

Abstract
Fluorescence correlation spectroscopy (FCS) measurements have been carried out on the intestinal fatty acid binding protein (IFABP) to study microsecond dynamics of the protein in its native state as well as in pH-induced intermediates. IFABP is a small (15 kDa) protein that consists mostly of antiparallel β-strands enclosing a large central cavity into which the ligand binds. Because this protein does not contain cysteine, two cysteine mutants (Val60Cys and Phe62Cys) have been prepared and covalently modified with fluorescein. Based on fluorescence measurements, one of the mutants (Val60Flu) has the fluorescein moiety inside the cavity of the protein, whereas the fluorescein is exposed to solvent in the other (Phe62Flu). The protein modified at position 60 demonstrates the presence of a conformational event on the order of 35 μsec, which is not seen in the other mutant (Phe62Flu). The amplitude of this fast conformational event decreases sharply at low pH as the protein unfolds. Experiments measuring the diffusion as a function of pH indicate the formation of a compact state distinct from the native state at about pH 3.5. Steady state fluorescence and far-UV CD indicates that unfolding occurs at pH values below pH 3.
Keywords: protein folding, conformational dynamics, diffusion coefficient, unfolded state
The intestinal fatty acid binding protein (IFABP) belongs to a family of proteins that bind a variety of ligands (fatty acids, bile salts, and retinoids) into a large cavity located in the interior of the protein. The structure has been determined by both x-ray (1, 2) and NMR methods (3, 4). The x-ray structure shown in Fig. 1 is believed to represent the closed form of the apo protein, whereas the NMR structure suggests the open form (4). The structure of IFABP consists of 2 β-sheets, each containing 5 β-strands, and a small helical region. This protein has been an excellent model system for folding studies of β-sheet proteins because it is small (15 kDa), monomeric, and does not contain any proline or cysteine residues. Equilibrium unfolding transitions of IFABP monitored by steady state fluorescence or CD measurements can be fit by typical two-state model. However, two recent kinetic studies (5, 6) indicated the presence of intermediates, the first one forming within 200 μsec. A second intermediate, formed with a rate constant of ≈2,000 sec−1, contains the majority of the secondary structure (6). Previous 19F NMR studies as a function of urea concentration had indicated the presence of an intermediate state in addition to the folded and unfolded forms (7). Hodsdon and Frieden (8), monitoring urea denaturation by NMR, found several unidentified resonances in heteronuclear sequential quantum correlation (HSQC) spectra at low urea concentrations, which disappeared at higher urea concentrations, again indicating the presence of intermediates. In the present study, we have explored the possibility of using fluorescence correlation spectroscopy (FCS) as a tool to measure the conformational dynamics and diffusional properties of apo IFABP in its folded, unfolded, and intermediate states.
Fig 1.

Crystal structure of IFABP (2) showing the positions of Val-60, Phe-62, and Trp-82.
FCS is emerging as an important technique in chemistry, biophysics, and biochemistry for its applications in measuring diffusional properties and chemical kinetics at low molar concentrations (9, 10). The technique involves measuring fluorescence fluctuations resulting from the changes in the number of fluorophores due to diffusion or chemical reaction under conditions of thermodynamic equilibrium in a small observation volume.
There are generally two kinds of applications of FCS in protein biophysics. First, the diffusion time and hence the diffusion coefficient of a protein can be measured very accurately. Second, by suitably optimizing the measurement conditions, FCS can be used to study protein dynamics or conformational events in the microsecond time scale (11).
In earlier studies, we mutated specific residues of IFABP to cysteine and examined the properties of the mutated proteins (12) as well as proteins for which the fluorescent probe fluorescein was covalently attached to the cysteine residue (13). These studies showed that the probe attached to cysteine at position 60 (V60C) was located within the interior cavity and that the fluorescence properties of the probe were sensitive to the unfolding transition. In the present study, both the V60C and F62C mutants were covalently modified with fluorescein (called V60Flu and F62Flu). In contrast to the V60Flu, the fluorescence of the F62Flu mutant is not sensitive to the unfolding transition and thus it is assumed that the probe is not located in the cavity, but rather on the outer surface of the protein.
