

Abstract
We report the results of solid state nuclear magnetic resonance (NMR) and atomic force microscopy measurements on amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p (Ure2p10–39). Measurements of intermolecular 13C-13C nuclear magnetic dipole-dipole couplings indicate that Ure2p10–39 fibrils contain in-register parallel β-sheets. Measurements of intermolecular 15N-13C dipole-dipole couplings, using a new solid state NMR technique called DSQ-REDOR, are consistent with hydrogen bonds between sidechain amide groups of Gln18 residues. Such sidechain hydrogen bonding interactions have been called “polar zippers” by M.F. Perutz and have been proposed to stabilize amyloid fibrils formed by peptides with glutamine- and asparagine-rich sequences, such as Ure2p10–39. We propose that polar zipper interactions account for the in-register parallel β-sheet structure in Ure2p10–39 fibrils and that similar peptides will also exhibit parallel β-sheet structures in amyloid fibrils. We present molecular models for Ure2p10–39 fibrils that are consistent with available experimental data. Finally, we show that solid state 13C NMR chemical shifts for 13C-labeled Ure2p10–39 fibrils are insensitive to hydration level, indicating that the fibril structure is not affected by the presence or absence of bulk water.
Abbreviations: NMR, nuclear magnetic resonance; Aβ, β-amyloid peptide; Ure2p10–39, residues 10-39 of the Ure2p yeast prion protein; EM, electron microscopy; FMOC, 9-fluorenylmethoxycarbonyl; TFA, trifluoroacetic acid; AFM, atomic force microscopy; MAS, magic-angle spinning; fpRFDR-CT, constant-time finite-pulse radiofrequency-driven recoupling; REDOR, rotational echo double resonance; DSQ, double single-quantum; TPPM, two-pulse phase modulation; CSA, chemical shift anisotropy; MD, molecular dynamics
One goal of current efforts to elucidate the molecular structures of amyloid fibrils (1–35) is to identify the intermolecular interactions that determine the details of these structures and make amyloid fibrils a stable structural state for many peptides and proteins despite their diversity of amino acid sequences (36–41). Recent studies by solid state nuclear magnetic resonance (NMR) of fibrils formed by the β-amyloid (Aβ) peptide associated with Alzheimer’s disease (5,13,16,18,24,30,31), by various Aβ fragments (1,3,4,6–8,12,21,25), and by other amyloid-forming peptides (22) indicate that the β-sheets in amyloid fibrils have structures that tend to maximize contacts among hydrophobic residues when the component peptides contain continuous hydrophobic segments. Electrostatic interactions appear to play a secondary role, dictating the choice between parallel and antiparallel β-sheet structures when either type of structure could maximize hydrophobic contacts (6,21,23) and dictating the precise registry of intermolecular hydrogen bonds in antiparallel β-sheets (1,25). Electrostatic repulsions in the low-dielectric core of an amyloid fibril could in principle overwhelm the favorable hydrophobic interactions and thus destabilize the amyloid structure (16). However, amyloid-forming peptides have been found to adopt conformations and supramolecular organizations that avoid electrostatic repulsions in the core (6,16,23,25). These conclusions from solid state NMR data are supported by structural data from other sources, including electron paramagnetic resonance (17,19,42), proline-scanning mutagenesis (28), disulfide crosslinking (26), chemical derivatization (21), and x-ray diffraction (29).
Amyloid fibrils formed by peptides and proteins that lack hydrophobic segments may be stabilized by different sets of interactions. Peptides and protein segments that are rich in glutamine or asparagine residues are of particular interest, both because of their involvement in neurodegenerative disorders such as Huntington’s disease (43,44) and spinocerebellar ataxias (45) and because of their occurrence in amyloid-forming yeast prion proteins (46–50). The propensity for glutamine- and asparagine-rich peptides and proteins to aggregate as amyloid fibrils in aqueous solutions (which may be surprising, given that glutamine and asparagine are not generally considered to be highly hydrophobic (51,52)) led Perutz to propose that these fibrils are stabilized by linear chains of hydrogen bonds among the amide groups of glutamine and asparagine sidechains on neighboring β-strands within a single β-sheet layer. Perutz coined the phrase “polar zippers” to describe the putative sidechain hydrogen bonding interactions (43,53–56). Molecular modeling indicates the feasibility of polar zipper formation in amyloid fibrils (45,53); sidechain hydrogen bonds among asparagine residues in neighboring β-strands have been observed in protein crystal structures (57–59); experimental constraints on the conformations of polyglutamine peptides in amyloid fibrils have been reported (60). However, direct experimental evidence for sidechain hydrogen bonds among glutamine or asparagine residues in amyloid fibrils has not been presented previously.
