


Abstract
Owing to the markedly increased reactivity of amino functional groups versus hydroxyls, the 5′-amino-5′-deoxy nucleoside and nucleotide analogs have proven widely useful in biological, pharmaceutical and genomic applications. However, synthetic procedures leading to these analogs have not been fully explored, which may possibly have limited the scope of their utility. Here we describe the synthesis of the 5′-amino-2′,5′-dideoxy analogs of adenosine, cytidine, guanosine, inosine and uridine from their respective naturally occurring nucleosides via the reduction of 5′-azido-2′,5′-dideoxy intermediates using the Staudinger reaction, and the high yield conversion of these modified nucleosides and 5′-amino-5′-deoxythymidine to the corresponding 5′-N-triphosphates through reaction with trisodium trimetaphosphate in the presence of tris(hydroxymethyl)aminomethane (Tris). We also show that each of these nucleotide analogs can be efficiently incorporated into DNA by the Klenow fragment of Escherichia coli DNA polymerase I when individually substituted for its naturally occurring counterpart. Mild acid treatment of the resulting DNA generates polynucleotide fragments that arise from specific cleavage at each modified nucleotide, providing a sequence ladder for each base. Because the ladders are generated after the extension, the corresponding products may be manipulated by enzymatic and/or purification processes. The potential utility of this extension–cleavage procedure in genomic sequence analysis is discussed.
INTRODUCTION
Chemically modified nucleotides have been extensively used in the study of many complicated biological systems. In particular, they have proven indispensable in the analysis of protein–nucleic acid interactions, the sequencing of nucleic acids and, more recently, the determination of genotypes. In general, these applications rely on differences in the chemical reactivity or steroelectronic properties of the modified nucleosides as compared to the naturally occurring counterpart. Owing to its markedly increased reactivity, 5′-amino-5′-deoxythymidine (NH2-dT) has been employed in a variety of studies, including model studies on DNA replication (1), template-directed chemical amplification (2), mechanistic studies on polymerases and reverse transcriptases (3) and the construction of combinatorial peptide–DNA hybrids (4) and their libraries (5,6) for antisense applications. Although 5′-amino-5′-deoxythymidine-5′-N-triphosphate (NH2-dTTP) was initially described in the 1970s (7,8), interest in 5′-amino-2′,5′-dideoxynucleotides (NH2-dNTPs) has revived recently due to their potential utility in genomic analysis. Shchepinov et al. (9) have reported a method for the high throughput detection of single nucleotide polymorphisms (SNPs) by MALDI-TOF mass spectrometry that involves the matrix-assisted fragmentation of DNA containing NH2-dT or NH2-dC. Recently, we have developed a novel dinucleotide cleavage method involving polymerase incorporation of a ribonucleotide and a 5′-amino-2′,5′-dideoxynucleotide that may be utilized for SNP discovery by MALDI-TOF mass spectrometry (Wolfe,J.L., Wang,B.H., Kawate,T., DeMaria,C. and Stanton,V.P., submitted for publication). In order to take full advantage of these novel technologies and to provide additional tools for model studies that utilize 5′-amino-nucleosides (1,3–6), it is necessary to develop a simple and reliable method for generating 5′-amino-2′,5′-dideoxynucleosides (NH2-dNs) and corresponding 5′-N-triphosphate nucleotides (NH2-dNTPs).
The present report describes the synthesis and characterization of 5′-amino-2′,5′-dideoxy-5′-N-triphosphate nucleotide analogs for adenosine, cytidine, guanosine, thymidine, uridine and inosine. Each of these analogs is readily incorporated into DNA by the Klenow fragment of DNA polymerase I and can partially or completely replace its naturally occurring counterpart. The phosphoramidate bonds formed within the resulting polynucleotides can be specifically cleaved with dilute acetic acid, thereby generating a high quality sequencing ladder for the corresponding base. Furthermore, we demonstrate that when NH2-dTTP is partially incorporated into a 7.2 kb polynucleotide the resulting DNA duplex is a good substrate for restriction endonucleases such as MscI. We propose that these unique properties may render the NH2-dNTPs exceptionally well suited for genomic sequence analysis.
MATERIALS AND METHODS
Reagents and solvents were purchased from Sigma-Aldrich (St Louis, MO) or J. T. Baker (Phillipsburg, NJ). The 1H and 31P NMR spectra were recorded on a Bruker DPX-400 NMR spectrometer using either tetramethylsilane (TMS) as an internal standard (1H spectra) or 80% H3PO4 as an external standard (31P spectra) (Dr Jin Hong, 77 Pine Ridge Drive, Ayer, MA). Chemical shifts (δ) are reported in p.p.m. downfield from TMS or H3PO4. Column chromatography was performed with silica gel 60 (230–400 mesh, Merck 9385) using standard flash methods. Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel 60 F254 pre-coated plates. Oligonucleotides were purchased from Sigma-Genosys (Woodlands, TX). Nucleotide dNTPs were from Amersham Biosciences (Piscataway, NJ).
