

Abstract
The voltage-dependent and Ca2+-activated K+ channel (MaxiK, BK) and the cellular proto-oncogene pp60c-Src (c-Src) are abundant proteins in vascular smooth muscle. The role of MaxiK channels as a vasorelaxing force is well established, but their role in vasoconstriction is unclear. Because Src participates in regulating vasoconstriction, we investigated whether c-Src inhibits MaxiK as a mechanism for agonist-induced vasoconstriction. Functional experiments in human and rat show that inhibitors of Src (Lavendustin A, PP2) but not inactive compounds (Lavendustin B, PP3) induce a pronounced relaxation of coronary or aortic smooth muscle precontracted with 5-hydroxytriptamine, phenylephrine, or Angiotensin II. Iberiotoxin, a MaxiK blocker, antagonizes the relaxation induced by Lavendustin A or PP2, indicating that c-Src inhibits the Iberiotoxin-sensitive component, likely MaxiK channels. In agreement, coronary muscle MaxiK currents were enhanced by Lavendustin A. To investigate the molecular mechanism of c-Src action on MaxiK channels, we transiently expressed its α subunit, hSlo, with or without c-Src in HEK293T cells. The voltage sensitivity of hSlo was right-shifted by ≈16 mV. hSlo inhibition by c-Src is due to channel direct phosphorylation because: (i) excised patches exposed to protein tyrosine phosphatase (CD45) resulted in a partial reversal of the inhibitory effect by ≈10 mV, and (ii) immunoprecipitated hSlo channels were recognized by an anti-phosphotyrosine Ab. Furthermore, coexpression of hSlo and c-Src demonstrate a striking colocalization in HEK293T cells. We propose that MaxiK channels via direct c-Src-dependent phosphorylation play a significant role supporting vasoconstriction after activation of G protein-coupled receptors by vasoactive substances and neurotransmitters.
The tone of vascular myocytes, a key determinant of blood flow, is highly regulated by a variety of extracellular signals that induce vascular contraction or relaxation. At the cellular level, large-conductance voltage-dependent and Ca2+-activated K+ (MaxiK, BK) channels play a critical role in maintaining arterial tone (1). MaxiK acts as a steady vasorelaxing (hyperpolarizing) force in response to spontaneous increases in local Ca2+ (2), or as a negative feedback mechanism to limit Ca2+ entry by hyperpolarizing the plasma membrane and closing voltage-dependent Ca2+ channels (3) previously opened by pressure (4) or vasoconstrictors like 5-hydroxytriptamine (5-HT) and Angiotensin II (AngII) (5, 6). In vitro evidence suggests that MaxiK also may play a role in vasoconstriction as it is inhibited by the potent constrictors AngII (7) and thromboxane A2 (8) in bilayers. However, the functional role of MaxiK channels in agonist-induced contraction has not been demonstrated.
Pharmacomechanical and biochemical evidence indicate that one mechanism of agonist-induced contraction may involve tyrosine phosphorylation/dephosphorylation with phosphorylation associated with vasoconstriction (9). However, most studies have been performed by using inhibitors with broad actions (e.g., tyrphostin and genistein) (10, 11). Using more selective inhibitors for Src-family tyrosine kinases, PP1 and PP2 (12) recent studies in rat aorta (13) and mesenteric arteries (14) show that 5-HT and AngII contractions involve a Src tyrosine kinase, likely c-Src. However, the downstream effector(s) of c-Src promoting vasoconstriction are unknown.
We hypothesized that MaxiK may be a potential downstream effector of c-Src favoring vasoconstriction. This is based on the facts that both c-Src tyrosine kinase and MaxiK are particularly abundant in smooth muscles including the vasculature (15–18), and that Lavendustin A (LavA), a c-Src and Lck inhibitor (10, 19), increases the activity of rat tail artery MaxiK (20). Here, we provide evidence showing that agonist-induced vasoconstriction by 5-HT, AngII, and phenylephrine involves inhibition of MaxiK channels by c-Src via direct phosphorylation of the channel protein. This new signaling pathway has a significant role in human and rat vasoconstriction, providing a link between electromechanical and pharmacomechanical coupling (21). Furthermore, the results indicate that MaxiK channels can function as a rheostat controlling both vasoconstriction and vasorelaxation.
Experimental Procedures
Tissue.
Human coronary arteries were obtained from explanted hearts (University of California, Los Angeles, Medical Center). Male 3-mo-old F344 rats were used. Protocols received institutional approval.
Isometric Contraction.
Arterial rings (2.0- to 3.0-mm internal diameter, 3 mm long) without endothelium were stretched to an optimal resting tension (2 g, human coronaries; 1.2 g, rat aorta) and equilibrated for 60 min in Krebs solution. Percentage relaxation after drug application was calculated for tonic contractions from: percentage relaxation = (max – min)/(max – basal) × 100; where max = maximal steady-state tension after constricting agonist stimulation, min = minimal tension attained after drug application, basal = basal tension prior constricting agonist stimulation. For phasic contractions, percentage relaxation = (cycles/hbefore drug application/cycles/hafter drug application) × 100. Because IbTx treatment abolished phasic contractions, in this case percentage relaxation was calculated as for tonic contractions.
Transient Transfection of HEK293T Cells.
Cells were cotransfected by using the DEAE–dextran method with c-myc-hSlo-pcDNA3 (22) ± chicken c-Src-pcDNA3 (GenBank accession no. J00844), which shares 97% homology with and is 94% identical to human c-Src (GenBank accession no. K03218). Cells were used within 2–4 days.
Single Cell Isolation.
Rat coronary myocytes were isolated in Ca2+-free Krebs solution (mM): 119 NaCl/4.7 KCl/1.17 MgSO4/22 NaHCO3/1.18 KH2PO4/8 Hepes/5.5 glucose, pH 7.4. Vessels were incubated 17 min with papain (1.5 mg/ml), DTT (1 mM), and BSA (2 mg/ml), and 10 min with collagenase F (1 mg/ml), collagenase H (0.3 mg/ml), and BSA (2 mg/ml) at 37°C. Dispersed cells were plated and stored at 4°C until use.
Patch Clamp.
Cell-attached or inside-out patches (transfected cells) and whole-cell configuration (coronary myocytes) were performed at room temperature. Transfected cells were identified by using anti-CD8 Ab-coated magnetic beads (23) or by cotransfecting GFP. For transfected cells the pipette and bath solutions contained (mM): 105 potassium methanesulfonate, 5 KCl, 10 Hepes, and 5 HEDTA (CaCl2 was added to reach the desired free Ca2+ concentration and measured with a Ca2+ electrode), pH 7.0. For cell-attached patches, the pipette contained 60 nM free Ca2+ and the bath contained 10 μM free Ca2+.
