

Abstract
Flavin mononucleotide and flavin adenine dinucleotide are essential coenzymes in redox reactions. For example, flavin adenine dinucleotide is a coenzyme for both glutathione reductase and enzymes that mediate the oxidative folding of secretory proteins. Here we investigated short-term effects of moderately riboflavin-deficient culture medium on flavin-related responses in HepG2 hepatocarcinoma cells. Cells were cultured in riboflavin-deficient (3.1 nmol/L) medium for up to six days; controls were cultured in riboflavin-sufficient (532 nmol/L) medium. The activity of glutathione reductase decreased by 98% within four days of riboflavin-deficient culture. Transport rates of riboflavin increased in response to riboflavin depletion, whereas expression of enzymes mediating flavocoenzyme synthesis (flavokinase and flavin adenine dinucleotide synthetase) decreased in response to depletion. The oxidative folding and synthesis of plasminogen and apolipoprotein B-100, respectively, was impaired within four days of culture in riboflavin-deficient medium; this is consistent with impaired processing of secretory proteins in riboflavin-deficient cells. Riboflavin depletion was associated with increased DNA-binding activities of transcription factors with affinity for endoplasmic reticulum stress elements and NF-κB consensus elements, suggesting cell stress. Moreover, the abundance of the stress-induced protein GADD153 was greater in riboflavin-deficient cells compared with controls. Riboflavin deficiency was associated with decreased rates of cell proliferation caused by arrest in G1 phase of the cell cycle. These studies are consistent with the hypothesis that HepG2 cells have a great demand for riboflavin, and that cell stress develops rapidly if riboflavin supply is marginally low.
Keywords: cell cycle, cell stress, deficiency, protein folding, riboflavin
1. Introduction
Riboflavin is converted to flavocoenzymes to attain biological activity [1]. In a first step of bioactivation, riboflavin is phosphorylated by flavokinase to generate flavin mononucleotide (FMN)4. In a second step, FMN is adenylated by flavin adenine dinucleotide (FAD) synthetase to generate FAD. Both FMN and FAD are essential coenzymes in numerous redox reactions. For example, FAD is a coenzyme for glutathione reductase, which mediates regeneration of reduced glutathione [1]. Moreover, FAD is a coenzyme for Ero1 and sulfhydryl oxidases, which mediate the oxidative folding (formation of disulfide bonds) of secretory proteins; oxidative folding of proteins in the endoplasmic reticulum is essential for their subsequent secretion into the extracellular space [2,3].
Consistent with a role for FAD in glutathione recycling and protein folding, riboflavin deficiency causes cell stress. First, evidence has been provided that cellular concentrations of reduced glutathione decrease in response to riboflavin deficiency [4,5]. Reduced glutathione is a scavenger of free radicals and reactive oxygen species [6,7] and also regulates protein function by S-glutathionylation [8]. Consistent with these observations, riboflavin-deficient HepG2 hepatocarcinoma cells exhibit an increased incidence of oxidative damage to proteins and DNA compared with riboflavin-sufficient controls (K. C. Manthey and J. Zempleni, unpublished observation). Second, evidence has been provided that riboflavin deficiency in human Jurkat (lymphoid) cells and HepG2 cells impairs the oxidative folding of secretory proteins [4,5]. Impaired folding is associated with a decrease of global translational activity, activation of ubiquitin-dependent degradation of misfolded proteins, and cell cycle arrest and apoptosis. These events are hallmarks of a cell stress response named the Unfolded Protein Response [9,10].
Some tissues are more susceptible to developing riboflavin deficiency than others. For example, Jurkat cells developed mild symptoms of riboflavin deficiency only if cultured in severely riboflavin-deficient (0.85 nmol/L) medium for extended periods of time, i.e., for about five weeks [4]. This suggests that lymphoid cells are unlikely to develop riboflavin deficiency in free-living humans consuming moderately riboflavin-deficient diets for short periods of time. In contrast, HepG2 cells developed severe riboflavin deficiency if cultured in medium containing moderately low concentrations of riboflavin (3.1 nmol/L) for eight days [5]. This is consistent with the hypothesis that moderately low riboflavin supply may impair liver cell function.
Here we investigated effects of moderately riboflavin-deficient culture medium on flavin-related responses in HepG2 cells. Specifically, (i) we quantified time courses of glutathione reductase activities and riboflavin transport rates in HepG2 cells cultured in riboflavin-deficient medium; and (ii) we determined whether short-term riboflavin deficiency affects the following flavin-dependent variables: expression of genes coding for flavokinase and FAD synthetase; oxidative folding and synthesis of the secretory proteins plasminogen and apolipoprotein B-100; activation of stress-dependent transcription factors; and cell cycle progression.
2. Methods and materials
2.1. Cell culture
HepG2 cells (ATCC; Manassas, VA) were cultured in customized RPMI-1640 containing 3.1 nmol/L riboflavin (denoted “deficient”) for up to six days; control cells were cultured in medium containing 532 nmol/L [5]. Media contained 10% dialyzed, riboflavin-depleted bovine growth serum [4]. Riboflavin concentrations in media were chosen based on the following lines of reasoning. First, previous studies provided evidence that HepG2 cells develop severe riboflavin deficiency if cultured in medium containing 3.1 nmol/L riboflavin for eight days [5]; this concentration represents the level of riboflavin observed in plasma from moderately deficient pregnant women [11]. Second, previous studies provided evidence that a concentration of 532 nmol/L riboflavin in culture medium is sufficient to prevent riboflavin deficiency in HepG2 cells [5]; this concentration represents the level of riboflavin observed in plasma from individuals taking riboflavin supplements [12] and in normal portal vein blood [5,13].