Synthesis of 5′-azido-2′,5′-dideoxyadenosine (7)
To a solution of 599 mg (1.6 mmol) of 5′-azido-N6-benzoyl-2′,5′-dideoxyadenosine (10) in 4 ml of methanol was added 6 ml of concentrated NH4OH. The resulting mixture was stirred at 58–60°C for 15 h and then concentrated to dryness in vacuo. The residue was dissolved in H2O (60 ml) and extracted with CHCl3 (5 × 5 ml). The aqueous layer was collected and the organic solutions were combined and back-extracted with H2O (2 × 5 ml). Aqueous solutions from both extractions were combined and lyophilized. The residue was fractionated on silica gel (CH2Cl2:MeOH, 49:1–9:1) to afford 350 mg 7 (80%) as a light yellow foam. 1H NMR (DMSO-d6): δ 2.30 (1H, ddd, J = 3.7, 6.4, 13.3 Hz, 2′a), 2.92 (1H, td, J = 6.7, 13.4 Hz, 2′b), 3.48 (1H, ddd, J = 3.9, 12.9 Hz, 5′a), 3.65 (1H, ddd, J = 7.3, 12.9 Hz, 5′b), 3.96 (1H, m, 4′), 4.41 (1H, m, 3′), 5.48 (1H, d, J = 4.0 Hz, 3′OH), 6.37 (1H, t, J = 6.9 Hz, 1′), 7.29 (2H, s, NH2), 8.14 (1H, s, 2), 8.33 (1H, s, 8).
Synthesis of 5′-azido-2′,5′-dideoxycytidine (8)
Compound 5′-azido-N4-benzoyl-2′,5′-dideoxycytidine (11) (454 mg, 1.27 mmol) was dissolved in a mixture of 5 ml of pyridine and 5 ml of concentrated NH4OH and the resulting solution was stirred at 60°C for 8.5 h. The mixture was cooled on ice and subjected to rotary evaporation. The residue was partitioned between H2O (30 ml) and EtOAc (3 ml) and the aqueous layer was washed with Et2O (3 × 3 ml) and EtOAc:Et2O (1:2, 2 × 5 ml). Lyophilization of the aqueous layer gave 316 mg of 8 (98%). 1H NMR (DMSO-d6): δ 2.07 (2H, m, 2′), 3.51 (1H, dd, J = 4.4, 13.1 Hz, 5′a), 3.56 (1H, dd, J = 6.2, 13.1 Hz, 5′b), 3.83 (1H, m, 4′), 4.12 (1H, m, 3′), 5.38 (1H, d, J = 4.3 Hz, OH), 5.73 (1H, d, J = 7.4 Hz, 5), 6.21 (1H, t, J = 6.8 Hz, 1′), 7.16 (1H, brs, NH), 7.20 (1H, brs, NH), 7.60 (1H, d, J = 7.4 Hz, 6).
Synthesis of 5′-azido-2′,5′-dideoxyguanosine (9)
To a solution of 409 mg (1.13 mmol) 5′-azido-N2-isobutyryl-2′,5′-dideoxyguanosine (10) in 4 ml of methanol was added 6 ml of concentrated NH4OH. The mixture was stirred at 58–60°C for 13 h and then concentrated to dryness in vacuo. The residue was dissolved in H2O and extracted with EtOAc. The aqueous layer was collected and lyophilized to afford 340 mg of 9 (100%) as a white powder. 1H NMR (DMSO-d6): δ 2.22 (1H, ddd, J = 3.2, 6.1, 13.2 Hz, 2′a), 2.70 (1H, td, J = 6.3, 13.5 Hz, 2′b, 3.46 (1H, dd, J = 4.2, 13.0 Hz, 5′a), 3.61 (1H, dd, J = 7.2, 13.0 Hz, 5′b), 3.91 (1H, m, 4′), 4.29 (1H, m, 3′), 5.44 (1H, br, OH), 6.14 (1H, t, J = 6.9 Hz, 1′), 6.53 (2H, brs, NH2), 7.90 (1H, s, 8), 9.48 (1H, brs, NH).
Synthesis of 5′-amino-2′,5′-dideoxyadenosine (12)
A solution of 34 mg (0.12 mmol) of 7 and 100 mg triphenylphosphine (Ph3P; 0.38 mmol) in 1 ml of pyridine was stirred at room temperature for 7 h. Concentrated aqueous NH4OH (0.2 ml) was added and the mixture stirred at room temperature overnight. After additional stirring at 55°C for 1 h, 5 ml of H2O was added and the resulting precipitate removed by filtration and washed with H2O. After extraction by EtOAc (5 × 3 ml), the combined filtrate was lyophilized to give 63 mg of crude 12, which was used without further purification. 1H NMR (DMSO-d6): δ 2.23 (1H, ddd, J = 3.3, 6.1, 13.1 Hz, 2′a), 2.80 (3H, m, 2′b and 5′), 3.82 (1H, m, 4′), 4.42 (1H, m, 3′), 5.70 (2H, br, NH2), 6.32 (1H, t, J = 6.6 Hz, 1′), 7.29 (2H, brs, NH2), 8.14 (1H, s, 2), 8.35 (1H, s, 8).
Synthesis of 5′-amino-2′,5′-dideoxycytidine (13)
A solution of 80 mg of 8 (0.31 mmol) and 245 mg Ph3P (0.37 mmol) in pyridine (2 ml) was stirred at room temperature for 7 h. Concentrated NH4OH (0.2 ml) was added and the mixture was stirred at room temperature for an additional 15 h. Water (5 ml) was added and the resulting precipitate was removed by filtration. The filtrate was extracted with EtOAc and the resulting aqueous layer was lyophilized to afford 104 mg of crude 13, which was used without further purification. 1H NMR(DMSO-d6): δ 1.95 (1H, td, J = 6.6, 13.4 Hz, 2′a), 2.09 (1H, ddd, J = 3.9, 5.7, 13.2 Hz, 2′b), 2.75 (2H, brs, 5′), 3.69 (1H, m, 4′), 4.15 (1H, m, 3′), 5.14 (3H, br, NH2 and OH), 5.73 (1H, d, J = 7.3 Hz, 5), 6.14 (1H, t, J = 6.7 Hz, 1′), 7.31 (2H, br, NH2), 7.72 (1H, d, J = 7.3 Hz, 6).
