


Abstract
Ski interacting protein (Skip) plays an important role in the transforming activity of both v-Ski and EBNA2 (Epstein–Barr virus encoded latency protein) and is involved in EBNA2 and NotchIC activation of CBF1-repressed promoters. We have previously shown that Skip acts as a transcriptional co-activator on a number of cellular and viral promoters. Here, we report that Skip also interacts with pRb and, in cooperation with Ski, can overcome pRb-induced transcriptional repression. We show a strong and direct interaction between pRb and Skip, and we map the site of interaction to amino acid residues 171–353 of the evolutionarily conserved SNW domain of Skip. Furthermore, the combination of Skip and Ski can successfully overcome the G1 arrest and flat cell phenotype induced by pRb. Taken together, these studies suggest that one potential function of the Skip–Ski complex is to overcome the growth-suppressive activities of pRb.
INTRODUCTION
Skip, the human homolog of Drosophila melanogaster nuclear protein Bx42, was originally discovered as Ski interacting protein (1,2). Ski is a proto-oncogene and was discovered through its presence in the genome of an avian acutely transforming retrovirus (3). Its overexpression in avian fibroblasts leads to an anchorage-independent growth and morphological transformation (4). Cellular Ski binds DNA in a mammalian nuclear extract, in association with other proteins, and regulates transcription (5). Ski can function either as a transcriptional activator or as a repressor depending on the cellular and promoter context. Cellular Ski was found to be a component of the histone deacetylase (HDAC1) complex through binding to nuclear hormone receptor co-repressor (N-CoR) and mSin3A, thereby mediating transcriptional repression by the thyroid hormone receptor, by Mad and by pRb (6,7). In contrast, the v-Ski protein, which lacks a 292 amino acid region from the C-terminus of c-Ski, inhibits pRb-mediated transcriptional repression in a dominant negative fashion. This leads to the intriguing question of whether the c-Ski and v-Ski oncoproteins can transform cells through similar mechanisms (7,8). In addition, the Ski–Skip interaction is also thought to be important for Ski’s transforming activity, since Skip interacts with the domain of Ski required for the induction of transformation (2).
Skip is a nuclear protein with a broad tissue distribution. Skip homologs have been identified from many diverse species (1,9–11). A large amount of evidence in recent years suggested that human Skip may also have a role in transcriptional regulation, similar to its Drosophila homolog Bx42. Skip expression enhanced vitamin D-responsive transient gene expression and also enhanced retinoic acid-, estrogen- and glucocorticoid-mediated gene expression (12). In addition, Skip has been shown to have a role in EBNA2- and Notch-mediated transcriptional activation (13,14). A significant contribution of EBNA2 to immortalization is attributable to EBNA2-mediated activation of CBF1-repressed cellular gene expression, and contact of EBNA2 with Skip is required for the effective EBNA2 targeting to DNA (13). In the case of Notch, the activated intracellular form, NotchIC, activates the expression of target genes by overcoming the CBF1-mediated repression (15), an activity that also requires Skip (14).
We recently reported that Skip can activate the expression of many different promoters and is capable of cooperating with known transcription factors in promoter activation (16). In agreement with our data, most recent studies indicate that Skip interacts with Smad proteins to augment TGF-β-dependent transcription, and interacts with poly(A)-binding protein 2 to synergistically activate E-box-mediated transcription through MyoD (17,18). However, the mechanism of transcriptional activation mediated by Skip is not clear. Therefore, we sought to investigate the probable mechanism behind the transactivation capabilities of Skip. As v-Ski can inhibit pRb-induced transcriptional repression, it was of interest to see whether Skip can also modulate pRb activity. Here, we show that Skip also interacts with the pRb tumor suppressor and abrogates its ability to repress gene expression. Skip is also able to inhibit the transcriptional repression mediated by another pocket protein, p130. We also demonstrate that the combination of Skip and Ski can inhibit pRb-induced G1 arrest and the flat cell phenotype. These results suggest that one means by which Skip can act as a general transcriptional activator is through abrogation of pocket protein transcriptional repression.
MATERIALS AND METHODS
Plasmids
Various forms of Skip used in the study were cloned in pcDNA 3.1 at BamHI–EcoRI by PCR amplification from the Skip expression plasmid, pCGSP-Skip (2,16). For making His6-Skip 1–353, the gene was cloned in pET7His6 at BamHI–EcoRI sites. These constructs were verified by DNA sequencing using the dideoxy chain termination method (19) and were used both for in vitro translations and for expression in cells. Cellular Ski expression plasmid, pMT2-Ski, has been described previously (2) and the GST-Ski expression plasmid was a kind gift from Shunsuke Ishii (6). pCMV-Rb has been described previously (20), as has the CAT reporter constructs AdE2CAT and 5X GAL4TK-CAT (21,22). GAL4-Rb was a gift from Tony Kouzarides (23).
Cells
U2OS and Saos-2 cells were grown in DMEM supplemented with 10% fetal calf serum. Transfections were performed using the calcium phosphate precipitation method.