The present paper reports FCS measurements on these two mutants. At pH 7.3, a conformational fluctuation on the time scale of 30–45 μsec has been observed in case of V60Flu, the amplitude of which decreases on unfolding. Experiments with F62Flu do not report this dynamic process. Although it is possible to perform FCS measurements in guanidine hydrochloride (Gdn) and urea solutions, measurements in these solutions are complicated because of changes in the viscosity and the refractive index (14). Experiments as a function of pH, which avoid these problems, show a small increase in the diffusion coefficient around pH 3.5, indicating the formation of a compact intermediate state before unfolding at lower pH values. Even though the acid unfolding is accompanied by a significant decrease in diffusion coefficient, the acid unfolded state of the protein appears not to be a random coil. Surprisingly, in the unfolded state, we observe a conformational fluctuation with smaller amplitude but in the same time range as observed in the folded state. To the best of our knowledge, this is the first example of application of FCS in protein folding studies.
Materials and Methods
Mutagenic primers were obtained from Integrated DNA Technologies (Coralville, IA). The QuickChange site-directed mutagenesis kit was obtained from Stratagene. Fluorescein and Alexa488Maleimide were purchased from Molecular Probes. Urea and Gdn were obtained from ICN. All other reagents used were analytical grade.
Expression and purification of the V60C mutant of IFABP were as described elsewhere (12), and those of the F62C mutant were performed by using an identical procedure. The reaction with fluorescein or with Alexa488Val60Cys was carried out by using a published procedure (13). For acid unfolding experiments, a series of solutions of different pH values were prepared by using 2 mM citrate buffer. For experiments as a function of urea or Gdn, a series of protein solutions at different denaturant concentrations were prepared in 20 mM phosphate buffer at pH 7.3.
Steady state fluorescence experiments were performed at 20°C by using a PTI Alphascan fluorometer (Photon Technology International, South Brunswick, NJ) using protein concentrations of ≈1 μM. For fluorescein- and Alexa488-modified proteins, the excitation wavelength was 488 nm and emission spectra were recorded between 500 and 600 nm. The fluorescence intensity at 517 nm was plotted against denaturant concentrations. The unfolding transition was fit to a simple two-state model (15). CD experiments were performed on a Jasco J-715 using a 0.1-cm path length cell.
FCS Experiments.
The laser beam from a titanium sapphire laser cavity (Mira 900 Coherent Radiation, Palo Alto, CA), pumped with a diode pumped Verdi V-10 (Coherent Radiation) was used in the experiments. The repetition rate of the laser was 80 MHz and the pulse width 120 femtoseconds. The laser light was directed to the side of an Olympus IX 70 microscope using gold-coated mirrors. A glass filter (RG715, Chroma Technology, Battleboro, VT) was used to eliminate the pump beam contamination from the laser. A ×5 beam expander, mounted on the side of the microscope, expanded the beam to over-fill the back aperture of an Olympus ×60 water immersion objective. The IR laser beam was reflected toward the objective using a dichroic mirror (725dcspxr, Chroma Technology). The fluorescence of the sample was collected by using the same objective passing through the same dichroic mirror. A band pass filter (e700SP special, Chroma Techology) was used to filter out the remaining scattered laser light. The fluorescence of the sample was collected from the image plane on the side port of the microscope and was then collimated by using a simple convex lens. The collimated fluorescence was then passed through a 50/50 beam splitter and focused on to two avalanche photo diode units (Photon counting Module SPCM-QC, Perkin–Elmer Optoelectronics). The TTL pulses generated were then collected by a correlator card (http://correlator.com, FLEX01-08D), which enables both the storage of photon arrival times and real time correlation function calculation. A test rhodamine sample in alcohol was always used for calibration of the beam profile.