Synthesis of 5′-amino-2′,5′-dideoxyguanosine (14)
A mixture of 40 mg (0.14 mmol) of 9 and 111 mg (0.43 mmol) of Ph3P was added to 1 ml of pyridine and stirred at room temperature for 5 h. Concentrated aqueous NH4OH (0.3 ml) was added and the mixture was stirred for 17 h at room temperature followed by 1 h at 55°C. Water (6 ml) was added to the mixture and the resulting precipitate was removed by filtration and washed with H2O. The combined filtrate was extracted with EtOAc. The aqueous solution was lyophilized to afford 50 mg of crude 14, which was used without further purification. 1H NMR (DMSO-d6): δ 2.19 (1H, m, 2′a), 2.57 (1H, td, J = 6.6, 13.4 Hz, 2′b), 2.82 (2H, brs, 5′), 3.79 (1H, brs, 4′), 4.37 (1H, brs, 3′), 5.76 (3H, brs, NH2 and OH), 6.11 (1H, td, J = 6.9 Hz, 1′), 6.90 (2H, brs, NH2 and OH), 7.88 (1H, s, 8).
Synthesis of 5′-amino-2′,5′-dideoxyuridine (15)
A solution of 5′-azido-2′,5′-dideoxyuridine (10) (11) (50 mg, 0.20 mmol) and Ph3P (160 mg, 0.61 mmol) in pyridine (1 ml) was stirred at room temperature for 6 h. Concentrated NH4OH (0.2 ml) was added and the mixture was stirred at room temperature for 19.5 h followed by 55°C for 2 h. Water (5 ml) was added and the resulting precipitate was removed by filtration. The filtrate was washed with EtOAc and lyophilized to give 59 mg of crude 15, which was used without further purification. 1H NMR (DMSO-d6): δ 2.10 (2H, m, 2′), 2.72 (2H, m, 5′), 3.70 (1H, dd, J = 5.1, 8.5 Hz, 4′), 4.19 (1H, m, 3′), 5.62 (1H, d, J = 8.1 Hz, 5), 6.00 (3H, br, NH2 and OH), 6.13 (1H, t, J = 6.9 Hz, 1′), 7.82 (1H, d, J = 8.1 Hz, 6).
Synthesis of 5′-amino-2′,5′-dideoxyinosine (16)
Compound 16 was prepared from 5′-azido-2′,5′-dideoxyinosine (11) by contract synthesis using hydrogenation catalyzed by Pd (GLSynthesis Inc., Shrewsbury, MA). Compound 11 was made using procedures similar to that described by Yamamoto et al. (11).
Synthesis of 5′-amino-2′,5′-dideoxynucleoside-5′-N-triphosphates 1–6
A mixture of 250 µmol trisodium trimetaphosphate (TMP) and 50 µmol 12, 13, 14, 15, 16 (described above, the amounts of 12–15 were based on assuming 100% conversion from 5′-azido-nucleosides) or 17 (obtained from Sigma-Aldrich) was dissolved in 0.5 ml of 0.5 M aqueous Tris (measured pH ∼ 11). The resulting solution was allowed to incubate at room temperature for 5–7 days. The progress of the reaction was monitored by reversed phase HPLC. The resulting solutions were used as 100 mM 1, 2, 3, 4, 5 or 6 for enzymatic reactions without purification.
Reversed phase HPLC analysis of nucleotides 1–6
The reaction mixtures were analyzed on an HPLC instrument (Waters, Marlboro, MA) that consisted of a 515 HPLC pump, a 2700 sample manager, a 996 photodiode array detector and a temperature control module. A Waters Nova-pak® C18 (3.9 × 150 mm, 4 µm) column was used with mobile phase buffer A (0.1 M TEAA, 10 mM Tris pH 9) and buffer B (25% MeOH, 75% buffer A) at a flow rate of 1.00 ml/min. The buffer gradient was varied from 98% A:2% B to 100% B to achieve good separation of each nucleoside from its triphosphate: 0–8 min, 98% A to 70% A; 8–9 min, 70% A to 0% A; 9–10 min, 0% A; 10–11 min, 0% A to 98% A; 11–15 min, 98% A. The extent of conversion from each starting nucleoside to the corresponding product was determined to be 85.6% for 1, 78.9% for 2, 83.2% for 3, 80.0% for 4, 89.1% for 5 and 91.5% for 6 (Fig. 1). The photodiode array detector also provided UV absorption spectra of each compound from which λmax for each compound was obtained: λmax(1) = 259 nm; λmax(2) = 271 nm; λmax(3) = 252 nm; λmax(4) = 263 nm; λmax(5) = 249 nm; λmax(6) = 267 nm.
Figure 1.

HPLC chromatograms of reaction mixtures containing NH2-dATP, NH2-dCTP, NH2-dGTP, NH2-dUTP, NH2-dITP and NH2-dTTP. The conversion yield was determined by comparing the areas of the late eluting peak (nucleotide 5′-N-triphosphate) versus the early eluting peak (nucleoside) in each chromatogram.
