPG1 in E.coli
Since the expression obtained with the baculovirus system was low, constructs were also generated for expression in E.coli. Escherichia coli strain BK3004 (fpg::kan) was used as a host to avoid background activity of bacterial Fpg. The hFPG1 protein was followed by assaying for faPy DNA glycosylase activity and purified to apparent physical homogeneity from 10 l of cell culture by a three-step procedure involving HiTrapQ, ResourceQ and MonoQ chromatography. The purified 43 kDa protein released faPy with an efficiency close to E.coli Fpg and human OGG1 (Fig. 4B).
Several hFPG2 bacterial expression plasmids were also constructed, including pT7 and plac derivatives. However, we have not been successful in recovering an active hFPG2 DNA glycosylase by expression in E.coli. Nevertheless, the baculovirus insect cell extracts consistently showed hFPG2 faPy DNA glycosylase activity significantly above background levels, implying that such activity is inherent in hFPG2 as well as hFPG1. However, it could be that hFPG2 requires some post-translational modification to be active and that this will only take place in mammalian systems and to some extent also in the insect cells.
hFPG1 represents an alternative enzyme for removal of 8-oxoG in human cells
In view of the minimal phenotypic effects exerted by the targeted deletion of the OGG1 gene in mice (30,31), we were particularly interested in whether the newly discovered DNA glycosylases were able to remove 8-oxoG from DNA and thus serve as a back-up for removal of 8-oxoG in mammalian cells. Using oligonucleotide substrates with a single 8-oxoG at a defined position it was found that hFPG1 was indeed able to remove 8-oxoG from DNA (Fig. 5). Human FPG1 cleaved DNA containing 8-oxoG opposite C, G or T but not opposite A. Relative to 8-oxoG:C, the efficiency was 85% for 8-oxoG:G, 50% for 8-oxoG:T and <2% for 8-oxoG:A (Fig. 5B). Human OGG1 also discriminates strongly against A in the opposite strand; however, it also acts weakly on 8-oxoG:G and 8-oxoG:T (Fig. 5A) (18). Kinetic experiments showed that hOGG1 is 10-fold more efficient than hFPG1 on the 8-oxoG:C substrate, but this is sequence context- dependent and varies between 5- and 15-fold for other oligonucleotides (data not shown). Nevertheless, the 8-oxoG DNA glycosylase activity of hFPG1 is significant and has the right preference for 8-oxoG:C versus 8-oxoG:A to prevent mutations from being formed from misincorporation at 8-oxoG in the template strand and therefore will represent an alternative mechanism in mammalian cells for 8-oxoG repair.
Figure 5.

8-OxoG DNA glycosylase activity of hFPG1. (A) An aliquot of 30 ng of purified E.coli Apn1, Fpg, hOGG1, hFPG1 or no enzyme was incubated with 100 fmol of a 24 bp duplex oligodeoxyribonucleotide containing a single 8-oxoG residue opposite A, C, G or T. Strand cleavage was analysed by 20% PAGE and phosphorimaging. (B) Quantification of the strand cleavage reactions. Results represent the averages of three independent experiments and error bars indicate standard deviation.
Lack of expression in E.coli and low expression in the baculovirus system made it difficult to analyse possible 8-oxoG DNA glycosylase activity of hFPG2. However, in the baculovirus extracts with low hFPG2 expression we were unable to detect any 8-oxoG DNA glycosylase activity above background levels (data not shown). These results suggest that hFPG2 does not have significant ability to remove 8-oxoG; however, further experiments with more active enzyme preparations are required to draw firm conclusions on this point.
Excision of 5-ohC by hFPG1
5-Hydroxycytosine is another frequent and stable oxidation product in DNA, which also has strong premutagenic properties (42,43). This lesion has been shown to be a substrate for Nei and Fpg as well as Nth from E.coli, thus illustrating the redundancy of repair enzymes for removal of this kind of damage. We also tested if this oxidized pyrimidine lesion would be a substrate for hFPG1. DNA containing 5-ohC:G was cleaved by hFPG1 whereas no appreciable cleavage was observed for the same amount of hOGG1 (Fig. 6A). Positive controls with the three E.coli enzymes were included for comparison (Fig. 6A and B). Bifunctional DNA glycosylases with associated AP lyase activity normally use an amino group as a nucleophile, resulting in a covalent imino enzyme–DNA substrate complex intermediate (44). These complexes can be trapped with NaCNBH3 and seen as DNA band shifts on denaturing SDS–polyacrylamide gels (45). Trapping experiments with hFPG1 and E.coli Fpg, Nei and Nth showed the formation of Schiff base intermediates of different sizes corresponding to the molecular weight differences between these proteins, Nth being the smallest and hFPG1 the largest (Fig. 6B). Assays with increasing amounts of enzymes showed that hFPG1 removed 5-ohC more efficiently than Fpg and less efficiently than Nei (Fig. 6C) and with about the same efficiency as 8-oxoG (data not shown).