Transfections and CAT assays
Cells were transfected with 1 µg of reporter plasmids (unless otherwise stated) along with indicated amounts of expression plasmids. Where titrations were performed, DNA input was balanced with empty vector DNA. After 48 h, the cells were harvested in 100 µl of CAT buffer (40 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA) and subjected to three cycles of freeze–thawing, followed by incubation at 65°C for 10 min. Samples were clarified by centrifugation at 14 000 r.p.m. in an Eppendorf microfuge for 2 min and the protein concentration of the supernatant was measured by the Bio-Rad protein assay. CAT assays were routinely performed with 5–10 µg of protein incubated with 2.5 µl of acetyl-CoA (33.3 mg/ml) and 1.5 µl of [14C]chloramphenicol (50 mCi/mol; Amersham) in a final volume of 50 µl at 37°C for 1 h. Following extraction with ethyl acetate, samples were analyzed by thin-layer chromatography and quantified with a Packard Instant Imager (Packard, Meriden, CT).
Western blotting
Proteins were separated by SDS–PAGE and transferred to nitrocellulose. His6-Skip was detected by using a polyclonal antibody raised against the His-tagged Skip 1–353, and then developed using the ECL detection system according to the manufacturer’s instructions (Amersham). For the analysis of pRb expression, U2OS cells were transfected with various combinations of the plasmids pCMV-Rb, pCDNA3-HA-Skip and pCMV-Ski. The β-galactosidase (β-Gal)-expressing plasmid, pCMV-βGal, was also included as a transfection control. After 48 h, the cells were harvested, lysed in SDS sample buffer and subjected to SDS–PAGE and western blotting. The proteins were detected using anti-pRb monoclonal antibody (Becton Dickinson), anti-HA monoclonal antibody (Roche) and anti-β-Gal monoclonal antibody (Promega), and then developed using the ECL detection system (Amersham).
In vitro translations and GST-pull-down assays
35S-labeled proteins were produced in vitro by using a coupled transcription–translation system (Promega TNT) according to the manufacturer’s instructions. For fusion protein production and purification, 100 ml of an overnight culture of Escherichia coli strain BL-21 containing the GST-Ski expression plasmid was inoculated into 1 l of Luria broth containing ampicillin and incubated at 37°C for 1 h. Recombinant protein expression was induced for 3 h with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma). The cells were harvested by centrifugation, disrupted by sonication and cell debris was removed by centrifugation. The levels of protein induction were then determined by SDS–PAGE and Coommassie brilliant blue R staining. For the direct binding assay, His-tagged Skip protein was purified as described previously (24).
Equal amounts of GST and GST-Rb fusion protein bound to glutathione-linked agarose (Sigma) were incubated with the in vitro translated proteins for 1 h at room temperature in a buffer containing 50 mM Tris–HCl pH 7.5, 100 mM NaCl and 2.5 mM EDTA. Bound proteins were washed extensively in PBS containing 0.5% NP-40 before analysis by SDS–PAGE and autoradiography.
Cell-cycle analysis
Determination of the percentage of cells in the G1 phase of the cell cycle was done using fluorescence-activated cell sorting (FACS). One microgram of green fluorescent protein (GFP)-expressing plasmid was co-transfected with 1 µg of pRb expression plasmid together with 5 µg each of Skip and Ski expression plasmids or empty vector as indicated. Forty-eight hours post-transfection the cells were trypsinized and pelleted. Cells were fixed in 5% paraformaldehyde in PBS for 1 h on ice. After washing in PBS containing 1% fetal calf serum, the cells were then treated with 70% ethanol for 1 h on ice. Prior to FACS analysis the cells were incubated with 200 µg of RNase A for 1 h and then stained with propidium iodide (20 µg/ml). GFP-expressing cells were sorted and analyzed for DNA content (FACSCalibur; Beckton Dickinson). Estimation of the cell-cycle distribution from the DNA content was analyzed using the MODFIT LT program.
Flat cell assay
The flat cell assay was done as described previously (25). Saos-2 cells were co-transfected with pRb-expressing plasmid and pBabe-puro together with Skip and Ski expression plasmids. Transfected cells were selected on puromycin for 2 weeks, cell morphology was assessed and β-Gal staining was done as described before (26). Briefly, cells were washed in PBS, fixed for 3–5 min at room temperature in 2% formaldehyde/0.2% glutaraldehyde, washed, and incubated at 37°C with fresh senescence-associated (SA) β-Gal stain solution: 1 mg/ml 5-bromo-4-chloro-3-indolyl-d-galactosidase (X-Gal), 40 mM citric acid/sodium phosphate pH 6.0, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl2. Staining was evident in 2–4 h and maximal in 12–16 h.
