

Abstract
One of the most serious consequences of cytotoxic cancer therapy is the development of therapy-related acute myeloid leukemia (t-AML), a neoplastic disorder arising from a multipotential hematopoietic stem cell. To gain insights into the molecular basis of this disease, we performed gene expression profiling of CD34+ hematopoietic progenitor cells from t-AML patients. Our analysis revealed that there are distinct subtypes of t-AML that have a characteristic gene expression pattern. Common to each of the subgroups are gene expression patterns typical of arrested differentiation in early progenitor cells. Leukemias with a –5/del(5q) have a higher expression of genes involved in cell cycle control (CCNA2, CCNE2, CDC2), checkpoints (BUB1), or growth (MYC), and loss of expression of the gene encoding IFN consensus sequence-binding protein (ICSBP). A second subgroup of t-AML is characterized by down-regulation of transcription factors involved in early hematopoiesis (TAL1, GATA1, and EKLF) and overexpression of proteins involved in signaling pathways in myeloid cells (FLT3) and cell survival (BCL2). Establishing the molecular pathways involved in t-AML may facilitate the identification of selectively expressed genes that can be exploited for the development of urgently needed targeted therapies.
Therapy-related myelodysplastic syndrome (t-MDS) and acute myeloid leukemia (t-AML) are late complications of cytotoxic therapy (radiation and/or chemotherapy) used in the treatment of both malignant and nonmalignant diseases (1–3). These neoplasms are thought to be the direct consequence of mutational events induced by cytotoxic therapy. Several distinct cytogenetic and clinical subtypes of t-MDS/t-AML are recognized that are closely associated with the nature of the preceding treatment. Survival times of t-AML patients are short, because current forms of therapy are largely ineffective, and new therapeutic approaches are needed (1–4).
The most common subtype of t-AML (≈75% of cases) develops after exposure to alkylating agents and is characterized by loss of a whole chromosome 5 and/or 7 or a deletion of the long arms of these chromosomes [−5/del(5q), −7/del(7q)] (4). Patients who develop t-MDS/t-AML in this setting typically show a latency of 3–7 years from alkylating agent exposure and present with cytopenias and myelodysplasia. The median time until progression to t-AML is 6 mo. Frequently, all three hematopoietic cell lineages (erythroid, myeloid, and megakaryocytic) are involved in the dysplastic process, suggesting that this subtype of t-AML arises in a hematopoietic stem/progenitor cell. The prognosis is poor, with a median survival of 5 mo. In the University of Chicago's series of 306 patients with t-MDS/t-AML, 64 (21%) patients had abnormalities of chromosome 5; 85 (28%) patients had abnormalities of chromosome 7; and 65 (21%) patients had abnormalities of both chromosomes 5 and 7 (3).
A second, and distinctly different, form of t-AML arises after therapy with topoisomerase II inhibitors (1–4). These patients are younger, rarely present with MDS, and have a more favorable response to remission induction therapy (1, 2). Balanced translocations involving MLL at 11q23 or RUNX1/AML1 at 21q22 are common in this subgroup (4).
t-AML represents an important model for cancer for several reasons. First, the incidence of t-AML is rising, as a result of the increasing number of cancer survivors at risk of developing this disorder and the changes in therapeutic trends. Second, t-AML provides a unique opportunity to examine the effects of mutagens on carcinogenesis in humans, as well as the role of genetic susceptibility to cancer. Third, the mechanisms of leukemogenesis that are uncovered in t-AML will likely apply to those subtypes of AML de novo, which share the same cytogenetic abnormalities, e.g., AML de novo with abnormalities of chromosome 5 or 7.
Analysis of the expression profiles of tumors with microarray technology has been used to identify new subtypes of cancer and to classify the diseases more accurately (5–8). To expand our understanding of the molecular basis of t-AML, we performed expression profiling of this disease. Our analysis revealed that there are distinct subtypes of t-AML with unique expression profiles. A global perspective on gene expression in t-AML provides new insights into the underlying biology of this disease and may facilitate the identification of molecular targets for therapeutic approaches.
Methods
Bone Marrow Samples.
Bone marrow aspirates were obtained from t-AML patients at diagnosis or relapse (patients 5–6 and 1–3) and were cryopreserved in liquid nitrogen. All patients gave written informed consent under Institutional Review Board-approved protocols for the use of their leukemia cells. Cytogenetic analysis was performed by using standard techniques. The clinical features of the patients and cytogenetic pattern and immunophenotype of the leukemias are published as Table 3 in supporting information on the PNAS web site (www.pnas.org). Mononuclear cells were purified from the bone marrow by density gradient centrifugation in NycoPrep 1.077 (Nycomed, Oslo). CD34+ cells were labeled with magnetic microbeads conjugated with anti-CD34+ antibodies and isolated by using miniMACS magnetic cell separation columns (Miltenyi Biotec, Auburn, CA). Normal CD34+ progenitor cells from bone marrow of two donors were purchased from Cambrex (East Rutherford, NJ). CD34+ progenitor cells from a third donor were isolated from bone marrow of an individual with breast cancer that was not metastatic to the marrow (control 1–1). The UoC-M1 cell line was derived from the malignant cells of a 68-year-old male with acute myeloid leukemia de novo characterized by both a del (5)(q12q34) and –7 (9).