Figure 6.
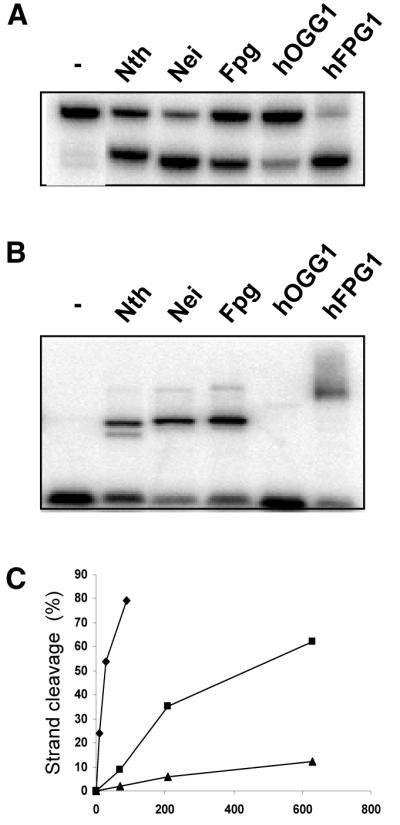
5-ohC DNA glycosylase activity of hFPG1. (A) Incision and (B) probing for covalent hFPG1 DNA intermediates by NaCNBH3 of 5-ohC-containing DNA by hFPG1. An aliquot of 30 ng of purified E.coli Nth, Nei, Fpg, hOGG1, hFPG1 or no enzyme was incubated with 100 fmol of a 40 bp duplex oligodeoxyribonucleotide containing a single 5-ohC residue opposite G. Strand cleavage was analysed by 20% denaturing PAGE and bands detected by phosphorimaging. Protein–DNA complexes were separated by 10% Tricine–SDS–PAGE and detected by phosphorimaging. (C) Increasing amounts of purified E.coli Nei (diamonds), E.coli Fpg (triangles) and hFPG1 (squares) were incubated with 100 fmol of a 40 bp duplex oligodeoxyribonucleotide containing a single 5-ohC residue opposite G, and strand cleavage was quantified by 20% PAGE followed by phosphorimaging.
hFPG1 cleaves abasic sites by a β,δ-elimination mechanism independent of the base in the complementary strand
To examine the incision mechanism of hFPG1, purified E.coli Nei, Nth, Fpg and Apn1, as well as hOGG1 and hFPG1 were incubated with duplex oligonucleotides containing a single abasic site and their cleavage products were analysed by denaturing PAGE (Fig. 7A). Cleavage of DNA duplexes containing a single AP site by either hydrolysis (Apn1), β-elimination (Nth, Nei, Fpg and hOGG1) or δ-elimination (Nei and Fpg) yields products that have different mobilities on denaturating PAGE gels. Incision of 5′-labelled AP DNA by hFPG1 resulted in labelled products that corresponded to β- and δ-elimination (Fig. 7A). Trapping experiments with AP DNA produced a similar pattern for the different enzymes as for the 5-ohC DNA except that hOGG1 also produced a clear band consistent with its activity on AP DNA (Fig. 7B). The incision of abasic sites by hFPG1 was further studied on duplex DNA containing a single AP site opposite each of the four normal bases in the complementary strand. Cleavage of AP sites by hFPG1 is independent of the base positioned in the opposite strand (Fig. 7C). In contrast, human hOGG1 has a clear preference for AP sites opposite C, as previously demonstrated (18).
Figure 7.