31P NMR analysis of 1–3, 5 and 6
The above solutions containing nucleotide triphosphates 1–3, 5 and 6 and an aqueous solution of a mixture of Tris and TMP were diluted with D2O and analyzed by 31P NMR. The most prominent peak was at –21 p.p.m. and was observed for all samples, corresponding to TMP. A doublet around –1 p.p.m. corresponding to a phosphoramidate (Pα-N) resonance was observed for samples containing 1, 2, 3, 5 and 6, but not for the mixture of Tris and TMP. Due to the presence of a large excess of TMP and its breakdown products, the resonances corresponding to Pβ and Pγ were not assigned. NH2-dATP (1), –0.99 p.p.m. (d, J = 20.8 Hz); NH2-dCTP (2), –0.94 p.p.m. (d, J = 19.5 Hz); NH2-dGTP (3), –1.00 p.p.m. (d, J = 20.8 Hz); NH2-dITP (5), –1.03 p.p.m. (d, J = 20.8 Hz); NH2-dTTP (6), –0.97 p.p.m. (d, J = 20.1 Hz).
Preparation of single-stranded DNA template
A biotinylated forward primer 5′-biotin-d(TCGGAGAAACTGGACAGCAC) was paired with reverse primer 5′-d(TTGAGATCCAGCCTCACGAGG) or 5′-d(AACAATGGGAAATTTAGTCTG) to produce biotin-tagged 87 and 480 bp amplification fragments by PCR, using a plasmid containing the human transferrin receptor gene as template (12,13). Each PCR product was immobilized to streptavidin–agarose beads in 5 mM Tris pH 7.5, 1 M NaCl, 0.5 mM EDTA, 0.05% Tween-20 overnight. After removal of the supernatant, the beads were washed with H2O, then treated twice with 0.3 ml of 0.2 M NaOH for 15 min. After washing with 150 µl of H2O, all three supernatants were combined and cooled on ice before being neutralized to pH 8 with 1 M HCl (∼120 µl). The solution was concentrated on a SpeedVac (Savant, Farmingdale, NY) to ∼50 µl and purified using a Quick-Spin G25 (TE) column (Roche Biosciences, Oakland, CA). The collected solutions of non-biotinylated, single-stranded DNA templates (87 or 480 nt) were quantified and stored at –20°C. The amount of the 480 nt fragment was estimated by UV analysis using the formula [OD260/(480 × 10 000)] × 106 = concentration (µM) (14). The concentration of the 87 nt solution was estimated by capillary electrophoresis analysis by comparing peak sizes with an oligonucleotide of known concentration.
Primer extension with complete analog substitution
The primer 5′-d(TCGGAGAAACTGGACAGCAC) was labeled at the 5′ end using T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and [γ-32P]ATP (Perkin-Elmer, Boston, MA) and was purified using a Sephadex G50 column. The radiolabeled primer (∼1.6 pmol) was annealed to the single-stranded 87 nt DNA template (∼2.4 pmol) in 20 mM MgCl2 and 50 mM NaOAc. A typical extension reaction (10 µl) contained ∼0.01 µM primer–template duplex, 45 mM Tris pH 9.5, 10 mM DTT, 20 mM MgCl2, 4 mM NH2-dNTP, 0.1 mM each of the other three dNTPs and 5 U Klenow (exo–) polymerase (New England Biolabs). After incubation at 37°C for 1 h, 30 µl of TE (10 mM Tris, 1 mM EDTA, pH 8) was added and the mixture was purified using a Sephadex G50 column.
Primer extension with partial (statistical) analog substitution
The radiolabeled primer 5′-[32P]dp(TCGGAGAAACTGGACAGCAC) (∼4 pmol) was annealed to the single-stranded 480 nt DNA template (4 pmol) in 20 mM MgCl2 and 50 mM NaOAc. A typical extension reaction (10 µl) contained ∼0.02 µM primer–template duplex, 50 mM Tris pH 9.5, 5 mM DTT, 22 mM MgCl2, 5 mM NaOAc, 4 mM NH2-dNTP and 0.016–0.046 mM of the corresponding dNTP (0.04 mM dATP, 0.026 mM dCTP, 0.016 mM dGTP, 0.046 mM TTP, 0.026 mM dITP or 0.02 mM dUTP), 0.4 mM each of the other three dNTPs and 5 U Klenow (exo–). After incubation at 37°C for 1 h, each extension reaction mixture was supplemented with additional d(ACGT)TPs (1 µl of 1 mM) and incubated for another 15 min. The solutions were stored at –20°C after dilution with TE pH 8.
Acid cleavage of extension products
To each of the Sephadex purified 87 nt extension products (8 µl) was added 2 µl of 1% acetic acid (HOAc), followed by incubation at 40°C for 30 min. Deionized water (100 µl) was added to each sample and the diluted solutions were dried in vacuo at room temperature. To each of the unpurified, TE diluted 480 nt extension mixtures (2 µl) was added 1 µl of 10 mM EDTA, 1 µl of 10% acetic acid and 6 µl of dH2O. The resulting mixtures were incubated at 37°C for 10 min then diluted with 100 µl of dH2O. The resulting mixtures were dried in a SpeedVac evaporator.
Restriction digestion of M13mp18 DNA duplex containing P-N linkers
A 32P-labeled M13 universal primer (5′-GTAAAACGACGGCCAGT, 5 pmol) was annealed to 1 µg single-stranded M13mp18 plasmid (New England Biolabs) in TE, and ∼13% of the resulting duplex (2 of 15 µl) was used for each primer extension reaction. The extension reaction was carried out at 37°C for 1 h in 50 mM Tris pH 9, 5 mM DTT, 10 mM MgCl2, 0.4 mM d(ACG)TPs, 4 mM NH2-dTTP and 0.08 mM dTTP with 5 U Klenow (exo–). The product was purified using a G50 column and the eluate was concentrated to a final volume of 20 µl. For the cleavage reaction, 4 µl of the duplex was treated with 4% HOAc at 37°C for 10 min and analyzed by 5% PAGE. For restriction digestion, 2 µl of the duplex was digested with 3 U of MscI (New England Biolabs) in 50 mM KOAc, 20 mM Tris–OAc pH 7.9, 10 mM Mg(OAc)2 and 1 mM DTT at 37°C for 1 h. The product was ethanol precipitated, redissolved in formamide loading buffer and analyzed on a 0.6% TBE–agarose gel. The gel was dried and subjected to autoradiography.