In this paper, we report the results of solid state NMR measurements on amyloid fibrils formed by the peptide Ure2p10–39, which represents residues 10–39 of the 354-residue Ure2p prion protein of Saccharomyces cerevisiae. Aggregation of Ure2p into amyloid fibrils is responsible for the self-propagating [URE3] phenotype in S. cerevisiae (47,61,62), in which the cells’ ability to stop utilization of ureidosuccinate for pyrimidine biosynthesis in the presence of ammonia is suppressed (63). Residues 97–354 of Ure2p form a globular domain with a glutathione S-transferase fold (64). Residues 1–80 or shorter segments thereof constitute the “prion domain” of Ure2p, responsible for aggregation of the full-length protein (47) and capable of forming amyloid fibrils that can seed fibrillization of the full-length protein (65,66). Ure2p10–39 is the most highly conserved segment in the prion domain of Ure2p and spans the portion of the prion domain that has been shown to interfere with normal Ure2p activity when coexpressed with full-length Ure2p (49). Electron microscopy (EM), electron diffraction, and x-ray diffraction measurements have shown that synthetic Ure2p10–39 forms fibrils with the morphology and cross-β structure characteristic of amyloid (67,68).
With amino acid sequence SNLSNALRQV NIGNRNSNTT TDQSNINFEF, Ure2p10–39 contains no hydrophobic segments longer than two residues and contains 33% glutamine and asparagine residues. The data reported below demonstrate that Ure2p10–39 fibrils contain in-register parallel (not antiparallel) β-sheets and provide direct evidence for polar zipper interactions involving sidechain amide groups of Gln18 in Ure2p10–39 fibrils. These are the first constraints from solid state NMR on the molecular structure of amyloid fibrils formed by a peptide that both lacks a hydrophobic segment and contains a high percentage of glutamine and asparagine residues. In addition, we present NMR data that argue against recent proposals that amyloid fibrils formed by glutamine- and asparagine-rich peptides are water-filled, tubular structures (55,69). Finally, we present two structural models for Ure2p10–39 fibrils that are consistent with available constraints but differ in certain respects from a recent model proposed by Kajava et al. (67)
Materials and Methods
Sample preparation and characterization
Ure2p10–39 samples were synthesized on an Applied Biosystems 433A peptide synthesizer, using a Wang resin (0.59 meq/g substitution level; Anaspec, Inc.), 9-fluorenylmethoxycarbonyl (FMOC) chemistry with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate activation, and acetic anhydride capping after each coupling step (except for the coupling of Ser10, producing a free amino terminus). The synthesis scale was 0.1 mmol, with a ten-fold excess and 20 min coupling time for each unlabeled amino acid and a three-fold excess and 45 min coupling time for each labeled amino acid. Two samples were prepared for solid state NMR, one with 13C labels at the methyl carbon of Ala15, the α-carbon of Gly22, and the carbonyl carbon of Phe37 (Ure2p10–39-AGF), the other with a 13C label at the methyl carbon of Ala15 and with uniform 15N and 13C labeling of Gln18 (Ure2p10–39-AQ). Labeled FMOC-amino acids were obtained from Cambridge Isotope Laboratories and Isotec. FMOC and trityl sidechain protecting groups were attached to labeled L-glutamine according to published methods (70). Crude Ure2p10–39 was cleaved from the synthesis resin using standard protocols (100 min reaction in 95% triflouroacetic acid (TFA) with phenol, ethanedithiol, and thioanisole scavengers), precipitated in t-butyl methyl ether, and dried. The crude material was purified by high-performance liquid chromatography, using a water/acetonitrile gradient with 0.1% TFA and a preparative scale Vydac C4 reverse-phase column. Samples were dissolved in TFA and then diluted to 50% TFA/47.5% water/2.5% acetonitrile before injection onto the column. Fractions containing Ure2p10–39 were frozen in liquid nitrogen immediately after collection and subsequently lyophilized. Peptide purity was at least 95% by electrospray mass spectrometry. The yield of purified peptide, relative to the 0.1 mmol synthesis scale, was approximately 50%.