For coronary myocytes, the pipette contained (mM): 140 potassium methanesulfonate, 0.1 CaCl2, 2 MgCl2, 0.145 EGTA, 10 Hepes, and 1 Na2ATP, pH 7.4; the external solution contained (mM): 135 sodium methanesulfonate, 5 potassium methanesulfonate, 0.1 CaCl2, 2 MgCl2, 10 Hepes, and 5 glucose, pH 7.4. The holding potential (Vh) was 0 mV to inactivate other voltage-dependent K+ currents.
Data were filtered at one-fourth to one-fifth of the sampling frequency (10 kHz) and fitted to Boltzmann distributions: FPo = G/Gmax = 1/{1 + exp[(V − V)zδF/RT]}, where FPo = fractional open probability, G = conductance, Gmax = limiting maximum conductance, V
− V)zδF/RT]}, where FPo = fractional open probability, G = conductance, Gmax = limiting maximum conductance, V = potential of 50% limiting open probability or one-half activation potential, zδ = effective valence, and F,R,T have their usual thermodynamic meanings.
= potential of 50% limiting open probability or one-half activation potential, zδ = effective valence, and F,R,T have their usual thermodynamic meanings.
Immunoprecipitation of hSlo Proteins.
Cells were solubilized in radioimmunoprecipitation assay solution (mM): 100 NaCl, 50 Tris⋅HCl, 10% sucrose, 0.1% SDS, 1% Nonidet P-40, and 0.5% sodium deoxycholate (pH 7.4), supplemented with 2 EDTA, 50 NaF, 40 β-glycerophosphate, 1 Na3V04, 2 PMSF, 1 iodoacetamide, 0.1 benzamidine, 0.001 leupeptin, 0.15 aprotinin, and 0.15 pepstatin. Lysates were centrifuged at 15,000 × g (10 min, 4°C) and immunoprecipitated with anti-c-myc Ab (1 mg/ml, 3 h) followed by overnight incubation with Protein A-Sepharose 4 Fast Flow beads (Pharmacia) in radioimmunoprecipitation assay. Beads were washed three times with (mM): 100 NaCl, 10 EDTA, and 100 Tris (pH 8) supplemented with 2 mM PMSF and 1% Triton X-100, and once without Triton X-100. Samples were eluted by boiling with Laemmli buffer (three times) containing 10% β-mercaptoethanol for 3 min and centrifuged at 18,500 × g. The immunoprecipitated material was sized fractionated by using 7.5% SDS/PAGE, elecrotransferred onto nitrocellulose membranes, and used for immunoblotting.
Immunoblotting.
Cell lysates or immunoprecipitated proteins were immunoblotted with antiphosphotyrosine (pY) mAb (clone 4G10), anti-c-Src mAb (clone GD11) (Upstate Biotechnology, Lake Placid, NY), or affinity purified anti-hSlo913–926 polyclonal Ab. Secondary Abs were anti-rabbit or anti-mouse peroxidase-conjugated. Immunoreactive bands were detected by using enhanced chemiluminescence and quantified by densitometry.
Immunocytochemistry.
Live cells were incubated with 2 μg/ml anti-c-myc mAb (1 h, 4°C), fixed with ice-cold 4% paraformaldehyde (30 min), and permeabilized with 0.2% Triton X-100/PBS. Nonspecific binding was blocked with 5% donkey serum. Cells were double labeled with 0.5 μg/ml rabbit polyclonal anti-cSrc (sc-18; Santa Cruz Biotechnology) (overnight, 4°C), and incubated with FITC anti-mouse and rhodamine red-X succinimidyl ester-conjugated donkey anti-rabbit Abs (7 μg/ml) for visualization. Images were acquired every 0.25 or 0.5 μm in the z-plane with a confocal microscope. Analysis was performed by using fluoview (Olympus, New Hyde Park, NY) or METAMORPH5 (Universal Imaging, Media, PA) software. For deconvolution, images were acquired with a ×60 objective (1.40 numeric aperture); resolution was 0.0574 μm per pixel in the x–y plane.
Statistics.
A two-tail Student's t test was used. P < 0.05 was considered significantly different.
Results
Functional Coupling of MaxiK Channels and Src in 5-HT-Induced Contraction of Human Coronary Arteries.
To test the possibility that agonist-induced contraction of human coronary arteries may involve Src pathway and MaxiK channels, we performed functional experiments by using inhibitors of Src tyrosine kinases (LavA) (10, 19) and of MaxiK [iberiotoxin (IbTx)] (24). LavA is 500 times more potent in inhibiting c-Src than cAMP-dependent protein kinase, PKC, or Ca2+-calmodulin dependent protein kinase II that have IC50s of >100 μM (10). Human coronary arterial rings were from seven subjects with idiopathic dilated cardiomyopathy with no known coronary artery disease. Ages were: 7, 24, 29, 47, 55, 59, and 63 years old. When contracted with 2 μM 5-HT, coronary rings displayed two patterns of contraction, phasic (Fig. 1) and tonic (not shown). From 56 arterial rings, 34 exhibited oscillatory contractions and 22 were sustained. In both cases, subsequent application of 10 μM LavA produced ≈50% relaxation. In rings with phasic contractions (Fig. 1A), their frequency was reduced from 6.6 ± 0.4 cycles/h (n = 12) to 3.3 ± 0.4 cycles/h (n = 12) with a 50 ± 3% relaxation. Similarly, coronary rings with tonic contractions attained 52 ± 7% relaxation (n = 10) on treatment with LavA (not shown). As expected for a specific action of LavA, Fig. 1B shows that its inactive analog LavB did not cause any reduction of the oscillatory contractions induced by 5-HT (7 ± 0.7 vs. 7 ± 0.7 oscillations per hour, n = 4). Similar lack of relaxation was obtained by using PP3 (n = 4) (Fig. 1C), an inactive analog of the Src tyrosine kinase inhibitors, PP1 and PP2. The fact that LavA is able to counteract by ≈50% the 5-HT induced contraction indicates that Src activation is a significant player in human coronary contractile response to 5-HT. In the following experiments, we demonstrate that this effect is mediated via MaxiK channels.
Fig 1.