RESULTS
Abrogation of pRb-induced transcriptional repression by Skip and Ski
Since v-Ski has been shown to inhibit pRb transcriptional repression (7), we wanted to investigate the effects of Ski and Skip on pRb transcriptional activity. To do this, first we used the AdE2 promoter in Saos-2 cells, where the promoter is constitutively active due to the absence of pRb (21,27). Cells were transfected with the reporter plasmid together with pRb. In addition, c-Ski and Skip were also included, either alone or in combination. The results are shown in Figure 1A. It is clear that, as expected, pRb represses transcription from the AdE2 promoter. Addition of increasing amounts of either c-Ski or Skip alone has only minimal effects on the ability of pRb to repress transcription from the AdE2 promoter. Interestingly, however, when increasing amounts of c-Ski and Skip are added in combination, it is clear that pRb-induced repression is readily overcome. We next wanted to investigate whether the effects of Skip and Ski were directed specifically towards pRb transcriptional suppressor activity or to a more generalized effect of affecting E2F inhibition. To do this, we investigated the effects of expression of Skip and Ski on Gal4-Rb-induced transcription of a 5xGal4TK-CAT reporter. As expected, Gal4-Rb strongly repressed transcription from the Gal4 site-containing reporter (Fig. 1B). However, it is clear that this Gal4-Rb-induced repression was abrogated by co-expression of Skip and Ski. None of the above effects were due to a decrease in the levels of pRb as a result of Skip and Ski overexpression, since western blot analysis of pRb expression in the presence of increasing amounts of Skip and Ski revealed no major differences in the levels of pRb protein expression (Fig. 1C).
Figure 1.

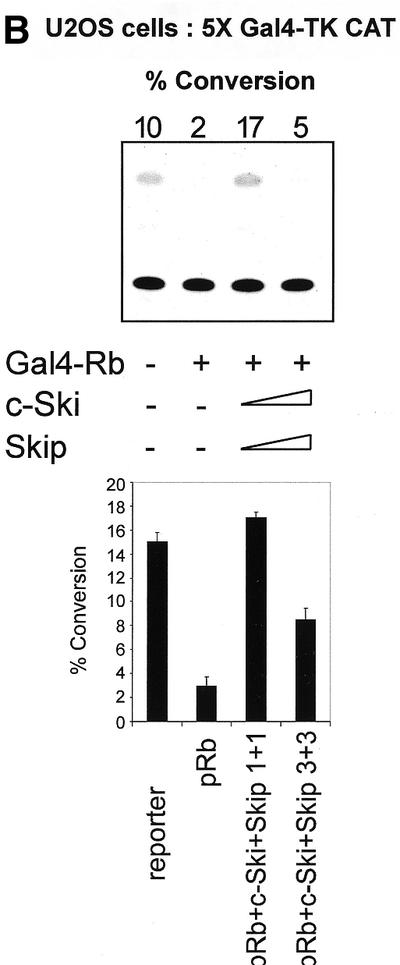
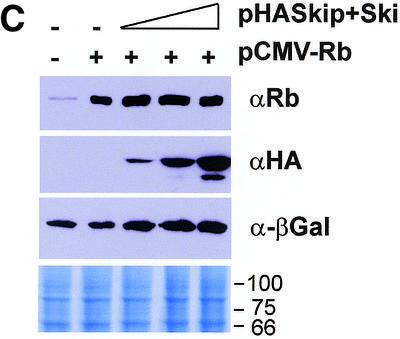
(A) Skip and c-Ski synergistically overcome pRb-mediated repression of the AdE2 promoter. pRb null Saos-2 cells were transfected with 3 µg of the AdE2 CAT reporter construct together with 0.1 µg of pRb expression plasmid as indicated. Effects of Skip and c-Ski were assessed by transfecting them in increasing amounts either alone or in combination as shown (concentrations are in µg). After 48 h the cells were harvested and CAT assays performed. The upper panel shows the results from a representative assay and the lower panel shows the collated results from at least three independent transfections. Standard deviations are shown. (B) Skip and c-Ski together inhibits transcriptional repression by pRb. A mixture of Gal4 site containing TK CAT reporter, the Gal4-Rb and the plasmid expressing Skip and c-Ski were transfected into U2OS cells as indicated, and after 48 h the cells were harvested and CAT assays performed. The upper panel shows the results from a representative assay and the lower panel shows the collated results from at least three independent transfections. Standard deviations are shown. (C) Analysis of pRb expression in the presence of ectopic Skip and Ski expression. U2OS cells were transfected with the plasmids pCMV-Rb (0.1 µg), pCDNA3-HA-Skip/pCMV-Ski (0.5, 1 and 2 µg of each as shown) and pCMV-βGal (0.1 µg). Cell extracts were analyzed by western blotting for Rb (αRb), Skip (αHA), βGal (α-βGal). The Coomassie protein stain of an SDS–PAGE run in parallel with an aliquot of the extract used for the western blot is shown in the bottom panel and the numbers indicate the position of molecular weight markers.
Taken together, these results demonstrate that c-Ski and Skip can act in concert to overcome the transcriptional repression mediated by pRb.
Skip interacts with pRb
Having found that Skip and Ski can together inhibit pRb transcriptional repression, it was of interest to investigate the underlying mechanisms. Since c-Ski has been found to interact with the ‘B’ pocket of pRb (7), we first investigated whether Skip, the binding partner of Ski, can also associate with pRb. Bacterially expressed GST alone or GST-Rb fusion proteins were immobilized on glutathione–agarose (Sigma) and were then mixed with in vitro translated radiolabeled Skip protein. After extensive washing, the bound proteins were analyzed by SDS–PAGE and autoradiography. As shown in Figure 2, over 20% of the input of Skip protein was retained on the pRb-conjugated agarose beads. It can also be seen from Figure 2 that the interaction was stable in 0.5 M NaCl, suggesting that pRb and Skip can form a highly stable complex in vitro. These results suggest that the ability of Skip and Ski to overcome pRb transcriptional repression most likely depends on the physical interaction of these three proteins.