2.2. Flavin homeostasis
The activity of glutathione reductase was quantified in lysed HepG2 cells as described [14] with minor modifications [4]. Rates of riboflavin transport into HepG2 cells were quantified using a physiological concentration of [3H]riboflavin (10 nmol/L) as described [15].
2.3. Reverse transcriptase polymerase chain reaction (RT-PCR)
The abundance of mRNA coding for flavokinase and FAD synthetase was quantified using RT-PCR as described [16,17]; mRNA coding for glyceraldehyde-3-phosphate dehydrogenase was used as a control. The following gene-specific primers were used: 5′-AGA TGG TGG TGA GCA TAG GA-3′ and 5′-CCA CTG CAC TTG GCC TTA AT-3′ for flavokinase; 5′-GTT TGC CGA GTC TCA GTT GT-3′ and 5′-AAT GCC TGG GAA GAG GTA GA-3′ for FAD synthetase; and 5′-ACC ACA GTC CAT GCC ATC AC-3′ and 5′-TCC ACC ACC CTG TTG CTG TA-3′ for glyceraldehyde-3-phosphate dehydrogenase. Only values from within the exponential phase of PCR amplification were considered for analysis by gel densitometry [17].
2.4. Detection of sulfhydryl groups in plasminogen
Polyethylene oxide-iodoacetyl biotin (Pierce, Rockford, IL) is a sulfhydryl-reactive biotin derivative. Theoretically, polyethylene oxide-iodoacetyl biotin reacts with sulfhydryl groups in unfolded proteins from cell extracts in vitro, increasing the molecular weight of the protein and causing a mobility shift in gel electrophoresis. In contrast, proteins that are properly folded (=low abundance of free sulfhydryl groups) do not react with polyethylene oxide-iodoacetyl biotin. As a marker for protein folding we used plasminogen from HepG2 cell extracts as described below. Plasminogen has a molecular weight of 91 kDa and contains 21 disulfide bonds. As a control we used 98% pure plasminogen from human plasma (MP Biomedicals, Aurora, OH); some samples of chemically pure plasminogen were pre-treated with 1 mmol/L dithiothreitol for 1 hour in order to convert disulfide bonds to free sulfhydryl groups before incubation with polyethylene oxide-iodoacetyl biotin.
Extranuclear proteins were released from HepG2 pellets by suspending cells in 10 mmol/L HEPES (pH 7.9), containing 50 mmol/L sodium chloride, 0.5 mol/L sucrose, 0.1 mmol/L EDTA, 5 mL/L Triton X-100, 0.5 mmol/L sodium orthovanadate, 10 mmol/L disodium pyrophosphate, 100 mmol/L sodium fluoride, 17.5 mmol/L β-glycerophosphate, 1 mmol/L phenylmethylsulfonyl fluoride, and 0.04 mL/L protease inhibitor cocktail (catalog no. P-8340, Sigma, St. Louis, MO). Cell nuclei were removed by centrifugation (3000 g, 10 min). Proteins in the supernatant were precipitated with 1.2 mol/L trichloroacetic acid on ice for 10 min, and protein pellets were washed twice with cold acetone. Proteins were re-suspended in 50 mmol/L Tris-HCl (pH 8.3), containing 35 mmol/L lauryl sulfate sodium salt, 1 mmol/L phenylmethylsulfonyl fluoride, and 90 mmol/L polyethylene oxide-iodoacetyl biotin; samples were incubated at room temperature for 90 minutes. Equal amounts of proteins were resolved using 3-8% Tris acetate gels (Invitrogen, Carlsbad, CA) and electroblotted onto polyvinylidene difluoride membranes [18]. A (Sigma) was used as secondary antibody. Bands were visualized by chemiluminescence [18].
2.5. Western blot analyses
GADD153 in extranuclear extracts was analyzed by Western blot analysis as described [5]. For analysis of apolipoprotein B-100, extranuclear extracts were prepared, resolved by gel electrophoresis, and electroblotted as described in section 2.4. Apolipoprotein B-100 on blots polyclonal anti-human β-actin antibody and a mouse monoclonal anti-goat/sheep IgG peroxidase conjugate. Bands were visualized by chemiluminescence as described [18].
2.6. Electrophoretic mobility shift assays (EMSAs)
The nuclear abundance of transcription factors was quantified by EMSA and gel densitometry as described [19,20]. Transcription factors binding to endoplasmic reticulum stress elements were probed using the following oligonucleotides, which contain consensus binding sites for the proteins of interest: 5′-GAG GGC CTT CAC CAA TCG GCG GCC TCC ACG ACG GGG CTG G-3′ and 5′-CCA GCC CCG TCG TGG AGG CCG CCG ATT GGT GAA GGC CCT C-3′ [21]. Nuclear factor κB (NF-κB) was probed as described before [20]. As a control we quantified the nuclear abundance of the transcription factor Oct-1 by using the following oligonucleotide probes: 5′- TGT CGA ATG CAA ATC ACT AGA A-3′ and 5′-TTC TAG TTT GCA TTC GAC A-3′. Some samples were assayed in the presence of a 100-fold molar excess of unlabeled probe or in the absence of nuclear extract (specificity controls) as described [19,20].
2.7. Cell proliferation and cell cycle analysis
Proliferation rates of HepG2 cells were quantified by measuring the cellular uptake of [3H]thymidine as described with minor modifications [22]. For analysis of cell cycle distribution, DNA was labeled with propidium iodide and samples were analyzed using flow cytometry [23]. Flow cytometry experiments were conducted at the Cell Analysis Facility Center (University of Nebraska Medical Center, Omaha, NE).
