

Abstract
Infants born prematurely risk significant life-long cognitive disability, representing a major pediatric health crisis. The neuropathology of this cohort is accurately modeled in mice subjected to sublethal postnatal hypoxia. Massively parallel transcriptome analysis using cDNA microchips (9,262 genes), combined with immunohistochemical and protein assays, reveals that sublethal hypoxia accentuates genes subserving presynaptic function, and it suppresses genes involved with synaptic maturation, postsynaptic function, and neurotransmission. Other significantly affected pathways include those involved with glial maturation, vasculogenesis, and components of the cortical and microtubular cytoskeleton. These patterns reveal a global dysynchrony in the maturation programs of the hypoxic developing brain, and offer insights into the vulnerabilities of processes that guide early postnatal cerebral maturation.
Despite the development of sophisticated perinatal care techniques, the incidence of neurodevelopmental handicap among the very low birth weight survivors of newborn intensive care remains high (1). Infants born at <1,000 g represent 1% of all United States live births, and 85% survive. Children born in this condition are now a major pediatric health problem (2). By 8 years of age, half of these children require special education, one in five has repeated a grade, and 10–15% suffer serious motor handicaps (1, 3). The genesis of these cognitive and motor deficits in otherwise normal infants remains poorly understood.
A failure in oxygen delivery to the developing brain is a major problem for such infants, often as a result of immature lung development (4). Over 60% of very low birth weight infants experience such injury (2, 5). Thus, moderate sublethal hypoxia, per se, presumably irreversibly alters key developmental programs of the newborn brain, abrogating its developmental and cognitive potential. Yet, insight into the molecular and cellular events that define this vulnerability to sublethal hypoxia has been limited, because infant studies are restricted to in vivo imaging and rare postmortem evaluations (6). We have therefore developed a rodent model of this condition (7, 8). This model involves a period of moderate postnatal hypoxia in the rodent, during a period (in the rodent) that corresponds closely to late prenatal human brain development. Specifically, as in the preterm infant at the end of the second trimester, neuronal generation in the newborn rodent brain is largely complete, whereas axonal and dendritic branching is ongoing and synaptogenesis is just beginning (9). In newborn rats, the first 20 postnatal days represent the period of rapid differentiation of axons and dendrites (9–12), and between the first and third postnatal week the cortex doubles in volume (9). These data suggest that the newborn rodent provides a good model for the developing preterm human brain during the third trimester of gestation. This model also accurately reproduces the morphologic features characteristic of very low birth weight preterm infants, including reductions in cortical volume, decreases in central white matter, and cerebral ventriculomegaly. With this model, we have now sought a global molecular understanding of this complex condition. By using massively parallel transcriptome analysis, we have identified key developmental pathways that are most severely impacted by postnatal hypoxia and have validated these findings by protein and morphologic assessment.
Methods
Animal Model.
C57BL/6 litters, fostered by CD-1 dams, were reared under hypoxic (Hx; ambient O2 = 9.5 ± 1.0%) or normoxic (Nx; ambient O2 = 22 ± 1.0%) conditions from postnatal day 3 (P3). Gas levels were monitored with a Cameron dual channel oxygen monitor (Cameron Instrument Co., Port Aransas, TX). The chamber was opened 10 min biweekly for routine care.
Microarray Protocol and Data Analysis.
Experimental animals and age-matched controls were killed at postnatal days 3, 4, 6, and 11 (P3, P4, P6, and P11). Three sets of animals were individually measured for each condition. RNA was extracted from the whole brain, less the cerebellum, by using Trizol (Invitrogen). Poly(A)+ RNA was extracted by using Oligotex (Qiagen, Valencia, CA). Reference RNA was pooled mRNA from all of the experimental samples. Probe labeling, hybridization, and scanning were performed by Incyte Genomics (Palo Alto, CA). The mouse UniGene 1 cDNA microarrays contained 9,596 elements mapped to 9,262 unique annotated genes and EST clusters (Incyte Genomics). Annotation was based on Incyte Genomics' UniGene Mus musculus build no. 86. Each array was hybridized with an experimental cDNA sample (Cy3-labeled) and a reference cDNA sample (Cy5-labeled). The scanned data were analyzed with gemtools software (Incyte Genomics) that allowed inspection and quantitation of both the reference and sample channels of the array. Data were further analyzed as below, and hierarchical clustering was used to display the expression pattern for selected genes (13).