RNA Isolation and Oligonucleotide Microarray Analysis.
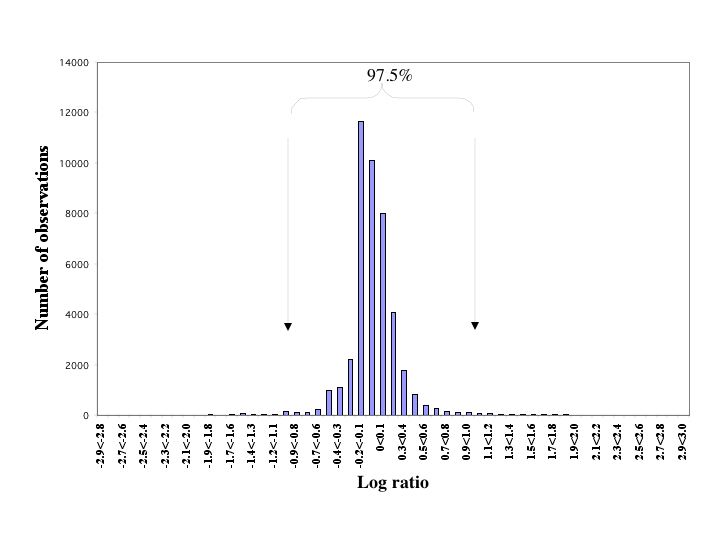
Total RNA was extracted from CD34+ cells by using Trizol (Invitrogen) according to the manufacturer's procedures. Detailed protocols for generating biotinylated cRNA, and microarray processing were provided by Affymetrix (Santa Clara, CA). Briefly, 2–5 μg of good-quality total RNA was used to produce single-strand cDNA by reverse transcription by using a T7-(dT)24 primer containing the T7 polymerase promoter (Genset, La Jolla, CA) and Superscript II reverse transcriptase (Invitrogen). After generating second-strand DNA, the DNA was transcribed in vitro into cRNA and labeled with biotin-modified ribonucleotides by using an RNA transcript labeling kit (T7) (Enzo Diagnostics). The high-density oligonucleotide-based Affymetrix arrays (U95A V2) containing probes representing 12,600 genes and expressed sequence tags were used for hybridization following the manufacturer's protocol. After hybridization, the chips were washed, stained on the Affymetrix Fluidics Station, and scanned on an Affymetrix scanner. Data analysis was performed by using genespring software, Ver. 4.0 (Silicon Genetics). To ensure the reproducibility of the data, we performed four replicate analyses of RNA extracted from the KG-1 myeloid leukemia cell line. An exceedingly high reproducibility was observed with >99% of genes designated as “no change in intensity” by using the Affymetrix software. For 97.5% of genes, the signal log ratio was within +/− 1.0 (see Supporting Methods, Fig. 6, and Table 4, which are published as supporting information on the PNAS web site).
Real-Time RT-PCR.
The cDNAs prepared for the microarray analysis were used in a TaqMan assay by using the LightCycler System (Roche). The Standard Curve Method was used for the relative quantitation of expression for each gene. Expression of the housekeeping form of porphobilinogen dehydrogenase gene (HMBS) was used to normalize the expression data (10). TaqMan probes containing a 5′ FAM reporter and a 3′ TAMRA quencher were purchased from Synthegen (Houston) (Table 1).
Table 1.
Analysis of gene expression by real-time RT-PCR
| Gene | Forward primer | Reverse primer | TaqMan probe |
|---|---|---|---|
| HMBS | ACTTTCCAAGCGGAGCCAT | CGAATCACTCTCATCTTTGG | CGGCTGCAACGGCGGAAGAAAAC |
| ICSBP | GTTTACCGAATTGTTCCTGAGGA | CATTCACCGCAGCCAGCAG | AGCAAAAATGCAAACTAGGCGTGGCA |
| TAL1 | ATGAGATGGAGATTACTGATG | GCCCCGTTCACATTCTGCT | ACCAACAGCCGGGAGCGATGG |
| FLT3 | AGCCTGCGGAGAGAGTAGCC | GGAATGTCCTCACACCTACCAAA | TCCATCTCTCTGCTGAAAGGTCGCCTG |
Results
Gene Expression Profiling of CD34+ Progenitor Cells in t-AML Patients.