2.8. Statistics
Homogeneity of variances among groups was confirmed using Bartlett's test [24]. Significance of differences among groups was tested by one-way ANOVA. Fisher's Protected Least Significant Difference procedure was used for posthoc testing [24]. Paired t-test was used for paired comparisons [24]. StatView 5.0.1 (SAS Institute; Cary, NC) was used to perform all calculations. Differences were considered significant if P < 0.05. Data are expressed as mean ± SD.
3. Results
3.1. Flavin homeostasis
Activities of glutathione reductase in HepG2 cells decreased significantly from day 2 to day 3 in riboflavin-deficient medium (Fig. 1). Four days after transfer into riboflavin-deficient medium, glutathione reductase activity had decreased by 98 ± 2 % compared with riboflavin-sufficient controls (day 0). Six days after transfer into riboflavin-deficient medium, glutathione reductase activity was below limits of detection.
Fig. 1.
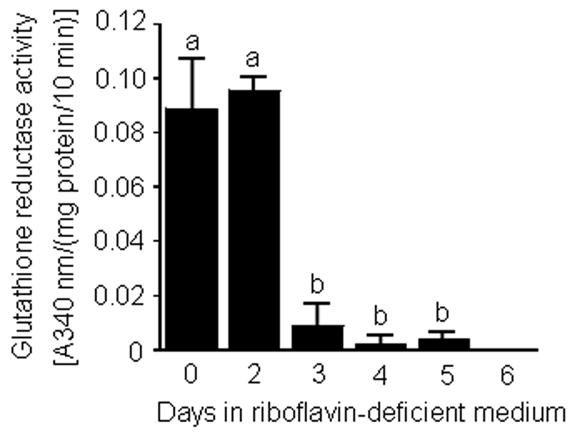
Time course of glutathione reductase activity in HepG2 cells in response to transfer "from riboflavin-sufficient medium (532 nmol/L, day 0) to riboflavin-deficient medium (3.1 nmol/L). a,bBars not sharing the same letter are significantly different (P < 0.05; n = 4). Means ± SD are shown.
HepG2 cells responded to riboflavin deficiency by increasing riboflavin transport rates. Transport rates equaled 38 ± 4 fmol riboflavin/(106 cells × 10 min) in riboflavin-sufficient cells (day 0). Transport rates increased by about two times after cells were transferred into riboflavin-deficient medium; riboflavin transport rates peaked at about 3 – 4 days after transfer into riboflavin-deficient medium. The following temporal pattern of riboflavin transport was observed [units = fmol riboflavin/(106 cells × 10 min)]: 53 ± 8 for day 2; 88 ± 29 for day 3; 86 ± 24 for day 4; 73 ± 10 for day 5; and 76 ± 24 for day 6 (P < 0.05 vs. riboflavin-sufficient controls, day 0; n = 3). Collectively, data from experiments with glutathione reductase and transport rates suggest that riboflavin deficiency developed in HepG2 cells within 3 – 4 days of culture in riboflavin-deficient medium. Hence, for subsequent studies we used cells cultured in riboflavin-deficient and riboflavin-sufficient media for four days.
Riboflavin deficiency was associated with decreased abundance of mRNA coding for flavokinase and FAD synthetase (Fig. 2). After four days in riboflavin-deficient medium, the abundance of mRNA coding for flavokinase and FAD synthetase decreased to <20% of control values. The abundance of mRNA encoding glyceraldehyde-3-phosphate dehydrogenase (control) was not affected by riboflavin (data not shown).
Fig. 2.
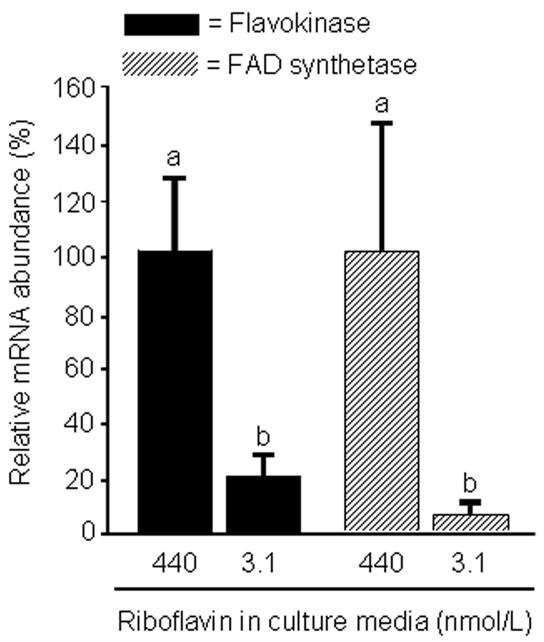
The abundance of mRNA coding for flavokinase and FAD synthetase decreases in response to riboflavin deficiency in HepG2 cells. Cells were cultured in riboflavin-deficient (3.1 nmol/L) and riboflavin-sufficient (532 nmol/L) media for 4 d. mRNA abundance was quantified using RT-PCR. Data are expressed in units of percent mRNA abundance, using cells cultured in medium containing 532 nmol/L riboflavin as the reference. a,bBars not sharing the same letter are significantly different (P < 0.05; n = 4). Means ± SD are shown.