To determine the significance of the changes induced by hypoxia, an unbalanced two-way ANOVA was conducted by using the logarithmic ratio of the balanced intensity of each experimental sample to the intensity of the reference sample for each gene. Specifically, i = 1 and 2 to index the Hx and control conditions, respectively, j = 1, 2, and 3 to index P4, P6, and P11, respectively, and k indexes the replicate samples. Genes without a replicate at any combination of condition by postnatal day were excluded from the analysis. Let Yijk be the logarithmic ratio of the intensity from any specified gene, and consider the following two-way ANOVA model:
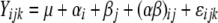 |
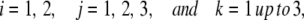 |
where μ is the overall mean logarithmic ratio for both conditions across all days, αi is the main effect for condition i reflecting the deviation of the mean from the overall mean under condition i, βj is the main time effect, (αβ)ij is the interaction effect between condition and day, and ɛijk is the random error for the kth replicate that refers to the individual mouse under condition i and occasion j. Normality for the logarithmic ratios was not assumed because their empirical distribution did not conform to a normal distribution. Thus, the P values were calculated by using a nonparametric approach (see below). The F statistics for the two effects were computed for each gene. A standard random-permutation procedure was used to generate the empirical distribution for the F values, similar to the one-way ANOVA (14). Under the null hypothesis (i.e., no true effect of condition or time) the logarithmic ratios of any combination of conditions by day can be randomly permuted, and the corresponding F values for the two effects after each permutation recomputed. This procedure was repeated 10,000 times, and the resulting 10,000 F values were obtained for the two effects for each gene. We chose the 99th percentiles from the empirical distributions of the F statistics as the critical values. If the F statistic of the effect from the original data set for a gene was larger than its critical value, the gene was regarded as significantly differentially expressed between the two conditions.
Immunohistochemistry and Histology.
Brains were perfusion fixed and postfixed at 4°C in 4% paraformaldehyde overnight, then stored in 30% sucrose/PBS at −20° as 60-μm cryosections. Before use, sections were blocked with 4% BSA in PBS for 1 h. Antibodies [vascular endothelial growth factor (VEGF), 1:200, and Flk-1, 1:100, Santa Cruz Biotechnology; myelin-associated glycoprotein (MAG), 1:100, Chemicon; synapsin 1, 1:500, Calbiochem] were applied overnight and visualized by fluorescent microscopy with the appropriate Cy3- or Cy5-labeled secondary antibodies. Digital images at a resolution of 0.5 μm per pixel were recorded by using a Photometrics (Tucson, AZ) 300 charge-coupled device (CCD) camera coupled to a Nikon microscope equipped with a DeltaVision platform under software control (SOFTWORX 2.5, Applied Precision, Issaquah, WA). Alternatively, high-resolution images were obtained by using an Olympus (New Hyde Park, NY) AX70 epifluorescent microscope.
Other.
Northern blots used poly(A)+ RNA (1 μg) separated on 1% formaldehyde/agarose, blotted, and hybridized with either a random-primed and labeled 250-bp EcoRI/HindIII VEGF cDNA fragment or a 1.2-kb cDNA fragment of the 18S RNA (Ambion, Austin, TX). For Western blotting, cortical lysates (20 μg) were separated on Bistris 4–12% polyacrylamide gels (Invitrogen) and probed with primary antibody to arylhydrocarbon receptor nuclear translocator 1 (ARNT 1) (1:500, Santa Cruz Biotechnology), hypoxia-inducible factor 1α (HIF-1α) (1:500, Santa Cruz Biotechnology), or β-actin (1:1,000, Sigma).
Results
Transcriptome Analysis of the Hx Newborn Brain.
The experimental protocol using Hx C57BL/6 mice retarded growth and reduced brain weight and size (Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). Ventricular volumes were increased, and the cortices thinned. To evaluate the overall transcriptional response of the newborn brain to Hx stress, 21 cDNA microarray analyses were performed, representing triplicate samples analyzed at P4, P6, and P11 for control and Hx animals and triplicate Nx animals killed at P3. Data were available for 9,115 (98%) of the clones. Adequate triplicate determinations were achieved for 7,743 genes (84%). Unlike conventional cDNA array experiments in which a comparison is made between just two experimental conditions, the present investigation required a study over time under normoxia vs. hypoxia. Of interest was how Hx conditions would alter brain development. An analysis was thus sought that would detect significant deviations in the developmental profile. To accomplish this result, the ratio of the intensity of each experimental sample to the intensity of the reference sample was obtained for each gene at each time point. These raw expression data were then recompiled for every sample as a Hx/Nx ratio, a task possible because all samples shared a common reference. Significant changes were identified by using a two-way ANOVA as described in Methods. A complete summary of the genes analyzed, and their expression profiles, organized into functional or pathway hierarchies as annotated by Incyte Genomics, is presented as Table 1, which is published as supporting information on the PNAS web site. Cluster analysis revealed several patterns of response (Fig. 1A). Of the 7,743 genes with complete data, 1,109 (14%) were significantly changed (P < 0.01) (Table 1 and Fig. 1A). For these genes, the annotations provided by Incyte Genomics (www.incyte.com) were verified by manual comparison with the published literature and gene databases and grouped into functional categories. Whereas the available genes could be sorted in many ways, the criteria used required that each category must contain >12 genes and that all members of a category must contribute to a subset of related cellular or neural processes.