β,δ-elimination cleavage at abasic site DNA by hFPG1. (A) Incision and (B) probing for covalent hFPG1 DNA intermediates by NaCNBH3 of AP DNA by hFPG1. An aliquot of 30 ng of purified E.coli Apn1, Nei, Nth, Fpg, hOGG1, hFPG1 or no enzyme was incubated with 100 fmol of a 25 bp duplex oligodeoxyribonucleotide containing a single abasic site opposite C. Strand cleavage was analysed by 20% PAGE and phosphorimaging. Protein–DNA complexes were separated by 10% Tricine–SDS–PAGE. (C) Opposite base-independent AP DNA cleaving activity of hFPG1. An aliquot of 30 ng of purified hOGG1 (white) or hFPG1 (grey) was incubated with 500 fmol of a 24 bp duplex oligodeoxyribonucleotide containing a single abasic site opposite A, C, G or T as indicated. Strand cleavage was quantified by 20% denaturing PAGE and phosphorimaging. The results shown represent the averages of three independent experiments.
HFPG1 and HFPG2 genomic organisation
The current release of the human genome sequence indicates that HFPG1 is located on chromosome 15q22.33 and is flanked on the telomeric side by LOC112998 (a hypothetical gene supported by AL136876; NM_006715) and on the centromeric side by LOC95355 (similar to ribosomal protein L44). The HFPG1 gene spans ∼8 kb and is composed of 10 exons that vary in size from 28 to 456 bp. The sizes of introns range from 92 bp to ∼2.7 kb (Fig. 8). Sequence analysis identified one CpG island (between nucleotides –329 and 1667) containing exon 1 and a second CpG island (between nucleotides 1954 and 2374) containing exon 2. HFPG2 is located on chromosome 4q34.2 and is flanked on the telomeric end by the AGA (aspartylglucosaminidase) gene involved in the catabolism of N-linked oligosaccharides of glycoproteins. On the centromeric end HFPG2 is flanked by FLJ22649 (a hypothetical protein similar to signal peptidase SPC22/23). The gene spans ∼55 kb consisting of 10 exons that vary in size from 74 to 557 bp, and those of the introns ranged from 286 bp to 13 kb (Fig. 8). Analysis of the sequence around exon 1 revealed a CpG island located between nucleotides –289 and 556.
Figure 8.

Genomic structures of HFPG1 and HFPG2. (A) The regions around chromosomes 15q22.33 and (B) 4q34.2 are shown to scale. The sizes (in bp) of exons (striped boxes, untranslated exons; black boxes, translated exons) are shown above the line; the sizes of introns (in kb) are shown below the line. The positions of neighbouring genes are shown.
DISCUSSION
Until recently, it was assumed that the family of DNA glycosylases typified by Fpg/MutM in E.coli only existed in bacterial species. Instead, yeast and mammalian cells are equipped with OGG1, a protein of the HhH superfamily that performs much of the same functions as Fpg in bacteria, including its role in the removal of 8-oxoG. However, as the larger eukaryotic genome projects have been completed, it has been clear that genes for such enzymes are also found in plants, and more recently have also been detected as ESTs in mammalian cells. We have cloned two full-length human cDNAs of this type and shown that both express proteins with faPy DNA glycosylase activity and hence are enzymes involved in the repair of oxidative DNA damage in humans. FaPy lesions are also substrates for the human DNA glycosylases hNTH1 (17) and hOGG1 (18), thus implicating four different enzymes in the removal of this type of lesion in mammalian cells.
In bacteria, a prime role for Fpg is to remove 8-oxoG from DNA. To test if this would also be true for the Fpg-like enzymes described in this report, one of the proteins, hFPG1, was purified and characterised, and shown to be active against 8-oxoG. Like Fpg and hOGG1, it discriminates against removal of 8-oxoG across A, as would be expected for an enzyme involved in the repair of 8-oxoG in vivo. During DNA replication, 8-oxoG:A mispairs may be formed by misincorporation of A across unrepaired 8-oxoG in the template and a true repair enzyme should not remove 8-oxoG in this context. The presence of hFPG1 can thus account for the limited effects of knocking out OGG1 in mice. Homozygous ogg1–/– mice have an essentially normal phenotype without any obvious neurological abnormalities or increased cancer incidence. They only show a small increase in the steady-state level of 8-oxoG in the DNA and then only in proliferating tissues, implying that some back-up mechanism must exist for 8-oxoG removal (30,32). This back-up mechanism could well involve the hFPG1 enzyme, which, like hOGG1, is ubiquitously expressed and enriched in nucleoli (41). Whether hFPG1 is involved in TCR remains to be investigated.