3.2. Protein synthesis and folding
Riboflavin deficiency impaired the oxidative folding of plasminogen. Protein extracts from riboflavin-sufficient and riboflavin-deficient HepG2 cells were treated with the sulfhydryl-reactive reagent polyethylene oxide-iodoacetyl biotin. Incubation with polyethylene oxide-iodoacetyl biotin caused a noticeable increase of the apparent molecular weight of plasminogen from riboflavin-deficient cells compared with controls (Fig. 3). The increased molecular weight is consistent with the availability of free sulfhydryl groups for chemical biotinylation in unfolded plasminogen. The apparent molecular weight of commercial, chemically pure plasminogen was similar to the mobility observed for plasminogen from riboflavin-sufficient cells (Fig. 3). In contrast, if disulfide bonds in commercial plasminogen were reduced using dithiothreitol before incubation with polyethylene oxide-iodoacetyl biotin, the apparent molecular weight was similar to plasminogen from riboflavin-deficient cells.
Fig. 3.
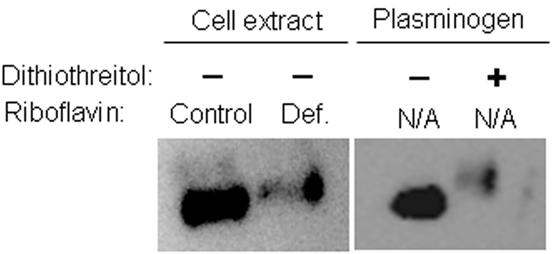
The abundance of free sulfhydryl groups is greater in proteins from riboflavin-deficient HepG2 cells (“Def.” = 3.1 nmol/L riboflavin) compared with riboflavin-sufficient controls (“Control” = 532 nmol/L riboflavin). Cells were cultured in riboflavin-defined media for 4 d. Sulfhydryl groups in proteins from cell extracts were derivatized with polyethylene oxide-iodoacetyl biotin; plasminogen was visualized by Western blot analysis using an antibody to human plasminogen. Commercial, chemically pure plasminogen (with and without pretreatment with dithiothreitol) was used as a control. N/A = not applicable.
Previous studies suggested that HepG2 cells respond to severe riboflavin deficiency with decreased expression of the apolipoprotein B-100 gene [5]. The present study suggested that the abundance of cellular apolipoprotein B-100 also decreased after short-term riboflavin deficiency. Specifically, if HepG2 cells were cultured in riboflavin-deficient medium for 4 d, the abundance of apolipoprotein B-100 decreased by 85 ± 4% compared with riboflavin-sufficient controls (Fig. 4). The abundance of β-actin (control) was not affected by riboflavin deficiency (data not shown). Collectively, these studies suggest that culturing HepG2 cells in riboflavin-deficient medium for only four days is sufficient to impair the oxidative folding of secretory proteins, and that impaired protein folding attenuates protein synthesis.
Fig. 4.
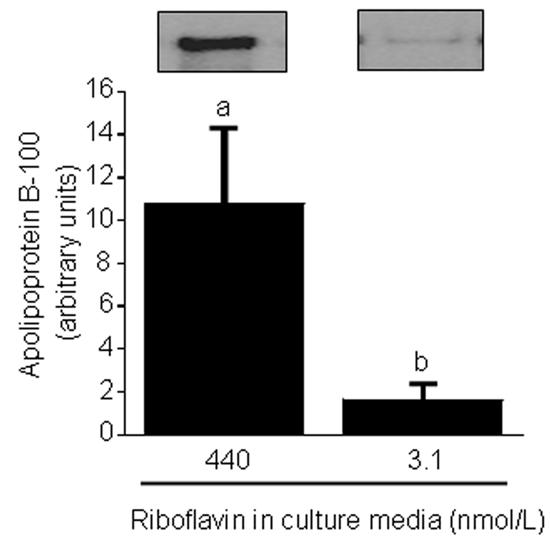
Riboflavin deficiency decreases the abundance of apolipoprotein B-100 in HepG2 cells. Cells were cultured in riboflavin-deficient (3.1 nmol/L) and riboflavin-sufficient (532 nmol/L) media for 4 d. Apolipoprotein B-100 was quantified by Western blot analysis and gel densitometry. Inserts depict representative Western blot images. a,bBars not sharing the same letter are significantly different (P < 0.05; n = 4). Means ± SD are shown.
3.3. Cell stress
The binding activity of nuclear transcription factors with affinity for endoplasmic reticulum stress elements was greater in riboflavin-deficient cells than in riboflavin-sufficient controls, as judged by EMSA (Fig. 5, lanes 1 and 2). No bands were detectable if samples were incubated in the presence of a 100-fold molar excess of unlabeled probe (lane 3) or in the absence of nuclear extract (lane 4); DNA-binding activity of Oct-1 (specificity control) was not affected by riboflavin (lanes 5 and 6). These observations suggest that binding of transcription factors to endoplasmic reticulum stress elements increased specifically in response to riboflavin deficiency. Binding to endoplasmic reticulum stress elements was 99 ± 38 arbitrary units in riboflavin-deficient cells and 31 ± 36 arbitrary units in riboflavin-sufficient controls, as judged by using gel densitometry (P < 0.01; n = 5). In previous studies we provided evidence that ATF6 accounts for some of the binding to the endoplasmic reticulum stress element [5]. Activation of ATF6 is consistent with endoplasmic reticulum stress caused by accumulation of unfolded proteins [25,26].
Fig. 5.