Amyloid fibrils were formed by dissolution of purified Ure2p10–39 at a peptide concentration of 50 μM in 10 mM phosphate buffer, pH 7.4. Dissolution was assisted by sonication. Fibrils formed immediately, appearing as a visible precipitate.
For atomic force microscopy (AFM), a 50 μl aliquot of the fibrillized solution was acidified to pH 4 by addition of 10 μl of 1% acetic acid, in order to promote adhesion of fibrils to the mica substrate. Approximately 10 μl of the solution was adsorbed on freshly cleaved mica for 5 min. Excess solution was then poured off and the surface was dried in a gentle air stream. Images were obtained in air with a MultiMode AFM system (Veeco Instruments) in tapping mode, using etched silicon tips (Veeco RTESP probes, 10 nm nominal radius of curvature).
Solid state NMR measurements
For solid state NMR measurements, fibrillized solutions were lyophilized and packed into 3.2 mm magic-angle spinning (MAS) rotors. Sample quantities were 3–5 mg. Measurements were performed at room temperature in Varian MAS probes operating in double-resonance 1H/13C or triple-resonance 1H/13C/15N configurations, using Varian InfinityPlus-400 and Infinity-600 spectrometers. 13C and 15N NMR chemical shift values in Fig. 2 and the discussion below are relative to tetramethylsilane and liquid NH3, respectively. Measurements of intermolecular 13C-13C nuclear magnetic dipole-dipole couplings were carried out on the Ure2p10–39-AGF sample at a 13C NMR frequency of 100.4 MHz and an MAS frequency of 20.0 kHz, using the constant-time, finite-pulse radio-frequency-driven recoupling (fpRFDR-CT) technique (13,71). Pulse sequence conditions for fpRFDR-CT measurements were identical to those in earlier studies of Aβ fibrils (13,30).
Figure 2.
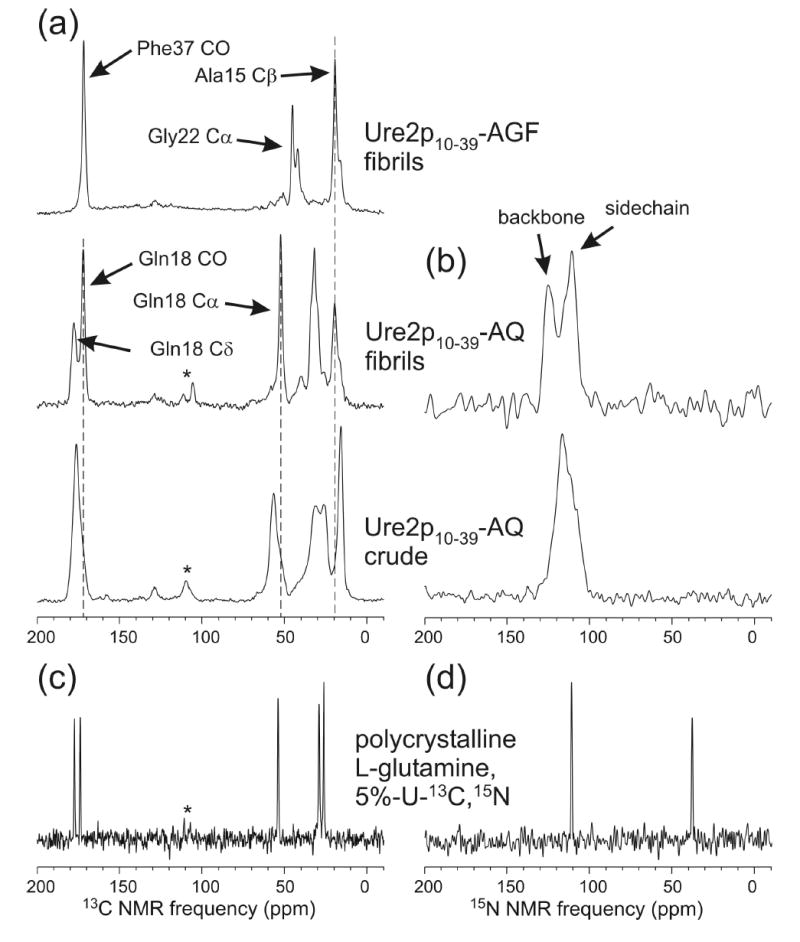
(a) Solid state 13C NMR spectra of Ure2p10–39 in fibrillar and nonfibrillar (crude) form. The peptide is 13C-labeled at the carbonyl carbon of Phe37, the α-carbon of Gly22, and the β-carbon of Ala15 (AGF), or 13C-labeled at the β-carbon of Gly22 and uniformly 15N,13C-labeled at Gln18 (AQ). (b) Solid state 15N NMR spectra of Ure2p10–39-AQ. (c,d) Solid state 13C and 15N NMR spectra of polycrystalline L-glutamine, in which 5% of the molecules are uniformly 13C,15N-labeled. All spectra are recorded with cross-polarization, magic-angle spinning, and proton decoupling. Asterisks indicate spinning sidebands. Signals near 130 ppm in 13C NMR spectra are probe background signals.
