

Abstract
We have used luminescence resonance energy transfer between regulatory light chains (RLC) to detect structural changes within the dimeric myosin molecule in contracting muscle fibers. Fully functional scallop muscle fibers were prepared such that each myosin molecule contained a terbium-labeled (luminescent donor) RLC on one head and a rhodamine-labeled (acceptor) RLC on the other. Time-resolved luminescence energy transfer between the two heads increased upon the transition from relaxation (ATP) to contraction (ATP plus Ca) and increased further in rigor (no ATP). Combined with experiments on mutant RLCs labeled specifically at other sites, these results support a model in which the force-generating weak-to-strong transition causes one myosin LC domain to tilt through a 30° angle toward the other, thus acting as a coordinated lever arm.
Most myosins, including those from vertebrate skeletal and scallop muscle, exist as dimers: each myosin molecule consists of a homodimer of heavy chains, each of which binds an essential light chain (ELC) and a regulatory light chain (RLC). Although many experiments have focused on the molecular motions of individual myosin heads, few have addressed directly the movement of the two heads within the myosin molecule relative to each other. In the present study, we have directly detected head–head movement within the myosin molecule by using luminescence resonance energy transfer (LRET). We have done this in the native environment of the muscle fiber to include interactions of the heads with both thick and thin filaments and to detect structural changes during muscle contraction.
Paramagnetic probes of myosin in contracting muscle have shown that force generation is correlated with a dynamic disorder-to-order transition in the tilting of the catalytic domain and a distinct rotation of the light chain domain (1, 2). Recent support for this mechanism has come from electron microscopy, which confirms that the catalytic domain motions largely precede the light-chain domain motions (3). Previous electron paramagnetic resonance (EPR) studies using spin labels on the myosin RLC have shown that the transition from relaxation to contraction is accompanied by a large rotation (≈36°) of the light chain (LC) domain in a small fraction of myosin heads, which is consistent with the LC domain's action as a lever arm (2). Fluorescence polarization data are consistent with this model but do not provide sufficient resolution to detect clearly the change in orientation between relaxation and contraction (4, 5). The EPR data (2, 6) show that there are two equally populated orientations of RLC in relaxed muscle, suggesting that the two heads of myosin have LC domains with distinct orientations. This result suggests that the dimeric structure of myosin plays a significant role, supporting previous evidence from x-ray diffraction (7) and electron microscopy (8).
Although previous experiments detected the movement of individual LC domains with respect to the actin filament, they did not provide information about movement of heads relative to one another in the myosin molecule. In the present study, we have addressed this problem directly by detecting interhead structural changes within the myosin molecule in intact muscle fibers, focusing on distance changes between the two LC domains. We labeled each myosin molecule in a skinned muscle fiber bundle with a donor probe on one head and an acceptor probe on the other, detected resonance energy transfer to determine distances between the probes, and thus detected changes in this distance during contraction. This specific labeling was made possible by the negative cooperativity of RLC binding in scallop muscle (9, 10); the reliability and precision of the measurements was greatly enhanced by the use of Tb3+ as a luminescent donor (11, 12).
Materials and Methods
Reagents, Solutions, Labels, and Muscle Sources.
Live scallops were obtained from Marine Biological Laboratories (Woods Hole, MA). Chicken gizzards were obtained directly from the Gold'n Plump processing plant in Cold Spring, MN. Mg wash (MW) consisted of 2 mM MgCl2, 40 mM NaCl, and 10 mM Mops, pH 7. Extraction buffer (EB) contained 15 mM EDTA, 40 mM NaCl, and 10 mM Mops, pH 7. Labeling buffer (LB) contained 50 mM NaCl, 2 mM EDTA, and 20 mM EPPS, pH 8. Urea buffer (UB) contained 8 M urea and 20 mM EPPS, pH 8.6. Rigor buffer (RB), pH 7, contained 20 mM Mops (or 20 mM EPPS for pH 8), 5 mM MgCl2, 1 mM EGTA, 20 mM creatine phosphate, and 0.25 mg/ml creatine phosphokinase. Ionic strength was adjusted to 200 mM with potassium propionate, and pH was adjusted with potassium hydroxide. Relaxation was induced by the addition of 5 mM MgATP or 5 mM MgATP plus 1 mM sodium vanadate to RB. Contraction was induced by adding 2.5 mM CaCl2 (resulting in pCa 4.5). The probes used were a terbium chelate (cysteine-reactive and UV-absorbing chelator, DTPA-cs124-EMPH, prepared as described in ref. 12) as luminescent donor and tetramethylrhodamineiodoacetamide (TMRIA, from Molecular Probes) as acceptor.
