

Abstract
Large conductance voltage- and Ca2+-dependent K+ (MaxiK) channels show sequence similarities to voltage-gated ion channels. They have a homologous S1-S6 region, but are unique at the N and C termini. At the C terminus, MaxiK channels have four additional hydrophobic regions (S7-S10) of unknown topology. At the N terminus, we have recently proposed a new model where MaxiK channels have an additional transmembrane region (S0) that confers β subunit regulation. Using transient expression of epitope tagged MaxiK channels, in vitro translation, functional, and “in vivo” reconstitution assays, we now show that MaxiK channels have seven transmembrane segments (S0-S6) at the N terminus and a S1-S6 region that folds in a similar way as in voltage-gated ion channels. Further, our results indicate that hydrophobic segments S9-S10 in the C terminus are cytoplasmic and unequivocally demonstrate that S0 forms an additional transmembrane segment leading to an exoplasmic N terminus.
High conductance voltage- and Ca2+-sensitive K+ (MaxiK) channels are expressed in a wide variety of tissues. Mammalian MaxiK channels are characterized by their high sensitivity to blockade by iberiotoxin (IbTx) and charybdotoxin. In neurons and skeletal muscle they may be responsible for fast repolarization of the action potential and modulate transmitter release (1–3). In smooth muscles they are crucial in regulating contractility (4, 5). The MaxiK channel was first cloned from Drosophila melanogaster (6, 7). Subsequently, it was cloned from several mammalian species including humans (8–14) and recently from Caenorhabditis elegans (15).
The N-terminal third of MaxiK channels shows sequence homology to the voltage sensor and the pore region of voltage-dependent ion channels. However, we have recently given evidence that MaxiK channels carry a unique N-terminal transmembrane segment (S0) that leads to an exoplasmic N terminus. This additional transmembrane segment (S0) is critical for β subunit modulation (16).
The C-terminal region (after S6) carries four additional hydrophobic, possibly membrane spanning regions (S7, S8, S9, and S10). This region comprises about two-thirds of the total length of the primary amino acid sequence. The last one third (also called “tail”), containing hydrophobic regions S9 and S10, shows the highest sequence conservation among species. This motif can be expressed as a separable domain, and has been suggested to determine the Ca2+ sensitivity (17). A series of negative charges just before S10 is believed to participate in Ca2+ sensing and has been called the “Ca2+ bowl” (15).
Voltage-dependent ion channels form a large family of related structures that include K+, Na+, and Ca2+ channels and also cyclic nucleotide-gated channels, despite the fact that the latter are not voltage activated (18, 19). Based on their sequence similarity it is thought that all of them have the same membrane topology: six membrane spanning regions with intracellular N and C termini, extracellular linkers between S1-S2 and S3-S4, and a pore loop between transmembrane regions S5 and S6 that dips into the membrane from the external side (20, 21). This membrane topology has been confirmed in many studies (22–28).
Sequence analysis (7, 16) and the fact that MaxiK channels possess an intrinsic voltage sensor that opens the channel in the practical absence of Ca2+ (29–31) support the view that MaxiK channels have a close functional and structural relationship with voltage-gated ion channels. We have recently shown that MaxiK channels share some of the conserved charged residues critical in voltage-dependent gating (sensing and structural residues) (16), not only in the S4 segment but also in S3 region, with voltage-gated ion channels (32–34).
In this study, we have used several experimental approaches to analyze the membrane topology of MaxiK channels. We expressed epitope tagged channels and used, in addition to fluorescent labeled antibodies (Abs), Ab-coated magnetic beads as a new method to map extracellular regions. To test the cytosolic nature of the tail region, we performed in vitro translation experiments and employed an “in vivo” reconstitution approach. We were able to record MaxiK currents after a patch containing the membrane-bound “core” region was introduced into the cytoplasm of an oocyte expressing the tail region alone. The results demonstrate that MaxiK channels are structurally distinct members of the voltage-gated ion channel family; their topology is highlighted by an extracellular N terminus, and a seventh transmembrane domain at the N terminus (S0). They also demonstrate that hydrophobic segments S9 and S10 are intracellular.
MATERIALS AND METHODS
Molecular Biology.
Numbering in humanMaxiK (Hslo) and Drosophila MaxiK (Dslo) is according to GenBank accession numbers U11058 and JH0697, respectively (7, 13). However, Dslo variant used is A1C2E1G3I0 (7). GCG programs (35) were used for sequence analysis.
The c-myc epitope (AEEQKLISEEDL) was introduced either with two complementary phosphorylated oligos at unique restriction sites (HF1, NcoI; HF4, BsrGI), or by introducing the c-myc sequence into overlapping PCR primers (HF2 and HF3) (36, 37). For expression in COS-M6 cells, constructs were cloned into the pcDNA3 vector (Invitrogen).