Fig 1.

cDNA microarray analysis of newborn mice subjected to sublethal hypoxia. Of the 9,262 genes analyzed, complete triplicate data for P4, P6, and P11 animals under Nx and Hx conditions were obtained for 7,743 genes. (A) Cluster analysis indicates that most were up-regulated (red) vs. down-regulated (green) by P11; some demonstrated biphasic responses even after triplicate analysis (see Table 1). The significance of this phased response is unknown. In total, 1,109 (14%) genes demonstrated significant (P < 0.01) changes over the course of the experiment. All genes were grouped into functional categories and ranked by the fraction of genes changed in each category. The total in each category is as indicated. Note that hypoxia most significantly impacted genes involved with synaptic function and neural transmission, followed by protein synthesis, apoptosis, stress response, and the membrane and microtubule cytoskeleton. (B) Cluster analysis revealed a striking divergence in the coordinate expression of genes involved with presynaptic function vs. genes involved in synaptic maturation, postsynaptic function, or neural transmission. Analysis for apoptosis genes and genes involved with the membrane and microtubule-associated cytoskeleton revealed a more variable pattern.
Of greatest interest were the changes identified in four categories: (i) genes involved with synaptic function, maturation, and neural transmission (38% altered); (ii) genes involved with apoptosis (30% altered); (iii) genes involved with stress responses (25%); and (iv) genes involved with the membrane cytoskeleton and microtubules (22% altered). These data are summarized in Table 2, which is published as supporting information on the PNAS web site, and Fig. 1. Postnatal hypoxia also yielded an overall elevation in transcription, and the machinery needed for this was increased (e.g., protein synthesis, translation, and transcription genes). Other categories that were less dramatically affected included metabolism, adhesion, secretion, and DNA/RNA processing, among others. Of the 4,869 genes with unknown function that were on the array, 597 (12%) were significantly altered.
Genes Involved with Synaptic Function, Maturation, and Neural Transmission Reveal a Dysynchrony in Expression That Correlates with Delayed Synapse Maturation.
Twenty-three of the 59 genes on the array (38%) subserving synaptic function, maturation, and neural transmission were significantly altered by chronic sublethal hypoxia (Table 2 and Fig. 1A). Cluster analysis of these genes revealed a striking divergence between those involved with presynaptic function vs. those marking neurite outgrowth, synaptic maturation, and postsynaptic or perisynaptic roles (Fig. 1B). The loss of transcription of postsynaptic and synaptic maturation genes typically occurred in two waves, an initial fall at P4, followed by recovery at P6, and a more profound fall at P11. Conversely, the bulk of the up-regulation of presynaptic genes occurred late in the time course of the Hx period (P11). Because these analyses directly compare the Hx vs. the Nx response during development, these results signal that late in the period of perinatal hypoxia there is a profound loss of coordinate regulation of neuronal gene transcription.
Examples of the significantly altered synaptic function genes include snapin (SNAP-25 binding protein), synapsin 1, reelin (Rln), and various glutamate receptors. The synaptic vesicle-associated proteins snapin and synapsin 1 were both up-regulated. In contrast, reelin and the NMDA1 and kainate glutamate receptors were down-regulated. Reelin is an extracellular matrix protein located in perineuronal nets and concerned with the modulation of neuronal plasticity; reelin deposition is associated with increasing synaptic integrity in the developing nervous system and may mark the sites of synapse formation (15–17). Inspection of other genes on the array that were less significantly altered but still probably important (P < 0.05) provided additional evidence of widespread alterations in pre- and postsynaptic pathways and a failure of synapse formation. Bassoon (Bsn), a cytomatrix protein associated with the formation of active synapses (18), was down-regulated. The gene for disabled-1 (Dab1), a tyrosine kinase adapter protein that modulates cytoskeletal protein expression by relaying the reelin signal in target neurons (17, 19, 20), was up-regulated, a result consonant with in vivo studies demonstrating that loss of reelin yields compensatory increases in disabled-1 in the developing mouse brain (21).