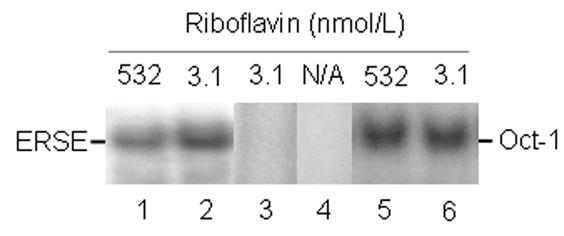
Riboflavin deficiency enhances binding of transcription factors to endoplasmic reticulum stress elements in HepG2 cells. Cells were cultured in riboflavin-deficient (3.1 nmol/L) and riboflavin-sufficient (532 nmol/L) media for 4 d. Binding to endoplasmic reticulum stress elements was visualized using EMSA; binding to an Oct-1 site was used as a control. N/A = not applicable. Lanes 1 and 2 = binding to an endoplasmic reticulum stress element by nuclear extracts from control cells and riboflavin-deficient cells, respectively; lane 3 = 100-fold molar excess of unlabeled endoplasmic reticulum stress element; lane 4 = endoplasmic reticulum stress element in the absence of nuclear extract; lanes 5 and 6 = binding to a consensus binding sequence for Oct-1 by nuclear extracts from control cells and riboflavin-deficient cells, respectively.
Previous studies suggested that cell stress causes nuclear translocation of the transcription factor NF-κB; translocation of NF-κB is associated with transcriptional activation of anti-apoptotic genes, mediating survival of stressed cells [27]. In the present study, DNA-binding activity of NF-κB in nuclear extracts was assessed using EMSA. DNA-binding activity was greater in extracts from riboflavin-deficient cells compared with riboflavin-sufficient controls (Fig. 6, lanes 1 and 2). No bands were detectable if samples were incubated in the presence of a 100-fold molar excess of unlabeled probe (lane 3) or in the absence of nuclear extract (lane 4); DNA-binding activity of Oct-1 (specificity control) was not affected by riboflavin (lanes 5 and 6). These observations suggest that nuclear activity of NF-κB increased specifically in response to riboflavin deficiency. Binding to NF-κB was 270 ± 23 arbitrary units in riboflavin-deficient cells and 165 ± 47 arbitrary units in riboflavin-sufficient controls, as judged by using gel densitometry (P < 0.05; n = 3). In previous studies we used Western blot analysis, reporter-gene constructs, and enzyme-linked immunosorbent assay to confirm the validity of EMSA with regard to NF-κB activity [20].
Fig. 6.
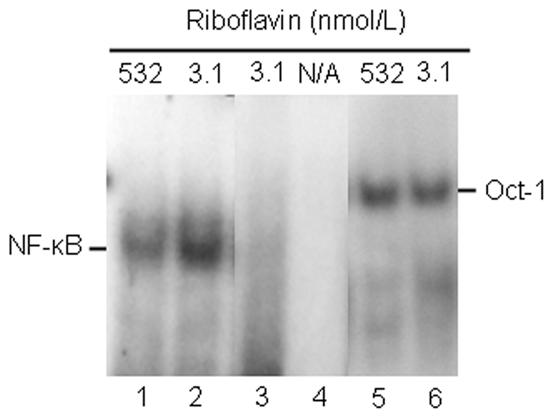
The DNA-binding activity of NF-κB increases in response to riboflavin deficiency in HepG2 cells. Cells were cultured in riboflavin-deficient (3.1 nmol/L) and riboflavin-sufficient (532 nmol/L) media for 4 d. Binding of NF-κB to response elements was visualized using EMSA; binding to an Oct-1 site was used as a control. N/A = not applicable. Lanes 1 and 2 = binding to a consensus sequence for NF-κB by nuclear extracts from control cells and riboflavin-deficient cells, respectively; lane 3 = 100-fold molar excess of unlabeled consensus binding site for NF-κB; lane 4 = consensus binding site for NF-κB in the absence of nuclear extract; lanes 5 and 6 = binding to a consensus binding sequence for Oct-1 by nuclear extracts from control cells and riboflavin-deficient cells, respectively.
The transcription factor GADD153 plays a role in cell growth arrest and apoptosis [28], decreasing the proliferation rate of stressed cells. Increased expression of the GADD153 gene in response to stress is mediated by endoplasmic reticulum stress elements [10,28]. In the present study, the abundance of GADD153 increased by about three times within four days of transfer of HepG2 cells into riboflavin-deficient medium, as judged by gel densitometric analysis of Western blots (arbitrary units): 16 ± 7 in riboflavin-sufficient controls (day 0) versus 52 ± 9 in riboflavin-deficient cells (day 4) (Fig. 7). The abundance of β-actin was not affected by riboflavin (control).
Fig. 7.
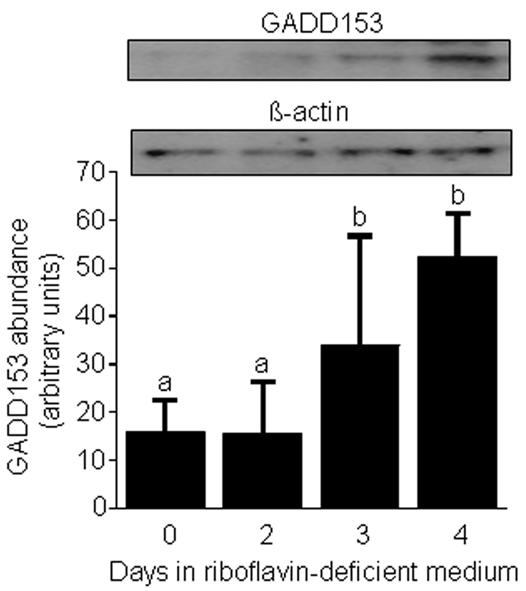
The abundance of GADD153 increases in response to riboflavin deficiency in HepG2 cells. Cells were transferred from riboflavin-sufficient medium (532 nmol/L, day 0) to riboflavin-deficient medium (3.1 nmol/L). At timed intervals, GADD153 and β-actin (control) were quantified by Western blot analysis and gel densitometry. Inserts depict representative Western blot images. a,bBars not sharing the same letter are significantly different (P < 0.05; n = 4). Means ± SD are shown.