In the nucleus DNA metabolic processes such as transcription and replication will be particularly vulnerable to damage residing in the template DNA. Directing DNA repair activities towards sites of ongoing DNA metabolism increases the efficiency of these processes and at the same time ensures efficient repair. For example, studies have shown that DNA glycosylases such as the human MutY DNA glycosylase homologue (hMYH) and the human uracil DNA glycosylase (UNG2) are localized to replication foci. These enzymes have been shown to interact with PCNA and RPA and to initiate post-replicative removal of misincorporated A and U, respectively (46–48). Alignment of the hFPG2 sequence with known PCNA- and RPA-binding proteins suggests putative PCNA- (residues 80–96) and RPA-binding (residues 30–45) motifs. RPA is required for multiple processes, including DNA replication, DNA repair and recombination (reviewed in 49). The observed co-localisation of hFPG2 with RPA suggests that hFPG2 participates in the repair of oxidative DNA damage residing at replication foci or occurring at other sites of ongoing DNA metabolism in which RPA is involved.
The Fpg superfamily of DNA glycosylases also comprises Nei from E.coli. Nei is mostly involved in the removal of oxidized pyrimidine base lesions and primarily serves as a back-up for Nth. One of its substrates is 5-ohC, which is a stable cytosine oxidation product that is strongly premutagenic, producing C→T transitions and C→G transversions, and is formed in amounts in DNA comparable to that of 8-oxoG (42,43). hFPG1 also excises 5-ohC with an efficiency similar to 8-oxoG and therefore could serve as a back-up for hNTH1 as well as hOGG1 in human cells. We have tried to classify hFPG1 and hFPG2 as Fpg- or Nei-like proteins based on sequence alignments with homologous groups of proteins from different species. However, such a comparison does not give a clear cut answer; if anything, hFPG2 is more like Nei and hFPG1 is more like Fpg. Fpg and hFPG1 both contain the RRFG motif (residues 118–121 in hFPG1), which is highly conserved among the Fpg enzymes and probably makes contact with the cytosine base in the opposite strand.
The sequence alignments further show that the homology between mammalian and bacterial Fpg/Nei is confined to certain domains and residues. In particular, the N-terminal (residues 1–6) and the H2TH motif are highly conserved. The H2TH motif has a function similar to the HhH motif of the Nth/hOGG1/hNTH1 superfamily. Structural studies of bacterial Fpg/Nei show that the H2TH motifs participate in DNA recognition by interaction with phosphate and oxygen atoms of the DNA backbone (50–52). The zinc finger motif of Fpg/Nei appears not to be conserved in hFPG1. The β-hairpin loop extending from the zinc finger of bacterial Fpg/Nei protrudes into the minor groove of DNA and is proposed to interact with the H2TH domain in DNA binding and damage recognition. Similar types of interactions involving different loop structures have been reported in other DNA glycosylases; i.e. the aspargine loop of human hOGG1 and the Fe-S cluster loop of E.coli Nth intercalate into the minor groove of DNA. Since intercalation of DNA by loop structures appears to be a general mechanism for damage recognition by DNA glycosylases it would be expected that hFPG1 also contains a loop/hairpin structure extending into the active site pocket in spite of missing the zing finger. Both hFPG1 and hFPG2 have a C-terminal part with a high density of prolines and charged residues that could be involved in protein–DNA or protein– protein interactions. The proposed catalytic residues of Nei and Fpg are conserved in hFPG1 (Pro2, Glu3, Glu6 and Lys54), suggesting a similar mechanism for the DNA glycosylase/AP lyase/δ-elimination reaction (Fig. 1). Lys54/61 (hFPG1/hFPG2) is proposed to act as a proton donor of the N-glycosylic cleavage reaction. The N-terminal amine of the primary catalytic residue Pro2 forms a Shiff base intermediate with the C1′ carbonyl of the deoxyribose at the abasic site, and the carboxylic group of Glu6 and Glu3 act as proton acceptors for the β- and δ-elimination reactions, respectively.