The distribution of synapsin 1 in the Hx postnatal brain also signaled marked alterations in synapse development (Fig. 2). In the developing fetal and early postnatal brain, synapsin 1 expression proceeds from the ventral white matter and the cortical plate into what will become layers 2 to 6 of the cortical gray matter (22). By P11 of Nx development, cortical synapses are abundant (23, 24). This pattern was evident in the Nx animals examined here, as measured by the diffuse appearance of a very fine punctate staining of the cortical neuropil with synapsin 1 by P11, with little residual staining of soma, axons, or white matter. Under Nx conditions, cortical structures also achieved a mature synapsin 1 pattern before limbic structures, recapitulating the pattern of fetal neurogenesis (25, 26). Synapses in the cingulate gyrus were less mature, as signified by a coarser pattern of synapsin 1 staining and a preponderance of synapsin 1 concentrated in the soma and axons; in the hippocampus, only coarse staining was evident in the dentate gyrus and CA3 and CA4 regions at P11. In contrast, the Hx brain displayed a developmentally inappropriate pattern of synaptic maturation in all regions. Whereas the coarse synapsin 1 staining of the cortical plate, typical of early postpartum animals (22), was gone, significant residual synapsin remained in the white matter, as well as in the soma and axonal processes of the cortical neurons. The density of mature synapses, as measured by the abundance of fine punctate synapsin 1-positive boutons, was also significantly reduced by hypoxia (Fig. 2). Overall, these changes suggest that hypoxia caused a loss of synchronization in the rate of synapse formation in different brain regions, changes consistent with the observed alterations in gene expression.
Fig 2.

Hypoxia disrupts synapse formation in the cortex and hippocampus. In the P11 Nx brain (Left), synapsin 1 stains a diffuse cortical zone throughout the neuropil characteristic of mature synapses (double arrowheads). An immature pattern of cell body and axonal staining is evident in the cingulate gyrus and in the hippocampus. In the P11 Hx brain (Right), an immature pattern of synapsin 1 staining (arrows) is evident, largely confined to the neuronal cell body and axons in cortical layer 5. Under Hx conditions, synapsin 1 is inapparent at P11 in the hippocampus but increased in the optic tract and internal capsule. High-resolution images of synapsin 1 (red, Middle and Bottom) reveal a finely punctate pattern of synaptic boutons in the Nx cortex, whereas, in the same region of the Hx cortex, synapsin 1 is largely confined to the soma and coarse neurites, a pattern characteristic of more immature neurons (44). DG, dentate gyrus. Nuclei are stained blue with 4′,6-diamidino-2-phenylindole (DAPI). The faint grid pattern evident in the composite micrographs is a technical artifact arising from an imperfect juxtaposition of high-resolution images.
Genes Marking Oligodendrocyte Differentiation Are Down-Regulated.
Other genes affecting synapse development and neural transmission involve central myelination. The oligodendrocyte genes encoding proteolipid protein, cyclic nucleotide phosphodiesterase, platelet-derived growth factor receptor-α (PDGF-α), and MAG are down-regulated (Table 1), highlighting the exceptional vulnerability of glia to Hx stress (27, 28). Most changed was MAG, a protein associated with the modulation of axonal growth and the establishment of the nodes of Ranvier (29, 30). MAG is also present in nonmyelin-forming perisynaptic Schwann cells that envelop axonal terminals and detect and modulate synaptic vesicle release (31). As such, MAG is a marker of mature oligodendroglia, and its loss is a surrogate marker of the loss of mature glia under hypoxia. At the protein level, as measured by immunofluorescence, MAG appeared to be reduced throughout the developing corpus callosum and fiber pathways of the P11 Hx brain (Fig. 3).
Fig 3.

Hypoxia induced a loss of myelin-associated proteins. In Hx P11 brains there was a down-regulation of MAG transcription and other myelin-associated genes. MAG was lost from the developing corpus callosum and myelinated fiber pathways. Hypoxia also induced an unusual MAG investiture of discrete clusters of cortical microvessels.
Membrane and Microtubule Skeleton Genes Are Significantly Altered.
When membrane-associated skeletal proteins and microtubule-associated proteins were considered as a group, 9 of 40 genes (22%) were changed (Table 2 and Fig. 1A). Cluster analysis of 16 significantly altered genes within this category (including 5 genes with P < 0.05) revealed no clear pattern of change (Fig. 1B). Because many of these genes are involved with neurite outgrowth and synapse formation and function because of their participation in vesicular transport and membrane organization (32, 33), some of these changes may simply parallel the alterations noted in the processes that guide synaptic maturation. Examples would include the changes in dynamin, ankyrin (Ank), and spectrin. Dynamin is a small G protein required for receptor-mediated endocytosis. Its loss correlates well with the reduced synapse maturity and complexity. In the CNS, ankyrins establish and maintain organized clusters of voltage-gated sodium channels at the nodes of Ranvier (34), a process initiated by the binding of MAG to the L1 cell adhesion molecule (L1CAM, both down-regulated by hypoxia, Table 1). The product of the ANK2 gene, AnkB, is typically expressed before the other ankyrins during early postnatal development, is required for the correct localization of certain voltage-gated sodium channels in other tissues (35), and may be required for dendritic maturation in the brain (30). Thus, the suppression of ANK2 transcription is consistent with a pattern of delayed synapse completion. Taken together with the loss of spectrin transcription (Fig. 1B), these results suggest widespread disruption of the mechanisms underlying receptor organization as effected by the spectrin–ankyrin skeleton.