Preparation and Labeling of RLC.
RLC was purified from chicken gizzard myosin and designated cgRLC (13). Mutant cgRLC was expressed and purified as described in (14). Before labeling, RLC was dialyzed into LB. The probe (dissolved in DMF for TMRIA or DMSO for Tb3+ chelate) was incubated at a concentration of 150–300 μM with 100 μM RLC in LB for 2 h at room temperature and then overnight on ice (12, 15, 16). Unreacted probe was removed by dialysis.
Scallop Fiber Preparation and RLC Exchange.
Striated adductor muscle was dissected from the bay scallop, Placopecten magellanicus, and skinned as described, producing fiber bundles ≈0.5 mm in diameter (2). For partial extraction, these skinned fiber bundles were washed with EB at 10°C for 30 min, which removes half of the RLC (2). For reconstitution, partially extracted fibers were incubated in MW that contained 2 mg/ml of donor-labeled cgRLC for 2 h at 4°C, followed by a 15-min wash with MW to remove excess labeled cgRLC. Partial extraction was repeated to remove the remaining native RLC; reconstitution was repeated with acceptor-labeled cgRLC, producing myosin molecules with donor on one RLC and acceptor on the other (demonstrated in Results).
Characterization of Labeled Fibers.
To determine RLC content, fibers were finely minced and incubated in UB for 2 h at room temperature, then loaded on a 12.5% (wt/vol) polyacrylamide gel containing UB. After electrophoresis, the gel was stained with Coomassie brilliant blue R-250 and analyzed with a densitometer to quantitate protein-band density. The RLC value was divided by the ELC value in the same lane, and this value was divided by that observed for untreated fibers to obtain the number of RLCs per myosin head (9, 17). To assess the functionality of muscle fibers at various stages of labeling, calcium sensitivity of isometric tension development was measured at 25°C by using an Akers force transducer, as described (6). At each step in the extraction and reconstitution procedure, tension (force per cross-sectional area) was measured in the presence and absence of Ca, and Ca sensitivity was calculated as [1 − (−Ca)/(+Ca)].
Optical Spectroscopy and Data Analysis.
Fiber bundles were tied with string on either end and tied to a mounting stand that was placed in a quartz cuvette. Experiments were performed at 20°C. Time-resolved luminescence decays were recorded by using an instrument described (18). The terbium donor was excited with a 10-ns pulse of light from a XeCl excimer laser (308 nm, 55 Hz repetition rate). The terbium emission was monitored at 546 nm or 488 nm by using interference filters from Corion (Holliston, MA) with a range of ± 10 nm. The luminescence decay was analyzed by fitting it, using the nonlinear least-squares Levenberg-Marquardt algorithm in the ORIGIN 6 program, to the expression
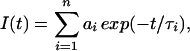 |
where τi is the excited-state lifetime for component i and ai is the mole fraction of probes in that state. To eliminate effects of gating the TMR prompt fluorescence, the fits did not include the first 50 μs. The number of exponentials n was increased until no further improvement was seen in the fit, as evaluated by χ2 and the residual plot. For donor-only and donor/acceptor data in rigor and relaxation, the fit was optimal with n = 2. For the donor/acceptor contraction data, we found good fits with n = 2 and 3.
The energy transfer efficiency, E, was calculated from the fractional decrease in donor lifetime in the absence (τD) and presence (τDA) of acceptor by using the relation
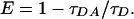 |
Probe separation (R) was then calculated by
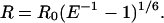 |
R0 is the Förster distance specific to the dye pair used and was calculated from
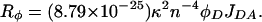 |
JDA was determined from the overlap of the donor emission and acceptor excitation spectra, n is the index of refraction (nwater = 1.33), φD (0.57) is the quantum yield of the donor, and κ2 is the orientation factor, assumed to be 2/3. The quantum yield was determined as the ratio of τD (1.53 ms) to its value in D2O (2.63 ms; ref. 16). Here, we found R0 = 59 Å, which is consistent with previous calculations (16).