RESULTS AND DISCUSSION
Syntheses of 5′-amino-2′,5′-dideoxynucleosides
Although there are several reports on the preparation of NH2-dU (15) (15) and NH2-dT (17) (6,15–17), the isolation and characterization of other 5′-amino analogs (12–14 and 16) have not been reported. The Staudinger reaction, which converts azides into primary amines through Ph3P treatment followed by hydrolysis of the iminophosphinimine intermediates, had previously been used to prepare NH2-dT (17) from N3-dT (17). Using a similar strategy (Scheme 1), we synthesized 5′-amino-2′,5′-dideoxynucleosides 12–16 (NH2-dA, NH2-dC, NH2-dG, NH2-dU and NH2-dI) from the corresponding azides 7–11 (N3-dA, N3-dC, N3-dG, N3-dU and N3-dI). This conversion was virtually quantitative for each nucleoside analog that we examined. Compounds N3-dU (10) and N3-dI (11) were synthesized according to previously described procedures (10,11), whereas N3-dA (7), N3-dC (8) and N3-dG (9) were prepared from derivatives containing protected exocyclic amino groups (10) followed by an intermediate step of NH4OH aminolysis. We first attempted to carry out the Staudinger reaction directly on the protected form of 5′-azido-N2-isobutyryl-2′,5′-dideoxyguanosine, hoping that the NH4OH treatment would also remove the isobutyryl group to yield NH2-dG (14). However, the apparently redundant conversion to N3-dG (9) prior to the Staudinger reaction turned out to be essential. Direct treatment of 5′-azido-N2-isobutyryl-2′,5′-dideoxyguanosine with Ph3P in pyridine followed by hydrolysis/deprotection with NH4OH yielded a mixture of products that could not be fully converted to 9, even after prolonged heating in concentrated ammonia. We suspect that the major side-product comes from transamidation of the isobutyryl group; the resulting 2′,5′-dideoxyguanosine-5′-isobutyrylamide is much less reactive to NH4OH than the starting material.


Scheme 1. (a) (i) Ph3P, pyridine; (ii) NH4OH. (b) TMP (18), Tris, H2O.
Preparation of 5′-amino-2′,5′-dideoxynucleoside 5′-N-triphosphates
In early reports, the synthesis of NH2-dTTP was accomplished by reacting NH2-dT (17) with TMP (18), with moderate conversion yield (8). Attempted purification by HPLC fractionation failed to yield pure NH2-dTTP because the isolated NH2-dTTP appeared to slowly degrade until the ratio NH2-dTTP:NH2-dT reached 40:60. In addition, NH2-dTTP was reported to be labile to both acidic and alkaline conditions, yielding either NH2-dT or its 5′-N-diphosphate (NH2-dTDP), respectively, although the identity of the latter was supported only by circumstantial evidence (8). To our knowledge, there have been no prior reports on the synthesis and characterization of any other NH2-dNTPs.
We set out to apply and, we hoped, improve upon this elegant approach in the synthesis of other 5′-N-triphosphates from corresponding 5′-amino-nucleosides. In our attempts to improve the TMP reaction, we reasoned that a more careful regulation of the pH of the reaction mixture might result in higher yields of the desired 5′-N-triphosphates. To accomplish this we included tris(hydroxymethyl)aminomethane (Tris) in the reaction mixture. Tris is a commonly used buffering agent with a pKa of 8.1 and, because it is a sterically hindered primary amine, it should not critically interfere with the desired reaction. An added benefit is that Tris is frequently used as a buffer for polymerization reactions, which should allow the direct use of the TMP reaction mixtures without purification. Six nucleotide analogs (1–6) were successfully prepared from the corresponding nucleosides (12–17) using this strategy (Scheme 1). Typically, five molar equivalents of TMP (18) and 0.5 M Tris (pH ∼ 11) were used in each reaction, which was monitored by HPLC until the ratio of NH2-dNTP to NH2-dN reached a plateau. The HPLC chromatograms (Fig. 1) showed that each NH2-dNTP is well separated from its nucleoside and 80–90% conversion was achieved in every case. When stored at –20°C these products are stable for at least several months. Attempts to purify the 5′-N-triphosphates by HPLC consistently yielded mixtures of NH2-dN and NH2-dNTP, even when alkaline HPLC buffers were used. This observation is in agreement with previous reports on the stability of NH2-dTTP (8) and may imply greater proportions of NH2-dNTP in each reaction mixture, since some degradation might have occurred during the HPLC analysis. Additional characterization of each product by 31P NMR spectroscopy showed the appearance of a single phosphoramidate resonance, which is indicative of the formation of a P-N bond. The absence of additional P-N signals in the spectra confirms the purity of each sample as seen by HPLC analysis.