LavA but not inactive tyrosine kinase inhibitors (LavB, PP3) relax human coronary arteries precontracted with 5-HT via an IbTx-sensitive component. Isometric contractions of human coronary arterial rings were triggered with 2 μM 5-HT (arrow in A–D). (A) Relaxation by 10 μM LavA. (B) No effect by 10 μM LavB. (C) No effect by 10 μM PP3. (D) IbTx (100 nM) prevented the LavA-relaxing effect. (E) Summary of IbTx prevention of LavA-induced relaxation in arteries responding with phasic and tonic contractions to 2 μM 5-HT. Bars indicate the point in time and duration of drug application. *, P < 0.001.
Incubation with 100 nM IbTx for 25–30 min before application of LavA almost completely blocked its relaxing effect (Fig. 1D) with a maximum relaxation of only 11 ± 2% (n = 9). In rings with sustained contraction, IbTx also prevented LavA-induced relaxation by ≈83% from 52 ± 7% relaxation (n = 10) in the absence to 9 ± 3% relaxation (n = 12) in the presence of IbTx (Fig. 1E). These results strongly support the idea that MaxiK channels are coupled to Src activation after 5-HT receptor stimulation. One possible explanation is that diminished c-Src activity by LavA decreases c-Src mediated phosphorylation of MaxiK enhancing their activity and thus, producing vasorelaxation. In other words, 5-HT receptor stimulation would lead to c-Src activation, MaxiK phosphorylation with diminished channel activity, and consequently to vasoconstriction; the inhibition of MaxiK channels by c-Src would promote depolarization, activation of voltage-dependent Ca2+ channels, and constriction, analogous to MaxiK blockade by charybdotoxin (1). This hypothesis was tested with the following strategy: (i) additional pharmacomechanical experiments in the rat model, (ii) MaxiK current measurements with LavA in coronary myocytes, (iii) MaxiK α subunit, hSlo, modulation by c-Src by using heterologous expression, (iv) direct phosphorylation of hSlo by c-Src, and (v) colocalization of hSlo and c-Src.
MaxiK Channels and c-Src Participate in 5-HT-, AngII-, and Phenylephrine-Induced Contraction of Rat Aorta.
To investigate whether MaxiK and c-Src play a role in 5-HT-induced contraction of rat arteries, aortic rings precontracted with 2 μM 5-HT were exposed to cumulative concentrations of PP2 ± IbTx (Fig. 2). PP2 effectively reversed 5-HT induced tonic contractions (Fig. 2A), and IbTx greatly reduced the ability of PP2 to relax aortic rings (Fig. 2B), indicating that MaxiK channels are coupled to the Src pathway in rat aorta as well. The mean dose–response curve in the absence (control) and presence of IbTx was fitted to a Hill function: percentage relaxation = max/(1 + (K /[PP2])N), where max = maximum relaxation, K
/[PP2])N), where max = maximum relaxation, K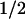 = concentration needed for 50% relaxation, [PP2] = PP2 concentration, and N = Hill coefficient (Fig. 2C). The fitted curves gave N ≈ 1 in both cases, and a K
= concentration needed for 50% relaxation, [PP2] = PP2 concentration, and N = Hill coefficient (Fig. 2C). The fitted curves gave N ≈ 1 in both cases, and a K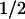 = 300 ± 60 nM PP2 (n = 8) in control experiments, which corresponds well to the IC50 reported for c-Src and PP1 (170 nM) a closely related pyrazolopyrimidine (12). Both PP1 and PP2 are virtually inactive (IC50 of >50–100 μM) for inhibition of other nonreceptor tyrosine kinases ZAP-70 and JAK2, and cAMP-dependent protein kinase, and inhibit with high affinity Src-tyrosine kinases, Lck and Fyn with IC50s of ≈5 nM (12). Thus, the K
= 300 ± 60 nM PP2 (n = 8) in control experiments, which corresponds well to the IC50 reported for c-Src and PP1 (170 nM) a closely related pyrazolopyrimidine (12). Both PP1 and PP2 are virtually inactive (IC50 of >50–100 μM) for inhibition of other nonreceptor tyrosine kinases ZAP-70 and JAK2, and cAMP-dependent protein kinase, and inhibit with high affinity Src-tyrosine kinases, Lck and Fyn with IC50s of ≈5 nM (12). Thus, the K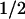 obtained for PP2 in aorta support the view that c-Src is the member of the Src-tyrosine kinase family mediating aortic 5-HT contraction. In agreement, PP2 inhibits AngII-induced c-Src activity of human cultured arterial myocytes (14).
obtained for PP2 in aorta support the view that c-Src is the member of the Src-tyrosine kinase family mediating aortic 5-HT contraction. In agreement, PP2 inhibits AngII-induced c-Src activity of human cultured arterial myocytes (14).
Fig 2.

Blockade of MaxiK channels decreases the vasorelaxing effect of PP2 in rat aorta. (A) Cumulative dose response of 5-HT-precontracted rings with PP2 (numbers are PP2 concentration in micromolar). (B) Preincubation with 100 nM IbTx for ≈25 min diminished the efficacy of PP2 to relax aortic rings. (C) Dose–response curves of PP2-induced relaxation ± 100 nM IbTx. Continuous lines are the best fit to a Hill function. (D) Mean percentage relaxation by 10 μM PP2 with or without (control) 100 nM IbTx in rings precontracted with 2 μM 5-HT (110 ± 3, n = 8 vs. 37 ± 6% relaxation, n = 6), 100 nM AngII (58 ± 10, n = 6 vs. 10 ± 4% relaxation, n = 5), or 2 μM phenylephrine (Phe) (56 ± 8, n = 9 vs. 13 ± 3% relaxation, n = 7); *, P < 0.005.
Pretreatment with 100 nM IbTx decreased the PP2-induced relaxation K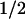 near 10-fold to 2 ± 0.4 μM (n = 6) and reduced its efficacy by approximately one-half (maxcontrol = 115 ± 4% relaxation, n = 8 to maxIbTx = 50 ± 2% relaxation, n = 6). IbTx was also able to prevent PP2-induced relaxation of aortas precontracted with AngII or phenylephrine (Fig. 2D). Because c-Src may activate smooth muscle L-type Ca2+ channels (25, 26), we investigated whether 50 nM diltiazem (IC50 for Ca2+ currents in vascular myocytes; ref. 27) affects 10 μM PP2-induced relaxation. Diltiazem produced ≈50% relaxation of 5-HT contractions but did not modify the extent of PP2 induced relaxation (percentage relaxationPP2 110 ± 3, n = 8; percentage relaxationdiltiazem+PP2 = 91 ± 8%, n = 7), nor of its inhibition by the MaxiK-specific blocker, IbTx (100 nM) (percentage relaxationPP2+IbTx = 37 ± 6%, n = 6; percentage relaxationdiltiazem+PP2+IbTx = 23 ± 6%, n = 7). Thus, blocking 50% of Ca2+ channels was a scaling factor without modifying the relative actions of PP2 and IbTx, supporting the idea that PP2-induced relaxation is mainly because of MaxiK channel activation.