Although the decays fit best to two or three components, only the lifetime of the longer component was used in calculation of energy transfer efficiency. The decay always contained a short lifetime component of <0.5 ms. Changes in this component were qualitatively similar to changes in the longer component (lifetime >1 ms), but the detection and analysis of this component was less precise, so only the longer lifetimes were used in energy transfer calculations. This approach is standard in LRET experiments involving Tb3+ (16).
The emission of Tb3+ is unpolarized, so the approximation of κ2 = 2/3 has less uncertainty in LRET than in FRET with a polarized donor. The anisotropy of TMR when bound to RLC in scallop fibers is A = 0.278 ± 0.004, resulting in limits on κ2 of 0.388 < κ2 < 1.225 (19, 20), which limits the uncertainty in distance as R −8%/+11%. This uncertainty in the absolute distance measured is probably much greater than the uncertainty in a distance change, e.g., upon addition of ATP.
Modeling of LC Domain Rotation.
Chicken gizzard RLC homology was modeled onto the scallop heavy chain and ELC and verified by EPR spectroscopy (see Fig. 3; ref. 14). The coordinate system defining the rotational changes (see Fig. 4) is defined with the origin at Gly-825 of the hook in the heavy chain, the x axis passing through residue 777 along the long axis, and the y axis in the direction of residue 836. Gly-825 was selected as a plausible origin (hinge) because of its small size. Residue 777 was selected for the direction of the lever arm axis because myosin is easily cleaved there, which is consistent with a possible pivot point. Placement of the y axis is not critical because the LC domain is allowed to rotate about the x axis. Modeling of LC domain rotation was done by using INSIGHT II. LC domain separation and angles of rotation were fit by using the constraints of the experimentally measured distances by minimization of χ2. One LC domain was assumed to remain fixed, and the motion of the other LC domain was modeled as a rigid body axial tilt of one LC domain about its y axis (in the direction required for axial force production) and a twist about its x axis. For simplicity of illustration, the two LC domain orientations are shown in the same plane in Fig. 4, but the actual modeling (and measurement of distances and axial angles) took place starting with the helical geometry of the rigor complex derived previously from modeling of data from electron microscopy and x-ray crystallography (21), which is consistent with the modeling of previous EPR data on scallop fibers (14).
Fig 3.

(A) Crystal structure of LC domain with heavy chain (blue), ELC (green), RLC (red), and labeling sites (yellow). (B) FRET-detected distances (bars) and distance changes (brackets) between heads in the myosin molecule in relaxation (weak-binding, W) and rigor (strong-binding, S), as measured from probes attached to single-Cys mutants.
Fig 4.

Transition of myosin molecules from the weak-binding structure in relaxation to the strong-binding structure in rigor, resulting in a 30° axial tilt of one LC domain relative to the other. This model is based on LRET distance changes for four labeled sites on RLC (yellow). The distance change for Cys-108 is shown. Modeling and coordinate system are described in Materials and Methods.
Results
Labeling of Myosin in Scallop Muscle Fibers.
In scallop muscle, Ca sensitivity of force requires RLC-binding, RLC binds with negative cooperativity to the two heads of myosin, and cgRLC binds much more tightly to myosin than native scallop RLC and restores Ca sensitivity (9, 10, 22). We exploited these properties to label the two heads of each myosin molecule with donor and acceptor and to quantitate the extent and specificity of labeling. In native fibers, every myosin head contains one RLC, and force is calcium sensitive (Fig. 1, “Native” bar). Our extraction procedure removed half of the RLC (Fig. 1A, “Ex 1” bar, 44 ± 3% extracted) and abolishes nearly half of the Ca sensitivity (Fig. 1B, “Ex 1” bar), as observed previously by using a similar procedure (22). Doubling the extraction time resulted in no significant further extraction (<3%, not shown), confirming the negative cooperativity of RLC binding and indicating that each myosin molecule has lost one of its two RLCs, as shown (9, 10). After reconstitution with donor-labeled cgRLC, RLC content and Ca sensitivity were restored to native levels, as reported for unlabeled cgRLC (9, 10) and labeled cgRLC (Fig. 1B, “D” bar; ref. 2). When a second extraction was carried out on these donor-labeled fibers, nearly half of the RLC was extracted (Fig. 1A, “Ex 2” bar), but none (<3%, not shown) of the extracted RLC contained the luminescent donor. This result confirms that cgRLC binds to myosin much more tightly than native scallop RLC (9, 10) and implies that at least 94% of all RLC-depleted heads at this point are on myosin molecules containing a donor-labeled head. The subsequent reconstitution using acceptor-labeled cgRLC restored both RLC content and Ca-sensitive function (Fig. 1 A and B, “DA” bars), indicating that the other head of each myosin molecule had been labeled with acceptor. Thus, virtually every (>90%) myosin molecule in these fibers that contains a donor-labeled RLC on one head contains an acceptor-labeled RLC on the other head and is fully functional.