The predicted protein sequences of constructs used are as follows. HF1, MGAEEQKLISEEDLV followed by Met-1 to Leu-1113 from Hslo; HF2, GAEEQKLISEEDLG inserted between Ser-70 and Ser-71 of Hslo; HF3, PGAEEQKLISEEDLG inserted between Asn-136 and Pro-137 of Hslo; HF4, MYTGAEEQKLISEEDLV inserted between Glu-535 and Met-536 of Hslo; HDP: Asn-258 to Gly-300 of Hslo replaced by Asn-273 to Gly-314 from Dslo.
Hslo-tail and Dslo-tail constructs were made by PCR. Hslo-tail: Met-679 to Leu-1113. Dslo-tail: Met-690 to Ser-1175; His-691 was replaced by Asp to introduce an NcoI site at the translational start.
Hslo-core carries Met-1 to Gln-675 and an additional (random) 58 amino acids before the translation reaches a stop codon. HS7-S8: Met-Val followed by Arg-443 to Pro-583 from Hslo. DS7-S8: Met-Val followed by Met-456 to Pro-600 from Dslo. All constructs were analyzed by restriction digestions. Sequences amplified by PCR and ligation connections were confirmed by sequencing. cRNA was transcribed using the mMesSAGEmMACHINE kit (Ambion, Austin, TX).
Electrophysiology.
Oocytes were injected with 5–10 ng of cRNA and measured 2–5 days after injection. Currents were measured in the inside-out or outside-out configuration of the patch clamp technique in symmetrical 110 mM K+ (105 mM K+-methanesulfonate/5 mM KCl/10 mM Hepes, pH 7) (16, 29). The bath solution had in addition 5 mM N-(2-hydroxyethyl)ethylenediamine triacetic acid (HEDTA) and CaCl2, which was added to the desired free Ca2+. Free Ca2+ was measured with a Ca2+ electrode.
In Vitro Translation and Protein Gels.
cRNA was translated (0.5–1 μg in a 25 μl reaction) with rabbit reticulocyte lysate in presence of dog pancreatic microsomes (Promega) and [35S]-methionine. Aliquots (5–10 μl) were diluted with 100 μl of PBS (9 mM Na2HPO4/1.4 mM NaH2PO4/137 mM NaCl), 0.1 or 0.3 M Na2CO3 (pH 11), and kept on ice for 30 min (38). Microsomes bearing translated proteins were collected by centrifugation (1 hr at ≈20,000 × g at 4°C). Pellets were washed two times with PBS (100 μl). Supernatant proteins (100 μl) were precipitated with cold acetone (200 μl), centrifuged, and dissolved in sample buffer (0.125M Tris, pH 6.8/20% glycerol/4% SDS/2% 2-mercaptoethanol). SDS/PAGE was carried out on 12–15% gels. After electrophoresis, gels were stained in 30% methanol, 10% acetic acid supplemented with 0.1% Coomassie brilliant blue R-250 and destained in the same solution without dye before soaking for 30 min in Amplify (Amersham). Gels were dried and exposed to x-ray films.
Cell Transfections.
Cos-M6 cells were transfected using the DEAE-Dextran (Pharmacia) method as described (39). Cells were replated on poly-l-lysine-coated petri dishes 24 hr after transfection. For cell permeabilization the cells were fixed with 4% paraformaldehyde in PBS for 20 min at 4°C followed by three or four washes with 0.2% Triton X-100 in PBS at room temperature.
Immunostaining.
Cells were incubated with a 1:200 dilution of anti-c-myc (clone 9E10) mAb (PharMingen) for 1 hr. Excess primary antibody was removed by two washes with PBS. Cells were incubated with slight shaking for several minutes with 1–4 μl of sheep anti-mouse IgG coated Dynabeads (Dynal, Lake Success, NY) or 1:500 dilution of fluorescein isothiocyanate (FITC)-labeled secondary antibody. Excess secondary antibody (beads or FITC-labeled Ab) was removed by washing with PBS before imaging. Cells with beads were imaged with a microscope mounted video camera connected to a frame grabber board. Cells stained with FITC were imaged with fluorescence (Axiovert 135) and confocal microscopes (LSM 310; Zeiss).
RESULTS
Hydrophobicity Analysis and Topology Models of MaxiK Channels.
Transmembrane regions are usually characterized by the abundance of hydrophobic residues in stretches of ≈20 amino acids. Fig. 1A shows the hydrophobicity pattern of the mammalian and the Drosophila MaxiK channels. With respect to the C terminus, this analysis reveals that: (i) the overall hydrophobicity of the C-terminal regions S7–S10 is considerably lower than that of transmembrane regions S0, S1, and S6, and (ii) the hydrophobicity of regions S8 and S9 in Dslo is low and they may be too short to span the membrane. Therefore, assuming that Hslo and Dslo fold in the same way, it seems rather unlikely that these regions are membrane spanning. Similar to Dslo, the recently cloned C. elegans homolog (15) shows a low hydrophobicity in regions S8 and S9 and also carries the additional hydrophobic region S0 (not shown).
Figure 1.