Figure 2.
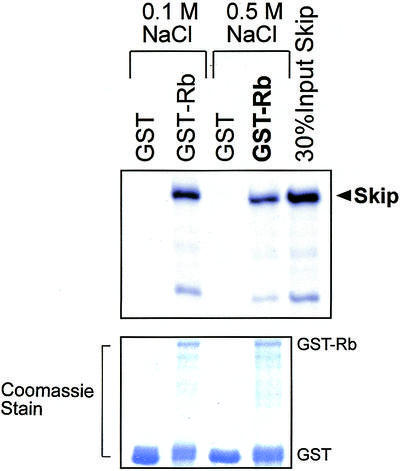
Skip interacts with pRb in vitro. Purified GST or GST-Rb on glutathione resin was incubated with equal amounts of 35S-labeled wild-type Skip. After extensive washing, bound proteins were resolved by SDS–PAGE and autoradiography. Lower panel shows the Coomassie blue stain of the gel used in the binding assay.
Identification of the pRb-binding domain on Skip
In order to map the site of the pRb interaction on Skip, we made use of a series of Skip deletion mutants, and these are shown schematically in Figure 3D. The Skip deletion mutants were translated in vitro and GST pull-down assays were performed using GST-Rb fusion protein bound to glutathione-coupled agarose. The results obtained are shown in Figure 3A. As can be seen, the wild-type Skip showed strong binding to pRb. In addition, mutant 1–353 showed wild-type levels of interaction, whereas the C-terminal mutant containing amino acid residues 350–536 failed to bind and only weak binding was obtained with the mutant spanning residues 1–179. None of the mutants spanning residues 1–292 retained wild-type binding with pRb (Fig. 3B). These results raise the possibility that the region of Skip required for interaction with pRb lies principally between residues 179 and 353. To confirm this, additional deletion constructs of Skip were used in GST pull-down assays, and the results obtained are shown in Figure 3C. As can be seen, the mutants 171–536 and 179–353 retained wild-type binding with pRb, indicating that residues 171–353, which comprise the highly conserved SNW domain of Skip (1,2,9–11,16) represents the interaction domain for pRb.
Figure 3.

Mapping the pRb-interaction domain on Skip. (A–C) The results from representative GST pull-down assays. Purified GST or GST-Rb on glutathione resin was incubated with the in vitro translated Skip deletion mutants for 1 h at room temperature. After extensive washing, bound proteins were analyzed by SDS–PAGE and autoradiography. (D) Schematic diagram of the Skip deletion mutants used in the analysis. Numbers refer to the amino acid residues and the central red-boxed region corresponds to the highly conserved SNW region where homology with Skip proteins derived from other species is >90%. The relative strength of binding of each protein to Rb is also shown, together with the mean percentage binding from at least three separate assays, where +++ indicates 20–30%, whereas ++ and +/– indicate 10–15% and 2–3% binding, respectively.
Obviously, the GST pull-down assay using in vitro translated Skip does not exclude the possibility that the interaction between pRb and Skip occurs via an intermediate protein. To investigate this, the 1–353 deletion mutant of Skip, which shows wild-type binding with pRb, was expressed in bacteria as a six histidine-containing fusion protein (His6) and was purified by Ni2+-NTA agarose column chromatography. A GST pull-down assay was then performed using recombinant His6-Skip: 1–353 with GST or GST-Rb. GST-c-Ski and GST-HDAC1 were also included in the assay to clarify the nature of the already known interactions between Skip and Ski and between Skip and HDAC1, respectively (2,13,16). Following extensive washing, the amount of Skip retained was determined by electrophoresis and western blotting with Skip-specific antibodies. The results obtained are shown in Figure 4. As can be seen, quantitative recovery of the Skip protein was obtained on the GST-Rb agarose resin (lanes 2), whereas no binding to GST alone (lane 1), GST-Ski (lane 3) or GST-HDAC1 (lane 4) was detected. These results demonstrate that the interaction between Skip and pRb is direct and is not mediated by any other protein. These results also suggest that the Skip and Ski interaction (2,16) is indirect and is probably mediated by pRb.
Figure 4.
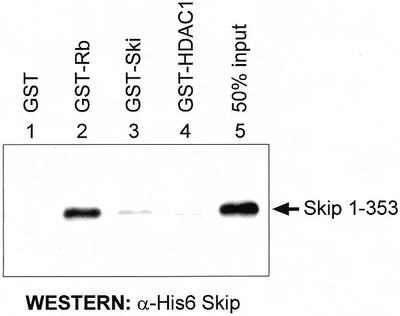
The interaction between Skip and pRb is direct. GST-fusion proteins and GST alone were purified on glutathione–agarose columns. These were then incubated with 40 ng of recombinant purified His6-Skip 1–353 at room temperature for 1 h. After extensive washing, bound Skip was assessed by SDS–PAGE and western blot analysis with an antiserum raised against Skip.