hFPG2 deviates somewhat from the consensus sequence of the Fpg family. Firstly, the Pro2 is missing. By analogy to bacterial Fpg this should result in a 90% reduction in enzyme activity (53). However, hFPG2 has another proline (188) that, from comparisons to solved structures, will be predicted to be very close to the active site. This proline is not found in any other member of the Fpg family and could serve the same function as Pro2 in the other enzymes. Secondly, the Glu6 is missing; however, a site-specific mutation at this position in Nei does not affect the enzyme activity (54). Thirdly, hFPG2 has a duplicated zinc ribbon domain at the C-terminal end. Interestingly, the same motif is also found in another BER enzyme, APE2, in topoisomerase IIIα and in an RNA polymerase II termination factor of the SWI2/SNF2 DNA helicase domain family. It has been speculated that RNA polymerase II terminator protein represents the transcription repair coupling factor in mammalian cells (55); however, more recent experiments indicate that this factor simply dissociates RNA polymerase II stalled at the site of DNA damage (56). Nevertheless, one might speculate that such a conserved domain (Fig. 1B) specifies a particular function, or a DNA or protein binding motif, which links the function of these proteins together.
The expression of HFPG1 in a broad range of tissues suggests a caretaker function of hFPG1 whereas the limited expression of HFPG2 indicates a specialised function restricted to certain tissues, or simply that expression below the detection level is sufficient to exert its function (Fig. 2). Interestingly, the expression pattern of HFPG1 revealed two additional transcripts in which the largest one corresponds to the size of the unprocessed HFPG1 gene (7.5 kb). These transcripts could possibly serve as storage for mature transcripts that are formed in response to some type of induction mechanism, thus up-regulating hFPG1 when needed.
In conclusion, the discovery of mammalian DNA glycosylases of the Fpg family has expanded the repertoire of repair pathways for removal of oxidative DNA damage in mammalian cells, thus explaining the mild phenotypes of single knock-outs of mnth1 (57) and mogg1 (30). Duplicate enzymes function to remove key premutagenic lesions such as 8-oxoG and 5-ohC and four enzymes are involved in the repair of the primary cytotoxic oxidative base lesion faPy. Similarly, four distinct DNA glycosylases have also been shown to be involved in the removal of uracil residues. Clearly, further mutant analysis is required before we can assess the importance of DNA glycosylases for normal existence and the price of not having such enzymes in mammalian cells.
During the preparation of this manuscript, a paper was published that also describes human DNA glycosylases of the Fpg family (58). In that paper, hFPG1 was also characterised in some detail (designated hNEH1), although the results appear to be somewhat different from those reported here, especially with respect to the activity on 8-oxoG-containing DNA. hFPG2 is not described in that report; however, they report, without further characterisation, yet another Fpg sequence, implying that at least three distinct Fpg homologues exist in human cells (58). During revision, yet another report appeared on the biochemical characterization of hFPG1 (59). It now appears that this function has been assigned NEIL1 in the genome database and with this nomenclature hFPG1/hFPG2 will be synonymous with NEIL1/NEIL3.
Acknowledgments
ACKNOWLEDGEMENTS
This research was supported by the Research Council of Norway and The Norwegian Cancer Society as well as by an EU contract (QRLT 1999-02002).
REFERENCES
- 1.Demple B. and Harrison,L. (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed] [Google Scholar]
- 2.Kasai H., Crain,P.F., Kuchino,Y., Nishimura,S., Ootsuyama,A. and Tanooka,H. (1986) Formation of 8-hydroxyguanine moiety in cellular DNA by agents producing oxygen radicals and evidence for its repair. Carcinogenesis, 7, 1849–1851. [DOI] [PubMed] [Google Scholar]
- 3.Kasai H. and Nishimura,S. (1991) Formation of 8-hydroxydeoxyguanosine in DNA by oxygen radicals and its biological significance. In Sies,H. (ed.), Oxidative Stress: Oxidants and Antioxidants. Academic Press, London, UK, pp. 99–116.