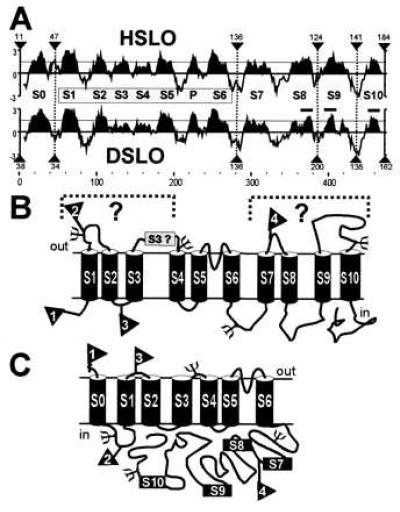
Hydrophobicity analysis and alternative Models for MaxiK channel topology. (A) Hydrophobicity analysis of Hslo and Dslo using GCG-program pepplot (window size of nine amino acids). Hydrophobic regions (S0 to S4) are according to the model in C (16). Numbers at arrows indicate amino acids that were removed from the protein sequence. Bars above S8, S9, and S10 in Dslo correpond to 16 amino acids length (minimum requirement for a protein α helix to span a lipid bilayer of 2.5 nm thickness). (B) Previous model with intracellular N terminus, and unknown topology of the C terminus (S7-S10). (C) Proposed membrane topology of MaxiK channels. (B and C) Flags, numbered from 1–4 in both models, mark positions where c-myc epitopes were introduced. ψ, consensus sites for N-linked glycosylation (NXS/T) in bovine slo, regardless of being extra- or intracellular.
The membrane topology of MaxiK channels has been proposed to have six transmembrane regions analogous to voltage-dependent ion channels, but with four additional hydrophobic regions at the C terminus with uncertain topology (Fig. 1B). Some authors (7, 21) have chosen to draw the C terminus (downstream from S6) as entirely cytoplasmic, presumably because of the relatively low hydrophobicity. In addition, this part is likely to contain the Ca2+ binding site(s) that has (have) to be on the cytoplasmic side of the membrane. Others have shown these regions as membrane spanning in their proposed models (40) (Fig. 1B). However, considering that each of these four additional hydrophobic regions may or may not span the membrane, there are many other alternative models. Fig. 1C shows our recently proposed model with seven transmembrane regions, an exoplasmic N terminus and an intracellular C terminus (16).
Glycosylation is generally an indication that the glycosylated part of the protein is exoplasmic, i.e., faces the lumen of vesicles or the extracellular face of the plasma membrane. Bovine MaxiK channel α-subunits are not glycosylated when purified from tissues (41). In the model shown in Fig. 1B, three consensus sequences for N-linked glycosylation in bovine MaxiK channel α-subunits and Hslo would be extracellular. In our model (Fig. 1C) only one of these sites, located in the short loop (only three amino acids) between transmembrane regions S3 and S4, is extracellular. Such sites residing in close proximity to the membrane are likely to be restricted in their accessibility for glycosylating enzymes. Therefore, the model in Fig. 1C is consistent with the finding that bovine MaxiK channel α-subunits are not glycosylated. Furthermore, this model is strongly supported by our previous experimental evidence showing that: (i) truncation clones of Hslo and Dslo, carrying only the hydrophobic segment S0, behave like integral membrane proteins and exhibit the expected glycosylation pattern and (ii) the addition of an N-terminal cleavable signal sequence to Hslo and Dslo did not alter their functional behavior (16).
MaxiK Channels Have an Extracellular N Terminus.
To unequivocally distinguish between the two possible folding models of MaxiK channels at the N terminus, we introduced c-myc epitope tags into critical positions in Hslo (flags 1–3 in Figs. 1 B and C and 2A) and tested for antibody binding under permeabilizing and nonpermeabilizing conditions. This approach exploits the fact that Abs cannot cross cell membranes. Therefore, Abs binding under nonpermeabilizing conditions is a demonstration that the epitope is extracellular. Intracellular epitopes become accessible only after cell permeabilization.
Epitope tagged constructs that showed functional expression were used for immunolabeling studies. Functional expression was considered as an indication that the epitope insertion did not lead to changes in the overall membrane topology. Constructs HF1, HF2 (Fig. 2A), and HF4 (see Fig. 4A) are similar to wild-type channels in their Ca2+ and voltage sensitivities (Fig. 2B). Only HF3 (Fig. 2A) deviates from the wild-type Hslo channels; it shows a lower voltage sensitivity at all Ca2+ concentrations tested (Fig. 2B). The behavior of HF3 (c-myc epitope between S1 and S2 in our model), is qualitatively similar to Shaker K+ channels with an epitope insertion between transmembrane segments S1 and S2, which also require higher voltages for activation (28). This similarity supports the view that this region in Shaker K+ and MaxiK channels is structurally and functionally equivalent.
Figure 2.