Skip interacts with other pocket proteins
pRb belongs to the pocket protein family, which also includes p107 and p130. Having seen the physical and functional interaction between Skip and pRb, it was of interest to determine whether Skip could also interact with the other pocket proteins, p130 and p107. To do this, a series of pull-down assays were performed using GST-Skip and in vitro translated radiolabeled p107 and p130. The results obtained are shown in Figure 5, and, as can be seen, Skip shows substantial binding with both p130 and p107.
Figure 5.
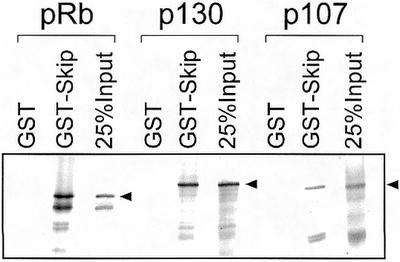
Interaction of Skip with pocket proteins. Purified GST or GST-Skip on glutathione resin was incubated with the in vitro translated pocket proteins: pRb, p130 and p107 for 1 h at room temperature. After extensive washing, bound proteins were analyzed by SDS–PAGE and autoradiography.
We then proceeded to investigate whether Ski and Skip could modulate the transcriptional activity of p130 in a manner similar to that seen with pRb. Saos-2 cells were transfected with the AdE2 reporter plasmid together with an expression plasmid for p130. In addition, c-Ski and Skip were also included either alone or in combination. The results are shown in Figure 6 and it is clear that p130 represses transcription from the AdE2 promoter. Addition of increasing amounts of either c-Ski or Skip alone has only minimal effects on the ability of p130 to repress transcription. Interestingly, however, when increasing amounts of c-Ski and Skip are added in combination, it is clear that the p130-induced repression of the AdE2 promoter is readily overcome. These results demonstrate that c-Ski and Skip can act in concert to overcome the transcriptional repression mediated by both pRb as well as p130.
Figure 6.
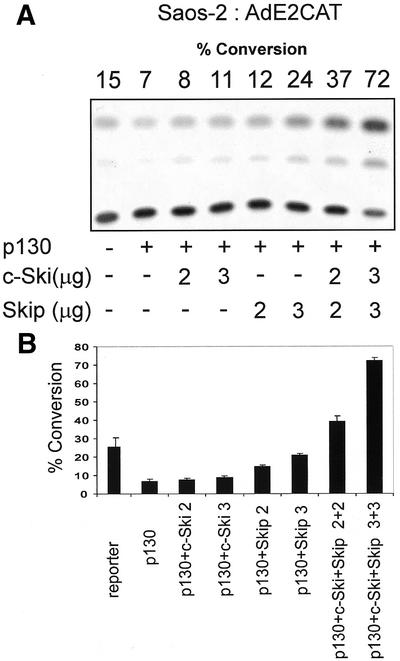
Skip and c-Ski synergistically overcome p130-mediated repression of the AdE2 promoter. Saos-2 cells were transfected with 3 µg of the AdE2 CAT reporter construct together with 0.5 µg of p130 expression plasmid as indicated. Effects of Skip and c-Ski were assessed by transfecting them in increasing amounts either alone or in combination as shown (concentrations are in µg). After 48 h, the cells were harvested and CAT assays performed. (A) The results from a representative assay and (B) the collated results from at least three independent transfections. Standard deviations are shown.
Inhibition of pRb-induced G1 arrest and flat cell phenotype by Ski and Skip
Introduction of the Rb gene into Saos-2 osteosarcoma cells, which lack functional pRb, prevents progression into the S phase of the cell cycle (20,25,28) and induces a G1 arrest (29,30) that is followed by marked cellular enlargement and flattening (25,31,32). In addition, constitutively expressed cyclins A and E can overcome the pRb-mediated cell-cycle block (25,33–35), and co-transfection of pRb with inactivators of pRb, such as Ad E1A and cyclin D, each prevent induction of the flat cell phenotype (25). Having found that Skip and Ski can abrogate pRb-mediated transcriptional repression, we then proceeded to investigate whether they could affect pRb-induced growth arrest. Saos-2 cells were co-transfected with pCMV-GFP together with Rb and Skip and Ski expression plasmids. After 48 h, the cells were trypsinized, fixed and the proportions of GFP-expressing cells in different phases of the cell cycle were determined by flow cytometry and the results obtained are shown in Figure 7A. As expected, pRb caused an increase in the G1 population (12–13%) compared with the vector-only control transfected cells, while co-expression of the Ski and Skip plasmids largely reversed the G1 block induced by pRb. This was not due to a general perturbation of the cell cycle, since Ski and Skip alone did not cause any changes in the cell-cycle distributions. We then proceeded to investigate the effects of expression of Skip and Ski on the ability of pRb to induce a flat cell morphology in Saos-2 cells. The cells were transfected with a pRb-expressing plasmid and a selectable marker (25,36) in the presence and absence of either wild-type or a non-pRb binding mutant of Skip, together with Ski, and after 2 weeks the cells were stained for SA-β-Gal. The results obtained are shown in Figure 7B. As can be seen, almost 70% of Saos-2 cells transfected with pRb had a flat cell phenotype. However, when pRb and wild-type Skip and Ski were co-transfected, there was a dramatic reduction in the percentage of flat cells to ∼12%. In contrast, inclusion of a mutant of Skip (350–536) that fails to bind pRb had no effect on the pRb flat cell phenotype, whilst the mutant of Skip (171–536) that retains the ability to bind pRb also retains the ability to overcome pRb activity. Taken together, these experiments demonstrate that the combination of Skip and Ski can successfully overcome both pRb-induced growth arrest and the flat cell phenotype.