Fig 1.

Characterization of RLC-labeled muscle fibers. (A) RLC content determined from urea/polyacrylamide gels (2). (B) Ca sensitivity [1−T(pCa 9)/T(pCa 4.5)], where T is isometric tension. Native fibers were partially extracted (Ex 1), reconstituted with donor-labeled RLC (D), extracted to remove the remaining unlabeled RLC (Ex 2), and then reconstituted with acceptor-labeled RLC (DA).
LRET of Probes Attached to Cys-108 on Native Gizzard RLC.
The luminescence decay of RLC-attached Tb3+ donor in scallop muscle fibers in rigor (no ATP) was nearly monoexponential (Fig. 2), which is consistent with previous results on myosin from skeletal muscle (16). The major component (88.7 ± 3% of the total amplitude) decayed with a lifetime (τD) of 1.53 ms, whereas the remainder decayed at 0.45 ms. Only changes in the longer lifetime were considered in distance calculations (Table 1). The donor lifetime τD, detected in the absence of acceptor, did not change upon addition of ATP or of ATP + Ca (Table 1, Relaxation or Contraction, respectively).
Fig 2.

Tb3+ luminescence decay in the absence (τD, results independent of ATP and Ca) and presence (τDA) of acceptor. Curves show best fit to two exponentials (Eq. 1). The longer of the two lifetimes is tabulated in Table 1.
Table 1.
Lifetimes and energy transfer calculations
| τ, ms | E, % | R, Å | XRIG | |
|---|---|---|---|---|
| Rigor (D) | 1.53 ± 0.01 | |||
| (n = 13) | ||||
| Rigor (DA) | 1.08 ± 0.02 | 29.4 ± 1.8 | 68.5 ± 1.0 | 1 |
| (n = 10) | ||||
| Relaxation (D) | 1.53 ± 0.01 | |||
| (n = 5) | ||||
| Relaxation (DA) | 1.38 ± 0.02 | 9.8 ± 1.8 | 85.0 ± 3 | 0 |
| (n = 7) | ||||
| Contraction (D) | 1.52 ± 0.01 | |||
| (n = 3) | ||||
| Contraction (DA) | 1.27 ± 0.01 | 16.5 ± 1.9 | 76.7 ± 1.8 | 0.36 ± 0.04 |
| (n = 5) |
D and DA indicate donor only and donor + acceptor, respectively. τ is the long-lifetime component of a two-exponential fit (Eq. 1), reported as mean ± SEM. E is calculated from Eq. 2 and R from Eq. 3. XRIG, the mole fraction of myosin in the rigor-like strong-binding state, was calculated for contraction from Eq. 5 (see Discussion). All changes in DA lifetimes are statistically significant (P < 0.01).
The acceptor increased the rate of decay in rigor (Fig. 2), resulting in a lifetime of τDA = 1.08 ms, corresponding to transfer efficiency E = 29.4% and a donor-acceptor distance R = 68.5 Å (Table 1). In relaxation (adding ATP in the absence of Ca), τDA = 1.38 ms, corresponding to a lower value of E (9.8%) and thus a longer distance (R = 85 Å). During contraction, τDA = 1.27 ms, a value intermediate between that of rigor and relaxation, corresponding to E = 16.5% and a distance of 76.7 Å. These results are summarized in Table 1.
Controls.