- 4.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxoG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 5.Maki H. and Sekiguchi,M. (1992) MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature, 355, 273–275. [DOI] [PubMed] [Google Scholar]
- 6.Grollman A.P. and Moriya,M. (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet., 9, 246–249. [DOI] [PubMed] [Google Scholar]
- 7.Wood M.L., Dizdaroglu,M., Gajewski,E. and Essigmann,J.M. (1990) Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry, 29, 7024–7032. [DOI] [PubMed] [Google Scholar]
- 8.Moriya M., Ou,C., Bodepudi,V., Johnson,F., Takeshita,M. and Grollman,A.P. (1991) Site-specific mutagenesis using a gapped duplex vector: a study of translesion synthesis past 8-oxodeoxyguanosine in E. coli. Mutat. Res., 254, 281–288. [DOI] [PubMed] [Google Scholar]
- 9.Cheng K.C., Cahill,D.S., Kasai,H., Nishimura,S. and Loeb,L.A. (1992) 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G→T and A→C substitutions. J. Biol. Chem., 267, 166–172. [PubMed] [Google Scholar]
- 10.Hollstein M., Shomer,B., Greenblatt,M., Souss,T., Hovig,E., Montesano,R. and Harris,C.C. (1996) Somatic point mutations in the p53 gene of human tumors and cell lines: updated compilation. Nucleic Acids Res., 24, 141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boiteux S. and Laval,J. (1983) Imidazole open ring 7-methylguanine: an inhibitor of DNA synthesis. Biochem. Biophys. Res. Commun., 110, 552–558. [DOI] [PubMed] [Google Scholar]
- 12.Seeberg E., Eide,L. and Bjørås,M. (1995) The base excision repair pathway. Trends Biochem. Sci., 20, 391–397. [DOI] [PubMed] [Google Scholar]
- 13.Dogliotti E., Fortini,P., Pascucci,B. and Parlanti,E. (2001) The mechanism of switching among multiple BER pathways. Prog. Nucleic Acid Res. Mol. Biol., 68, 3–27. [DOI] [PubMed] [Google Scholar]
- 14.Aspinwall R., Rothwell,D.G., Roldan-Arjona,T., Anselmino,C., Ward,C.J., Cheadle,J.P., Sampson,J.R., Lindahl,T., Harris,P.C. and Hickson,I.D. (1997) Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc. Natl Acad. Sci. USA, 94, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilbert T.P., Chaung,W., Boorstein,R.J., Cunningham,R.P. and Teebor,G.W. (1997) Cloning and expression of the cDNA encoding the human homologue of the DNA repair enzyme, Escherichia coli endonuclease III. J. Biol. Chem., 272, 6733–6740. [DOI] [PubMed] [Google Scholar]
- 16.Sarker A.H., Ikeda,S., Nakano,H., Terato,H., Ide,H., Imai,K., Akiyama,K., Tsutsui,K., Bo,Z., Kubo,K. et al. (1998) Cloning and characterization of a mouse homologue (mNthl1) of Escherichia coli endonuclease III. J. Mol. Biol., 282, 761–774. [DOI] [PubMed] [Google Scholar]
- 17.Luna L., Bjørås,M., Hoff,E., Rognes,T. and Seeberg,E. (2000) Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res., 460, 95–104. [DOI] [PubMed] [Google Scholar]
- 18.Bjørås M., Luna,L., Johnsen,B., Hoff,E., Haug,T., Rognes,T. and Seeberg,E. (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J., 16, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenquist T.A., Zharkov,D.O. and Grollman,A.P. (1997) Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 7429–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prieto Alamo M.J., Jurado,J., Francastel,E. and Laval,F. (1998) Rat 7,8-dihydro-8-oxoguanine DNA glycosylase: substrate specificity, kinetics and cleavage mechanism at an apurinic site. Nucleic Acids Res., 26, 5199–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demple B., Herman,T. and Chen,D.S. (1991) Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl Acad. Sci. USA, 88, 11450–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hadi M.Z. and Wilson,D.M. (2000) Second human protein with homology to the Escherichia coli abasic endonuclease exonuclease III. Environ. Mol. Mutagen., 36, 312–324. [PubMed] [Google Scholar]
- 23.Ikeda S., Biswas,T., Roy,R., Izumi,T., Boldogh,I., Kurosky,A., Sarker,A.H., Seki,S. and Mitra,S. (1998) Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J. Biol. Chem., 273, 21585–21593. [DOI] [PubMed] [Google Scholar]