In addition, many of the FCS experiments were repeated by using a ConfoCor 2 LSM combination instrument (Carl Zeiss-Evotec, Jena, Germany) with a 40× objective for water immersion. Alexa488 was excited at 488 nm by using an argon laser. Experiments were performed at room temperature with the protein solutions placed in Nunc chambers (Nalge Nunc). A 80-μm pinhole was used in one of the image plane of the microscope to further reduce the out of focus fluorescence.
All of the data analyses were performed by using ORIGIN 7.0 (Originlab, Northampton, MA). An average of multiple experiments (typically 30) was determined for any single set of data. The average was weighed with the standard deviation of all experiments and then fit to various models as discussed later.
Calculation of Theoretical Diffusion Time of the Protein.
According to the crystal structure, IFABP has a volume of 18,479 Å3 (2). Assuming that the protein is spherical, the radius of the protein (r) can be calculated to be 16.5 Å. The viscosity of water (η) at 25°C is 0.89 cP (1 P = 0.1 Pa⋅s), and hence the theoretical diffusion coefficient of IFABP can be calculated as 1.49 × 10−6 cm2/sec by using Eq. 1.
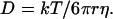 |
The beam radius (w0) of the detected focal volume can be related to the diffusion coefficient (D) using Eq. 2:
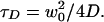 |
We have measured w0 of the present setup to be 0.26 μm using the known diffusion coefficient of Rhodamine Green (2.8 × 10−6 cm2/sec).
Results
Steady State Fluorescence Experiments.
Equilibrium unfolding experiments using different cysteine modified mutants of IFABP have been studied earlier (12, 13). Unfolding of mutant proteins with fluorescein attached to positions 60, 89, and 104 was accompanied by a significant increase in fluorescence intensity at 517 nm (excitation wavelength of 490 nm). It was concluded that the fluorescein group in these mutants was inside the cavity of the protein in the folded state and hence the fluorescence at 517 nm was quenched as a result of the interaction of the fluorescein with the side chains of the neighboring amino acids. Unfolding of the protein leads to the exposure of the fluorescein group to the solvent resulting in a large increase in fluorescence intensity. On the other hand, no significant change of fluorescence on unfolding was observed with other mutants with fluorescein attached to positions 23 and 72. For these mutants, the fluorescein group is not inside the cavity, but rather exposed to the solvent in the native state.
Fig. 2 shows the changes in fluorescence at 517 nm as a function of Gdn concentrations for both the V60Flu and F62Flu. The small fluorescence change observed for F62Flu did not yield a reliable measure of unfolding. Thus, these experiments were repeated by using circular dichroism (also shown in Fig. 2). The data can be fit to a model assuming a two state unfolding transition, and the parameters are listed in Table 1. Table 1 lists the ratio of fluorescence intensity (emission at 517 nm, excitation at 490 nm) at 2 M (unfolded protein) and 0 M Gdn (folded) for both the mutants reflecting the change in the fluorescence intensity on unfolding. The stability of both the mutants is similar with identical mid-points (Table 1). It can be seen that a large fluorescence change occurs with the V60Flu protein on unfolding which indicates that the fluorescein is inside the cavity consistent with earlier NMR data using V60Flu (13). The different behavior for F62Flu (Fig. 2) suggests that the fluorescein of this protein is solvent exposed in the native state.
Fig 2.

Equilibrium unfolding transitions of V60Flu (A) and F62Flu (B) monitored by the changes in CD at 216 nm (open circles) and by the changes in the fluorescence intensity at 517 nm (solid squares). The lines show the fit of the data using a two state model. Solutions of both the mutants were prepared in 20 mM potassium phosphate buffer (pH 7.3) at different Gdn concentrations.
Table 1.
Parameters obtained from the two state unfolding transitions of V60Flu and F62Flu
| Mutants | ΔG0, kcal⋅mol−1 | m, kcal⋅mol−1⋅M−1 | Mid-point, M | (F2/F0)517nm |
|---|---|---|---|---|
| V60Flu | 3.35 | −3.29 | 1.01 | 2.3 |
| F62Flu | 3.4 | −3.69 | 0.9 | 1.1 |
ΔG0 is the free energy of unfolding extrapolated to zero Gdn concentration.
m is the slope of the dependence of free energy of unfolding (ΔG) with Gdn concentration.