near 10-fold to 2 ± 0.4 μM (n = 6) and reduced its efficacy by approximately one-half (maxcontrol = 115 ± 4% relaxation, n = 8 to maxIbTx = 50 ± 2% relaxation, n = 6). IbTx was also able to prevent PP2-induced relaxation of aortas precontracted with AngII or phenylephrine (Fig. 2D). Because c-Src may activate smooth muscle L-type Ca2+ channels (25, 26), we investigated whether 50 nM diltiazem (IC50 for Ca2+ currents in vascular myocytes; ref. 27) affects 10 μM PP2-induced relaxation. Diltiazem produced ≈50% relaxation of 5-HT contractions but did not modify the extent of PP2 induced relaxation (percentage relaxationPP2 110 ± 3, n = 8; percentage relaxationdiltiazem+PP2 = 91 ± 8%, n = 7), nor of its inhibition by the MaxiK-specific blocker, IbTx (100 nM) (percentage relaxationPP2+IbTx = 37 ± 6%, n = 6; percentage relaxationdiltiazem+PP2+IbTx = 23 ± 6%, n = 7). Thus, blocking 50% of Ca2+ channels was a scaling factor without modifying the relative actions of PP2 and IbTx, supporting the idea that PP2-induced relaxation is mainly because of MaxiK channel activation.
Consistent with the view that Src inhibitors promote vascular relaxation through activation of MaxiK, perfusion of LavA promoted an increase in whole-cell current amplitude (Fig. 3 A and B) in unstimulated myocytes. As expected, the inactive analog LavB had no significant effect (Fig. 3 C and D). These results indicate that coronary MaxiK channels are partially phosphorylated by Src under basal conditions and that inhibition of Src tyrosine kinase activity by LavA displaces the equilibrium toward a tyrosine dephosphorylated more active MaxiK channel.
Fig 3.

LavA increases MaxiK currents in rat coronary myocytes. (A) Whole-cell MaxiK currents elicited to +80 mV from Vh = 0 mV before (control) and after ≈55% stimulation with 10 μM LavA (38 ± 9% stimulation, n = 3). (B) Time course of A. (C) Lack of current increase by 10 μM LavB (n = 3). (D) Time course of C.
Coexpression of c-Src and the Human Pore-Forming α Subunit of MaxiK Channels (hSlo) Inhibits Channel Activity in a Ca2+-Dependent Manner.
To directly assess whether c-Src can modulate MaxiK activity, we coexpressed the human α subunit of MaxiK channels (hSlo) ± c-Src in HEK293T cells. The resulting functional effects were assessed by measuring voltage activation curves (FPo-V curves) and determining hSlo voltage sensitivity (V ). In cell-attached patches, where [Ca2+]i is ≈100 nM, the V
). In cell-attached patches, where [Ca2+]i is ≈100 nM, the V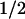 remained unchanged: V
remained unchanged: V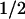 -hSlo = 166 ± 8 mV (n = 4) vs. V
-hSlo = 166 ± 8 mV (n = 4) vs. V -hSlo+c-Src = 169 ± 12 mV (n = 4). Upon patch excision (inside-out), at constant 1.5 μM Ca2+, V
-hSlo+c-Src = 169 ± 12 mV (n = 4). Upon patch excision (inside-out), at constant 1.5 μM Ca2+, V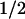 from cells expressing hSlo was also indistinguishable from cells expressing hSlo + cSrc (52 ± 9 mV, n = 4 vs. 56 ± 3 mV, n = 3) (Fig. 4C); current kinetics were practically indistinguishable and no currents were elicited at negative potentials in both cases (Fig. 4 A and B, top traces). However, at 7 μM [Ca2+]i, V
from cells expressing hSlo was also indistinguishable from cells expressing hSlo + cSrc (52 ± 9 mV, n = 4 vs. 56 ± 3 mV, n = 3) (Fig. 4C); current kinetics were practically indistinguishable and no currents were elicited at negative potentials in both cases (Fig. 4 A and B, top traces). However, at 7 μM [Ca2+]i, V becomes more positive in hSlo + c-Src (–10 ± 10 mV, n = 12) than in hSlo (–26 ± 6 mV, n = 46) (Fig. 4 C and D), demonstrating a 16 mV right shift by c-Src. However, the slope of the curves did not change (khslo = 1.66 ± 0.34; khslo+c-Src = 1.75 ± 0.24), indicating that the voltage sensor is not altered but rather its coupling with the opening of the pore. In the representative traces (Fig. 4 A and B; 7 μM), the decrease in the FPo is better seen between 0 and –50 mV where steady-state inward currents (arrows) are smaller in hSlo + cSrc than in hSlo. Also notice the slower activation kinetics induced by c-Src, indicating that c-Src also reduces hSlo voltage activation rate. Because c-Src affected hSlo voltage sensitivity in excised patches at 7 μM Ca2+ but not in cell-attached patches or at 1.5 μM Ca2+, we investigated the Ca2+ dependence of c-Src effect. Fig. 4D shows that c-Src also diminishes the V
becomes more positive in hSlo + c-Src (–10 ± 10 mV, n = 12) than in hSlo (–26 ± 6 mV, n = 46) (Fig. 4 C and D), demonstrating a 16 mV right shift by c-Src. However, the slope of the curves did not change (khslo = 1.66 ± 0.34; khslo+c-Src = 1.75 ± 0.24), indicating that the voltage sensor is not altered but rather its coupling with the opening of the pore. In the representative traces (Fig. 4 A and B; 7 μM), the decrease in the FPo is better seen between 0 and –50 mV where steady-state inward currents (arrows) are smaller in hSlo + cSrc than in hSlo. Also notice the slower activation kinetics induced by c-Src, indicating that c-Src also reduces hSlo voltage activation rate. Because c-Src affected hSlo voltage sensitivity in excised patches at 7 μM Ca2+ but not in cell-attached patches or at 1.5 μM Ca2+, we investigated the Ca2+ dependence of c-Src effect. Fig. 4D shows that c-Src also diminishes the V of hSlo at [Ca2+]i up to 100 μM. Representative traces (Fig. 4 A and B) show that similar to 7 μM Ca2+, the inward steady-state currents at 23 and 94 μM Ca2+ were smaller when c-Src was coexpressed.
of hSlo at [Ca2+]i up to 100 μM. Representative traces (Fig. 4 A and B) show that similar to 7 μM Ca2+, the inward steady-state currents at 23 and 94 μM Ca2+ were smaller when c-Src was coexpressed.