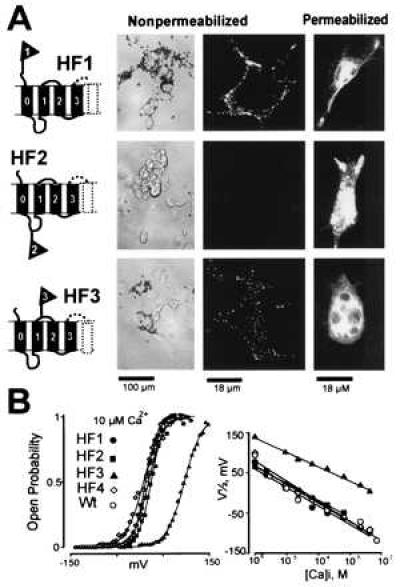
MaxiK channels have an exoplasmic N terminus with an additional membrane spanning segment (S0). (A) Immunocytochemistry of c-myc tagged MaxiK channels (HF1, HF2 and HF3) expressed in COS-M6 cells. Cells were incubated with anti-c-myc mAb under nonpermeabilizing and permeabilizing conditions. Antibody binding was visualized using beads coated with secondary antibodies or with FITC-labeled secondary antibodies. Confocal images (two right panels) are from sections at the middle of the cells. Experiments were performed at least three times for each construct (also in Fig. 4) with similar results. (B) Functional expression of c-myc tagged clones in oocytes measured in inside-out patches. (Left) Voltage activation curves at 10 μM intracellular Ca2+, [Ca2+]i. Values for half activation potentials (V1/2) in 10 μM [Ca2+]i are (in mV): 12 ± 18 (n = 64) for wild-type Hslo (Wt); 5 ± 9 (n = 3) for HF1; 14 ± 8 (n = 6) for HF2; 98 ± 7 (n = 4) for HF3; 13 ± 6 (n = 3) for HF4 (see Fig. 4). (Right) V1/2 as a function of [Ca2+]i.
Figure 4.
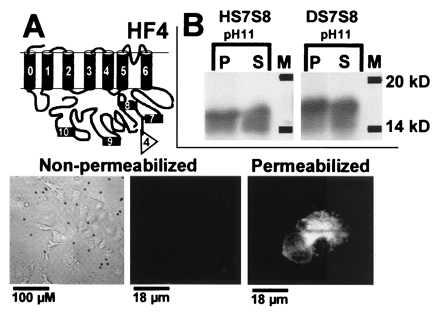
Topology of S7 and S8 regions. (A) Immunocytochemistry of Hslo epitope tagged between S7 and S8 (HF4). Visualization with both magnetic beads and FITC-labeled antibodies. (B) In vitro translation of HS7S8 and DS7S8 containing hydrophobic regions S7 and S8. P, pellet or microsomal fraction; S, soluble fraction; M, protein marker.
We used two different methods to detect primary (anti c-myc) antibody binding: (i) FITC-labeled antibodies (42), and (ii) magnetic beads coated with secondary antibodies. The latter is a modification of a procedure developed for visual identification of individual transfected cells for electrophysiological experiments (43). The beading method has some advantages: it is inexpensive, sensitive and does not require a fluorescence microscope. Labeling of extracellular epitopes with beads under nonpermeablizing conditions was more sensitive and gave clearer results for clone HF3 (see Fig. 2) than using fluorescent-labeled Ab. However, under permeabilizing conditions detection of Ab binding with beads was difficult to distinguish from background staining. This may be due to restricted accessibility to Ab binding caused by the beads.
Fig. 2A shows representative images of antibody-labeled cells expressing constructs HF1, HF2, and HF3. Cells expressing HF1 and HF3 are decorated with the beads under nonpermeabilizing conditions. In contrast, no binding of beads was observed in cells expressing HF2, which carries the epitope tag in the intracellular linker between S0 and S1 of our model. Similar results were observed with fluorescent-labeled secondary Ab. Confocal images did not show any labeling of nonpermeabilized cells expressing HF2. Labeling of cells expressing HF1 and HF3 was restricted to the cell surface in nonpermeabilizing conditions; whereas under permeabilizing conditions, cells expressing all three constructs were profusely labeled.
Although we did not notice any differences in functional expression levels between clones HF1 and HF3, the antibody labeling either with beads or fluorescein was always stronger in cells expressing HF1. After the permeabilizing treatment, the staining intensities of HF1 and HF3 were similar. Therefore, the difference in antibody binding to nonpermeabilized cells may be due to steric hindrance by the lipid bilayer that disappears when the membrane is dissolved by the permeabilizing treatment. A similar observation has been reported for epitope tags introduced close to the transmembrane regions of the N-methyl-d-aspartate receptor (42), and of Shaker K+ channels (44).
The fact that HF2 was only labeled after permeabilization is consistent with the view that this epitope is intracellular. We deem unlikely that the HF2 epitope is extracellular, and hindered to Ab-labeling due to closeness to the membrane, because the epitope was inserted well away from flanking transmembrane segments (S0 and S1).
The labeling of HF1 and HF3 under nonpermeablizing conditions proves that MaxiK channels possess an exoplasmic N terminus and that the linker between homology region S1 and S2 is outside, as in Fig. 1C. The inaccessibility of the epitope in clone HF2 is consistent with the notion that this region is intracellular. These results are incompatible with the previous model (Fig. 1B), in which the epitopes in HF1 and HF3 should be accessible only after cell permeabilization, and the epitope in HF2 should be labeled without permeabilization.