(F2/F0)517nm is the ratio of fluorescence intensity (emission at 517 nm, excitation at 490 nm) at 2 M (unfolded state) and at 0 M (folded) Gdn concentrations.
FCS Experiments.
A typical correlation function observed in the FCS measurements on V60Flu in the native condition at pH 7.3 is shown in Fig. 3. The data were analyzed by different models as described below. The values for the diffusion times obtained in each model are shown in Table 2.
Fig 3.

(A) A typical correlation function data obtained in the FCS experiments with V60Flu in 20 mM potassium phosphate buffer, pH 7.3. The data were fit to different models as described in the text, and the residual distribution for the fit using Eq. 3 (model 1) is shown in B. (C) The residual distribution for the fit using Eq. 4 (model 2).
Table 2.
FCS parameters obtained using different models
| Mutant | Fitting model | A, % | τR, μsec | τD, μsec | Dexp, cm2/sec | Dtheo, cm2/sec |
|---|---|---|---|---|---|---|
| V60Flu | 1Diff | NA | NA | 71 | 2.4 × 10−6 | 1.49 × 10−6 |
| V60Flu | 1DiffExp | 30 | 35 | 120 | 1.4 × 10−6 | 1.49 × 10−6 |
| V60Alexa | 1DiffExp | 20 | 20 | 120 | 1.4 × 10−6 | 1.49 × 10−6 |
| V62Flu | 1Diff | – | – | 117 | 1.45 × 10−6 | 1.49 × 10−6 |
| Fluorescein in solution | 1Diff | NA | NA | 55 | 2.8 × 10−6 |
NA, not applicable.
A, τR, and τD are defined in Eqs. 3 and in 4 (see text).
Dexp is the diffusion coefficients of IFABP obtained from τD values using Eq. 2.
Dtheo is the diffusion coefficient of IFABP calculated using crystal structure volume.
1Diff assumes the presence of a single diffusing species (model 1 in text, Eq. 3).
1DiffExp assumes the presence of an exponential component (reflecting an isomerization) with a single diffusing species (model 2 in text, Eq. 4).
Model 1: Simple Diffusion.
Assuming only one diffusing species with the diffusion time, τD, gives Eq. 3:
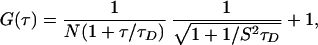 |
where N is the number of protein molecules in the observation volume and S is the depth to diameter ratio of the three-dimensional Gaussian volume element. A nonlinear least-squares fit of the correlation function data of V60Flu (shown in Fig. 3B) leads to a nonrandom residual distribution, indicating that this model is not adequate to explain the correlation function data of V60Flu under these experimental conditions. Moreover, the fit of the correlation function data to Eq. 3 results in a diffusion time of 71 μsec. For a protein of the size of IFABP, the diffusion coefficient can be calculated theoretically (Dtheo) to be 2.4 × 10−6 cm2/sec (Table 2), which does not agree with the diffusion coefficient calculated from the diffusion time, τD, using Eq. 2 (1.49 × 10−6 cm2/sec). Thus, a model with a single diffusing species does not adequately fit the correlation data observed for V60Flu.
Model 2: Simple Isomerization Reaction with Diffusion (Both Isomers Have Same τD).
This model adds an exponential component to reflect possible dynamic quenching of the fluorescein molecules as a result of internal structural fluctuations of the protein (Eq. 4) (11). The correlation function data for V60Flu at pH 7.3 can be fit successfully by
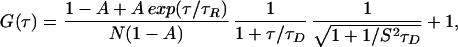 |
where τR is the relaxation time for an exponential component with an associated amplitude, A (Fig. 3). The diffusion coefficient (D, 1.4 × 10−6 cm2/sec) is close to the expected value for a protein of the size of IFABP (Table 2), which justifies use of this model.
Although our experiments clearly indicate the presence of an extra exponential component in addition to a diffusion component in the correlation function, a number of control experiments were carried out to make sure that τR is not an artifactual component. It was noted earlier that imperfect approximation of the focal volume as a three-dimensional Gaussian model, on which the equations used to analyze the correlation function data are based, can lead to the appearance of extra diffusion components (16, 17).