Fiber bundles retained both their ability to contract and their calcium sensitivity after prolonged exposure to the laser. One millimolar vanadate, which slows the ATPase cycle and ensures the predominance of the weak-binding state, had no effect on the data in relaxation, showing that our data in relaxation is truly representative of the weak-binding state. In fibers labeled with both donor and acceptor, a two-exponential analysis (fixing one lifetime at 1.53 ms) showed that no more than 5% of the donors decayed with the original 1.53 ms lifetime, confirming that the exchange procedure was complete so that at least 95% of the donor RLCs had an acceptor RLC on the adjoining head in the same myosin molecule. To test this point even more thoroughly, we performed a single partial extraction (removing half of the native RLC) and reconstituted with an equal mixture of donor- and acceptor-labeled cgRLC. No component (5% mol fraction or greater) was observed that had a significantly (5%) different lifetime from the donor-only lifetime of 1.53 ms. This result proves two important points. First, it proves that the original partial extraction removed RLC quite specifically from only one head per myosin molecule. If the extraction had removed RLC randomly from heads, and the reconstitution had restored them randomly, this experiment would have given 25% of the donors the same lifetime as in the presence of acceptor (1.08 ms instead of 1.53 ms). Second, it shows that there is no significant energy transfer between heads on different myosin molecules. This result is not surprising, because only a very small fraction of myosin heads from different myosin molecules would be expected to be attached to adjacent actin subunits, even in rigor, based on the large molar ratio of actin to myosin heads (six) and the helical mismatch between myosin and actin filaments in scallop muscle (23).
LRET of Probes Attached to Cys on Mutant Gizzard RLC.
We performed experiments complementary to the above by using mutant chicken gizzard RLCs, in which the native Cys residue (C108) was mutated to Ala and a different residue was mutated to Cys: M72C, M129C, and T96C (Fig. 3A). These mutant RLCs were used to label each myosin molecule in scallop muscle fibers with donor and acceptor, thus producing labeled fibers that were fully functional and calcium sensitive. LRET was used to measure the head–head distance as a function of the physiological state. All of the sites showed significantly shorter distances in rigor than in relaxation, except for M72C, which showed no significant difference (Fig. 3B). Experiments on these mutant RLCs also were performed in contraction, but (i) M72C showed no sensitivity to ATP or Ca, and (ii) the longer distances observed for T96C and M129C decreased precision substantially (see error bars in Fig. 3B), so that only for C108 was the precision sufficient to distinguish contraction from relaxation. Therefore, we focused on C108 in contraction.
These results suggest that the LC domains tilt relative to each other upon the transition from weak to strong binding, with residue 72 near a fixed pivot point. We used these four distance changes as constraints, assuming that the two LC domains have the same orientation in rigor, as shown previously by EPR (2), and that one LC domain rotates axially relative to the other as a rigid body. The best fit is a tilt of 30 ± 5° (Fig. 4). Small changes in relative twisting of the LC domains (24) relative to each other upon activation cannot be ruled out, but could only account for a small portion (1–2 Å) of each of the three significant distance changes.
Discussion
We have directly detected the distance between specific sites on the two heads of myosin in muscle fibers. This was made possible by the special properties of RLC in molluscan muscle, which allowed us to achieve complete and specific labeling of each myosin molecule with a donor-acceptor pair and to verify reconstitution of function. Because this labeling was accomplished in the muscle fiber, the observed structural changes reflect accurately the constraints of the filament lattice and are directly relevant to the physiological transitions between weak- and strong-binding crossbridge states. The observed changes in distance between the two LC domains are consistent with the coordinated movement of the two heads within the myosin molecule.
Previous studies using fluorescence RET to detect structural changes of the myosin molecule in solution found the two LC domains to be >50 Å apart with no distance changes in the presence of MgATP, Ca, or actin (25). In the current study, the use of a Tb3+ chelate as the donor probe increased the range of measurable distance (larger R0) and eliminated most of the uncertainty caused by the orientation-dependence (see Materials and Methods). Because of the long lifetime of Tb3+, myosin dynamics could enhance energy transfer and thus decrease the apparent distance measured (26, 27). A complete description of LC domain structural states must include the dynamic disorder that characterizes each state, because previous studies have shown that both catalytic and LC domains have substantial rotational disorder, with time constants in the microsecond range, and that a transition from disorder to order plays a crucial role in the force-generating weak-to-strong transition (1). Studies on scallop muscle with RLC labeled with spin (2, 17) or phosphorescent (15) probes have shown that the LC domain undergoes restricted microsecond rotational motion, with the amplitude of motion greater in relaxation than in rigor. These motions should enhance energy transfer in relaxation more than in rigor, so our observation that the energy transfer is greater in rigor than in relaxation cannot be because of dynamics. This observation strengthens our conclusion that the distance between the two RLCs of myosin is less in rigor than in relaxation.