Figure 7.


Release of pRb-induced effects by Skip and Ski. (A) The effects of Skip and Ski on the G1 block from pRb. pRb null Saos-2 cells were co-transfected with GFP and pRb expression plasmids (1 µg), together with Skip and Ski (5 µg). After 48 h, the cells were assessed by flow cytometry. The results are plotted as the percentage change in the G1 fraction relative to cells transfected with vector alone. The results show collated results from at least three independent transfections. Standard deviations are shown. (B) The effects of Skip and Ski on the flat cell phenotype induced by pRb: Saos-2 cells were transfected with 1 µg of pBabe-Puro expression plasmid and 5 µg each of the indicated plasmids. Transfected cells were subjected to puromycin selection for 7–10 days, and the percentage of flat cells was determined by SA-β-Gal assay. Percent flat cells indicates the total number of SA-β-Gal-positive cells divided by the total number of cells counted, and represents the average from three independent experiments. Standard deviations are shown.
DISCUSSION
In this communication we show that overexpression of Skip and Ski can effectively release pRb-induced repression of transcription and cell growth. We also show that this is most likely mediated via a direct interaction between pRb and Skip and map this interaction site to the evolutionarily conserved SNW domain of Skip, which has been shown previously to represent an interface for several protein–protein interactions (12,16,17,37).
Previous studies have shown that v-Ski complexes with pRb and inhibits pRb transcriptional repression (7). In addition, Skip has also been found in complex with the muscle cell differentiation-specific transcription factor, MyoD, which, together with PABP2, binds to hypophosphorylated pRb (18,38). These observations lead us to investigate the potential effects of Skip upon pRb transcriptional activity. Using both the AdE2 promoter and an artificial 5xGal4TK-CAT reporter in conjunction with a Gal4-Rb fusion protein, we found that Skip in cooperation with Ski could readily overcome pRb-induced repression of transcription. Based on the strong and direct interaction found between pRb and Skip it is tempting to suggest that this is the mechanism by which Skip exerts this activity. A mutational analysis of Skip was then performed in order to map the site of interaction with pRb. Our data provides evidence that the region of Skip required for binding pRb lies between residues 171 and 353. Interestingly, this region of Skip encompasses the evolutionarily conserved SNW domain, which is also required for Skip transcriptional activation (16). At present, we have been unable to define the region of pRb that is bound by Skip and further studies are currently in progress. In addition, it will also be interesting to determine whether differences in the phosphorylation status of pRb affect the interaction with Skip, although the fact that Skip binds strongly to GST-pRb suggests that Skip may preferentially recognize the hypophosphorylated form of the protein. Interestingly, Skip was also found to interact with the other members of the pocket protein family, p107 and p130, albeit not as strongly as it does with pRb. In the case of p130, Skip and Ski also appear to be capable of overcoming p130-mediated transcriptional repression. Taken together, these results suggest that one of the consequences of the combination of Skip and Ski is to overcome pocket protein transcriptional repression, and hence their growth-suppressive activity. Further support for this notion came from studies to investigate the effects of Skip and Ski upon the growth-suppressive activities of pRb. Using two different biological assays of pRb function, induction of G1 arrest and induction of the SA flat cell phenotype, we found that the combination of Skip and Ski was effective in reversing both aspects of pRb function. These results, therefore, suggest that the ability of Skip and Ski to abolish pRb transcriptional repression also results in abolition of the growth-suppressive activities of pRb.
There is mounting evidence that Skip can transcriptionally activate a variety of target promoters (12–14,16–18). However, Skip also forms part of a co-repressor complex whose components include Skip, SMRT, SAP30, Sin3A, CIR, HDAC1 and HDAC2 (6,13,15). Thus, Skip would appear to have a dual role whereby it is present in both activation and repression complexes, both of which are mutually exclusive due to competition for promoter units and specific components to form the functional complexes (13,14,15). It is interesting to note that the viral EBNA2 protein has been shown to displace co-repressor complexes by recruiting co-activators such as p300, CBP and PCAF to the promoter (39) and that it also requires the ability to interact with Skip in order to do this (13). Interestingly, the human papillomavirus (HPV) E7 oncoprotein has also been shown to interact with Skip and modulate its transcriptional activity (37). Therefore, it is tempting to speculate that these two divergent viral oncoproteins, EBNA2 and HPV E7, may have evolved a similar mechanism for perturbing the function of co-repressor and co-activator complexes, via the common intermediary of Skip.