We have carried out similar FCS experiments with the second mutant, F62Flu. It was noted above there is no significant quenching observed in the folded state and the fluorescein residue in this mutant resides outside the protein cavity. The correlation function data for this mutant can be well fit using Eq. 3 (a single diffusing species). No additional exponential component was required (Table 2). The diffusion time of the protein (τD) for F62Flu is identical to that observed for V60Flu (Table 2).
pH-Induced Unfolding of the Protein.
Measurements of unfolding using FCS in presence of urea and Gdn (not shown) are complicated by the effects of viscosity and refractive indices of the solutions (14). Therefore, FCS measurements at different pH values have been carried out to monitor the unfolding transition of the protein because they should be easier to interpret. Fluorescein fluorescence, however, has large pH dependence; and therefore, Alexa488Maleimide was used for these experiments. This dye is analogous to fluorescein except that it is more photostable with higher quantum yield. Data using V60Alexa were fit to Eq. 4, a model containing a diffusion component (with diffusion time τD) and an exponential component (τR) as described earlier. A large decrease in diffusion coefficient (D) was observed between pH 3 and pH 2 (Fig. 4) corresponding to the pH-induced unfolding transition of the protein observed by far UV CD and steady state fluorescence (data not shown). In addition, a small increase in the diffusion coefficient was observed between pH 5 and pH 3.2 before the unfolding transition, suggesting the formation of an intermediate state in equilibrium (Fig. 4). A similar intermediate state with a diffusion coefficient different from the native protein has been observed by FCS measurements at pH 2 in the presence of 100 mM KCl (data not shown).
Fig 4.

Dependence of diffusion coefficient (D) and the amplitude of τR (A) as a function of pH. The pH was adjusted by using 2 mM citrate buffer. The error bars of the measurements were obtained by using several measurements.
Fig. 4 also shows that the amplitude of the exponential component decreases sharply at low pH to ≈10% as a consequence of unfolding. Although the decrease in the amplitude of the native like dynamics is expected on unfolding, it is surprising to note that it remains with low amplitude in the unfolded state at pH 2. The relaxation time, τR, however, does not change (data not shown).
Discussion
Significance of τR.
The model described by Eq. 4 assumes (i) an inter-conversion of two states of the protein with a chemical relaxation time, τR, and (ii) that the fluorescence of one state is significantly quenched compared with the other. We have shown elsewhere that fluorescein fluorescence is quenched in the native state of V60Flu as a result of being inside the cavity (13). It is important, however, to understand the quenching of fluorescein in the native protein to gain further insight about what the microsecond dynamic measurement represents.
As Fig. 2 shows, the fluorescence of V60Flu changes markedly as it unfolds. One factor that may affect the fluorescence of the fluorescein moiety of V60Flu is the presence of a tryptophan (Trp-82) inside the cavity, which is close to Val-60 (≈7 Å). Solution studies indicate that tryptophan is an effective quencher of fluorescein fluorescence through both static and dynamic quenching mechanisms (18, 19). Steady state and time resolved fluorescence experiments on a fluorescein antifluorescein antibody complex indicated nearly complete quenching (≈90%) of the fluorescein fluorescence by tryptophan (18, 20). IFABP contains two tryptophan residues, namely Trp-6 and Trp-82, and energy transfer between Trp-82 and fluorescein has been detected earlier (13). Interaction of fluorescein with Trp-82 could fluctuate as a consequence of a conformational change. If the conformational fluctuation and the resulting fluorescence fluctuations occur on a sufficiently fast time scale, the process can be monitored by FCS. Therefore, the observation of an exponential component (τR, 35 μsec, k = 2.8 × 105 s−1) suggests the possible involvement of a conformational fluctuation of the native protein in the microsecond time regime.