Complete substitution and site-specific cleavage
Based upon the previously reported ability of DNA polymerase I to incorporate NH2-dTTP, we chose to examine the ability of the exonuclease-deficient Klenow fragment of DNA polymerase I to incorporate our newly generated 5′-N-triphosphates. Each of the analogs NH2-dATP (1), NH2-dCTP (2), NH2-dGTP (3) and NH2-dTTP (6) was readily incorporated in polymerization reactions in place of dATP, dCTP, dGTP and dTTP, respectively (Fig. 2A, lanes 1, 3, 5 and 7). The best extension was achieved when the pH of the reaction mixture was kept at >9, presumably because both the NH2-dNTPs and the DNA containing P-N linkers are more stable under basic conditions (8). The acid lability of the phosphoramidate linkages was demonstrated by PAGE of acetic acid-treated extension products containing NH2-dA, NH2-dC, NH2-dG or NH2-dT (Fig. 2A, lanes 2, 4, 6 and 8), in agreement with previous reports on NH2-dT-containing oligonucleotides (7,17). The site-specific cleavage occurred at every modified base, yielding distinct oligonucleotide bands that represent sequence ladders for each base. We should note that following the completion of this work, a newly reported study demonstrated the ability of Sequenase v.2.0 to incorporate NH2-dTTP and NH2-dCTP (nucleotide synthesis was not described) (9). In our hands, Sequenase v.2 was not as efficient at incorporating these analogs as Klenow (exo–).
Figure 2.

Denaturing PAGE (12%) analysis of polymerization products with dATP, dCTP, dGTP or dTTP completely substituted by the corresponding NH2-dNTPs. The sequence of the full-length products are listed with modified bases shown in coded colors: A, red; C, green; G, yellow; T, blue. (A) Primer extension reaction mixtures (E) and corresponding cleavage products (C) elicited by treatment with 0.2% acetic acid at 40°C for 30 min. Specific cleavage bands at each NH2-dN are highlighted with dots in coded colors. (B) Cleavage products generated by heating at 40°C with 5% acetic acid for 2 h. Non-specific cleavage bands are highlighted with gray boxes.
Surprisingly, the first cleavage band observed above the primer is {primer+AGp}, not the expected {primer+Ap} (Fig. 2A, lanes 6 and 2). A closer inspection of lane 2 revealed an extra band just below the 20 nt primer, which is likely to be the ‘missing’ {primer+Ap} fragment. This fragment runs aberrantly ∼0.5 nt below the primer, presumably because the additional 3′-phosphate group alters its charge versus mass ratio. This band is also visible in lane 1 of Figure 2B, where it resides between the 20 nt primer and a 19 nt band (a side-product of the synthetic 20 nt primer). Thus, in Figure 2A the sequence ladders are represented by oligonucleotides migrating ∼1.5 nt faster than the expected length due to the extra 3′-phosphate. Fragments up to 81 nt long (cleaved at the 82nd base, NH2-dT) were observed, as well as the full-length 87 nt extension product.
After prolonged heating at 40°C with a higher concentration (5%) of acetic acid, the full-length products were completely degraded and the shorter fragments enriched (Fig. 2B, lanes 1–4), indicating more extensive, but still incomplete, cleavage at each modified nucleotide. Whereas the relative extent of cleavage can be roughly estimated as NH2-dC > NH2-dT > NH2-dG ≈ NH2-dT, some non-specific cleavage bands were observed and are highlighted with gray boxes in Figure 2B (lanes 1 and 4). The mobility of these extra bands is less than one base slower than the specific cleavage bands, a mobility difference that was mentioned above as arising from loss or gain of a phosphate group. We speculate that the non-specific cleavage products come from a hydrolysis reaction catalyzed by the harsher acidic conditions, resulting in the formation of oligonucleotides without the 3′-phosphate group (Scheme 2). Whatever the cause, the results in Figure 2B suggest that complete and specific cleavage at P-N linkages in modified DNA is not easily achieved and that the most fruitful applications of phosphoramidate chemistry may be those that require only partial cleavage, such as DNA sequencing.

Scheme 2.
Partial substitution and site-specific cleavage: application in sequencing
Partial substitution of a designated base by its 5′-amino analog may be achieved by using a mixture of NH2-dNTP and dNTP in a polymerase reaction, resulting in a statistical representation of the analog at each matching site in a DNA molecule. This statistical distribution permits the generation of a sequence ladder through site-specific cleavage at the modified nucleotides. The use of partial incorporation of each of the NH2-dNTPs (1–6) allowed the full-length replication of a 480 nt DNA template (Fig. 3). The addition of extra dNTPs at the end of the primer extension reactions was found to minimize incomplete extension products (18). Direct treatment (without purification) of the polymerization mixtures with acetic acid resulted in sequencing ladders of A (lane 2, from NH2-dA), C (lane 3, from NH2-dC), G (lanes 4 and 6, from NH2-dG and NH2-dI) and T (lanes 5 and 7, from NH2-dT and NH2-dU). The distribution of the band intensities is reasonably uniform and can be adjusted by utilizing different ratios of native and modified nucleotides (Supplementary Material, Fig. A) and/or different cleavage conditions (see Fig. 2). Under the conditions shown, NH2-dA, NH2-dC, NH2-dI and NH2-dU provided readable band intensities up to full length. There are several bands in lane 1 (dNTPs) that are less than full length, which indicate stops or positions where the polymerase had difficulty proceeding. These artifacts are common in DNA sequencing reactions and can often impede sequence readout. Fortunately, with the incorporation–cleavage sequencing scheme that is outlined here, such artifacts can be eliminated by simple purification of the full-length extension product prior to the cleavage reaction.
Figure 3.