Implications of Myosin Head-to-Head Structural Changes.
The simplest interpretation of the LRET measurements would be to assume that the dimeric myosin molecule has a distinct structure in each physiological state, corresponding to a single donor-acceptor distance, which decreases from relaxation (85 Å) to contraction (76.7 Å) to rigor (68.5 Å; Table 1). The observation that the decays are each well fit by a single exponential does not prove that there is only one distance. We can conclude that there is no significant population with a distance less than R0 (59 Å) in any of our measurements, but the resolution of our technique is not enough to rule out two distances greater than R0. However, the existence of distinct structures in relaxation and rigor is consistent with x-ray and electron microscopic studies of scallop muscle that show evidence that myosin heads are helically ordered and splayed apart axially in relaxation and are helically oriented on actin in rigor (7, 8). EPR analysis of spin-labeled RLC in scallop muscle showed clearly that both heads are uniformly oriented in rigor and that there are two distinct and equally populated orientations in relaxation, separated by 36° (2). EPR detects the orientations of individual myosin heads but does not directly reveal information about the conformation of the myosin dimer. The current LRET results solve this problem by directly detecting the relationship of one head with respect to the other, showing that the two LC domains are farther apart in relaxation than in rigor, providing direct support for the model shown in Fig. 4. The use of Cys mutants provided enough distance constraints (Fig. 3) to construct plausible models for the LC domain structures in rigor and relaxation (Fig. 5). A key result of this analysis is that the angle between the two heads changes by 30° in the transition from relaxation to rigor. This finding is in excellent agreement with the EPR result of 36°, which was obtained by a completely independent method (2), thus lending strong support to the model in Fig. 4.
Fig 5.

Structural model for the force-generating structural transition during muscle contraction, in which the myosin molecule cycles between weak-binding (Left) and strong-binding (Right) structures to result in lever arm motion and movement of actin.
However, for muscle fibers in isometric contraction, biochemical and structural data strongly suggest that more than one myosin structural state is significantly populated, with the majority of myosin molecules in one or more weak-binding states and a significant fraction (20–40%) in strong-binding states. In particular, spin-label EPR studies on this same scallop muscle system (2) have shown that the orientation distribution of the LC domain in contraction is a linear combination of the orientations observed in rigor (strong-binding) and relaxation (weak-binding). Similar conclusions have resulted from EPR experiments on spin-labeled catalytic domain in vertebrate muscle, in which contraction exhibits a linear combination of an oriented strong-binding population and a dynamically disordered weak-binding population (28). To test whether our LRET data are consistent with EPR-based models, we have analyzed our energy transfer data in contraction with a two-component model.
We assume that the luminescence decay in contraction is related to the decays in relaxation and rigor by
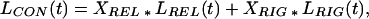 |
where XREL and XRIG are the mole fractions of myosin molecules in relaxation and rigor conformations, respectively, and L(t) are the luminescence decays of Fig. 2. We obtained an excellent fit of the contraction data to this model, with XRIG = 0.36 ± 0.04; i.e., 36% of myosin is in the rigor-like structural state. Although this fit was no better than the single-component fit, it is remarkable that the 36% value for XRIG agrees closely with the value of 35–40% obtained for the orientation states of the LC domain by both conventional EPR (2, 6) and saturation transfer EPR (17). The consistency in values supports the two-state model, in which the transition between weak- and strong-binding states constitutes the working stroke, and XRIG is the fraction of time each myosin molecule spends in the strong-binding (rigor-like) state. A value of XRIG < 0.5 is consistent with myosin being a low duty-cycle motor. This two-state model suggests that the weak-binding state in contracting muscle resembles the relaxation state more than it resembles the disordered state that has been observed for isolated scallop myosin filaments (29, 30) or heavy meromyosin (31) in the presence of ATP and calcium.