There have been a significant number of studies devoted to the structural and conformational dynamics of different proteins. Molecular dynamics simulations in the nanosecond time scale provide important information on the structure and dynamics of the water molecules in the apo and ligand bound conformation of IFABP (21). Measurements of slow backbone dynamics by NMR and ligand binding by stopped flow fluorescence revealed a global conformational exchange process between an open and a closed form of the protein with a rate constant of 580 sec−1 at 15°C (D. Cistola, personal communication). Similar binding experiments at 25°C lead to a higher rate of ≈1,000 sec−1 for this step (22). However, the global conformational change observed by NMR and stopped flow experiments is much slower than the conformational event observed by the FCS measurements. On the other hand, the conformational change observed by using FCS is slower than the nanosecond motions investigated by NMR and molecular dynamics simulations (21, 23). Recent continuous flow kinetics data on several turn mutants of IFABP suggested presence of a fast folding step occurring at times faster than 200 μsec. This event could not be followed because of the dead time limitation of the continuous flow measurements, but the data indicated a significant structure formation associated with that step. The present results show that FCS experiments can monitor structural and conformational events with a considerably faster time frame.
Possible Artifacts.
A model with two diffusion components (instead of one diffusion and one exponential component) can also fit V60Flu data well (not shown), which argues that the faster component could come from the presence of free dye in the solution. However, this possibility can be ruled out for two reasons. First, a diffusion time of 30 μsec for the second component has been obtained by this model, which does not match with the diffusion time of the free dye (55 μsec). Second, the amplitude and the time scale of τR was found to be unaffected when further purification steps of exhaustive dialysis and gel filtration column chromatography were used to remove any free dye present. It is also possible that an exponential term might arise from an imperfect Gaussian approximation of the light beam (17), though no such artifact has been reported for two-photon excitation. To further test this, experiments have been carried out with the V60C coupled with a fluorescein analogue, Alexa488Maleimide. Alexa488Maleimide is analogous to fluorescein except it is more photo-stable with higher quantum yield. An exponential component (20 μsec with an amplitude of 20%; Table 2) in addition to the diffusion has been observed. The observation of a nondiffusional component with a substantially more stable dye suggests that τR represents a true conformational event. Further, the experiment with F62Flu is an additional control. A conformational fluctuation would not be expected for this mutant with a fluorescence probe located outside the protein (and distant from Trp-82). Indeed, no similar chemical relaxation term is observed (Table 2).
Size of the Folding Intermediate.
Assuming a spherical molecule, one can calculate the radius of IFABP from its diffusion coefficient using Eq. 1 or from the crystal structure volume. The calculated radius in the absence of denaturant is 16.5 Å. From the crystal structure, IFABP has a volume of 18,479 Å and the radius obtained from this volume (16.4 Å) is thus comparable to that observed from diffusion coefficient data.
As Fig. 4 shows, there is a slight increase in the diffusion coefficient between pH 3 and 4. Calculation of the hydrodynamic radii at pH 3 indicates a small decrease in size of the protein molecule in the intermediate state compared with the native state, suggesting the formation of a compact intermediate state before unfolding. A similar intermediate state has also been formed at pH 2 in presence of salts (Table 3), indicating that the driving force for the formation of this intermediate is neutralization of repulsive charge and clustering of the hydrophobic residues. Because the protein encloses a large cavity, it may be that the increase in diffusion coefficient reflects a collapse of the cavity. The presence of an intermediate state at low denaturant concentrations had been suggested by 19F NMR experiments on IFABP at pH 7 (7) to rationalize unknown peaks in the heteronuclear sequential quantum correlation (HSQC) NMR spectrum. Submillisecond continuous flow kinetics measurements (6) have also indicated a partial collapse of the protein faster than 200 μs that could be due to hydrophobic clustering.
Table 3.
Calculated radii for the native, intermediate, and unfolded states
| State | Radius, Å |
|---|---|
| Native protein | 16.5 |
| Intermediate state (pH 3) | 15 |
| Intermediate state (pH 2+ 40 mM KCl) | 14.2 |
| Unfolded (at pH 2) | 22 |
Experiments performed in 20 mM phosphate buffer at room temperature.