The hallmark of t-AML induced by alkylating agents is trilineage dysplasia, suggesting that this disorder is the consequence of acquired somatic mutations in hematopoietic progenitor cells (1). The CD34 antigen is expressed on hematopoietic stem cells but not in fully differentiated cells. CD34+ cells account for ≈1–2% of normal human bone marrow cells. Thus, CD34+ mononuclear cells are highly enriched in hematopoietic progenitor cells (11). To avoid “pseudopositive” results due to varying proportions of specific cell populations in normal and leukemia samples (12), we purified the CD34+ progenitor cells from cryopreserved bone marrow samples from 14 t-AML patients. Of these, 11 (79%) had clonal chromosomal abnormalities. Three patients had −7, four patients had a del(5q) or an unbalanced translocation leading to loss of 5q, one patient had +13 as the sole abnormality, and one patient had a t(7;11)(p15;q23) that did not involve MLL. Two patients had recurring translocations involving 3q26.2 together with loss of 5q and 17p [t(3;21)(q26.2;q22), dic(5;17)(q11.2;p11.2); and t(3;3)(q21;q26.2), del(5)(q15q34),–17)].
To define the biological characteristics of t-AML, we compared the gene expression profiles of CD34+ progenitor cells from t-AML patients and normal individuals by using Affymetrix arrays, which contain probes for 12,600 genes/transcripts. These genes were filtered to 2,945 genes, which were expressed in CD34+ progenitor cells from three control samples and had relatively stable expression levels (≤2-fold). A two-dimensional hierarchical clustering algorithm was used to analyze the gene expression data. Although many of the leukemias contained multiple cytogenetic abnormalities, we identified two major groups (A and B) (Fig. 1). t-AML patients with abnormalities of chromosomes 5 or 7 have similar clinical and morphological features. Surprisingly, these patients did not cluster into the same group. Group A included all three patients with –7 but no abnormality of chromosome 5. Four of six patients with an abnormality of chromosome 5 clustered into one group (Group B). All four of these patients had complex karyotypes. The exceptional patients, samples 5–4 and 2–4, were the only patients we analyzed who had recurring translocations [t(3;3)(q21;q26.2) or t(3;21)(q26.2;q22)]. A possible explanation is that the recurring translocations may have defined the gene expression profile, a phenomenon identified with other translocations in leukemias (7, 8). The remainder of the patients with other abnormalities or with a normal karyotype clustered into Group A. Two patients with a normal karyotype, and one patient with +13 clustered into a subgroup (Group A1) (Fig. 1). Because there are a limited number of patients with each chromosome abnormality, we were unable to separate the remaining Group A patients into additional subgroups. However, expression profiling of large groups of patients suggests that this analysis can accurately identify each of the known cytogenetic and prognostic subgroups of childhood acute lymphoblastic leukemia (ALL) (8). Thus, it is likely that Group A may be subdivided further, e.g., −7/del(7q) subgroup, by future analyses of additional patients.
Fig 1.

Two-dimensional hierarchical clustering of gene expression data for 2,945 genes. Each row represents a leukemia sample and each column represents a single gene. The figure illustrates the ratio of hybridization signal intensity of cRNA prepared from each experimental mRNA sample to that of control sample 1-1. Expression levels greater than the normal control value are shown in red, and levels less than this value are shown in blue. Increasing distance from the control value is depicted by increasing color intensity. Recurring cytogenetic abnormalities for the corresponding samples are listed on the right.
We identified 61 genes whose expression pattern is significantly altered in t-AML, and whose expression best distinguishes the unique groups of t-AML. Twenty-nine genes are overexpressed in Group A leukemias, whereas the remaining 32 are overexpressed in Group B leukemias (Fig. 2). Forty-six genes were expressed in ≥50% of the leukemias, but not in the normal control samples (see Tables 5 and 6, which are published as supporting information on the PNAS web site).
Fig 2.

The expression level of 61 genes distinguishes Group A from Group B t-AML. The upper 29 genes have relatively high expression levels in Group A leukemias but not in Group B leukemias. In contrast, the lower 32 genes have relatively high expression levels in Group B leukemias, characterized by a −5/del(5q). Each column represents a leukemia sample, and each row represents the gene indicated by the gene accession numbers and gene symbol or DNA sequence names on the right (note that four genes are represented twice on the array with different oligonucleotides). Red indicates up-regulation and blue depicts down-regulation (see color bar in Fig. 1).
CD34+ Progenitor Cells Are Blocked at an Early Stage of Differentiation in t-AML.
To define the biological features that are common to CD34+ stem cells from t-AML patients, we searched for genes whose expression pattern differed between the control and leukemia samples. Ten genes that are essential for the function of mature neutrophils, platelets, and eosinophils were expressed in normal CD34+ stem cells from all three control samples but were not expressed in CD34+ stem cells from most of the t-AML patients (12/14) (Fig. 3). Among these genes, PF4 (platelet factor 4) and PPBP (proplatelet basic protein) encode proteins that are involved in wound repair, inflammation, and coagulation and are released from the α granules of activated platelets (13). The serine proteases, cathepsin G (CTSG), neutrophil elastase (ELA2), proteinase 3 (PRTN3), and azurocidin (AZU1) are antibiotic proteins of neutrophilic granules that contribute to the destruction of ingested microorganisms (14). Myeloperoxidase is a lysosomal enzyme in neutrophils. The genes encoding eosinophil cationic protein (RNASE3) and Charcot–Leyden crystal protein are expressed in the eosinophil lineage (15). These data provide molecular evidence that the initiating somatic mutation(s) occurs in early progenitor cells, which results in the dysregulation of genes essential for the differentiation of multiple hematopoietic lineages.