Genes Involved with Apoptosis and Stress Response Are Altered.
Of the 60 genes most relevant to apoptosis, 18 (30%) were changed at P < 0.01 (Fig. 1B and Table 2). Cluster analysis revealed no dominant pattern of pro- or antiapoptotic gene expression. For example, while there was marked enhancement of caspase 3, a major effector protease, and other proapoptotic genes such as metaxin 2 [a mitochondrial protein necessary for tumor necrosis factor α (TNFα)-mediated apoptosis], an equal number of gene changes suggested antiapoptotic action. This latter group included the down-regulation of Bcl-2 antagonist killer (Bak), the loss of apoptosis-associated tyrosine kinase, and the enhancement of the FAS (CD95) apoptotic inhibitor. Finally, genes involved with stress response pathways were generally enhanced. Of the 244 such genes on the array, 61 (25%) exhibited significant (P < 0.01) alterations (Table 2). Such changes are not unexpected (36).
Genes Regulated by VEGF and Hypoxia-Inducible Factors Are Stimulated.
VEGF and its receptor, FMS-like tyrosine kinase 1 (Flk-1), were up-regulated in the Hx mouse brain (Table 1, Fig. 4). Examination by indirect immunofluorescence revealed enhanced staining throughout most of the cortical layers, as well as in the pyramidal cell layer and CA1 region of the hippocampus, and the granule cells of the dentate gyrus. Interestingly, both VEGF and Flk-1 were expressed in the neurons themselves, based on the appearance of VEGF-positive cells, and by earlier reports of a paracrine and autocrine role for this signaling system in the control of neuronal differentiation and development (8). These alterations in VEGF expression were also detected by Northern blot analysis, yielding quantitative correspondence with the microarray data (Fig. 4A).
Fig 4.

Hypoxia induced VEGF-regulated pathways in both neurons and the microvasculature. (A) Composite micrographs of representative sections of P11 brain harvested from Nx and Hx animals. VEGF and its receptor Flk-1 were increased in neurons throughout the cortex, with marked concentrations in the dentate gyrus and in the CA1 region of the hippocampus. Microarray analysis indicated a >1.4-fold enhancement (Hx/N) at P11 for VEGF (Table 1). This same result was also obtained by conventional Northern blot analysis (Inset: dark bars, Hx/N by blot; light bars, microarray results), by using freshly extracted mRNA standardized against 18S ribosomal RNA. (B) Western blot analysis, standardized against actin, of ARNT and HIF-1α. Note that although ARNT levels remain nearly unchanged, the upstream inducer of VEGF expression, HIF-1α, is up-regulated at P11.
Other genes involved with the Hx response include HIF-1α and genes downstream of this transcriptional pathway (Fig. 7, which is published as supporting information on the PNAS web site). ARNT 1 may synergize HIF-1α by enhancing its rate of translocation to the nucleus (37). HIF-1α is also degraded by a ubiquitin proteosome pathway that is blocked by hypoxia (38). The steady-state level of HIF-1α thus represents a combination of hypoxia-induced transcription and hypoxia-induced prolongation of its lifetime. Western blot analysis of the P3 to P11 Hx cortex revealed selectively increased HIF-1α (Fig. 4B). Curiously, in contrast to the mature brain (39), nearly a week was required after the onset of hypoxia for HIF-1α to be induced (Table 1, Fig. 4B). The genesis of this prolonged delay in the HIF-1α response appears not to arise from an absolute deficiency in constitutively synthesized HIF-1α, because this protein was detectable by Western blot as early as P3 (Fig. 4B). Change in other genes involved in HIF-1α signaling was also delayed (Table 1 and Fig. 7) and included recognized downstream effectors, including VEGF, Flk-1, connective tissue growth factor, and the angiogenesis-associated genes encoding procollagen type 1 α2 and tissue inhibitor of metalloproteinase.
Finally, it should be noted that several other genes associated with differentiation of the developing brain were altered by the Hx stress (Table 1). Examples include reductions in myeloid-associated differentiation marker gene (MYADM), a gene not previously recognized in neural tissue, and α2-HS-glycoprotein (AHSG), a protein found in the developing rat cortex between P5 and P10 that plays a role in early neocortical cerebral differentiation.