3.4. Cell cycle arrest
Riboflavin deficiency arrested HepG2 cells in G0/G1 phases of the cell cycle. Analysis by flow cytometry suggested that 74% of cells arrested in G0/G1 phases of the cell cycle after four days in riboflavin-deficient medium, whereas only 61% of riboflavin-supplemented controls were in G0/G1 phase (Fig. 8). The relative abundance of cells in G2/M phases of the cycle decreased from 6.7% to “not detectable” in response to riboflavin deficiency. Consistent with these findings, proliferation rates (as judged by thymidine uptake) decreased by about 30% in response to riboflavin deficiency: 20,288 ± 3,841 cpm thymidine/(105 cells × 6 h) in riboflavin-deficient cells versus 28,378 ± 2,978 cpm thymidine/(105 cells × 6 h) in riboflavin-sufficient controls (P < 0.05; n = 5).
Fig. 8.
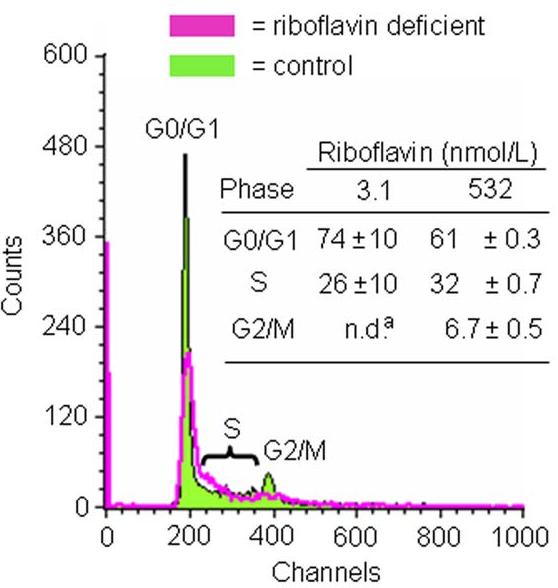
Riboflavin deficiency causes G0/G1 phase arrest in HepG2 cells. Cells were cultured in media containing 3.1 and 532 nmol/L riboflavin for 4 d. Cell cycle phase distribution was quantified by flow cytometry. Representative flow cytometry charts are depicted. The insert provides a table with percent cell cycle phase distributions. n.d. = not detectable. aSignificantly different from riboflavin-sufficient controls (P < 0.01; n = 3).
4. Discussion
The present study is consistent with the hypothesis that HepG2 cells have a great demand for riboflavin, and that cell stress develops rapidly if riboflavin supply is marginally low. Here we made the following observations. The activity of glutathione reductase decreased significantly three to four days after transfer of HepG cells into riboflavin-deficient medium. This was paralleled by increased riboflavin transporter activities and decreased expression of genes coding for flavokinase and FAD synthetase. The oxidative folding and synthesis of plasminogen and apolipoprotein B-100, respectively, was impaired within four days of culture in riboflavin-deficient medium; this is consistent with impaired processing of secretory proteins in riboflavin-deficient cells. Riboflavin depletion was associated with increased DNA-binding activities of transcription factors with affinity for endoplasmic reticulum stress elements and NF-κB consensus elements, suggesting cell stress. Moreover, the abundance of GADD153 was greater in riboflavin-deficient cells compared with controls. Riboflavin deficiency was associated with decreased rates of cell proliferation caused by arrest in G0/G1 phases of the cell cycle.
These observations are physiologically important, based on the following lines of reasoning. First, riboflavin-deficient HepG2 cells exhibit an increased incidence of oxidative damage to proteins and DNA compared with riboflavin-sufficient controls (K. C. Manthey and J. Zempleni, unpublished observation). Second, decreased secretion of proteins by riboflavin-deficient HepG2 cells might impair various processes in the extracellular space. For example, apolipoprotein B-100 and plasminogen play essential roles in lipid transport and blood clotting, respectively. Effects of riboflavin status on protein secretion in vivo remain to be determined. Third, G0/G1 phase arrest of riboflavin-deficient cells is likely to decrease the renewal of tissues that depend on rapid cell proliferation.
These findings have practical implications for nutritionists and health care professionals. Evidence has been provided that plasma concentrations of riboflavin may decrease in pregnancy [11] and in response to treatment with antimalarial drugs [1]. Moreover, riboflavin deficiency has been observed in preterm newborns treated with phototherapy [29] and in patients with cystic fibrosis [30]. Finally, hypothyroidism impairs the conversion of riboflavin to its coenzyme forms [31,32]. Afflicted individuals are likely to develop signs of riboflavin deficiency as described here. Consistent with this notion, the riboflavin-deficient medium used in the present study contained a concentration of riboflavin (3.1 nmol/L) similar to that observed in plasma from moderately deficient pregnant women [11]. However, we shall point out that liver cells are supplied with water-soluble nutrients through the portal vein in vivo, and that riboflavin concentrations are greater in portal blood compared with peripheral blood [13]. Hence, the applicability of the findings reported here to in-vivo situations is somewhat unclear. Notwithstanding this uncertainty, the current study provides proof of principle, i.e., moderate riboflavin deficiency causes cell stress after only four days in riboflavin-defined medium.