Fig 3.

Ten hematopoietic-specific genes were significantly down-regulated in CD34+ cells from t-AML patients. Red indicates up-regulation and blue indicates down-regulation (see color bar in Fig. 1).
t-AML with −5/del(5q) (Group B) Has a Distinct Expression Profile.
The CD34+ progenitor cells from patients with –5/del(5q)/t(5q) (Group B) show a gene expression pattern that is markedly different from that of other t-AMLs (Group A) (Fig. 2). The UoC-M1 cell line, derived from a patient with AML de novo with a del(5q) and −7, showed a similar expression pattern to that of t-AMLs with −5/del(5q), suggesting that UoC-M1 maintains the characteristic features of leukemias with −5/del(5q). Genes involved in cellular proliferation were expressed at higher levels in Group B than in Group A, suggesting that CD34+ progenitor cells from t-AML with a −5/del(5q) may have an increased proliferative rate. This proliferation signature includes the cell cycle control genes encoding cyclin A2 (CCNA2), cyclin E2 (CCNE2), and CDC2, and the cell cycle checkpoint gene BUB1. CKS2 (CDC28 protein kinase 2) and the MYC oncogene were also up-regulated in Group B as compared with Group A leukemias. MYC induces CKS2, which modulates cyclin-dependent kinases thus regulating proliferation (16). We found that t-AML Group B and the UoC-M1 cell line lack expression of the gene encoding IFN consensus sequence-binding protein (ICSBP), a regulator of proliferation and differentiation in hematopoietic cells, in contrast to CD34+ progenitor cells from normal individuals and most other t-AML samples (Fig. 4A).
Fig 4.

Comparison of ICSBP (A), TAL1 (B), and FLT3 (C) gene expression data obtained by microarray analysis (open bars) and real-time RT-PCR analysis (shaded bar). The y-axis represents the log base ratio of fold-change in gene expression from each sample to the average of three control samples. The x-axis represents different samples.
A number of genes located on chromosome 5, including NR3C1 (glucocorticoid receptor gene), APC, CYFIP2, IQGAP2, DOCK2, ADRB2, CSPG2, and two putative genes (U30521, HSPC16), were down-regulated in all t-AML samples with −5/del(5q), suggesting that the loss of 5q may result in a gene-dosage effect for some of the genes on this chromosome arm.
t-AML Group A1 Has a Unique Expression Profile.
One of the clearest distinctions between the gene expression patterns of Groups A1 and B is the expression of several critical transcriptional regulators of differentiation for multiple hematopoietic lineages, namely the TAL1/SCL, GATA1, and EKLF transcription factors. All three genes are dramatically down-regulated in Group A1 leukemias (Fig. 5A), whereas Group B leukemias have normal or even high levels of expression of these genes. The expression levels of several genes encoding signaling proteins, FLT3/STK1 and PI3K (PIK3C2B), as well as the antiapoptotic gene, BCL2, are coordinately up-regulated in Group A1, but not in Group B leukemias (Fig. 5B). Expression of FLT3 was increased by 4- to 13-fold in Group A1 leukemias, when compared with controls and Group B leukemias (Fig. 5B). FLT3 plays a role in the maintenance of pluripotent stem cells, and the development of B cell progenitors and dendritic cells (17). The concurrent expression of FLT3, and proteins downstream of the FLT3 signaling pathway (PI3K and BCL2) suggests that FLT3 plays a role in a cell survival signaling pathway in these leukemias.
Fig 5.

Comparison of expression levels of (A) TAL1, GATA1, EKLF, and RUNX1/AML1 transcription factor genes or (B) FLT3, PIK3C2B, and BCL2 in CD34+ cells from t-AML patients. Each bar represents the fold change in gene expression relative to the average of the three controls.
Validation of Oligonucleotide Array Gene Expression Results.
To validate the expression data obtained from the oligo-array analysis by an independent method, we chose the TaqMan real time RT-PCR assay to analyze the expression level of several genes (ISCBP, TAL1, and FLT3). The relative expression levels obtained from both assays are in good agreement (Fig. 4).
Discussion
Expression Profiling Provides Molecular Evidence for a Block in Early Differentiation of Multiple Hematopoietic Lineages in t-AML.
To elucidate the molecular basis of t-AML, we performed expression profiling of CD34+ stem/progenitor cells. An advantage to our approach is that the initiating mutation(s) occurs in CD34+ progenitor cells, and the resultant leukemic blasts retain this marker. Based on clustering analysis of ≈3,000 genes, we identified distinct subtypes of t-AML that are characterized by unique expression profiles.