Localization of the Pore Region.
To confirm that the linker between homology regions S5 and S6 in MaxiK channels faces the extracellular milieu and forms part of the pore region, as in other K+ channels, we took advantage of the different “pore pharmacology” of mammalian (Hslo) and Drosophila (Dslo) MaxiK channels. Mammalian MaxiK channels are blocked by low concentrations (Kd ≈ 1 nM) of externally applied IbTx (Fig. 3A) (13). In contrast, Drosophila channels are completely insensitive to IbTx concentrations up to 100 nM (Fig. 3B). To test if amino acid differences in the S5-S6 linker are responsible for this difference, we constructed a chimera (HDP) where we replaced the S5-S6 linker of the human channel with the corresponding part of the Drosophila channel and measured IbTx block in the outside-out configuration of the patch clamp technique. The chimeric construct HDP (Hslo with Dslo pore) is completely insensitive to 100 nM IbTx (Fig. 3C). This result confirms the view that the S5-S6 linker determines IbTx binding and forms the MaxiK channel pore loop entering the membrane from the external side.
Figure 3.
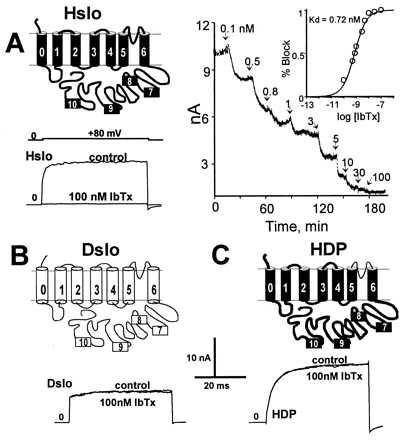
Linker between S5 and S6 forms the pore region of MaxiK channels. (A) IbTx sensitivity of Hslo. IbTx (100 nM) applied to the external side of an outside-out patch, completely blocks ionic currents. Holding potential = 0 mV; test potential = 80 mV. (Right): Time course of IbTx blockade at different IbTx concentrations. (Insert) Dose-response curve, Kd = 0.72 nM, Hill coefficient near one. (B) Dslo currents are insensitive to 100 nM IbTx. (C) Chimeric construct HDP carries the pore of Dslo in the Hslo backbone. Current traces show that the pore of Dslo makes Hslo insensitive to 100 nM IbTx.
Membrane Topology of the C-Terminal Regions S7 and S8.
MaxiK channels carry a long C terminus that has four additional hydrophobic, possibly membrane spanning regions, termed S7, S8, S9, and S10. However as shown in Fig. 1A, the hydrophobicity of these regions is relatively low. To determine if S7 and S8 are membrane spanning, we introduced a c-myc epitope tag between hydrophobic regions S7 and S8 (HF4) (Fig. 1B and Fig. 4). This construct was functional and had similar Ca2+ and voltage sensitivities when compared with wild-type Hslo (see Fig. 2B). In immunolabeling experiments under nonpermeablizing conditions, we could not detect beading or fluorescent staining. Only after permeabilization a strong labeling was observed (Fig. 4A). This result suggests that the epitope is intracellular, but is not conclusive since detergent treatment used for permeabilization could have exposed a previously hidden extracellular epitope. Therefore, to further test the intracellular nature of this region (S7 and S8), we carried out in vitro translation experiments in presence of microsomes of both Dslo and Hslo. Membrane spanning regions are inserted into microsomal membranes during the in vitro translation and can be separated from soluble proteins by centrifugation; high pH treatment is used to exclude a peripheral membrane association (38). Interestingly, upon in vitro translation of both Dslo (DS7-S8) and Hslo (HS7-S8) clones carrying S7 and S8 (including 40 N-terminal and 25 C-terminal adjacent amino acids) resulted in about 50% of the protein in the pellet fraction, and the residual 50% in the soluble fraction (Fig. 4B). These results are inconclusive and may be due to the limitations of this in vitro approach; proteins may assume proper folding and their native membrane orientation only in the context of a functional domain. Thus, at present the membrane topology of this region remains uncertain.
MaxiK Channels C-Terminal Tail Region (Containing S9 and S10) Is Cytosolic.
Wei et al. (17) showed that the tail region can be functionally expressed as a separable domain in Xenopus oocytes. We have reproduced these results (Fig. 5B), and tested the tail region clones of Hslo and Dslo for membrane insertion by in vitro translation in the presence of microsomes (Fig. 5A). The majority of the protein was found in the soluble fraction (Fig. 5A), suggesting that the tail region containing S9 and S10 segments is cytosolic. The slight amount of protein associated with the microsomal fraction is probably not due to membrane association since it was also observed for luciferase, a soluble protein that was used as a control (not shown). The decrease in signals in the soluble fraction after high pH treatment (0.1 and 0.3 M Na2CO3) seems to be due to the increased tendency of protein aggregation (seen as a smear at higher molecular weight) after Na2CO3 treatment.
Figure 5.