Discussion
Preterm birth remains a major cause of neurodevelopmental disability, and the impact of this insult on the developing brain involves the spectrum of genetic and epigenetic processes involved in corticogenesis during the last trimester of gestation. By using an established animal model that faithfully recapitulates the pathology of extremely premature birth (7, 8), we have explored the molecular basis of this pathology. Of 9,262 genes examined, significant changes (P < 0.01) were detected in 1,109 genes. Beyond an assortment of predictable stress and metabolic responses, the most coherent pattern that emerged was a global disregulation in the coordination of genes required to effect synapse formation and neural transmission. We thus term this loss of coordination in the expression of genes required to assemble mature synaptic structures dysynchrony, and we consider it to be the most significant pathology of the extremely premature brain (as modeled by moderate postnatal hypoxia). This conclusion is supported by several lines of data. Specifically: (i) genes involved in synaptic function and neural transmission are among the most frequently altered; (ii) these alterations are almost perfectly distributed such that most presynaptic genes are up-regulated, and postsynaptic genes or genes marking synaptic maturation (whether pre- or postsynaptic) are suppressed relative to the normal developmental program; (iii) genes involved in myelination and cytoskeletal organization and that contribute to synapse maturation and neural transmission are significantly altered; and (iv) hypoxia-responsive pathways are activated that not only stimulate angiogenesis and altered vascular permeability but also control neural differentiation (e.g., VEGF).
A detailed analysis of the altered genes strengthens the argument for dysynchrony as a key factor underlying the pathology of this disorder. For example, synapsin 1 and snapin are both up-regulated in Hx animals by P11. Both are synaptic vesicle-trafficking proteins differentially expressed in the developing brain. Synapsin 1 is the downstream effector of the brain-derived neurotrophic factor (BDNF) tyrosine kinase cascade pathway up-regulated in mice after spatial learning (40, 41). Snapin is enriched in neurons and exclusively located on the synaptic vesicle membrane. It operates downstream of a cAMP-dependent signal transduction pathway and modulates interactions between SNAREs [soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptors] and synaptotagmin (42). Their enhancement suggests that even under Hx conditions, the proteins needed to establish axons and competent presynaptic structures in cortical neurons are present. Conversely, under such conditions, postsynaptic effectors such as the ionotrophic glutamate receptors and proteins that stabilize synapses, such as Rln and Bsn, are reduced. There is also transcriptional up-regulation of disabled 1, a tyrosine kinase adapter in the pathway controlling Rln during fetal brain development. MAG, a marker of mature oligodendroglia, stimulates neurite outgrowth in models of the developing nervous system (43). MAG is also found in non-myelin-producing Schwann cells responsible for modulating synaptic transmission. Its reduction, along with the loss of other glial-associated proteins, is thus consistent with a hypoxia-mediated loss of glia and presumably, therefore, a consequential impairment of synapse formation. These changes are summarized in cartoon form in Fig. 5. Finally, there were changes in the genes controlling apoptosis and an overall increase in certain proapoptotic markers such as caspase 3. These changes, when taken in the context of the reduced cortical volumes and other studies examining the proapoptotic effects of more acute forms of hypoxia, suggest that waves of inappropriate apoptosis, possibly as a result of impaired synaptogenesis, may lead to the permanent loss of cortical complexity and the irreversibility of neonatal sublethal Hx injury.
Fig 5.

Cartoon summarizing the major response pathways of the brain during periods of perinatal sublethal hypoxia. Three major changes characterize the response of the neonatal brain to modest Hx insult. The most profound disturbance is a loss of the synchronized expression of the genes needed to complete the formation of mature synapses and to enable neural transmission (large arrows). Whereas the overall expression of these genes does increase with time, this increase is exaggerated in hypoxia for genes associated with presynaptic function (red), whereas genes associated with postsynaptic function or with the stabilization of mature and active synapses are diminished (green). Glial transcripts are also lost, and cytoskeletal systems needed to construct neuritic processes and synaptic structures are reduced. Together, these processes retard the formation of synapses and the machinery of neural transmission. The second consequence of hypoxia is the up-regulation of stress and hypoxia response pathways, particularly those driven by HIF-1α (purple). Whereas stress response genes may enhance neuronal survival, the hypoxia response pathways with neoangiogenesis (cortical microvessels) and the neuronal expression of VEGF and Flk-1 are pathologic consequences that presumably retard neuronal maturation through autocrine and paracrine pathways. Finally, there are widespread changes in glial maturation (white), with a loss of myelinated fiber tracts.