A common feature that we identified in all t-AMLs examined was the down-regulation of genes required for the function of differentiated hematopoietic cells, e.g., platelets, neutrophils, and eosinophils (Fig. 3). These include the serine proteases (CTSG, ELA2, PRTN3, AZU1) and myeloperoxidase involved in neutrophil function, and the platelet-specific proteins PF4 and PPBP. Proteinase 3 (PRTN3) regulates growth and differentiation of the myelomonocytic lineage (18). Platelet factor 4 (PF4) functions in hemostasis, in cell growth and differentiation, and in the survival of hematopoietic progenitor cells (19). The expression of these genes in normal CD34+ progenitor cells is consistent with their role in normal hematopoietic development; down-regulation in t-AML may be linked to an arrest of maturation in early progenitor cells and is consistent with the characteristic feature of trilineage dysplasia in t-AML. PF4 and PPBP are closely linked on chromosome 4 (20); similarly, the ELA3, PRTN3, and AZU1 genes are closely linked on chromosome 19 (21). The chromosomal location, together with their coordinated expression in hematopoietic cells (and down-regulation in t-AML), suggests a close evolutionary relationship.
Expression Profiling Reveals Distinct Molecular Profiles in t-AML.
Leukemias with −5/del(5q) have a distinct expression pattern that differs from t-AML with –7 alone, recurring translocations, other cytogenetic abnormalities, or a normal karyotype. Further analysis of the expression pattern revealed that Group B t-AMLs with –5/del(5q) have high expression of proliferation signature genes, such as the genes encoding cyclin A2, cyclin E2, CDC2, CKS2, BUB1, and MYC. In contrast, the expression of the ICSBP gene was significantly down-regulated in Group B leukemias when compared with that of normal individuals. ICSBP functions as a negative regulator of IFN-induced genes, and plays a role in regulating proliferation, differentiation, and apoptosis of hematopoietic cells (22, 23). Icsbp−/− mice develop a syndrome resembling chronic myelogenous leukemia, and the bone marrow is characterized by an increased number of myeloid progenitor cells as well as expanded populations of granulocytic, monocytic, and lymphoid lineages (23). Thus, the absence of ICSBP may play a key leukemogenic role in this subtype of t-AML.
Group B leukemias were also characterized by decreased expression of a number of genes on chromosome 5, including APC, a tumor suppressor gene mutated in sporadic colorectal carcinomas, as well as in familial adenomatous polyposis coli. A constitutional decrease in APC expression of 50% results in a predisposition to tumors (24). However, there is no evidence that haploinsufficiency of APC contributes to leukemogenesis. In previous studies, we mapped the commonly deleted segment (CDS) of 5q to a 1.5-Mb interval flanked by D5S479 and D5S500 (25). None of the genes located within the CDS are included on the expression arrays. Whether haploinsufficiency of a gene(s) within the CDS of 5q contributes to leukemogenesis in t-AMLs with −5/del (5q) is unknown.
In Group A1 leukemias, we observed loss of expression of TAL1, GATA1, and EKLF, which encode transcription factors that are essential for hematopoiesis. TAL1 is involved in the recurring t(1;14) in T cell leukemias, and a molecular deletion of this gene is the most common genetic alteration associated with T cell acute lymphoblastic leukemia (26). Homozygous deletion of Tal1 results in the absence of yolk sac hematopoiesis and embryonic lethality in mice, indicating that TAL1 is a key regulator of early hematopoiesis (27). Targeted mutagenesis in embryonic stem (ES) cells and in mice revealed that Gata1 is essential for the development of both erythroid and megakaryocyte lineages (28). Moreover, Tal1−/− ES cells have a defect in erythropoiesis that is similar to that observed in ES cells with loss of Gata1 (28). EKLF regulates expression of the β globin gene and is required for erythroid development. Homozygous Eklf−/− mice have defective fetal liver hematopoiesis, leading to a fatal anemia during early fetal development (29). Taken together, these results suggest that dysregulation of transcription factors critical to the development of hematopoietic stem cells may contribute to the pathogenesis of this subtype of t-AML.