Experiments performed using 2 mM citrate buffers to adjust the pH.
The Unfolded State.
Assuming the unfolded state of IFABP is truly an extended random coil, the radius of gyration can be calculated to be 46 Å (24). However, the hydrodynamic radius of the unfolded state of IFABP obtained at pH 2 is significantly smaller (around 22 Å), indicating that under this condition the unfolded state of IFABP is not as extended as expected for a random coil. Although the unfolded state of the protein has been envisioned as unstructured, recent experimental and theoretical studies suggest the importance of structures within the unfolded state that are necessary for efficient and rapid folding. Heteronuclear sequential quantum correlation (HSQC) experiments with IFABP indicate the presence of residual structure at very high urea concentrations (8). This residual structure has only 2–7% native like persistence involving the turn between β-strands and between the two short helices. Native-like spatial positioning, orientation of chain segments, and compact denatured states have been observed in other systems (25–27). The present paper suggests that the acid unfolded state of IFABP is compact and the hydrodynamic radius of IFABP in the unfolded state is only 35% larger than that of the native state.
FCS and Protein Folding.
The present study suggests that the application of FCS to protein folding studies can be twofold. First, FCS can give valuable information about the changes in diffusion coefficient under equilibrium conditions. Second, dynamic motions in time regimes of microseconds or longer can be explored by FCS under suitable experiment conditions. It should be noted that a number of recent protein folding kinetic studies, such as continuous flow measurements, have been devoted to studying fast reactions (6, 28–30). Recent experiments on IFABP folding revealed the presence of kinetic processes faster than 200 μsec (6), the dead time of a typical continuous flow mixer. FCS can be used to study reaction kinetics at faster time regimes. There are, however, several difficulties using FCS for such measurements. First, one needs a fluorescentally modified protein because tryptophan cannot be used for these experiments. Choosing a suitable dye is not easy because it has to be both bright and environmentally sensitive. Second, using either urea or Gdn as unfolding reagents affects both the viscosity and the refractive index of solution resulting in large artifactual changes in the diffusion time and determination of particle number (14).
Conclusions
FCS has been used to study both the diffusion and the conformational dynamics of the IFABP. FCS experiments indicate the presence of a conformational fluctuation in the native state of the protein in the time regime of ≈35 μsec likely to be a local fluctuation in the cavity involving the fluorescein moiety and Trp-82. pH-induced unfolding leads first to a compact intermediate state, identified by a small increase in the diffusion coefficient, and then to an apparent unfolded state accompanied by a decrease in the diffusion coefficient. The diffusion coefficient of this unfolded state is not consistent with that of a random coil, suggesting that some compact structure remains.
Acknowledgments
We thank Dr. David Cistola for helpful discussions. This work was supported by National Institutes of Health Grants DK13332 (to C.F.) and GM38838 (to E.L.E.).
Abbreviations
IFABP, rat intestinal fatty acid binding protein
FCS, fluorescence correlation spectroscopy