In summary, our data show that Skip and Ski can act in concert to overcome pRb-induced transcriptional repression and cell-cycle arrest. Previous studies had indicated that overexpression of wild-type c-Ski could partially abrogate transcriptional repression mediated by pRb (7). Our data suggest that c-Ski can completely achieve this in complex with Skip. This is consistent with the fact that overexpression of wild-type c-Ski also leads to cell transformation, and suggests that both v-Ski and c-Ski may transform cells through the common mechanism of inhibiting the function of the growth-suppressive activities of pRb via the interaction with Skip.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to P. Ramadevi and M. Thomas for critical reading of the manuscript. This work was supported in part by research grants from the Associazione Italiana per la Ricerca Sul Cancro and the EU Biomed 2 program.
REFERENCES
- 1.Folk P., Puta,F., Krpejsova,L., Blahuskova,A., Markos,A., Rabino,M. and Dottin,R.P. (1996) The homolog of chromatin binding protein Bx42 identified in Dictyostelium. Gene, 181, 229–231. [DOI] [PubMed] [Google Scholar]
- 2.Dahl R., Wani,B. and Hayman,M.J. (1998) The Ski oncoprotein interacts with Skip, the human homologue of Drosophila Bx42. Oncogene, 16, 1579–1586. [DOI] [PubMed] [Google Scholar]
- 3.Stavnezer E., Gerhard,D.S., Binari,R.C. and Balazs,I. (1981) Generation of transforming viruses in cultures of chicken fibroblasts infected with an avian leukosis virus. J. Virol., 39, 920–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colmenares C. and Stavnezer,E. (1989) The ski oncogene induces muscle differentiation in quail embryo cells. Cell, 59, 293–303. [DOI] [PubMed] [Google Scholar]
- 5.Nagase T., Mizuguchi,G., Nomura,N., Ishizaki,R., Ueno,Y. and Ishii,S. (1990) Requirement of protein co-factor for the DNA binding function of the human ski proto-oncogene product. Nucleic Acids Res., 18, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nomura T., Khan,M.M., Kaul,S.C., Dona,H.D., Wadhwa,R., Colmenares,C., Kohno,I. and Ishii,S. (1999) Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid hormone receptor. Genes Dev., 13, 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tokitou F., Nomura,T., Khan,M.M., Kaul,S.C., Wadhwa,R., Yasukawa,T., Kohno,I. and Ishii,S. (1999) Viral ski inhibits retinoblastoma protein (Rb)-mediated transcriptional repression in a dominant negative fashion. J. Biol. Chem., 274, 4485–4488. [DOI] [PubMed] [Google Scholar]
- 8.Liu X., Sun,Y., Weinberg,R.A. and Lodish,H.F. (2001) Ski/Sno and TGF-beta signaling. Cytokine Growth Factor Rev., 12, 1–8. [DOI] [PubMed] [Google Scholar]
- 9.Diehl B.R. and Pringle,J.R. (1991) Molecular analysis of Saccharomyces cerevisiae chromosome I: identification of additional transcribed regions and demonstration that some encode essential functions. Genetics, 127, 287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris S.D., Cheng,J., Pugh,T.A. and Pringle,J.R. (1992) Molecular analysis of Saccharomyces cerevisiae chromosome I on the number of genes and the identification of essential genes using temperature-sensitive-lethal mutations. J. Mol. Biol., 225, 53–65. [DOI] [PubMed] [Google Scholar]
- 11.Wieland C., Mann,S., von Besser,H. and Saumweber,H. (1992) The Drosophila nuclear protein Bx42, which is found in many puffs on polytene chromosomes, is highly charged. Chromosoma, 101, 517–525. [DOI] [PubMed] [Google Scholar]
- 12.Baudino T.A., Kraichely,D.M., Jefcoat,J.S.C., Winchester,S.K., Partridge,N.C. and MacDonald,P.N. (1998) Isolation and characterization of a novel coactivator protein, NcoA-62, involved in vitamin D-mediated transcription. J. Biol. Chem., 273, 16434–16441. [DOI] [PubMed] [Google Scholar]
- 13.Zhou S., Fujimuro,M., Hsieh,J.J., Chen,L. and Hayward,S.D. (2000) A role for SKIP in EBNA2 activation of CBF1-repressed promoters. J. Virol., 74, 1939–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou S., Fujimuro,M., Hsieh,J.J., Chen,L., Miyamoto,A., Weinmaster,G. and Hayward,S.D. (2000) SKIP, a CBF1-associated protein, interacts with the ankyrin repeat domain of NotchIC To facilitate NotchIC function. Mol. Cell. Biol., 20, 2400–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh J.J.-D., Henkel,T., Salmon,P., Robey,E., Peterson,M.G. and Hayward,S.D. (1996) Truncated mammalian Notch1 activates CBF1/RBPJk-repressed genes by a mechanism resembling that of Epstein–Barr virus EBNA2. Mol. Cell. Biol., 16, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prathapam T., Kühne,C., Hayman,M. and Banks,L. (2001) Ski interacts with the evolutionarily conserved SNW domain of Skip. Nucleic Acids Res., 29, 3469–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leong G.M., Subramaniam,N., Figueroa,J., Flanagan,J.L., Hayman,M.J., Eisman,J.A. and Kouzmenko,A.P. (2001) Ski-interacting protein (SKIP) interacts with Smad proteins to augment TGF b dependent transcription. J. Biol. Chem., 276, 18243–18248. [DOI] [PubMed] [Google Scholar]