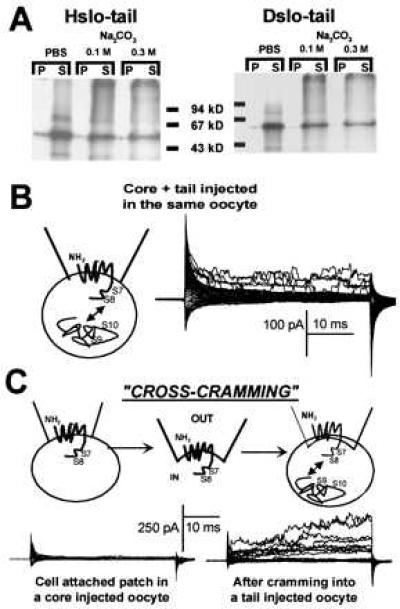
Cytosolic C terminus tail (containing S9-S10). (A) In vitro translation of the tail region containing S9 and S10. Hslo-tail and Dslo-tail cRNAs were in vitro translated (see Materials and Methods). The calculated molecular weights are: Hslo-tail, 48.2 kDa; Dslo-tail, 52.2 kDa. Pellet (P) or membrane fraction and soluble fraction (S). (B) Coexpression of core and tail in the same oocytes produce functional channels (17). Currents in cell-attached mode with test pulses from −50 mV to +136 mV every 6 mV (Vh = 0 mV). (C) Assembly of functional MaxiK channels by cross-cramming: (Left) Cell attached patch in a core injected oocyte does not show any current. (Right) Functional channels assemble within few minutes when the cell-attached patch was excised and crammed into a tail injected oocyte. Currents were elicited using the same protocol as in B. The larger K+ currents in the cross-cramming experiments compared with those in B, may be due to diffusion of external Ca2+ into the oocyte during cramming.
In addition, we performed experiments in an in vivo system. We injected cRNA for Hslo core (that includes S0-S8) and tail into separate oocytes to investigate the independent folding and localization of the tail protein by an electrophysiological technique that we named “cross-cramming” (Fig. 5C). The core region (Fig. 5C) as well as the tail region (not shown) by themselves do not form functional channels. However, functional channels assembled within minutes when patches excised from core expressing oocytes were crammed into tail injected oocytes (Fig. 5C, n = 11). This formation of functional channels was observed in all experiments performed. The efficiency of assembly seems to be low. It has to be kept in mind, however, that under these experimental conditions (voltage, cell-attached mode) the reconstituted channels are facing the low intracellular calcium concentration, where the open probability of wild-type channels is below 10% (29). Similar current levels were obtained when core and tail are coexpressed in the same oocyte (Fig. 5B). The above results suggest that the tail domain folds into its native conformation independently from the core region. Because, integral membrane proteins are restricted in their diffusion when studied with the patch clamp technique, the most natural way to explain these findings using cross-cramming is that the tail region forms a cytosolic protein in its native conformation. To our knowledge, the concept of testing the folding of a protein by electrophysiological methods is the first of its kind.
DISCUSSION
The fact that an epitope at the N terminus is accessible for antibody binding in nonpermeabilized cells (Fig. 2), unequivocally demonstrates that MaxiK channels have an exoplasmic N terminus. In our model (Fig. 1C) MaxiK channels have a unique transmembrane segment at the N terminus (S0) and regions S1-S6 similar to voltage-gated ion channels. These S1-S6 regions were first assigned based on sequence homology (7, 16); we now show experimental evidence supporting this view. We will be referring to S0-S6 as shown in our model (Fig. 1C).
Several lines of evidence indicate that the S1-S6 region of MaxiK has the same membrane topology (Figs. 1 A and C) of six membrane spanning regions and a pore loop between S5 and S6 as voltage-gated ion channels: (i) The accessibility of a tag introduced between regions S1 and S2 (HF3), under nonpermeabilizing conditions, shows that this region is extracellular. (ii) The inaccessibility of an epitope tag between transmembrane regions S0 and S1 (HF2) to antibody binding under nonpermeabilizing conditions is consistent with this region being intracellular. (iii) Transferring the linker between S5 and S6, carrying a signature sequence for a K+ selective pore, from Dslo into Hslo leads to a loss of IbTx blockade indicating that this region indeed forms the pore. (iv) MaxiK channels carry an intrinsic voltage sensor (29–31), which induces gating currents upon depolarization (30), in agreement with the conservation of charged residues in segment S4 (15) involved in voltage sensing (33, 34). Mutating positive charges in the MaxiK S4 region results in changes in their voltage-dependent gating (P.M. and L.T., unpublished results). All these findings are consistent with our suggested model in Fig. 1C, and epitope tagging experiments exclude the N-terminal folding of the alternative model shown in Fig. 1B.
Hydrophobicity analysis of the C-terminal regions (S7-S10) of Hslo reveals that their hydrophobicity is low when compared with S0, S1, or S6. This argues against, but does not exclude, the possibility that these regions are membrane spanning. For example, a low hydrophobicity is also observed for transmembrane regions S2, S3, and especially S4. In these cases, it seems that the charged residues in one membrane spanning segment are neutralized by oppositely charged residues in other membrane spanning regions (32). These charged residues, embedded in a hydrophobic environment, considerably diminish the overall hydrophobicity. Although unlikely, the same could be the case for these C-terminal regions.