This study provides evidence that riboflavin deficiency causes cell cycle arrest, but the mechanism mediating G0/G1 arrest remains uncertain. We speculate that one or more of the following mechanisms participate in mediating cell cycle arrest of riboflavin-deficient cells. First, progression through G1 phase of the cell cycle depends on D-type cyclins [33]. Translation of cyclin D1 mRNA decreases in response to accumulation of unfolded proteins [28] as observed in riboflavin-deficient cells [4,5]. Second, GADD153 is an important mediator of cell cycle arrest [34]; the present study provided evidence that the abundance of GADD153 increases in response to riboflavin deficiency. Third, riboflavin deficiency increases the incidence of DNA damage in HepG2 cells (K. C. Manthey and J. Zempleni, unpublished observation). DNA damage triggers signaling cascades mediating cell cycle arrest; GADD153 may participate in these events [35].
HepG2 cells responded to riboflavin-deficient culture conditions by increasing transport rates of riboflavin. The identity of the riboflavin transporter in human cells is unknown. Hence, we could not quantify transporter abundance by using Western blot analysis in the present study. Apparently, metabolic trapping of riboflavin by conversion to FMN and FAD does not play an important role in the sequestration of flavins in HepG2 cells. This notion is based on our observation that the abundance of mRNA coding for flavokinase and FAD synthetase decreased in response to riboflavin deficiency. The up-regulation of transporter activity was not sufficient to overcome riboflavin-deficient culture conditions, as judged by low activities of glutathione reductase in cells cultured in medium containing 3.1 nmol/L riboflavin.
Collectively, the present study is consistent with the hypothesis that HepG2 cells have a great demand for riboflavin. Signs of riboflavin deficiency and cell stress were observed within four days of transfer into moderately riboflavin-deficient medium. Previous studies suggested that HepG2 cells develop signs of severe deficiency within eight days of culture in riboflavin-deficient medium [5]. In contrast, lymphoid cells are relatively resistant to developing signs of riboflavin deficiency even if cultured in severely deficient medium for five weeks [4]. In previous studies we speculated that cells secreting large quantities of protein might have a greater demand for riboflavin compared with cells secreting small quantities of protein [5]. We further speculate that the increased demand for riboflavin in response to protein secretion is due to the role of FAD as a coenzyme for Ero1 and sulfhydryl oxidases [2,3]. Future studies of riboflavin requirements and deficiency should take into account that some tissues have a greater demand for riboflavin than others.
Footnotes
This work was supported by NIH grants DK 60447 and DK 063945, and by NSF EPSCoR grant EPS-0346476. This paper is a contribution of the University of Nebraska Agricultural Research Division, Lincoln, NE 68583 (Journal Series No. 14935).
Abbreviations: EMSA, electrophoretic mobility shift assay; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; NF-κB, nuclear factor κB; RT-PCR, reverse transcriptase polymerase chain reaction
References
- 1.Rivlin RS. Riboflavin. In: Present Knowledge in Nutrition. In: Bowman BA, Russell RM, editors. ILSI Press; Washington, DC: 2001. [Google Scholar]
- 2.Tu BP, Ho-Schleyer SC, Travers KL, Weissman JS. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science. 2000;290:1571–1574. doi: 10.1126/science.290.5496.1571. [DOI] [PubMed] [Google Scholar]
- 3.Thorpe C, Hoober K, Raje S, Glynn N, Burnside J, Turi G, Coppock D. Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch. Bioch. Biophys. 2002;405:1–12. doi: 10.1016/s0003-9861(02)00337-5. [DOI] [PubMed] [Google Scholar]
- 4.Camporeale G, Zempleni J. Oxidative folding of interleukin-2 is impaired in flavin-deficient Jurkat cells, causing intracellular accumulation of interleukin-2 and increased expression of stress response genes. J. Nutr. 2003;133:668–672. doi: 10.1093/jn/133.3.668. [DOI] [PubMed] [Google Scholar]
- 5.Manthey KC, Chew YC, Zempleni J. Riboflavin deficiency impairs oxidative folding and secretion of apolipoprotein B-100 in HepG2 cells, triggering stress-response systems. J. Nutr. 2005 doi: 10.1093/jn/135.5.978. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higuchi Y. Chromosomal DNA fragmentation in apoptosis and necrosis induced by oxidative stress. Biochem. Pharmacol. 2003;66:1527–1535. doi: 10.1016/s0006-2952(03)00508-2. [DOI] [PubMed] [Google Scholar]
- 7.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J. Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 8.Giustarini D, Rossi R, Milzani A, Colombo R, Dalle-Donne I. S-glutathionylation: from redox regulation of protein functions to human diseases. J. Cell. Mol. Med. 2004;8:201–212. doi: 10.1111/j.1582-4934.2004.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sidrauski C, Brickner JH, Walter P. The unfolded protein response. In: Dalbey RE, von Heijne G, editors. Protein Targeting, Transport & Translocation. Academic Press; San Diego, CA; 2002. [Google Scholar]