Intermolecular distances between Ala15 methyls and between Phe37 carbonyls could be measured separately in a single sample because of the 15.3 kHz difference in 13C NMR frequencies. The total experiment time for each fpRFDR-CT data set in Fig. 3 was 20 hr, using a pulse sequence recycle delay of 2.5 s. Measurements of intermolecular distances between Gly22 α-carbons in this sample were unsuccessful due to the relatively short transverse spin relaxation time for Gly22 α-carbons (approximately 15 ms under the conditions of fpRFDR-CT measurements), which led to a very low signal-to-noise ratio.
Figure 3.
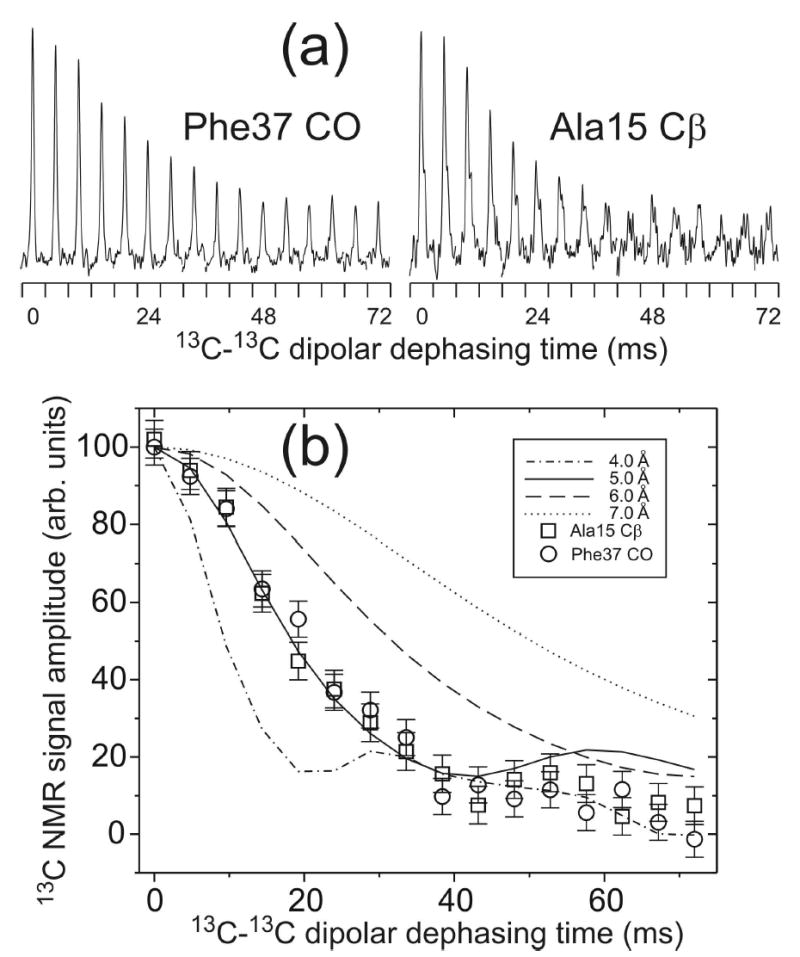
(a) Measurements of intermolecular 13C-13C dipole-dipole couplings in Ure2p10–39-AGF fibrils, using the fpRFDR-CT solid state NMR technique. Each Phe37 carbonyl or Ala15 β-carbon NMR peak is plotted in a 2.0 kHz spectral window. Peaks for dephasing times between 0 and 72 ms are concatenated to show the dipolar dephasing curves. (b) Comparison of experimental and simulated fpRFDR-CT curves, with intermolecular 13C-13C distances between 4.0 Å and 7.0 Å in the simulations. Error bars represent the root-mean-squared noise in the experimental spectra. Intermolecular distances are determined to be 5.0 ± 0.5 Å for both the Phe37 carbonyl and the Ala15 β-carbon, consistent with an in-register parallel β-sheet structure in the fibrils.