- 24.Eide L., Luna,L., Gustad,E.C., Henderson,P.T., Essigman,J.M., Demple,B. and Seeberg,E. (2001) Human endonuclease III acts preferentially on DNA damage opposite guanine residues in DNA. Biochemistry, 40, 6653–6659. [DOI] [PubMed] [Google Scholar]
- 25.van der Kemp P.A., Thomas,D., Barbey,R., de Oliveira,R. and Boiteux,S. (1996) Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine. Proc. Natl Acad. Sci. USA, 93, 5197–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boiteux S., O’Connor,T.R. and Laval,J. (1987) Formamidopyrimidine-DNA glycosylase of Escherichia coli: cloning and sequencing of the fpg structural gene and overproduction of the protein. EMBO J., 6, 3177–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tchou J., Kasai,H., Shibutani,S., Chung,M.H., Laval,J., Grollman,A.P. and Nishimura,S. (1991) 8-Oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc. Natl Acad. Sci. USA, 88, 4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melamede R.J., Hatahet,Z., Kow,Y.W., Ide,H. and Wallace,S.S. (1994) Isolation and characterization of endonuclease VIII from Escherichia coli. Biochemistry, 33, 1255–1264. [DOI] [PubMed] [Google Scholar]
- 29.Hazra T.K., Izumi,T., Venkataraman,R., Kow,Y.W., Dizdaroglu,M. and Mitra,S. (2000) Characterization of a novel 8-oxoguanine-DNA glycosylase activity in Escherichia coli and identification of the enzyme as endonuclease VIII. J. Biol. Chem., 275, 27762–27767. [DOI] [PubMed] [Google Scholar]
- 30.Klungland A., Rosewell,I., Hollenbach,S., Larsen,E., Daly,G., Epe,B., Seeberg,E., Lindahl,T. and Barnes,D.E. (1999) Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl Acad. Sci. USA, 96, 13300–13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minowa O., Arai,T., Hirano,M., Monden,Y., Nakai,S., Fukuda,M., Itoh,M., Takano,H., Hippou,Y., Aburatani,H. et al. (2000) Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl Acad. Sci. USA, 97, 4156–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osterod M., Hollenbach,S., Hengstler,JG., Barnes,D., Lindahl,T. and Epe,B. (2001) Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis, 22, 1459–1463. [DOI] [PubMed] [Google Scholar]
- 33.Le Page F., Kwoh,E.E., Avrutskaya,A., Gentil,A., Leadon,S.A., Sarasin,A. and Cooper,P.K. (2000) Transcription-coupled repair of 8-oxoguanine: requirement for XPG, TFIIH, and CSB and implications for Cockayne syndrome. Cell, 101, 159–171. [DOI] [PubMed] [Google Scholar]
- 34.Le Page F., Klungland,A., Barnes,D.E., Sarasin,A. and Boiteux,S. (2000) Transcription coupled repair of 8-oxoguanine in murine cells: the ogg1 protein is required for repair in nontranscribed sequences but not in transcribed sequences. Proc. Natl Acad. Sci. USA, 97, 8397–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rognes T. (2001) ParAlign: a parallel sequence alignment algorithm for rapid and sensitive database searches. Nucleic Acids Res., 29, 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Notredame C., Higgins,D.G. and Heringa,J. (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol., 302, 205–217. [DOI] [PubMed] [Google Scholar]
- 37.Nakai K. and Kanehisa,M. (1992) A knowledge base for predicting protein localisation sites in eukaryotic cells. Genomics, 14, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boiteux S., Belleney,J., Roques,B.P. and Laval,J. (1984) Two rotameric forms of open ring 7-methylguanine are present in alkylated polynucleotides. Nucleic Acids Res., 12, 5429–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eide L., Bjørås,M., Pirovano,M., Alseth,I., Berdal,K.G. and Seeberg,E. (1996) Base excision of oxidative purine and pyrimidine DNA damage in Saccharomyces cerevisiae by a DNA glycosylase with sequence similarity to endonuclease III from Escherichia coli. Proc. Natl Acad. Sci. USA, 93, 10735–10740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grishin N.V. (2000) C-terminal domains of Escherichia coli topoisomerase I belong to the zinc-ribbon superfamily. J. Mol. Biol., 299, 1165–1177. [DOI] [PubMed] [Google Scholar]