Fig 4.

Coexpression with c-Src induces a right-shift of hSlo voltage activation curves at Ca2+ > 1.5 μM. (A) hSlo currents in inside-out patches at different [Ca2+]i. Traces are from −100 to 145 mV every 5 mV for 1.5 μM [Ca2+]i; from −150 to 90 mV for 7, 23, and 94 μM [Ca2+]i. Vh = 0 mV. (B) hSlo + c-Src currents. [Ca2+]i and voltages are as in A. (C) Mean FPo-V curves of hSlo + c-Src were right shifted at 7 μM, but not at 1.5 μM [Ca2+]i. (D) V at [Ca2+]i > 1.5 μM in hSlo and hSlo + c-Src expressing cells (n = 3–6).
at [Ca2+]i > 1.5 μM in hSlo and hSlo + c-Src expressing cells (n = 3–6).
Smooth muscle MaxiK channels can be assembled by α (hSlo) and β1 subunits; thus, we examined the effect of MaxiK-β1 on c-Src inhibition of hSlo. Coexpression of β1 subunit enhanced the inhibition of hSlo by c-Src, measured at 7 μM Ca2+, by approximately 6-fold with a ≈100 mV shift (V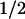 -hSlo+β1 = −69 ± 12 mV, n = 4 vs. V
-hSlo+β1 = −69 ± 12 mV, n = 4 vs. V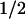 -hSlo+β1+c-Src = 30 ± 11 mV, n = 4). In summary, c-Src inhibits hSlo channel activity by decreasing its effectiveness in responding to voltage and Ca2+. Moreover, c-Src inhibition of hSlo is enhanced by its β1 subunit and switched on by micromolar Ca2+ (>1.5 μM).
-hSlo+β1+c-Src = 30 ± 11 mV, n = 4). In summary, c-Src inhibits hSlo channel activity by decreasing its effectiveness in responding to voltage and Ca2+. Moreover, c-Src inhibition of hSlo is enhanced by its β1 subunit and switched on by micromolar Ca2+ (>1.5 μM).
Protein Tyrosine Phosphatase (PTPase) Partially Reverses the Effect of c-Src.
Inhibition of the voltage sensitivity of hSlo by c-Src may involve a direct phosphorylation of the channel itself or a tightly associated protein. To test this possibility, inside-out patches from cells expressing hSlo (Fig. 5A) or hSlo + c-Src (Fig. 5B) were treated with 100 units/ml of the protein tyrosine phosphatase, CD45 (PTPase) (lower traces). Current traces were acquired every 10 min for up to 1 h. In hSlo alone (Fig. 5A), current amplitudes are slightly reduced after ≈1 h of recording because of rundown. The lack of effect of PTPase was confirmed by FPo-V curves, which were practically identical in control (V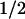 = −28 mV) and after PTPase treatment (V
= −28 mV) and after PTPase treatment (V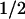 = −31 mV) (Fig. 5C). On the other hand, in hSlo + c-Src patches, PTPase treatment increased current amplitude despite the expected rundown (Fig. 5B). Indeed, FPo-V curves show that PTPase treatment facilitates channel opening by shifting the curve to more negative potentials (Fig. 5D) from V
= −31 mV) (Fig. 5C). On the other hand, in hSlo + c-Src patches, PTPase treatment increased current amplitude despite the expected rundown (Fig. 5B). Indeed, FPo-V curves show that PTPase treatment facilitates channel opening by shifting the curve to more negative potentials (Fig. 5D) from V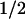 of −17 to –28 mV. The leftward shift by PTPase (ΔV
of −17 to –28 mV. The leftward shift by PTPase (ΔV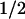 = 11 mV) represents ≈70% recovery when compared to the shift by c-Src (16 mV). The mean ΔV
= 11 mV) represents ≈70% recovery when compared to the shift by c-Src (16 mV). The mean ΔV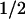 (Fig. 5E) after PTPase treatment shows no significant change in hSlo alone (−1.4 ± 0.8 mV; n = 5), indicating that the basal expression of c-Src in HEK293T cells (see Fig. 7) is not sufficient to significantly maintain a phosphorylated and inhibited hSlo channel. In contrast, PTPase induced a −10 ± 3 mV (n = 7) shift in hSlo + c-Src cells (Fig. 5E) which was attained with a half-time of 29 min (Fig. 5F). Overall these data show that PTPase is able to relieve the inhibition of hSlo by c-Src, strongly suggesting that hSlo (or a closely associated subunit) is directly phosphorylated by c-Src.
(Fig. 5E) after PTPase treatment shows no significant change in hSlo alone (−1.4 ± 0.8 mV; n = 5), indicating that the basal expression of c-Src in HEK293T cells (see Fig. 7) is not sufficient to significantly maintain a phosphorylated and inhibited hSlo channel. In contrast, PTPase induced a −10 ± 3 mV (n = 7) shift in hSlo + c-Src cells (Fig. 5E) which was attained with a half-time of 29 min (Fig. 5F). Overall these data show that PTPase is able to relieve the inhibition of hSlo by c-Src, strongly suggesting that hSlo (or a closely associated subunit) is directly phosphorylated by c-Src.
Fig 5.

PTPase partially reverses the inhibitory effect of c-Src on hSlo. (A) Inside-out recordings from the same cell expressing hSlo before (control) and after perfusion of 100 units/ml PTPase. (B) Currents from a cell coexpressing hSlo + c-Src before (control) and after perfusion of 100 units/ml PTPase. In A and B, traces are from −100 to +60 mV every 5 mV. The effect of buffer alone was tested before applying PTPase (control). (C and D) Corresponding FPo-V curves for hSlo and hSlo + c-Src, respectively. (E) Bar plot of mean ΔV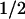 induced by PTPase in hSlo and hSlo + c-Src. *, P < 0.002. (F) Time course of PTPase effect (n = 6–7, hSlo; n = 7–9, hSlo + c-Src).
induced by PTPase in hSlo and hSlo + c-Src. *, P < 0.002. (F) Time course of PTPase effect (n = 6–7, hSlo; n = 7–9, hSlo + c-Src).
Fig 7.