Fig. 5 describes a model that takes into account both the distance changes between the LC domains observed in the present study by LRET, and the orientation changes of individual LC or catalytic domains detected previously by EPR (1, 2, 6, 28). During isometric contraction, the myosin dimer cycles between two states, with the majority (64 ± 4%) in the weak-binding structure (Fig. 5 Left), characterized by LC domains splayed apart by an angle of 30° (determined from LRET in the present study) to 36° (determined from previous EPR studies; refs. 2 and 6), with the catalytic domains dynamically disordered (1, 28). Force is generated when one of the myosin heads attaches to actin (Fig. 5 Right), causing the catalytic domain to become ordered in a rigor-like orientation (1, 2, 28) and its LC domain to tilt by 30–36° (2, 6), resulting in the strong-binding conformation of the myosin molecule with parallel LC domains (2, 6). In this strong-binding myosin molecule during contraction, our model shows only one head attached to actin with an ordered catalytic domain, because the fraction of well oriented catalytic domains in isometric contraction measured by EPR (19 ± 2%; refs. 1 and 28) is about half of the fraction of strong-binding myosin molecules measured by LC domain EPR (37 ± 3%; refs. 2, 6, and 17) or LRET (36 ± 4%, Fig. 5). This finding is consistent with other proposals that myosin attachment is primarily single-headed in contraction (3, 15, 28, 32). Note that, in both the LC domains and catalytic domains, the myosin molecule undergoes a disorder-to-order transition in the force-generating weak-to-strong transition, as suggested (1).
Conclusions
We have directly detected structural changes that indicate coordination of the two heads of myosin in contracting muscle. Labeling myosin molecules with site-directed RLC-bound donor and acceptor probes on each head, within intact skinned muscle fibers, allowed us to directly observe distance changes between the LC domains in the two heads during relaxation, rigor, and contraction. The use of mutant RLCs allowed us to label four specific sites in the LC domain, placing further constraints on the observed structural transitions. The changes in probe separation support a model in which the two LC domains of the myosin molecule are tilted apart axially relative to one another by about 30° in the weak-binding state, but tilt toward one another upon force generation to become nearly parallel in the strong-binding state.
Acknowledgments
We thank Wendy Nelson for providing mutant RLC. We thank Leslie LaConte, Vincent Voelz, Josh Baker, Dawn Lowe, Sarah Blakely, David Wacker, Paul Selvin, and Christine Karim for helpful discussions and technical assistance. This work was supported by National Institutes of Health Grant AR32961 (to D.D.T.), the Muscular Dystrophy Association, and the Minnesota Supercomputing Institute. D.S.L. was supported by National Institutes of Health predoctoral training grants and by the National Science Foundation.
Abbreviations
LC, light chain
ELC, essential LC
RLC, regulatory LC
LRET, luminescence resonance energy transfer
EPR, electron paramagnetic resonance
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Thomas D. D., Ramachandran, S., Roopnarine, O., Hayden, D. W. & Ostap, E. M. (1995) Biophys. J. 68, 135S-141S. [PMC free article] [PubMed] [Google Scholar]
- 2.Baker J. E., Brust-Mascher, I., Ramachandran, S., LaConte, L. E. & Thomas, D. D. (1998) Proc. Natl. Acad. Sci. USA 95, 2944-2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor K. A., Schmitz, H., Reedy, M. C., Goldman, Y. E., Franzini-Armstrong, C., Sasaki, H., Tregear, R. T., Poole, K., Lucaveche, C., Edwards, R. J., et al. (1999) Cell 99, 421-431. [DOI] [PubMed] [Google Scholar]