Intermolecular interactions among Gln18 sidechain amide groups were measured in the Ure2p10–39-AQ sample, using a newly devised solid state NMR technique represented by the radio-frequency (rf) pulse sequences in Fig. 4a. This technique is an extension of the well-established rotational echo double resonance (REDOR) (72) and frequency-selective REDOR (73) techniques for measuring 15N-13C dipole-dipole couplings in noncrystalline solid materials. The new technique permits the measurement of relatively weak intermolecular 15N-13C dipole-dipole couplings between Gln18 sidechains (approximately 3.4–5.0 Å nearest-neighbor 15N-13C distance in an in-register parallel β-sheet, corresponding to a 25–78 Hz coupling constant) in the presence of stronger intraresidue 15N-13C dipole-dipole couplings (1.338 Å chemical bond distance (74), corresponding to a 1280 Hz coupling constant). In Fig. 4a, a short frequency-selective REDOR pulse train (72,73) is applied during the pulse sequence period τ1 to prepare intraresidue, heteronuclear “double single-quantum” (DSQ) coherences between directly-bonded 13C/15N pairs in sidechain amide groups. DSQ coherences are represented by spin density operator terms of the form IxSx in the standard product operator formalism (75). The intraresidue DSQ coherences are unaffected by intraresidue 15N-13C couplings during the longer frequency-selective REDOR dephasing period τd, but can decay due to intermolecular 15N-13C couplings pairs if such couplings are present (because IxSx commutes with the effective intraresidue coupling dISIzSz but does not commute with the effective interresidue couplings dJSJzSz, where dIS and dJS are the effective coupling constants and Jz is the z-component of spin angular momentum for a third spin). DSQ coherences at the end of τd are converted to observable carbonyl 13C nuclear spin polarization by the final frequency-selective REDOR pulse train during period τ2. 13C NMR signals that arise from the DSQ coherences are selected by appropriate phase cycling of rf pulses in τ1 and τ2. We refer to the technique depicted in Fig. 4a by the acronym DSQ-REDOR.
Figure 4.
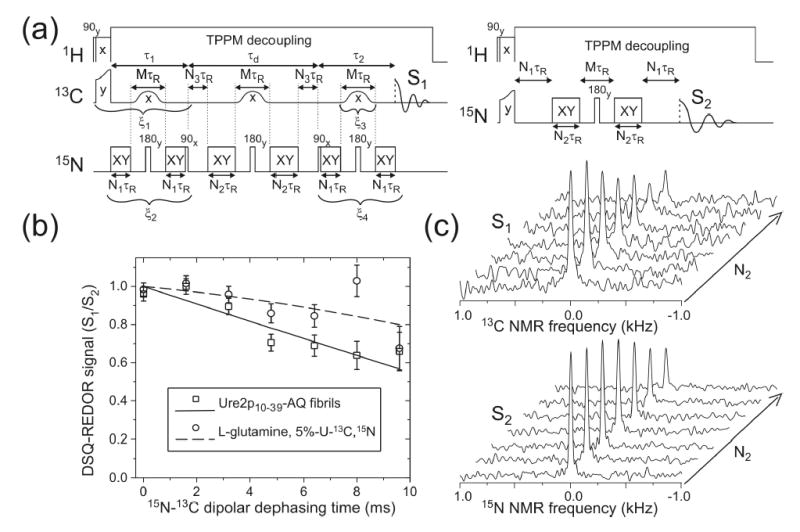
(a) Radio-frequency pulse sequences for measurements of S1 and S2 in DSQ-REDOR experiments. The magic-angle spinning period τR is 100 μs. XY represents a train of 15N π pulses with XY-16 phase patterns (76). TPPM represents two-pulse phase modulation(77). In these experiments, M = N1 = 4, N2 + N3 = 48, and N2 is incremented from 0 to 48 to produce effective dephasing times from 0 to 9.6 ms. Signals arising from intraresidue 15N-13C double single-quantum (DSQ) coherence are selected by phase cycling, with ξ1 = 0, π, 0, π, 0, π, 0, π, 0, π, 0, π, 0, π, 0, π; ξ2 = 0, 0, π, π, 0, 0, π, π, 0, 0, π, π, 0, 0, π, π; ξ3 = 0, 0, 0, 0, π, π, π, π, 0, 0, 0, 0, π, π, π, π; ξ4 = 0, 0, 0, 0, 0, 0, 0, 0, π, π, π, π, π, π, π, π. Signals