FLT3, PI3K/PIK3C2B, and BCL2 were highly expressed in Group A1 leukemias. The role of FLT3 in early hematopoiesis is complex and includes the regulation of diverse cellular responses, such as mitogenesis, chemotaxis, cell survival, and differentiation. FLT3 is expressed primarily in human CD34+ stem/progenitor cells, and encodes a member of the type III receptor tyrosine kinase family (17, 30). Constitutively activating mutations in FLT3 occur in the malignant cells of 25–30% of AML patients (31). The majority of these result from internal tandem duplications within the juxtamembrane domain of the FLT3 gene (20–25%), with a small proportion (≈5%) resulting from a substitution mutation at Asp-835 in the FLT3 kinase domain (17). We examined these three samples for internal tandem duplications of FLT3 and for Asp-835 substitution mutations, and determined that they did not contain FLT3 mutations (data not shown). Surprisingly, overexpression of FLT3 has been reported in acute lymphoblastic leukemia characterized by MLL translocations (7); thus, we examined these cases for MLL rearrangements. None had a MLL translocation, but one patient had an internal tandem duplication of MLL exons 2–8 (patients 5–7; data not shown). Within Group A1, leukemia 4–6 with +13, had the highest expression of FLT3 (Fig. 5B); notably the FLT3 gene maps to this chromosome. Whether the additional copy of FLT3 contributes to the overexpression of FLT3 in CD34+ progenitor cells from this patient is unknown but raises the possibility that +13, a recurring abnormality in 5% of AML, may represent an additional mechanism resulting in up-regulation of FLT3 function in AML. In a murine transplantation assay, mutant FLT3 with internal tandem duplications, but not wild-type FLT3, induces a myeloproliferative disorder (32). In this regard, overexpression of the FLT3 ligand predisposes mice to the development of leukemia, suggesting that, in some contexts, increased signaling through FLT3 contributes to leukemogenesis.
BCL2, an antiapoptotic gene, is up-regulated in t-AML Group A1 but not in Group B. Overexpression of BCL2 interferes with programmed cell death and has been reported to protect CD34+ AML blasts from chemotherapy-induced apoptosis, leading to drug resistance (33). Downstream targets of FLT3 tyrosine kinase activity include the p85 subunit of PI3K, which phosphorylates the AKT serine/threonine kinase, resulting in expression of antiapoptotic proteins, such as BCL-XL and BCL2. The coordinated overexpression of FLT3, PI3K, and BCL2 suggests that FLT3 may play a role as a survival factor by regulating the expression of BCL2 through the PI3K pathway.
Models for the Pathogenesis of t-AML.
An important aspect of leukemia biology is the elucidation of the spectrum of mutations that cooperate in the pathways leading to leukemogenesis. There is growing evidence that a limited number of molecular pathways may be involved. Gilliland and colleagues have described an emerging paradigm in AML, namely, the cooperation between constitutively activated tyrosine kinase molecules, such as FLT3, and transcription factor fusion proteins (34). In this model, the activated tyrosine kinase confers a proliferative and/or antiapoptotic activity, whereas the fusion protein impairs normal differentiation pathways but has a limited effect on cellular proliferation. In the context of this model, loss of TAL1, GATA1, and EKLF expression in Group A1 leukemias may result in impaired differentiation of hematopoietic cells, whereas overexpression of FLT3, PIK3C2B, and BCL2 result in a proliferative and survival advantage (Table 2). The initiating mutation(s) in these leukemias is unknown. In contrast, the initiating mutation in Group B leukemias is likely to be mutation/haploinsufficiency of a gene(s) on 5q. Loss of expression of ICSBP may have pleiotropic effects, leading to impaired differentiation and/or a proliferative and survival advantage, whereas increased expression of cell cycle regulatory proteins (CCNA2, CCNE2, CDC2) would be predicted to result in a proliferative advantage.
Table 2.
Model for the pathogenesis of t-AML
| Group | Initiating event | Impaired differentiation/ immortalization | Proliferative/ survival advantage |
|---|---|---|---|
| A1 | Unknown | ⇓ TAL1 | ⇑ FLT3 |
| ⇓ GATA1 | ⇑ PIK3C2B | ||
| ⇓ EKLF | ⇑ BCL2 | ||
| B | Mutation/ haploinsufficiency of a gene(s) on 5q | ⇓ ICSBP | ⇓ ICSBP |
| ⇑ TAL1 | ⇑ CCNA2 | ||
| ⇑ CCNE2 | |||
| ⇑ CDC2 | |||
| ⇑ MYC |
Pedersen-Bjergaard et al. (35) have proposed that t-AML can be subdivided into at least eight genetic pathways with different etiologies and biologic characteristics. The first class is characterized by abnormalities of chromosome 7, often as the sole abnormality, with NRAS mutations and CDKN2B/p15 methylation occurring as cooperating mutations. Class II is characterized by abnormalities of chromosome 5, complex karyotypes [often with –7/del(7q)], and TP53 (p53) mutations. Our data are consistent with this model in that the patients we examined with –7 and simple karyotypes clustered in Group A (samples 5–2 and 5–8), whereas patients with –5/del(5q) and complex karyotypes clustered into Group B. Nonetheless, our data extend this model and provide new insights into the underlying biology of t-AML.