c-Src tyrosine kinase directly phosphorylates hSlo. (A) Western blot with c-Src Ab of cell lysates of nontransfected cells (blank) or cells transfected with c-Src, hSlo, or hSlo + cSrc. (B) Western blot of immunoprecipitated proteins by using anti-c-myc Ab that recognizes c-myc-tagged hSlo, and probed with anti-pY Ab. Tyrosine phosphorylation of hSlo was clearly detected in hSlo + c-Src. (C) Same gel as in B stripped and reprobed with anti-hSlo913–922.
Colocalization and Direct Phosphorylation of hSlo by c-Src.
The compartmentalization of kinases and their substrates is a major determinant of their interaction under physiological conditions. Thus, it is expected that, if hSlo is direct target of c-Src, they should show some degree of colocalization. To test this possibility, HEK293T cells expressing both proteins were double labeled with specific Abs. As expected for the extracellularly c-myc tagged hSlo, hSlo is visualized in the membrane of nonpermeabilized cells (Fig. 6A). In the case of c-Src, which was labeled after permeabilization, its majority is targeted to the membrane with some staining in the cytosol (Fig. 6B). Overlapping confocal images (Fig. 6 C and D) demonstrate that c-Src targeted to the membrane colocalizes with hSlo (69 ± 6% colocalization; n = 10 membrane regions). A similar pattern was observed in 42 cells examined from three different cell transfections. These results are consistent with the view that inhibition of the voltage sensitivity of hSlo by c-Src (Fig. 4) and its partial reversal by PTPase (Fig. 5) involve a direct phosphorylation/dephosphorylation of the channel protein.
Fig 6.

Colocalization of hSlo and c-Src in HEK293T cells. Images were deconvoluted in the x–y plane by using the 3D Point Spread Function to compute and remove the contributions from out-of-focus planes. (A and B) hSlo (green) and c-Src (red) labeling at the middle of cells. (C) Overlap of A and B. (D) Zoom of area marked in C (arrow). (Inset) Colocalization measured as mean pixel intensity (PI) of area between bars in arbitrary units vs. distance across the membrane.
To test direct phosphorylation of the channel protein, we immunoprecipitated hSlo coexpressed with or without c-Src and examined its reactivity toward a pY-Ab. Lysates from nontransfected cells (blank) and cells transfected with hSlo alone show a slight basal expression of c-Src. In contrast, a strong c-Src labeling is observed in lysates of cells expressing c-Src alone or hSlo + c-Src (Fig. 7A). Fig. 7B shows that only immunoprecipitates of cells expressing hSlo + c-Src and a positive control lysate of EGF-stimulated A431 cells (pY-control) produced positive tyrosine phosphorylation labeling of bands with approximate molecular weight of hSlo (≈125 kDa), and an expected band >130 kDa for the pY-control. In contrast, no tyrosine phosphorylation signal was detected in the blank (nontransfected cells), c-Src, or hSlo immunoprecipitated fractions. The same gel was stripped and reprobed with anti-hSlo913–922. This Ab recognized a band at ≈125 kDa in hSlo and hSlo + c-Src (Fig. 7C), indicating that the phosphorylated band is indeed the hSlo channel protein. Note that no band was detected with anti-hSlo913–922 either in negative controls (blank), c-Src or in pY-control.
Discussion
Phosphorylation-dephosphorylation cycles play an important role in the regulation of MaxiK activity. Most of the studies have focused on MaxiK modulation by cGMP- and cAMP-dependent protein kinases possibly because of their roles in mediating vascular relaxation (28). Here, we have explored the possible role of phosphorylation and MaxiK channels in controlling vasoconstriction at the tissue, cellular, and molecular levels. Our results strongly indicate that activation of G protein-coupled receptors (GPCRs) may trigger c-Src-induced phosphorylation of MaxiK channel tyrosine residues promoting channel inhibition and vasoconstriction.
Besides PKC and Rho-kinase (29), a kinase family that may have a key role in vasoconstriction after activation of GPCRs is the tyrosine kinase superfamily (9, 15). Recent evidence and the present work indicate that a tyrosine kinase involved in GPCR-mediated vasoconstriction is the nonreceptor tyrosine kinase, c-Src. c-Src is abundant in vascular tissues (16), and its gene ablation dramatically decreases the ability of the constrictor peptide AngII to induce Ca2+ mobilization (14). Moreover, we found that contractions by 5-HT in human coronaries (Fig. 1) and by AngII and phenylephrine in rat aorta were reversed by inhibitors of c-Src tyrosine kinase, LavA, or PP2. PP1 was also able to relax aortic vessels precontracted with 5-HT but to a smaller degree (not shown). Although LavA, PP2, and PP1 can inhibit other Src tyrosine kinases besides c-Src, the K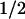 of PP2 in rat aorta (Fig. 2) argues in favor of the view that c-Src is the tyrosine kinase involved in regulating vasoconstriction. Additional data supporting a role for c-Src tyrosine kinase in regulating contraction is the observed protein tyrosil phosphorylation after phenylephrine or 5-HT stimulation of femoral artery cells (30) or aortic rings (31) and increased enzymatic activity of c-Src after AngII stimulation of human cultured arterial myocytes (14).
of PP2 in rat aorta (Fig. 2) argues in favor of the view that c-Src is the tyrosine kinase involved in regulating vasoconstriction. Additional data supporting a role for c-Src tyrosine kinase in regulating contraction is the observed protein tyrosil phosphorylation after phenylephrine or 5-HT stimulation of femoral artery cells (30) or aortic rings (31) and increased enzymatic activity of c-Src after AngII stimulation of human cultured arterial myocytes (14).