are coadded according to the pattern +, −, −, +, +, −, −, +, −, +, +, −, −, +, +, −. (b) Measurements of intermolecular 15N-13C dipole-dipole couplings for glutamine sidechain amide groups in Ure2p10–39-AQ fibrils (squares) and in polycrystalline 5%-U-13C,15N-Gln (circles), using the DSQ-REDOR solid state NMR technique. To correct for NMR signal decay due to rf pulse imperfections and nuclear spin relaxation, DSQ-REDOR points are obtained by dividing 13C NMR signal amplitudes from a constant-time DSQ-REDOR measurement (S1) by 15N NMR signal amplitudes (S2). The vertical scale is therefore in arbitrary units. Solid and dashed lines are least-squares fits to second-order polynomial functions. Error bars represent uncertainties derived from the root-mean-squared noise in the experimental spectra. (c) Experimental S1 and S2 spectra from which the DSQ-REDOR data for Ure2p10–39-AQ fibrils are obtained.
The DSQ-REDOR measurements reported below were carried out at a 13C NMR frequency of 150.65 MHz and an MAS frequency of 10.0 kHz, using 10.0 μs 15N π pulses with XY-16 phase patterns (76), 400 μs Gaussian-shaped 13C π pulses at the carbonyl 13C NMR frequency (i.e., M = 4), and 110 kHz proton decoupling fields with two-pulse phase modulation (TPPM) (77) in the frequency-selective REDOR periods. Pulsed spin-lock detection (not depicted in Fig. 4a) was used to enhance sensitivity (78). The τ1 and τ2 periods were 1.2 ms (i.e., N1 = 4), chosen to maximize signal amplitudes. The τd period was fixed at 10.0 ms (i.e., N2 + N3 = 48) and N2 was incremented from 0 to 48 in steps of 8, producing effective 15N-13C dephasing periods from 0.0 ms to 9.6 ms in 1.6 ms increments. The total experiment time for DSQ-REDOR measurements on Ure2p10–39-AQ was 52 hr, with a 2.0 s pulse sequence recycle delay.
We represent the DSQ-REDOR signals by S1(N2). Use of a fixed τd period eliminates decay of the DSQ-REDOR signals due to transverse 13C spin relaxation. Decay of DSQ-REDOR signals due to 15N rf pulse imperfections during the π pulse trains in τd was calibrated by directly measuring the dependence of amide 15N NMR signals on N2 under experimental conditions identical to those in the DSQ-REDOR measurements. We represent the 15N signal decay by S2(N2). Decay due to the intermolecular 15N-13C dipole-dipole couplings (i.e., the data plotted in Fig. 4b) is then S1(N2)/S2(N2).
To verify that experimental S1(N2)/S2(N2) decays were due to intermolecular 15N-13C dipole-dipole couplings as intended, rather than being due to intramolecular couplings, spin relaxation effects, or pulse sequence imperfections, we recorded DSQ-REDOR data for a 5 mg sample of polycrystalline L-glutamine in which uniformly 15N,13C-labeled molecules were diluted to 5% in unlabeled molecules by recrystallization from aqueous solution (5%-U-13C,15N-Gln). Data for polycrystalline 5%-U-13C,15N-Gln are shown in Figs. 2 and 4.