- 18.Kim Y.J., Noguchi,S., Hayashi,Y.K., Tsukahara,T., Shimizu,T. and Arahata,K. (2001) The product of an oculopharyngeal muscular dystrophy gene, poly (A)-binding protein 2, interacts with SKIP and stimulates muscle-specific gene expression. Hum. Mol. Genet., 10, 1129–1139. [DOI] [PubMed] [Google Scholar]
- 19.Sanger F., Nicklen,S. and Coulson,A.R. (1977) DNA sequencing with chain-terminating inhibitors. Proc. Natl Acad. Sci. USA, 74, 5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quin X.Q., Chittenden,T., Livingston,D.M. and Kaelin,W.G.,Jr (1992) Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev., 6, 953–964. [DOI] [PubMed] [Google Scholar]
- 21.Murthy S.C., Bhat,G.P. and Thimmappaya,B. (1985) Adenovirus EIIA early promoter: transcriptional control elements and induction by the viral pre-early E1A gene, which appears to be sequence independent. Proc. Natl Acad. Sci. USA, 82, 2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh J.J. and Hayward,S.D. (1995) Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein–Barr virus EBNA2. Science, 268, 560–563. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen S.J., Schneider,R., Bauer,U.M., Bannister,A.J., Morrison,A., O’Carroll,D., Firestein,R., Cleary,M., Jenuwein,T., Herrera,R.E. and Kouzarides,T. (2001) Rb targets histone H3 methylation and HP1 to promoters. Nature, 412, 561–565. [DOI] [PubMed] [Google Scholar]
- 24.Kühne C. and Banks,L. (1998) E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J. Biol. Chem., 273, 34302–34309. [DOI] [PubMed] [Google Scholar]
- 25.Hinds P.W., Mittnacht,S., Dulic,V., Arnold,A., Reed,S.I. and Weinberg,R.A. (1992) Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell, 70, 993–1006. [DOI] [PubMed] [Google Scholar]
- 26.Dimri G.P., Lee,X., Basile,G., Acosta,M., Scott,G., Roskelley,C., Medrano,E.E., Linskens,M., Rubelj,I., Pereira-Smith,O., Peacocke,M. and Campisi,J. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA, 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chellappan S.P., Hiebert,S., Mudryj,M., Horowitz,J.M. and Nevins,J.R. (1991) The E2F transcription factor is a cellular target for the Rb protein. Cell, 65, 1053–1061. [DOI] [PubMed] [Google Scholar]
- 28.Goodrich D.W., Wang,N.P., Qian,Y.W., Lee,E.Y. and Lee,W.H. (1991) The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell, 67, 293–302. [DOI] [PubMed] [Google Scholar]
- 29.Ewen M.E. (1994) The cell cycle and the retinoblastoma protein family. Cancer Metastasis Rev., 13, 45–66. [DOI] [PubMed] [Google Scholar]
- 30.Weinberg R.A. (1995) The retinoblastoma and the cell cycle control. Cell, 81, 323–330. [DOI] [PubMed] [Google Scholar]
- 31.Tiemann F. and Hinds,P.W. (1998) Induction of DNA synthesis and apoptosis by regulated inactivation of a temperature-sensitive retinoblastoma protein. EMBO J., 17, 1040–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexander K. and Hinds,P.W. (2001) Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol. Cell. Biol., 21, 3616–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen P.L., Scully,P., Shew,J.Y., Wang,J.Y. and Lee,W.H. (1989) Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell, 58, 1193–1198. [DOI] [PubMed] [Google Scholar]
- 34.Buchkovich K., Duffy,L.A. and Harlow,E. (1989) The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell, 58, 1097–1105. [DOI] [PubMed] [Google Scholar]
- 35.DeCaprio J.A., Ludlow,J.W., Lynch,D., Furukawa,Y., Griffin,J., Piwnica,W.H., Huang,C.M. and Livingston,D.M. (1989) The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell, 58, 1085–1095. [DOI] [PubMed] [Google Scholar]
- 36.Huang H.S., Yee,J.K., Shew,J.Y., Chen,P.L., Bookstein,R., Friedmann,T., Lee,E.Y. and Lee,W.H. (1998) Suppression of the neoplastic phenotype by replacement of the Rb gene in human cancer cells. Science, 242, 1563–1566. [DOI] [PubMed] [Google Scholar]
- 37.Prathapam T., Kühne,C. and Banks,L. (2001) The HPV-16 E7 oncoprotein binds Skip and suppresses its transcriptional activity. Oncogene, 20, 7677–7685. [DOI] [PubMed] [Google Scholar]
- 38.Gu W., Schneider,J.W., Condorelli,G., Kaushal,S., Mahdavi,V. and Nadal-Ginard,B. (1993) Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell, 72, 309–324. [DOI] [PubMed] [Google Scholar]
- 39.Wang L., Grossman,S.R. and Kieff,E. (2000) Epstein–Barr virus nuclear protein 2 interacts with p300, CBP and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl Acad. Sci. USA, 97, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]