- 41.Dantzer F., Luna,L., Bjørås,M. and Seeberg,E. (2002) Human OGG1 undergoes serine phosphorylation and associates with the nuclear matrix and mitotic chromatin in vivo. Nucleic Acids Res., 30, 2349–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraga C.G., Shigenaga,M.K., Park,J.W. and Ames,B.N. (1990) Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc. Natl Acad. Sci. USA, 87, 4533–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner J.R., Hu,C.C. and Ames,B.N. (1992) Endogenous oxidative damage of deoxycytidine in DNA. Proc. Natl Acad. Sci. USA, 89, 3380–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dodson M.L., Michaels,M.L. and Lloyd,R.S. (1994) Unified catalytic mechanism for DNA glycosylases. J. Biol. Chem., 269, 32709–32712. [PubMed] [Google Scholar]
- 45.Dodson M.L., Schrock,R.D. and Lloyd,S. (1993) Evidence for an imino intermediate in the T4 endonuclease V reaction. Biochemistry, 32, 8284–8290. [DOI] [PubMed] [Google Scholar]
- 46.Boldog I., Milligan,D., Lee,M.S., Bassett,H., Lloyd,R.S. and McCullough,A.K. (2001) hMYH cell cycle-dependent expression, subcellular localisation and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res., 29, 2802–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parker A., Gu,Y., Mahoney,W., Lee,S.H., Singh,K.K. and Lu,A.L. (2001) Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem., 276, 5547–5555. [DOI] [PubMed] [Google Scholar]
- 48.Otterlei M., Warbrick,E., Nagelhus,T.A., Haug,T., Slupphaug,G., Akbari,M., Aas,P.A., Steinsbekk,K., Bakke,O. and Krokan,H.E. (1999) Post-replicative base excision repair in replication foci. EMBO J., 18, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wold M.S. (1997) Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem., 66, 61–92. [DOI] [PubMed] [Google Scholar]
- 50.Sugahara M., Mikawa,T., Kumasaka,T., Yamamoto,M., Kato,R., Fukuyama,K. and Kuramitsu,S. (2000) Crystal structure of a repair enzyme of oxidatively damaged DNA, MutM (Fpg), from an extreme thermophile, Thermus thermophilus HB8. EMBO J., 19, 3857–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zharkov D.O., Golan,G., Gilboa,R., Fernandes,A.S., Gerchman,S.E., Kycia,J., Rieger,R.A., Grollman,A.P. and Shoham,G. (2002) Structural analysis of an Escherichia coli endonuclease VIII covalent reaction intermediate. EMBO J., 21, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilboa R., Zharkov,D.O., Golan,G., Fernandes,A.S., Gerchman,S.E., Matz,E., Kycia,J.H., Grollman,A.P. and Shoham,G. (2002) Structure of a formamidopyrimidine DNA glycosylase-DNA complex. J. Biol. Chem., 277, 19811–19816. [DOI] [PubMed] [Google Scholar]
- 53.Sidorkina O.M. and Laval,J. (2000) Role of the N-terminal proline residue in the catalytic activities of the Escherichia coli Fpg protein. J. Biol. Chem., 275, 9924–9929. [DOI] [PubMed] [Google Scholar]
- 54.Burgess S., Jaruga,P., Dodson,M.L., Dizdaroglu,M. and Lloyd,R.S. (2002) Determination of active site residues in Escherichia coli endonuclease VIII. J. Biol. Chem., 277, 2938–2944. [DOI] [PubMed] [Google Scholar]
- 55.Liu M., Xie,Z. and Price,D.H. (1998) A human RNA polymerase II transcription termination factor is a SWI2/SNF2 family member. J. Biol. Chem., 273, 25541–25544. [DOI] [PubMed] [Google Scholar]
- 56.Hara R., Selby,C.P., Liu,M., Price,D.H. and Sancar,A. (1999) Human transcription release factor 2 dissociates RNA polymerases I and II stalled at a cyclobutane pyrimidine dimer. J. Biol. Chem., 274, 24779–24786. [DOI] [PubMed] [Google Scholar]
- 57.Takao M., Kanno,S.I., Shiromoto,T., Hasegawa,R., Ide,H., Ikeda,S., Sarker,A.H., Seki,S., Xing,J.Z., Le,X.C. et al. (2002) Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J., 21, 3486–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hazra T.K., Izumi,T., Boldogh,I., Imhoff,B., Kow,Y.W., Jaruga,P., Dizdaroglu,M. and Mitra,S. (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl Acad. Sci. USA, 99, 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bandaru V., Sunkara,S. and Wallace,S.S. (2002) A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair, 1, 517–529. [DOI] [PubMed] [Google Scholar]