The role of MaxiK channel as downstream effector of c-Src contractile pathway is supported by tension and patch–clamp experiments by using its specific blocker, IbTx (Figs. 1–3). We show for the first time that relaxations produced by Src inhibitors in human coronary arteries or rat aorta pretreated with contracting agonists are readily prevented by IbTx to nearly 80% stressing the point that MaxiK channels are a major mechanism in this pathway. Our data can be interpreted as if Src inhibitors relax vascular muscle mainly by hampering the inhibition of MaxiK channels by c-Src (which would lead to depolarization, Ca2+ channel activation and contraction) after stimulation of GPCRs. Results supporting this view are: (i) Diltiazem did not alter the degree of IbTx-inhibition of PP2-induced relaxation, (ii) LavA increased MaxiK whole cell currents; (iii) coexpression with c-Src inhibited hSlo by decreasing its Ca2+/V sensitivities; and (iv) tyrosine phosphatase treatment reversed the inhibitory effect of c-Src coexpression. In agreement, stimulation of MaxiK by general tyrosine kinase inhibitors has been observed in the eye trabecular meshwork (32) and by LavA in myocytes from rat tail artery (20). Moreover the tyrosine phosphatase inhibitors, sodium orthovanadate and dephostatin, are able to inhibit MaxiK in mesenteric arterial myocytes (33). Thus, it is likely that under physiological conditions, the activity of MaxiK and the resulting vascular tension are tuned by its tyrosine phosphorylation/dephosphorylation state. A tyrosine phosphorylation state of arterial MaxiK channels would be enhanced by binding of 5-HT, AngII, or phenylephrine to their corresponding GPCRs activating Gi, Go, or Gq, G proteins known to activate c-Src (34, 35) possibly via protein–protein interactions (36). Activation of c-Src nearby MaxiK channels would then promote their tyrosine phosphorylation and inhibition leading to depolarization and vasoconstriction. It is important to note that Src pathway represents ≥50% of the agonist-induced vasoconstriction in human coronaries and rat aorta and that MaxiK contributes ≈70–80% (Figs. 1 and 2) to this response. Noticeable is the large Src contribution (≈100%) to 5-HT-induced contraction in rat aorta (Fig. 2D). Also relevant is the fact that the relaxation phase of 5-HT-induced phasic contractions in human coronaries was abolished by IbTx (Fig. 1), indicating that MaxiK channels are responsible for the relaxation phase of the contractile cycle. Thus, MaxiK channels are key determinants of vascular tone by controlling both the development of tension and its recovery to basal levels.
MaxiK channel activity can be ruled by other tyrosine kinases and in an opposite direction. In Chinese hamster ovary cells, the nonreceptor tyrosine kinase JAK2 activates endogenous MaxiK channels (37). Interestingly, an activation of MaxiK channels has been observed when active c-Src is coexpressed with its mouse homolog, mSlo (38). This may implicate that under oncogenic conditions the normal regulation of hSlo by c-Src may be reversed and stresses the importance to use nonconstitutively active c-Src to address its normal function.
The inhibitory regulation of c-Src on coexpressed MaxiK α-subunit is enhanced by its β1 subunit, is Ca2+ dependent and requires >1.5 μM [Ca2+]i (Fig. 4). This Ca2+ dependency is physiologically relevant because local Ca2+ near MaxiK is known to reach micromolar levels, allowing MaxiK channel openings (39) and thus permitting c-Src to regulate its activity. A similar Ca2+ dependency on channel modulation was also observed with the active c-Src (38). At present, it is not known whether Ca2+ is required for the channel to switch conformation and activity after c-Src mediated phosphorylation, as is the case for its functional coupling with its β1 subunit (40), or if c-Src activation requires Ca2+ as the cytoplasmic Pyk2 tyrosine kinase (41). In any case, as shown by our biochemical experiments, c-Src seems to mediate its inhibitory mechanism on hSlo via a direct phosphorylation of the channel protein (Fig. 7).
Confocal microscopy (Fig. 6) shows a striking colocalization of coexpressed hSlo and c-Src in the plasma membrane of HEK293T cells. These results support the emerging concept that compartmentalization of protein kinases and their substrates is a major determinant of their interaction under physiological conditions (42). Colocalization of c-Src and its effector(s) is supported by the fact that c-Src coimmunoprecipitates with Kv1.5 channel in myocardium likely through the SH3-binding domain of c-Src (43). Moreover, the Drosophila homolog of hSlo, dSlo, can also coimmunoprecipitate with c-Src; however, no functional effects were detected in this case (44). Consistent with the idea that c-Src colocalizes with hSlo, experiments in the trabecular meshwork show that tyrosine kinase inhibitors increase MaxiK channel activity after patch excision and presumed loss of the intracellular milieu (32). Thus, it is likely that in vascular myocytes c-Src is attached to the plasma membrane and in close proximity to the MaxiK channels forming a heteromultimeric complex.
Our results reveal that c-Src-MaxiK signaling pathway is an important mechanism not only in regulating rat but also human agonist-induced vasoconstriction. The fact that MaxiK channels are downstream effectors of AngII, 5-HT, and phenylephrine open the possibility that besides participating in vasoconstriction, MaxiK could also participate in the control of functions such as cell proliferation and migration known to be regulated by these peptides and neurotransmitters.
Acknowledgments
We thank Dr. Sarah J. Parsons for c-Src cDNA. This work was supported by National Institutes of Health Grants HL54970 (to L.T.), HL47382 (to L.T.), and HD38983 (to E.S.).
Abbreviations
5-HT, 5-hydroxytriptamine
AngII, angiotensin II
IbTx, iberiotoxin
pY, phosphotyrosine
MaxiK (BK), large conductance, voltage-dependent and Ca2+-activated K+ channel
hSlo, human MaxiK pore forming α-subunit