Gdn, guanidine hydrochloride
References
- 1.Sacchettini J. C., Scapin, G., Gopaul, D. & Gordon, J. I. (1992) J. Biol. Chem. 267, 23534-23545. [PubMed] [Google Scholar]
- 2.Scapin G., Gordon, J. I. & Sacchettini, J. C. (1992) J. Biol. Chem. 267, 4253-4269. [DOI] [PubMed] [Google Scholar]
- 3.Hodsdon M. E., Ponder, J. W. & Cistola, D. P. (1996) J. Mol. Biol. 264, 585-602. [DOI] [PubMed] [Google Scholar]
- 4.Hodsdon M. E. & Cistola, D. P. (1997) Biochemistry 36, 1450-1460. [DOI] [PubMed] [Google Scholar]
- 5.Yeh S. R., Ropson, I. J. & Rousseau, D. L. (2001) Biochemistry 40, 4205-4210. [DOI] [PubMed] [Google Scholar]
- 6.Chattopadhyay K., Zhong, S., Yeh, S.-R., Rousseau, D. L. & Frieden, C. (2002) Biochemistry 41, 4040-4047. [DOI] [PubMed] [Google Scholar]
- 7.Ropson I. J. & Frieden, C. (1992) Proc. Natl. Acad. Sci. USA 89, 7222-7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodsdon M. E. & Frieden, C. (2001) Biochemistry 40, 732-742. [DOI] [PubMed] [Google Scholar]
- 9.Hess S. T., Huang, S., Heikal, A. A. & Webb, W. W. (2002) Biochemistry 41, 697-705. [DOI] [PubMed] [Google Scholar]
- 10.Elson E. L. (1985) Annu. Rev. Phys. Chem. 36, 379-406. [Google Scholar]
- 11.Haupts U., Maiti, S., Schwille, P. & Webb, W. W. (1998) Proc. Natl. Acad. Sci. USA 95, 13573-13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang N. & Frieden, C. (1993) Biochemistry 32, 11015-11021. [DOI] [PubMed] [Google Scholar]
- 13.Frieden C., Jiang, N. & Cistola, D. P. (1995) Biochemistry 34, 2724-2730. [DOI] [PubMed] [Google Scholar]
- 14.Frieden C., Chattopadhyay, K. & Elson, E. L. (2002) Adv. Protein Chem. 62, 91-110. [DOI] [PubMed] [Google Scholar]
- 15.Santoro M. M. & Bolen, D. W. (1988) Biochemistry 27, 8063-8068. [DOI] [PubMed] [Google Scholar]
- 16.Rigler R., Mets, U., Widengren, J. & Kask, P. (1993) Eur. Biophys. J. 22, 169-175. [Google Scholar]
- 17.Widengren J. M., Mets, U. & Rigler, R. (1995) J. Phys. Chem. 99, 13368-13379. [Google Scholar]
- 18.Watt R. M. & Voss, E. W., Jr. (1977) Immunochemistry 14, 533-551. [DOI] [PubMed] [Google Scholar]
- 19.Templeton E. F. G. & Ware, W. R. (1985) Mol. Immunol. 22, 45-55. [DOI] [PubMed] [Google Scholar]
- 20.Klonis N., Clayton, A. H., Voss, E. W., Jr. & Sawyer, W. H. (1998) Photochem. Photobiol. 67, 500-510. [PubMed] [Google Scholar]
- 21.Bakowies D. & van Gunsteren, W. F. (2002) J. Mol. Biol. 315, 713-736. [DOI] [PubMed] [Google Scholar]
- 22.Cistola D. P., Kim, K., Rogl, H. & Frieden, C. (1996) Biochemistry 35, 7559-7565. [DOI] [PubMed] [Google Scholar]
- 23.Hodsdon M. E. & Cistola, D. P. (1997) Biochemistry 36, 2278-2290. [DOI] [PubMed] [Google Scholar]
- 24.Tanford C. (1968) Adv. Protein Chem. 23, 121-282. [DOI] [PubMed] [Google Scholar]
- 25.Shortle D. & Ackerman, M. S. (2001) Science 293, 487-489. [DOI] [PubMed] [Google Scholar]
- 26.Myers J. K. & Oas, T. G. (2001) Nat. Struct. Biol. 8, 552-558. [DOI] [PubMed] [Google Scholar]
- 27.Pollack L., Tate, M. W., Finnefrock, A. C., Kalidas, C., Trotter, S., Darnton, N. C., Lurio, L., Austin, R. H., Batt, C. A., Gruner, S. M. & Mochrie, S. G. (2001) Phys. Rev. Lett. 86, 4962-4965. [DOI] [PubMed] [Google Scholar]
- 28.Shastry M. C., Luck, S. D. & Roder, H. (1998) Biophys. J. 74, 2714-2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roder H. & Shastry, M. R. (1999) Curr. Opin. Struct. Biol. 9, 620-626. [DOI] [PubMed] [Google Scholar]
- 30.Chan C. K., Hu, Y., Takahashi, S., Rousseau, D. L., Eaton, W. A. & Hofrichter, J. (1997) Proc. Natl. Acad. Sci. USA 94, 1779-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]