If the combined goals of perinatal intensive care are to both promote survival and prevent handicap, then one must seek targets for intervention that ameliorate the neurologic and cognitive sequelae of preterm birth. Previous studies, including our own, have examined single genes or cascades but have not achieved a comprehensive perspective of the pathology of premature brain dysfunction. The present study offers our view of the molecular events underlying this complex condition and suggests that interventions aimed at preserving the coordination of synapse formation may be promising therapeutic goals.
Supplementary Material
Acknowledgments
We thank Dr. Giovanna Spinella and Dr. Robert Baughman for scientific advice, and K. W. Altshul, A. R. Duncan, D. R. Duncan, A. Fan, P. Lizardi, T. Solli, R. Camp, and Y. Kim for their assistance. This work was supported by National Institutes of Health (NIH) Grants NS 35476 and NS 27116 (to L.R.M.), NIH Grants NS42238, NS32578, and DK38979 (to J.S.M.), and NIH Training Grant T32-HL07778 (to S.M.C.).
Abbreviations
Pn, postnatal day n
VEGF, vascular endothelial growth factor
MAG, myelin-associated glycoprotein
ARNT 1, arylhydrocarbon receptor nuclear translocator 1
HIF-1α, hypoxia-inducible factor 1α
Hx, hypoxic
Nx, normoxic
References
- 1.Hack M., Taylor, H. G., Klein, N. & Mercuri-Minich, N. (2000) Pediatrics 106 554-560. [DOI] [PubMed] [Google Scholar]
- 2.Lemons J. A., Bauer, C. R., Oh, W., Korones, S. B., Papile, L.-A., Stoll, B. J., Verter, J., Temprosa, M., Wright, L. L., Ehrenkranz, R. A., et al. (2001) Pediatrics 107 E1. [DOI] [PubMed] [Google Scholar]
- 3.Taylor H. G., Klein, N. & Hack, M. (2000) Dev. Neuropsychol. 17 289-321. [DOI] [PubMed] [Google Scholar]
- 4.Inder T. E. & Volpe, J. J. (2000) Semin. Neonatol. 5 3-16. [DOI] [PubMed] [Google Scholar]
- 5.Huppi P. S., Murphy, B., Jaier, S. E., Zientara, G. P., Inder, T. E., Barnes, P. D., Kikinis, R., Jolesz, F. A. & Volpe, J. J. (2001) Pediatrics 107 455-460. [DOI] [PubMed] [Google Scholar]
- 6.Pape K. E., Bennett-Britton, S., Szymonowicz, W., Martin, D. J., Fitz, C. R. & Becker, L. E. (1983) J. Pediatr. (Berlin) 102 275-280. [DOI] [PubMed] [Google Scholar]
- 7.Ment L. R., Schwartz, M., Makuch, R. W. & Stewart, W. B. (1998) Brain Res. Dev. Brain Res. 111 197-203. [DOI] [PubMed] [Google Scholar]
- 8.Ogunshola O. O., Stewart, W. B., Mihalcik, V., Solli, T., Madri, J. A. & Ment, L. R. (2000) Brain Res. Dev. Brain Res. 119 139-153. [DOI] [PubMed] [Google Scholar]
- 9.Dobbing J. (1972) Bibl. Nutr. Dieta 17 35-46. [PubMed] [Google Scholar]
- 10.Olavarria J. & Van Sluyters, R. C. (1985) J. Comp. Neurol. 239 1-26. [DOI] [PubMed] [Google Scholar]
- 11.Rothblat L. A. & Hayes, L. L. (1982) Brain Res. 246 146-149. [DOI] [PubMed] [Google Scholar]
- 12.Malinak C. & Silverstein, F. S. (1996) Biol. Neonate 69 257-267. [DOI] [PubMed] [Google Scholar]
- 13.Eisen M. B., Spellman, P. T., Brown, P. O. & Botstein, D. (1998) Proc. Natl. Acad. Sci. USA 95 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hedenfalk I., Duggan, D. J., Chen, Y., Radmacher, M., Bittner, M., Simon, R. P., Meltzer, P., Gusterson, B., Esteller, M., Kallioniemi, O. P., et al. (2001) N. Engl. J. Med. 344 539-548. [DOI] [PubMed] [Google Scholar]
- 15.Pesold C., Liu, W. S., Guidotti, A., Costa, E. & Caruncho, H. J. (1999) Proc. Natl. Acad. Sci. USA 96 3217-3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borrell V., Del Rio, J. A., Alcantara, S., Derer, M., Martinez, A., D'Arcangelo, G., Nakajima, K., Mikoshiba, K., Derer, P., Curran, T. & Soriano, E. (1999) J. Neurosci. 19 1345-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rice D. S., Sheldon, M., D'Arcangelo, G., Nakajima, K., Goldowitz, D. & Curran, T. (1998) Development (Cambridge, U.K.) 125 3719-3729. [DOI] [PubMed] [Google Scholar]