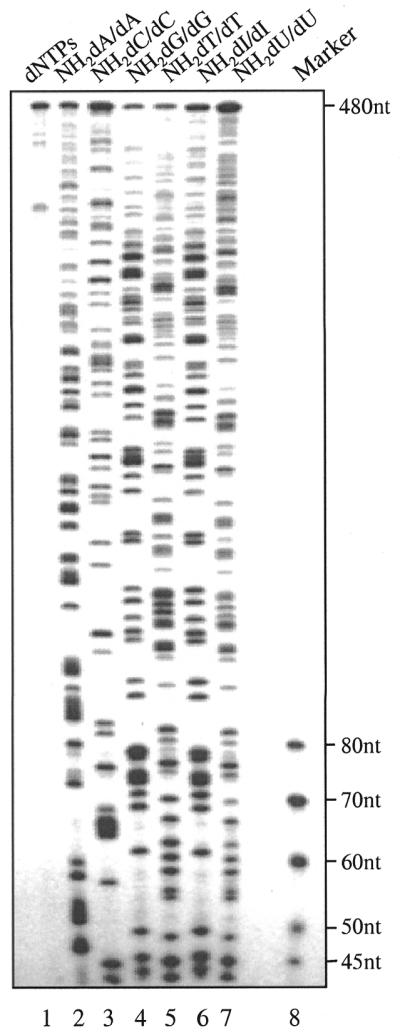
Sequence analysis utilizing statistical incorporation of NH2-dNTPs. The cleavage fragments from 480 nt DNA were separated by denaturing PAGE (6%).
To evaluate any length limits of this incorporation–cleavage sequencing method, a 7.2 kb single-stranded M13mp18 plasmid was used as a primer extension template. A 5′ radiolabeled primer was annealed to the plasmid and extended in the presence of NH2-dTTP:dTTP 100:1 and d(ACG)TPs using Klenow (exo–) (Fig. 4A). Upon treatment with dilute acetic acid, the extension product was cleaved to form DNA fragments well dispersed throughout the range ∼20 bp to 7.2 kb, indicating successful incorporation of NH2-dT (see Supplementary Material, Fig. B). The successful primer extension to 7.2 kb and the susceptibility of the extension product to restriction endonucleases is demonstrated in Figure 4. After treatment with the restriction enzyme MscI, which recognizes and cleaves at TGG/CCA and has a unique site within the 7.2 kb M13 sequence, the circular DNA (present as a mixture of supercoiled and non-supercoiled) was linearized and migrated as a 7.2 kb duplex (Fig. 4B, lanes 1 and 2). Heating of non-digested extension product prior to gel analysis resulted in two major bands near 8 kb, respectively corresponding to a single-stranded 7.2 kb fragment (upper band) and the duplex, probably as a result of incomplete heat denaturation. Heat denaturation of MscI-digested material liberated a single-stranded 1.2 kb fragment, which was the only product observed (Fig. 4A, and Fig. 4C, lane 2), as expected. These results indicate that primer extension by statistical incorporation of a NH2-dNTP is very efficient and the length limit for such polymerization may be well above 7.2 kb. Furthermore, despite the presence of P-N linkers in these polynucleotide products, they can be readily digested by restriction enzymes.
Figure 4.

(A) Schematic presentation of the primer extension, restriction digestion and heat denaturation procedures and corresponding products. (B) TBE–agarose gel (0.6%) analysis of the primer extension product containing statistically incorporated NH2-dT:dT using 7249 nt M13 plasmid as template and the corresponding MScI restriction digestion product. (C) Heat-denatured samples of (B) analyzed by 0.6% TBE–agarose gel electrophoresis.
Based on these observations, we envisioned a long-range sequencing approach (Scheme 3) that may be particularly useful for genome sequence assembly, a challenge faced by genome sequencing projects (19,20). Because the human genome is highly repetitive with >50% of repeated sequences (21), including multiple Alu repeats (consensus length 280 nt) and LINES (long interspersed DNA sequence elements; up to 7000 nt in length) (21), whereas the commonly used dideoxy sequencing method has an average read length of <600 nt (20), it is very difficult to assemble the sequence information thus obtained. Long-range primer extension reactions could be used to generate large duplex DNA containing statistically incorporated NH2-dNTPs, which could be digested with restriction endonucleases into shorter fragments (Scheme 3). These fragments could then be labeled with a fluorescent dye or radiolabel, then fractionated by HPLC. Each isolated fragment could be cleaved and sequenced. Digestion with different restriction endonucleases will produce complementary sets of sequencing data, which would facilitate assembly of the template sequence.
Scheme 3. Proposed long-range DNA sequencing method using polymerization, restriction digestion, fragment labeling, chemical cleavage, electrophoresis and sequence assembly.
CONCLUSIONS
We have developed reproducible synthetic procedures to prepare six 5′-amino-2′,5′-dideoxynucleotide analogs in high yield and purity, and have characterized each by UV, HPLC, 31P NMR and polymerase incorporation. The availability of these nucleotides will facilitate the development of new technologies that utilize them (9; Wolfe,J.L., Wang,B.H., Kawate,T., DeMaria,C. and Stanton,V.P., submitted for publication) and may inspire interests in additional applications. We also demonstrated that 5′-NH2-dNTPs can be efficiently incorporated by Klenow (exo–) polymerase to generate very long polynucleotides containing a modified nucleotide, and that these products can undergo site-specific cleavage at the phosphoramidate bonds under mild acidic conditions to generate sequence ladders. This incorporation– cleavage-based sequencing approach may provide an experimental means to improve genome sequence assembly.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Jeff Olson for providing transferrin receptor plasmids and to David Housman and Greg Verdine for helpful discussions. We thank Chuck Allerson and Ann Ferentz for critical reading of this manuscript.