Solid state NMR data analyses
Data were analyzed by comparison with numerical simulations. Simulations of fpRFDR-CT data were carried out as described previously (13,30), using a linear chain of six equally spaced 13C nuclei and with initial 13C spin polarization on the central two spins. Experimental fpRFDR-CT data in Fig. 3 were corrected for signal contributions from natural-abundance 13C nuclei by subtraction of a constant value equal to 25% (Phe37 carbonyl data) or 10% (Ala15 methyl data) of the signal at zero effective dephasing time (13,30). Simulations of DSQ-REDOR data were carried out with a C++ program running under Matlab™. A four-spin system (central 15N-13C pair plus the nearest 15N and 13C of flanking pairs) with the geometry shown in Fig. 5a was used, with initial spin polarization only on the central 13C nucleus. Six-spin simulations (all spins in Fig. 5a) gave nearly identical results in test simulations, but would have required prohibitively long times to generate the contour plot in Fig. 5b. DSQ-REDOR simulations included finite rf pulses, homonuclear and heteronuclear dipole-dipole couplings, 13C chemical shift anisotropies (CSA), and powder averaging over 2048 orientations. CSA tensor principal values and orientations relative to chemical bonds were taken from studies of model compounds (79).
Figure 5.
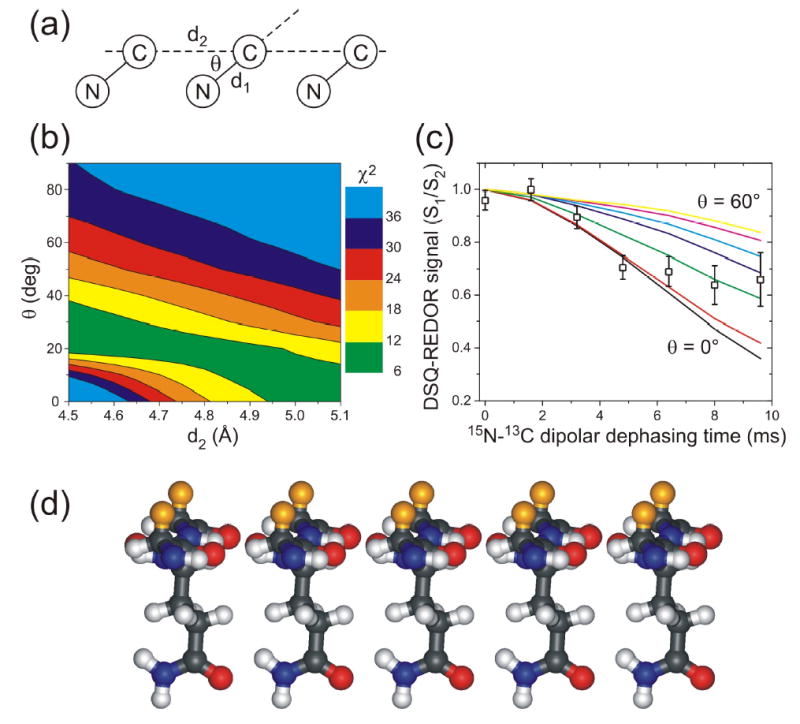
(a) Geometry for isotopically labeled glutamine sidechain amide groups in DSQ-REDOR simulations. Simulations assume a 15N-13C amide bond length d1 = 1.338 Å, a variable intermolecular distance d2, and a variable angle θ. (b) Contour plot of the χ2 deviation between experimental DSQ-REDOR data for Ure2p10–39-AQ fibrils and simulated DSQ-REDOR data as a function of the values of d2 and θ in the simulations. Good fits correspond to χ2 ≈ 10 or less. (c) Comparison of experimental and simulated DSQ-REDOR curves for d2 = 4.80 Å. The angle θ varies from 0° (black line) to 60° (yellow line). (d) Molecular model for glutamine sidechains pendent from an in-register parallel β-sheet in an amyloid fibril, with θ = 25°. Orange atoms are β-carbons of residues that immediately precede and follow the glutamine residue in each β-strand, showing that these β-carbons are on the opposite face of the β-sheet. The spacing between β-strands is 4.80 Å. This geometry permits polar zipper interactions among glutamine sidechains, as proposed by Perutz (43,53–56). (Model generated in MOLMOL (80).)
Molecular modeling
Ure2p10–39 peptides with strand-loop-strand conformations were created in MOLMOL (80) by assigning backbone torsion angles (φ,ψ) = (−140º,140º) to all residues and then changing these angles to (50°,50°) for Gly22 and (−70°,50°) for Ser26 (for the model in Fig. 8a) or to (−115°,35°) for Gly22 and Ser26 and (−110°,−155°) for Asn23 and Asn27 (for the model in Fig. 8b). Five copies of each peptide were combined, with 4.8 Å displacements along the intermolecular hydrogen bonding direction, to generate the starting structures for molecular dynamics (MD) and energy minimization simulations.