- 4.Sabido-David C., Hopkins, S. C., Saraswat, L. D., Lowey, S., Goldman, Y. E. & Irving, M. (1998) J. Mol. Biol. 279, 387-402. [DOI] [PubMed] [Google Scholar]
- 5.Hopkins S. C., Sabido-David, C., Corrie, J. E., Irving, M. & Goldman, Y. E. (1998) Biophys. J. 74, 3093-3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brust-Mascher I., LaConte, L. E., Baker, J. E. & Thomas, D. D. (1999) Biochemistry 38, 12607-12613. [DOI] [PubMed] [Google Scholar]
- 7.Wray J. S., Vibert, P. J. & Cohen, C. (1975) Nature 257, 561-564. [DOI] [PubMed] [Google Scholar]
- 8.Vibert P. (1992) J. Mol. Biol. 223, 661-671. [DOI] [PubMed] [Google Scholar]
- 9.Simmons R. M. & Szent-Gyorgyi, A. G. (1980) Nature 286, 626-628. [DOI] [PubMed] [Google Scholar]
- 10.Chantler P. D. & Szent-Gyorgyi, A. G. (1980) J. Mol. Biol. 138, 473-492. [DOI] [PubMed] [Google Scholar]
- 11.Selvin P. R. (2000) Nat. Struct. Biol. 7, 730-734. [DOI] [PubMed] [Google Scholar]
- 12.Li M. & Selvin, P. R. (1997) Bioconjug. Chem. 8, 127-132. [DOI] [PubMed] [Google Scholar]
- 13.Persechini A. & Hartshorne, D. J. (1983) Biochemistry 22, 470-476. [DOI] [PubMed] [Google Scholar]
- 14.LaConte L. E., Voelz, V., Enz, M., Neslon, W. D. & Thomas, D. D. (2002) Biophys. J. 83, 1854-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramachandran S. & Thomas, D. D. (1999) Biochemistry 38, 9097-9104. [DOI] [PubMed] [Google Scholar]
- 16.Burmeister Getz E., Cooke, R. & Selvin, P. R. (1998) Biophys. J. 74, 2451-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roopnarine O., Szent-Gyorgyi, A. G. & Thomas, D. D. (1998) Biochemistry 37, 14428-14436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ludescher R. D. & Thomas, D. D. (1988) Biochemistry 27, 3343-3351. [DOI] [PubMed] [Google Scholar]
- 19.Dale R. E., Eisinger, J. & Blumberg, W. E. (1979) Biophys. J. 26, 161-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lakowicz J. R., Gryczynski, I., Cheung, H. C., Wang, C. K., Johnson, M. L. & Joshi, N. (1988) Biochemistry 27, 9149-9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendelson R. & Morris, E. P. (1997) Proc. Natl. Acad. Sci. USA 94, 8533-8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simmons R. M. & Szent-Gyorgyi, A. G. (1985) J. Physiol. 358, 47-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Millman B. M. & Bennett, P. M. (1976) J. Mol. Biol. 103, 439-467. [DOI] [PubMed] [Google Scholar]
- 24.Corrie J. E., Brandmeier, B. D., Ferguson, R. E., Trentham, D. R., Kendrick-Jones, J., Hopkins, S. C., van der Heide, U. A., Goldman, Y. E., Sabido-David, C., Dale, R. E., et al. (1999) Nature 400, 425-430. [DOI] [PubMed] [Google Scholar]
- 25.Chantler P. D. & Tao, T. (1986) J. Mol. Biol. 192, 87-99. [DOI] [PubMed] [Google Scholar]
- 26.Thomas D. D., Carlsen, W. F. & Stryer, L. (1978) Proc. Natl. Acad. Sci. USA 75, 5746-5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stryer L., Thomas, D. D. & Meares, C. F. (1982) Annu. Rev. Biophys. Bioeng. 11, 203-222. [DOI] [PubMed] [Google Scholar]
- 28.Cooke R., Crowder, M. S. & Thomas, D. D. (1982) Nature 300, 776-778. [DOI] [PubMed] [Google Scholar]
- 29.Frado L. L. & Craig, R. (1989) J. Cell Biol. 109, 529-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vibert P. & Craig, R. (1985) J. Cell Biol. 101, 830-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stafford W. F., Jacobsen, M. P., Woodhead, J., Craig, R., O'Neall-Hennessey, E. & Szent-Gyorgyi, A. G. (2001) J. Mol. Biol. 307, 137-147. [DOI] [PubMed] [Google Scholar]
- 32.Fajer P. G., Fajer, E. A., Brunsvold, N. J. & Thomas, D. D. (1988) Biophys. J. 53, 513-524. [DOI] [PMC free article] [PubMed] [Google Scholar]