Despite a low sequence conservation, the hydrophobicity patterns of Shaker channels and other voltage-dependent ion channels (including S1 to S6 as assigned in Fig. 1C of MaxiK channels) look very much alike (not shown). In contrast, for regions S8 and S9, which show high sequence conservation between Hslo and Dslo, the hydrophobicity is not preserved and is considerably less pronounced in Dslo. Because of their similar function, it is highly unlikely that Dslo has a different membrane topology than Hslo. Therefore, this argues against the possibility that S7 and S8 are membrane spanning in both Dslo and Hslo.
Our experimental finding that the epitope tag (HF4) introduced between S7 and S8 cannot be labeled from the external side by an antibody is consistent with the idea that this region is intracellular. However, the results obtained by in vitro translation of a protein containing S7 and S8 suggests that one or both of these regions could be membrane spanning as 50% of the protein remains membrane associated even after high pH treatment. Therefore, the membrane topology of hydrophobic regions S7 and S8, remains an open question. Arguments in favor of a cytosolic nature of S7-S8 regions are: (i) their low hydrophobicity; (ii) the lack of antibody binding to intact cells expressing HF4; and (iii) that alternative splicing in this region results in profound changes in the “intracellular” Ca2+ sensitivity of Dslo channels (45). Therefore, in our model we have shown S7 and S8 hydrophobic regions as intracellular.
The cross-cramming experiments show that the tail region forms and behaves as a functional domain. This feature strengthens the conclusion, drawn from in vitro translation experiments, that the tail region is a soluble protein. Both cross-cramming and in vitro translation indicate that the tail region (containing S9-S10) is indeed cytosolic. The possibility that the tail region inserts into the membrane during cross-cramming experiments is remote because it does not show tendency to be incorporated into membranes during in vitro translation with microsomal membranes. In addition, a putative calcium binding site (Ca2+ bowl) (15) is located between S9 and S10 and should be cytosolic.
All voltage-dependent ion channels have two major discernible structural and functional motifs in common; the voltage sensor (roughly transmembrane segments S1-S4) and the conduction pathway (S5 to S6). MaxiK channels share these two motifs but have unique flanking parts appended to accommodate the modulatory effects of Ca2+ (C terminus) and the β subunit (N terminus). We have shown that the tail region folds and functions as an independent unit, presumably binding and mediating the facilitating effects of Ca2+. The N terminus mediates the facilitating effects of the regulatory β-subunit (16). Therefore, MaxiK channels constitute a remarkable example of a modular protein design.
Acknowledgments
We thank Drs. John Adelman and Armado Lagrutta for the generous gift of Dslo. This work was supported by National institutes of Health Grant HL54970 (L.T.). L.T. is an Established Investigator of the American Heart Association.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: MaxiK, large conductance voltage- and calcium-dependent K+ channel; Hslo, human MaxiK; Dslo, Drosophila MaxiK; IbTx, iberiotoxin; FITC, fluorescein isothiocyanate; Ab, antibody.A commentary on this article begins on page 13383.
References
- 1.Crest M, Gola M. J Physiol (London) 1993;465:265–287. doi: 10.1113/jphysiol.1993.sp019676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robitaille R, Garcia M L, Kaczorowski G J, Charlton M P. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- 3.Elkins T, Ganezky B, Wu C. Proc Natl Acad Sci USA. 1986;83:8415–8419. doi: 10.1073/pnas.83.21.8415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anwer K, Oberti C, Perez G J, Perez-Reyes N, McDougall J K, Monga M, Sanborn B M, Stefani E, Toro L. Am J Physiol. 1993;265:C976–C985. doi: 10.1152/ajpcell.1993.265.4.C976. [DOI] [PubMed] [Google Scholar]
- 5.Nelson M T, Cheng M R, Santana L F, Bonev A D, Knot H J, Lederer W J. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 6.Atkinson N S, Robertson G A, Ganetzky B. Science. 1991;253:551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- 7.Adelman J P, Shen K, Kavanaugh M P, Warren R A, Woo Y, Lagrutta A, Bond C T, North R A. Neuron. 1992;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- 8.Butler A, Tsunoda S, McCobb D P, Wei A, Salkoff L. Science. 1993;261:221–224. doi: 10.1126/science.7687074. [DOI] [PubMed] [Google Scholar]