REFERENCES
- 1.Luo P.Z., Leitzel,J.C., Zhan,Z.Y.J. and Lynn,D.G. (1998) Analysis of the structure and stability of a backbone-modified oligonucleotide: implications for avoiding product inhibition in catalytic template-directed synthesis. J. Am. Chem. Soc., 120, 3019–3031. [Google Scholar]
- 2.Zhan Z.Y.J. and Lynn,D.G. (1997) Chemical amplification through template-directed synthesis. J. Am. Chem. Soc., 119, 12420–12421. [Google Scholar]
- 3.Lutz M.J., Benner,S.A., Hein,S., Breipohl,G. and Uhlmann,E. (1997) Recognition of uncharged polyamide-linked nucleic acid analogs by DNA polymerases and reverse transcriptases. J. Am. Chem. Soc., 119, 3177–3178. [Google Scholar]
- 4.Bergmann F. and Bannwarth,W. (1995) Solid-phase synthesis of directly linked peptide-oligodeoxynucleotide hybrids using standard synthesis protocols. Tetrahedron Lett., 36, 1839–1842. [Google Scholar]
- 5.Koppitz M., Nielsen,P.E. and Orgel,L.E. (1998) Formation of oligonucleotide-PNA-chimeras by template-directed ligation. J. Am. Chem. Soc., 120, 4563–4569. [DOI] [PubMed] [Google Scholar]
- 6.Tetzlaff C.N., Schwope,I., Bleczinski,C.F., Steinberg,J.A. and Richert,C. (1998) A convenient synthesis of 5′-amino-5′-deoxythymidine and preparation of peptide-DNA hybrids. Tetrahedron Lett., 39, 4215–4218. [Google Scholar]
- 7.Letsinger R.L., Hapke,B., Petersen,G.R. and Dumas,L.B. (1976) Enzymatic synthesis of duplex circular phiX174 DNA containing phosphoramidate bonds in the (–) strand. Nucleic Acids Res., 3, 1053–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Letsinger R.L., Wilkes,J.S. and Dumas,L.B. (1976) Incorporation of 5′-amino-5′-deoxythymidine 5′-phosphate in polynucleotides by use of DNA polymerase I and a phiX174 DNA template. Biochemistry, 15, 2810–2816. [DOI] [PubMed] [Google Scholar]
- 9.Shchepinov M.S., Denissenko,M.F., Smylie,K.J., Worl,R.J., Leppin,A.L., Cantor,C.R. and Rodi,C.P. (2001) Matrix-induced fragmentation of P3′-N5′-phosphoramidate-containing DNA: high-throughput MALDI-TOF analysis of genomic sequence polymorphisms. Nucleic Acids Res., 29, 3864–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mag M. and Engels,J.W. (1989) Synthesis and selective cleavage of oligodeoxyribonucleotides containing non-chiral internucleotide phosphoramidate linkages. Nucleic Acids Res., 17, 5973–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamoto I., Sekine,M. and Hata,T. (1980) One-step synthesis of 5′-azido-nucleosides. J. Chem. Soc. Perkin Trans., 1, 306–310. [Google Scholar]
- 12.McClelland A., Kuhn,L.C. and Ruddle,F.H. (1984) The human transferrin receptor gene: genomic organization and the complete primary structure of the receptor deduced from a cDNA sequence. Cell, 39, 267–274. [DOI] [PubMed] [Google Scholar]
- 13.Wolfe J.L., Kawate,T., Sarracino,D.A., Zillmann,M., Olson,J., Stanton,V.P. and Verdine,G.L. (2002) A genotyping strategy based on incorporation and cleavage of chemically modified nucleotides. Proc. Natl Acad. Sci. USA, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 15.Horwitz J.P., Tomson,A.J., Urbanski,J.A. and Chua,J. (1962) Nucleosides. I. 5′-Amino-5′-deoxyuridine and 5′-amino-5′-deoxythymidine. J. Org. Chem., 27, 3045–3048. [Google Scholar]
- 16.Glinski R.P., Khan,M.S. and Kalamas,R.L. (1973) Nucleotide synthesis. IV. Phosphorylated 3′-amino-3′-deoxythymidine and 5′-amino-5′-deoxythymidine and derivatives. J. Org. Chem., 38, 4299–4305. [DOI] [PubMed] [Google Scholar]
- 17.Mungal W.S., Greene,G.L., Heavner,G.A. and Letsinger,R.L. (1975) Use of the azido group in the synthesis of 5′ terminal aminodeoxythymidine oligonucleotides. J. Org. Chem., 40, 1659–1662. [DOI] [PubMed] [Google Scholar]
- 18.Astatke M., Ng,K., Grindley,N.D. and Joyce,C.M. (1998) A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc. Natl Acad. Sci. USA, 95, 3402–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lander E.S., Linton,L.M., Birren,B., Nusbaum,C., Zody,M.C., Baldwin,J., Devon,K., Dewar,K., Doyle,M., FitzHugh,W. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 20.Venter J.C., Adams,M.D., Myers,E.W., Li,P.W., Mural,R.J., Sutton,G.G., Smith,H.O., Yandell,M., Evans,C.A., Holt,R.A. et al. (2001) The sequence of the human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 21.Martin-Gallardo A., McCombie,W.R., Gocayne,J.D., FitzGerald,M.G., Wallace,S., Lee,B.M., Lamerdin,J., Trapp,S., Kelley,J.M., Liu,L.I. et al. (1992) Automated DNA sequencing and analysis of 106 kilobases from human chromosome 19q13.3. Nature Genet., 1, 34–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.