LavA and B, lavendustin A and B
GPCR, G protein-coupled receptor
PTPase, protein tyrosine phosphatase
FPo, fractional open probability
G, conductance
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Brayden J. E. & Nelson, M. T. (1992) Science 256, 532-535. [DOI] [PubMed] [Google Scholar]
- 2.Knot H. J., Standen, N. B. & Nelson, M. T. (1998) J. Physiol. (London) 508, 211-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson M. T., Cheng, H., Rubart, M., Santana, L. F., Bonev, A. D., Knot, H. J. & Lederer, W. J. (1995) Science 270, 633-637. [DOI] [PubMed] [Google Scholar]
- 4.Knot H. J. & Nelson, M. T. (1998) J. Physiol. (London) 508, 199-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Worley J. F., Quayle, J. M., Standen, N. B. & Nelson, M. T. (1991) Am. J. Physiol. 261, H1951-H1960. [DOI] [PubMed] [Google Scholar]
- 6.Macrez N., Morel, J. L., Kalkbrenner, F., Viard, P., Schultz, G. & Mironneau, J. (1997) J. Biol. Chem. 272, 23180-23185. [DOI] [PubMed] [Google Scholar]
- 7.Toro L., Amador, M. & Stefani, E. (1990) Am. J. Physiol. 258, H912-H915. [DOI] [PubMed] [Google Scholar]
- 8.Scornik F. S. & Toro, L. (1992) Am. J. Physiol. 262, C708-C713. [DOI] [PubMed] [Google Scholar]
- 9.Hughes A. D. & Wijetunge, S. (1998) Acta Physiol. Scand. 164, 457-469. [DOI] [PubMed] [Google Scholar]
- 10.O'Dell T. J., Kandel, E. R. & Grant, S. G. (1991) Nature 353, 558-560. [DOI] [PubMed] [Google Scholar]
- 11.Barret J. M., Ernould, A. P., Ferry, G., Genton, A. & Boutin, J. A. (1993) Biochem. Pharmacol. 46, 439-448. [DOI] [PubMed] [Google Scholar]
- 12.Hanke J. H., Gardner, J. P., Dow, R. L., Changelian, P. S., Brissette, W. H., Weringer, E. J., Pollok, B. A. & Connelly, P. A. (1996) J. Biol. Chem. 271, 695-701. [DOI] [PubMed] [Google Scholar]
- 13.Banes A., Florian, J. A. & Watts, S. W. (1999) J. Pharmacol. Exp. Ther. 291, 1179-1187. [PubMed] [Google Scholar]
- 14.Touyz R. M., Wu, X. H., He, G., Park, J. B., Chen, X., Vacher, J., Rajapurohitam, V. & Schiffrin, E. L. (2001) J. Hypertens. 19, 441-449. [DOI] [PubMed] [Google Scholar]
- 15.Di Salvo J., Nelson, S. R. & Kaplan, N. (1997) Proc. Soc. Exp. Biol. Med. 214, 285-301. [DOI] [PubMed] [Google Scholar]
- 16.Oda Y., Renaux, B., Bjorge, J., Saifeddine, M., Fujita, D. J. & Hollenberg, M. D. (1999) Can. J. Physiol. Pharmacol. 77, 606-617. [PubMed] [Google Scholar]
- 17.Toro L., Vaca, L. & Stefani, E. (1991) Am. J. Physiol. 260, H1779-H1789. [DOI] [PubMed] [Google Scholar]
- 18.Giangiacomo K. M., Garcia-Calvo, M., Knaus, H. G., Mullmann, T. J., Garcia, M. L. & McManus, O. (1995) Biochemistry 34, 15849-15862. [DOI] [PubMed] [Google Scholar]
- 19.Ping P., Zhang, J., Zheng, Y. T., Li, R. C., Dawn, B., Tang, X. L., Takano, H., Balafanova, Z. & Bolli, R. (1999) Circ. Res. 85, 542-550. [DOI] [PubMed] [Google Scholar]
- 20.Xiong Z., Burnette, E. & Cheung, D. W. (1995) Eur. J. Pharmacol. 290, 117-123. [DOI] [PubMed] [Google Scholar]
- 21.Somlyo A. V. & Somlyo, A. P. (1968) J. Pharmacol. Exp. Ther. 159, 129-145. [PubMed] [Google Scholar]
- 22.Meera P., Wallner, M., Song, M. & Toro, L. (1997) Proc. Natl. Acad. Sci. USA 94, 14066-14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jurman M. E., Boland, L. M., Liu, Y. & Yellen, G. (1994) BioTechniques 17, 876-881. [PubMed] [Google Scholar]
- 24.Candia S., Garcia, M. L. & Latorre, R. (1992) Biophys. J. 63, 583-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wijetunge S., Aalkjaer, C., Schachter, M. & Hughes, A. D. (1992) Biochem. Biophys. Res. Commun. 189, 1620-1623. [DOI] [PubMed] [Google Scholar]
- 26.Seki T., Yokoshiki, H., Sunagawa, M., Nakamura, M. & Sperelakis, N. (1999) Pflügers Arch. 437, 317-323. [DOI] [PubMed] [Google Scholar]
- 27.Yamazaki M., Kamitani, K., Ito, Y. & Momose, Y. (1994) Br. J. Anaesth. 73, 209-213. [DOI] [PubMed] [Google Scholar]
- 28.Schubert R. & Nelson, M. T. (2001) Trends Pharmacol. Sci. 22, 505-512. [DOI] [PubMed] [Google Scholar]
- 29.Somlyo A. P. & Somlyo, A. V. (2000) J. Physiol. (London) 522, 177-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semenchuk L. A. & Di Salvo, J. (1995) FEBS Lett. 370, 127-130. [DOI] [PubMed] [Google Scholar]
- 31.Watts S. W., Yeum, C. H., Campbell, G. & Webb, R. C. (1996) J. Vasc. Res. 33, 288-298. [DOI] [PubMed] [Google Scholar]
- 32.Stumpff F., Que, Y., Boxberger, M., Strauss, O. & Wiederholt, M. (1999) Invest. Ophthalmol. Visual Sci. 40, 1404-1417. [PubMed] [Google Scholar]
- 33.Yokoshiki H., Seki, T., Sunagawa, M. & Sperelakis, N. (2000) Can. J. Physiol. Pharmacol. 78, 745-750. [PubMed] [Google Scholar]
- 34.Neves S. R., Ram, P. T. & Iyengar, R. (2002) Science 296, 1636-1639. [DOI] [PubMed] [Google Scholar]
- 35.Yamauchi J., Itoh, H., Shinoura, H., Miyamoto, Y., Tsumaya, K., Hirasawa, A., Kaziro, Y. & Tsujimoto, G. (2001) Biochem. Biophys. Res. Commun. 288, 1087-1094. [DOI] [PubMed] [Google Scholar]
- 36.Ma Y. C., Huang, J., Ali, S., Lowry, W. & Huang, X. Y. (2000) Cell 102, 635-646. [DOI] [PubMed] [Google Scholar]
- 37.Prevarskaya N. B., Skryma, R. N., Vacher, P., Daniel, N., Djiane, J. & Dufy, B. (1995) J. Biol. Chem. 270, 24292-24299. [DOI] [PubMed] [Google Scholar]
- 38.Ling S., Woronuk, G., Sy, L., Lev, S. & Braun, A. P. (2000) J. Biol. Chem. 275, 30683-30689. [DOI] [PubMed] [Google Scholar]
- 39.Perez G. J., Bonev, A. D., Patlak, J. B. & Nelson, M. T. (1999) J. Gen. Physiol. 113, 229-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meera P., Wallner, M., Jiang, Z. & Toro, L. (1996) FEBS Lett. 382, 84-88. [DOI] [PubMed] [Google Scholar]
- 41.Beitner-Johnson D., Ferguson, T., Rust, R. T., Kobayashi, S. & Millhorn, D. E. (2002) Cell. Signalling 14, 133-137. [DOI] [PubMed] [Google Scholar]
- 42.Levitan I. B. (1999) Adv. Second Messenger Phosphoprotein Res. 33, 3-22. [DOI] [PubMed] [Google Scholar]
- 43.Holmes T. C., Fadool, D. A., Ren, R. & Levitan, I. B. (1996) Science 274, 2089-2091. [DOI] [PubMed] [Google Scholar]
- 44.Wang J., Zhou, Y., Wen, H. & Levitan, I. B. (1999) J. Neurosci. 19, RC4., 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]