- 10.Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol. Rev. 1999;79:683–700. doi: 10.1152/physrev.1999.79.3.683. [DOI] [PubMed] [Google Scholar]
- 11.Zempleni J, Link G, Bitsch I. Intrauterine vitamin B2 uptake of preterm and full-term infants. Pediatr. Res. 1995;38:585–591. doi: 10.1203/00006450-199510000-00019. [DOI] [PubMed] [Google Scholar]
- 12.Zempleni J, Galloway JR, McCormick DB. Pharmacokinetics and utilization of orally and intravenously administered riboflavin in healthy humans. Am. J. Clin. Nutr. 1996;63:54–66. doi: 10.1093/ajcn/63.1.54. [DOI] [PubMed] [Google Scholar]
- 13.Feder S, Daniel H, Rehner G. In vivo kinetics of intestinal absorption of riboflavin in rats. J. Nutr. 1991;121:72–79. doi: 10.1093/jn/121.1.72. [DOI] [PubMed] [Google Scholar]
- 14.Sauberlich HE, Judd JH, Nichoalds GE, Broquist HP, Darby WJ. Application of the erythrocyte glutathione reductase assay in evaluating riboflavin nutritional status in a high school student population. Am. J. Clin. Nutr. 1972;25:756–762. doi: 10.1093/ajcn/25.8.756. [DOI] [PubMed] [Google Scholar]
- 15.Zempleni J, Mock DM. Uptake and metabolism of biotin by human peripheral blood mononuclear cells. Am. J. Physiol. Cell Physiol. 1998;275:C382–C388. doi: 10.1152/ajpcell.1998.275.2.C382. [DOI] [PubMed] [Google Scholar]
- 16.Zempleni J, Stanley JS, Mock DM. Proliferation of peripheral blood mononuclear cells causes increased expression of the sodium-dependent multivitamin transporter gene and increased uptake of pantothenic acid. J. Nutr. Biochem. 2001;12:465–473. doi: 10.1016/s0955-2863(01)00162-0. [DOI] [PubMed] [Google Scholar]
- 17.Wiedmann S, Eudy JD, Zempleni J. Biotin supplementation causes increased expression of genes encoding interferon-γ, interleukin-1β, and 3-methylcrotonyl-CoA carboxylase, and causes decreased expression of the gene encoding interleukin-4 in human peripheral blood mononuclear cells. J. Nutr. 2003;133:716–719. doi: 10.1093/jn/133.3.716. [DOI] [PubMed] [Google Scholar]
- 18.Griffin JB, Stanley JS, Zempleni J. Synthesis of a rabbit polyclonal antibody to the human sodium-dependent multivitamin transporter. Int. J. Vitam. Nutr. Res. 2002;72:195–198. doi: 10.1024/0300-9831.72.4.195. [DOI] [PubMed] [Google Scholar]
- 19.Griffin JB, Rodriguez-Melendez R, Zempleni J. The nuclear abundance of transcription factors Sp1 and Sp3 depends on biotin in Jurkat cells. J. Nutr. 2003;133:3409–3415. doi: 10.1093/jn/133.11.3409. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez-Melendez R, Schwab LD, Zempleni J. Jurkat cells respond to biotin deficiency with increased nuclear translocation of NF-κB, mediating cell survival. Int. J Vitam. Nutr. Res. 2004;74:209–216. doi: 10.1024/0300-9831.74.3.209. [DOI] [PubMed] [Google Scholar]
- 21.Roy B, Lee AS. The mammalian endoplasmic reticulum stress response element consists of an evolutionary conserved tripartite structure and interacts with a novel stress-inducible complex. Nucl. Acids Res. 1999;27:1437–1443. doi: 10.1093/nar/27.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zempleni J, Mock DM. Mitogen-induced proliferation increases biotin uptake into human peripheral blood mononuclear cells. Am. J. Physiol. Cell Physiol. 1999;276:C1079–1084. doi: 10.1152/ajpcell.1999.276.5.C1079. [DOI] [PubMed] [Google Scholar]
- 23.Vindelov LL. Flow microfluorometric analysis of DNA in cells from solid tumors and cell suspensions: A new method for rapid isolation and staining of nuclei. Virchows Arch. B Cell Path. 1977;24:227–242. [PubMed] [Google Scholar]
- 24.SAS Institute . StatView Reference SAS Publishing; Cary, NC: 1999. [Google Scholar]
- 25.Yoshida H, Okada T, Haze K, Yanaki H, Yura T, Negishi M, Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Stark GR. NFκB-dependent signaling pathways. Exp. Hematol. 2002;30:285–296. doi: 10.1016/s0301-472x(02)00777-4. [DOI] [PubMed] [Google Scholar]
- 28.Brewer JW, Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA. 2000;97:12625–12630. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lucas A, Bates C. Transient riboflavin depletion in preterm infants. Arch. Dis. Child. 1984;59:837–841. doi: 10.1136/adc.59.9.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCabe H. Riboflavin deficiency in cystic fibrosis: three case reports. J. Hum. Nutr. Dietet. 2001;14:365–370. doi: 10.1046/j.1365-277x.2001.00306.x. [DOI] [PubMed] [Google Scholar]
- 31.Rivlin RS. Hormonal regulation of riboflavin metabolism. In: Rivlin RS, editor. Riboflavin. Plenum Press; New York, NY: 1975. [Google Scholar]
- 32.Lee S-S, McCormick DB. Thyroid hormone regulation of flavocoenzyme biosynthesis. Arch. Biochem. Biophys. 1985;237:197–201. doi: 10.1016/0003-9861(85)90269-3. [DOI] [PubMed] [Google Scholar]
- 33.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 34.Igase M, Okura T, Nakamura M, Takata Y, Kitami Y, Hiwada K. Role of GADD153 (growth arrest- and DNA damage-inducible gene 153) in vascular smooth muscle cell apoptosis. Clin. Sci. 2001;100:175–281. [PubMed] [Google Scholar]
- 35.Murray A, Hunt T. The Cell Cycle Oxford University Press; New York, NY: 1993. [Google Scholar]