- 9.Tseng-Crank J, Foster C D, Krause J D, Mertz R, Godinot N, DiChiara T J, Reinhart P H. Neuron. 1994;13:1315–1330. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- 10.Dworetzky S I, Trojnacki J T, Gribkoff V K. Mol Brain Res. 1994;27:189–193. doi: 10.1016/0169-328x(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 11.Pallanck L, Ganetzky B. Hum Mol Genet. 1994;3:1239–1243. doi: 10.1093/hmg/3.8.1239. [DOI] [PubMed] [Google Scholar]
- 12.McCobb D P, Fowler N L, Featherstone T, Lingle C, Saito M, Krause J E, Salkoff L. Am J Physiol. 1995;269:H767–H777. doi: 10.1152/ajpheart.1995.269.3.H767. [DOI] [PubMed] [Google Scholar]
- 13.Wallner M, Meera P, Ottolia M, Kaczorowski G J, Latorre R, Garcia M L, Stefani E, Toro L. Recept Channels. 1995;3:185–199. [PubMed] [Google Scholar]
- 14.Vogalis F, Vincent T, Qureshi I, Schmalz F, Ward M W, Sanders K M, Horowitz B. Am J Physiol. 1996;271:G629–G639. doi: 10.1152/ajpgi.1996.271.4.G629. [DOI] [PubMed] [Google Scholar]
- 15.Wei A, Jegla R, Salkoff L. Neuropharmacology. 1996;35:805–829. doi: 10.1016/0028-3908(96)00126-8. [DOI] [PubMed] [Google Scholar]
- 16.Wallner M, Meera P, Toro L. Proc Natl Acad Sci USA. 1996;93:14922–14927. doi: 10.1073/pnas.93.25.14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei A, Solaro C, Lingle C, Salkoff L. Neuron. 1994;13:671–681. doi: 10.1016/0896-6273(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 18.Zagotta W N, Siegelbaum S A. Annu Rev Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]
- 19.Tang C Y, Papazian D M. J Gen Physiol. 1997;109:301–311. doi: 10.1085/jgp.109.3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Catterall W A. Annu Rev Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- 21.Jan L Y, Jan Y N. Annu Rev Neurosci. 1997;20:91–123. doi: 10.1146/annurev.neuro.20.1.91. [DOI] [PubMed] [Google Scholar]
- 22.Santacruz-Toloza L, Huang Y, John S A, Papazian D M. Biochemistry. 1994;33:5607–5613. doi: 10.1021/bi00184a033. [DOI] [PubMed] [Google Scholar]
- 23.Henn D K, Baumann A, Kaupp B. Proc Natl Acad Sci USA. 1995;92:7425–7429. doi: 10.1073/pnas.92.16.7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsson H P, Baker O S, Dhillon D S, Isacoff E Y. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 25.Yang N, George A L, Horn R. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- 26.Johannsson M, vonHeijne G. J Biol Chem. 1996;271:25912–25915. doi: 10.1074/jbc.271.42.25912. [DOI] [PubMed] [Google Scholar]
- 27.Swartz K J, MacKinnon R. Neuron. 1997;18:675–682. doi: 10.1016/s0896-6273(00)80307-4. [DOI] [PubMed] [Google Scholar]
- 28.Shih T M, Goldin A L. J Cell Biol. 1997;136:1037–1045. doi: 10.1083/jcb.136.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meera P, Wallner M, Jiang Z, Toro L. FEBS Lett. 1996;382:84–88. doi: 10.1016/0014-5793(96)00151-2. [DOI] [PubMed] [Google Scholar]
- 30.Stefani E, Ottolia M, Noceti F, Olcese R, Wallner M, Latorre R, Toro L. Proc Natl Acad Sci USA. 1997;94:5427–5431. doi: 10.1073/pnas.94.10.5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui J, Cox D H, Aldrich R W. J Gen Physiol. 1997;109:647–673. doi: 10.1085/jgp.109.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papazian D, Shao X M, Seoh S-A, Mock A F, Huang Y, Wainstock H. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 33.Seoh S A, Sigg D, Papazian D M, Bezanilla F. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 34.Aggarwal S K, MacKinnon R. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 35.Devereux J, Haeberli P, Smithies O. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 37.Horton R M, Hunt H D, Ho S N, Pullen J K, Pease L R. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 38.Fujiki Y, Hubbard A L, Fowler S, Lazarow P B. J Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu X, Birnbaumer L. Proc Natl Acad Sci USA. 1996;93:2827–2831. doi: 10.1073/pnas.93.7.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knaus H G, Eberhart A, Koch R O, Munujos P, Schmalhofer W A, Warmke J W, Kaczorowski G J, Garcia M L. J Biol Chem. 1995;270:22434–22439. doi: 10.1074/jbc.270.38.22434. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Calvo M, Knaus H-G, McManus O B, Giangiacomo K M, Kaczorowski G J, Garcia M L. J Biol Chem. 1994;269:676–682. [PubMed] [Google Scholar]
- 42.Hirai H, Kirsch J, Laube B, Betz H, Kuhse J. Proc Natl Acad Sci USA. 1996;93:6031–6036. doi: 10.1073/pnas.93.12.6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jurman M E, Boland L M, Liu Y, Yellen G. BioTechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- 44.Shih, T. M., Smith, R. D., Toro, L. & Goldin, A. (1998) Methods Enzymol., in press. [DOI] [PubMed]
- 45.Lagrutta A, Shen K, North R A, Adelman J P. J Biol Chem. 1994;269:20347–20351. [PubMed] [Google Scholar]