Global Gene Expression Analysis May Contribute to the Development of New Treatment Strategies for t-AML.
t-AML is highly resistant to current forms of remission induction therapy, and new therapeutic approaches are needed. The identification of unique expression profiles in t-AMLs provides insights into the molecular mechanism(s) of drug resistance. Loss of expression of ICSBP is observed in a high proportion of patients with chronic myelogenous leukemia (CML) (79%) or AML (66%) (36). In CML, treatment with IFN-α results in an increase in ICSBP gene transcription in the leukemia cells, and the development of resistance to IFN treatment is associated with loss of ICSBP expression (36). Furthermore, Icsbp-deficient myeloid cells undergo decreased spontaneous apoptosis, and overexpression of ICSBP in the human U937 monocytic leukemia cell line restores apoptosis and susceptibility to various chemotherapeutic agents (37). ICSBP is down-regulated in t-AMLs with −5/del(5q), raising the possibility that IFN treatment may be effective in this group of patients. BCL2 is highly expressed in Group A1 leukemias; thus, therapies targeting BCL2 may prove effective in these patients. Other potential targets in t-AML and AML de novo include FLT3 inhibitors currently in trial. The identification of unique expression profiles in t-AML lends further support that the individualization of cancer treatment based on the spectrum of mutations will provide successful new therapeutics.
Supplementary Material
Acknowledgments
We thank the technologists in the Cancer Cytogenetics Laboratory for expert technical assistance and Marjorie Isaacson and Ludmila Amerik for data management. We are grateful to Dr. Chris Dyanov in the Functional Genomics Core Facility (Univ. of Chicago) for assistance in performing the microarray analysis and to Dorie Sher, Dr. Wendy Stock, and Dr. Robert Anders for assistance in real-time RT-PCR assays. We are also grateful to members of the Le Beau laboratory and to Dr. Kevin Shannon for helpful discussions. This work was supported by Public Health Service Grant CA40046 (to M.M.L. and R.A.L.).
Abbreviations
AML, acute myeloid leukemia
t-AML, therapy-related AML
t-MDS, therapy-related myelodysplastic syndrome
ICSBP, IFN consensus sequence-binding protein
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Thirman M. J. & Larson, R. A. (1996) Hematol. Oncol. Clin. North Am. 10, 293-320. [DOI] [PubMed] [Google Scholar]
- 2.Smith M. A., McCaffrey, R. P. & Karp, J. E. (1996) J. Natl. Cancer Inst. 88, 407-418. [DOI] [PubMed] [Google Scholar]
- 3.Godley L. A. & Larson, R. A. (2001) in The Myelodysplastic Syndromes: Pathobiology and Clinical Management, ed. Bennett, J. M. (Dekker, New York), pp. 139–176.
- 4.Pedersen-Bjergaard J. & Rowley, J. D. (1994) Blood 83, 2780-2786. [PubMed] [Google Scholar]
- 5.Golub T. R., Slonim, D. K., Tamayo, P., Huard, C., Gaasenbeek, M., Mesirov, J. P., Coller, H., Loh, M. L., Downing, J. R., Caligiuri, M. A., et al. (1999) Science 286, 531-537. [DOI] [PubMed] [Google Scholar]
- 6.Alizadeh A. A., Eisen, M. B., Davis, R. E., Ma, C., Lossos, I. S., Rosenwald, A., Boldrick, J. C., Sabet, H., Tran, T., Yu, X., Powell, J. I., et al. (2000) Nature 403, 503-511. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong S. A., Staunton, J. E., Silverman, L. B., Pieters, R., den Boer, M. L., Minden, M. D., Sallan, S. E., Lander, E. S., Golub, T. R. & Korsmeyer, S. J. (2002) Nat. Genet. 30, 41-47. [DOI] [PubMed] [Google Scholar]
- 8.Yeoh E.-J., Ross, M. E., Shurtleff, S. A., Williams, W. K., Patel, D., Mahfouz, R., Behm, F. G., Raimondi, S. C., Relling, M. V., Patel, A., et al. (2002) Cancer Cell 1, 133-143. [DOI] [PubMed] [Google Scholar]
- 9.Allen R. J., Smith, S. D., Moldwin, R. L., Lu, M.-M., Giordano, L., Vignon, C., Suto, Y., Harden, A., Tomek, R., Veldman, T., et al. (1998) Leukemia 12, 1119-1127. [DOI] [PubMed] [Google Scholar]
- 10.Chretien S., Dubart, A., Beaupain, D., Raich, N., Grandchamp, B., Rosa, J., Goossens, M. & Romeo, P. H. (1988) Proc. Natl. Acad. Sci. USA 85, 6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krause D. S., Fackler, M. J., Civin, C. I. & May, W. S. (1996) Blood 87, 1-13. [PubMed] [Google Scholar]