- 18.Richter K., Langnaese, K., Kreutz, M. R., Olias, G., Zhai, R., Scheich, H., Garner, C. C. & Gundelfinger, E. D. (1999) J. Comp. Neurol. 408 437-448. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez M. A., Pesold, C., Liu, W. S., Kriho, V., Guidotti, A., Pappas, G. D. & Costa, E. (2000) Proc. Natl. Acad. Sci. USA 97 3550-3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang H., Jensen, P. & Goldowitz, D. (2002) J. Neurosci. 22 464-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howell B. W., Herrick, T. M., Hildebrand, J. D., Zhang, Y. & Cooper, J. A. (2000) Curr. Biol. 10 877-885. [DOI] [PubMed] [Google Scholar]
- 22.Chun J. J. & Shatz, C. J. (1988) Neuron 1 297-310. [DOI] [PubMed] [Google Scholar]
- 23.Warren R. A. & Jones, E. G. (1997) J. Neurosci. 17 277-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deans M. R., Gibson, J. R., Sellitto, C., Connors, B. W. & Paul, D. L. (2001) Neuron 31 477-485. [DOI] [PubMed] [Google Scholar]
- 25.Bayer S. A. & Altman, J. (1990) Exp. Neurol. 107 48-62. [DOI] [PubMed] [Google Scholar]
- 26.Levers T. E., Edgar, J. M. & Price, D. J. (2001) J. Neurobiol. 48 265-277. [DOI] [PubMed] [Google Scholar]
- 27.Back S. A., Luo, N. L., Borenstein, N. S., Levine, J. M., Volpe, J. J. & Kinney, H. C. (2001) J. Neurosci. 21 1302-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Back S. A., Gan, X., Li, Y., Rosenberg, P. A. & Volpe, J. J. (1998) J. Neurosci. 18 6241-6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shibata A., Wright, M. V., David, S., McKerracher, L., Braun, P. E. & Kater, S. B. (1998) J. Cell Biol. 142 191-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert S., Davis, J. Q. & Bennett, V. (1997) J. Neurosci. 17 7025-7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Georgiou J. & Charlton, M. P. (1999) Glia 27 101-109. [DOI] [PubMed] [Google Scholar]
- 32.De Matteis M. A. & Morrow, J. S. (2000) J. Cell Sci. 113 2331-2343. [DOI] [PubMed] [Google Scholar]
- 33.Hirai H. & Matsuda, S. (1999) Neurosci. Res. (N.Y.) 34 281-287. [DOI] [PubMed] [Google Scholar]
- 34.Bennett V. & Chen, L. (2001) Curr. Opin. Cell Biol. 13 61-67. [DOI] [PubMed] [Google Scholar]
- 35.Chauhan V. S., Tuvia, S., Buhusi, M., Bennett, V. & Grant, A. O. (2000) Circ. Res. 86 441-447. [DOI] [PubMed] [Google Scholar]
- 36.David J. C., Tanguay, R. M. & Grongnet, J. F. (2001) Brain Res. Dev. Brain Res. 128 91-99. [DOI] [PubMed] [Google Scholar]
- 37.Tomita S., Sinal, C. J., Yim, S. H. & Gonzalez, F. J. (2000) Mol. Endocrinol. 14 1674-1681. [DOI] [PubMed] [Google Scholar]
- 38.Salceda S. & Caro, J. (1997) J. Biol. Chem. 272 22642-22647. [DOI] [PubMed] [Google Scholar]
- 39.Halterman M. W. & Federoff, H. J. (1999) Exp. Neurol. 159 65-72. [DOI] [PubMed] [Google Scholar]
- 40.Gomez-Pinilla F., So, V. & Kesslak, J. P. (2001) Brain Res. 904 13-19. [DOI] [PubMed] [Google Scholar]
- 41.Fiumara F., Onofri, F., Benfenati, F., Montarolo, P. G. & Ghirardi, M. (2001) Neuroscience 104 271-280. [DOI] [PubMed] [Google Scholar]
- 42.Chheda M. G., Ashery, U., Thakur, P., Rettig, J. & Sheng, Z. H. (2001) Nat. Cell Biol. 3 331-338. [DOI] [PubMed] [Google Scholar]
- 43.Turnley A. M. & Bartlett, P. F. (1998) NeuroReport 9 1987-1990. [DOI] [PubMed] [Google Scholar]
- 44.Mason C. A. (1986) Neuroscience 19 1319-1333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.