- 12.Miyazato A., Ueno, S., Ohmine, K., Ueda, M., Yoshida, K., Yamashita, Y., Kaneko, T., Mori, M., Kirito, K., Toshima, M., et al. (2001) Blood 98, 422-427. [DOI] [PubMed] [Google Scholar]
- 13.Majumdar S., Gonder, D., Koutsis, B. & Poncz, M. (1991) J. Biol. Chem. 26, 5785-5789. [PubMed] [Google Scholar]
- 14.Lehrer R. I. & Ganz, T. (1990) Blood 76, 2169-2181. [PubMed] [Google Scholar]
- 15.Weller P. F., Goetzl, E. J. & Austen, K. F. (1980) Proc. Natl. Acad. Sci. USA 77, 7440-7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coller H. A., Grandori, C., Tamayo, P., Colbert, T., Lander, E. S., Eisenman, R. N. & Golub, T. R. (2000) Proc. Natl. Acad. Sci. USA 97, 3260-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reilly J. T. (2002) Br. J. Haematol. 116, 744-757. [DOI] [PubMed] [Google Scholar]
- 18.Bories D., Raynal, M. C., Solomon, D. H., Darzynkiewicz, Z. & Cayre, Y. E. (1989) Cell 59, 959-968. [DOI] [PubMed] [Google Scholar]
- 19.Han Z. C., Lu, M., Li, J., Defard, M., Boval, B., Schlegel, N. & Caen, J. P. (1997) Blood 89, 2328-2335. [PubMed] [Google Scholar]
- 20.Tunnacliffe A., Majumdar, S., Yan, B. & Poncz, M. (1992) Blood 79, 2896-2900. [PubMed] [Google Scholar]
- 21.Zimmer M., Medcalf, R. L., Fink, T. M., Mattmann, C., Lichter, P. & Jenne, D. E. (1992) Proc. Natl. Acad. Sci. USA 89, 8215-8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bovolenta C., Driggers, P. H., Marks, M. S., Medin, J. A., Politis, A. D., Vogel, S. N., Levy, D. E., Sakaguchi, K., Appella, E., Coligan, J. E., et al. (1993) Proc. Natl. Acad. Sci. USA 91, 5046-5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holtschke T., Lohler, J., Kanno, Y., Fehr, T., Giese, N., Rosenbauer, F., Lou, J., Knobeloch, K. P., Gabriele, L., Waring, J. F., et al. (1996) Cell 87, 307-317. [DOI] [PubMed] [Google Scholar]
- 24.Yan H., Dobbie, Z., Gruber, S. B., Markowitz, S., Romans, K., Giardiello, F. M., Kinzler, K. W. & Vogelstein, B. (2002) Nat. Genet. 30, 25-26. [DOI] [PubMed] [Google Scholar]
- 25.Zhao N., Stoffel, A., Wang, P. W., Eisenbart, J. D., Espinosa, R., III, Larson, R. A. & Le Beau, M. M. (1997) Proc. Natl. Acad. Sci. USA 94, 6948-6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia Y., Brown, L., Yang, C. Y.-C., Tsou Tsan, J., Siciliano, M. J., Espinosa, R., III, Le Beau, M. M. & Baer, R. J. (1991) Proc. Natl. Acad. Sci. USA 88, 11416-11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robb L., Lyons, I., Li, R., Hartley, L., Kontgen, F., Harvey, R. P., Metcalf, D. & Begley, C. G. (1995) Proc. Natl. Acad. Sci. USA 92, 7075-7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shivdasani R. A., Fujiwara, Y., McDevitt, M. A. & Orkin, S. H. (1997) EMBO J. 16, 3965-3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nuez B., Michalovich, D., Bygrave, A., Ploemacher, R. & Grosveld, F. (1995) Nature 375, 316-318. [DOI] [PubMed] [Google Scholar]
- 30.Small D., Levenstein, M., Kim, E., Carow, C., Amin, S., Rockwell, P., Witte, L., Burrow, C., Ratajczak, M. Z., Gewirtz, A. M., et al. (1994) Proc. Natl. Acad. Sci. USA 91, 459-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yokota S., Kiyoi, H., Nakao, M., Iwai, T., Misawa, S., Okuda, T., Sonoda, Y., Abe, T., Kahsima, K., Matsuo, Y., et al. (1997) Leukemia 11, 1605-1609. [DOI] [PubMed] [Google Scholar]
- 32.Kelly L. M., Liu, Q., Kutok, J. L., Williams, I. R., Boulton, C. L. & Gilliland, D. G. (2002) Blood 99, 310-318. [DOI] [PubMed] [Google Scholar]
- 33.Williams G. T. (1991) Cell 65, 1097-1098. [DOI] [PubMed] [Google Scholar]
- 34.Kelly L., Clark, J. & Gilliland, D. G. (2002) Curr. Opin. Oncol. 14, 10-18. [DOI] [PubMed] [Google Scholar]
- 35.Pedersen-Bjergaard J., Anderson, M. K., Christiansen, D. H. & Nerlov, C. (2002) Blood 99, 1909-1912. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt M., Nagel, S., Proba, J., Thiede, C., Ritter, M., Waring, J. F., Rosenbauer, F., Huhn, D., Wittig, B., Horak, I., et al. (1998) Blood 91, 22-29. [PubMed] [Google Scholar]
- 37.Gabriele L., Phung, J., Fukumoto, J., Segal, D., Wang, I. M., Giannakakou, P., Giese, N. A., Ozato, K. & Morse, H. C., III (1999) J. Exp. Med. 190, 411-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}