Figure 8.
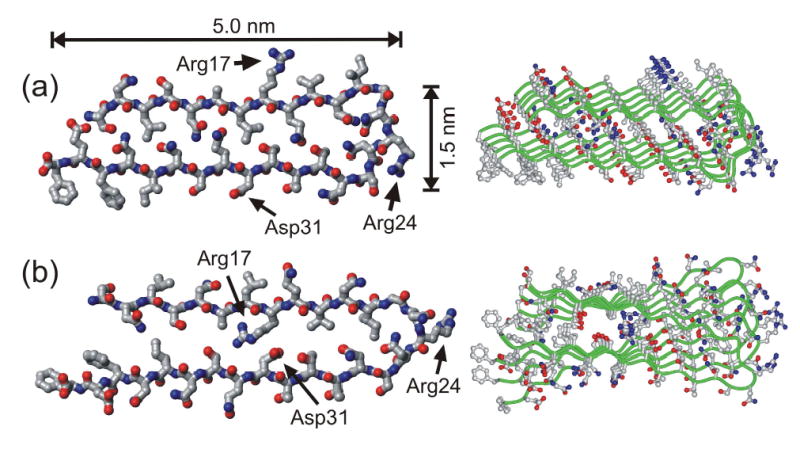
Structural models for Ure2p10–39 fibrils generated by restrained molecular dynamics and energy minimization simulations for a pentameric assembly of Ure2p10–39 molecules. Both the final energy-minimized structure of the pentamer (right) and the conformation of the central molecule in the pentamer (left) are shown. The two models differ in the relative orientation of the β-sheets formed by the two β-strand segments (residues 10–21 and 27–39), as dictated by the conformation of the intervening loop segment (residues 22–26). (a) Model in which all charged sidechains are outside the fibril core. (b) Model in which oppositely charged sidechains of Arg17 and Asp31 can form internal salt bridges in the fibril core.
MD and energy minimization simulations were carried out in TINKER, version 4.2, using the CHARMM27 force field and the Force Field Explorer interface on a notebook computer. All electrostatic interactions were turned off throughout these simulations. Backbone torsion angles of residues 10–21 and 27–38 were restrained to (φ,ψ) = (−140º,140º). Intermolecular backbone hydrogen bonds for these residues were enforced by 2.15 Å distance restraints between the appropriate carbonyl oxygens and amide protons. All other torsion angles were unrestrained. Intermolecular sidechain hydrogen bonds for Gln18 and Gln33 were also enforced by distance restraints. Close contacts between β-sheets formed by residues 10–21 and 27–38 were enforced by 9.0–11.0 Å distance restraints between α-carbons of residues 12, 14, 16, and 18 and α-carbons of residues 35, 33, 31, and 29, respectively. For the model in Fig. 8a, salt bridges between the sidechain carboxylate group of Glu38 and the N-terminal amino group were enforced by 3.0 Å distance restraints between carboxylate oxygens and amino nitrogens. For the model in Fig. 8b, salt bridges between the sidechain carboxylate group of Glu 31 and the sidechain guanidino group of Arg17 were enforced by 4.0–6.0 Å distance restraints between carboxylate carbons and guanidino carbons. Models in Fig. 8 result from energy minimization of the starting structures, followed by a 10 ps MD simulation at 400 K, followed by a final energy minimization.
Results
Atomic force microscopy indicates that Ure2p10–39 aggregates are amyloid fibrils
Fig. 1 shows a typical AFM image of Ure2p10–39 fibrils, obtained in air on a mica substrate. The fibrils have the characteristic straight, unbranched appearance of amyloid fibrils. In this and multiple other images, a uniform fibril height of 1.6 ± 0.1 nm was observed. Apparent fibril diameters were 12 ± 2 nm, limited by the AFM tip sharpness. In an earlier electron microscope (EM) study of Ure2p10–39 fibrils (67), the fibrils appeared as 10–50 nm wide bundles of finer protofilaments, with protofilament diameters of approximately 5 nm or less. The difference between the fibril height in our AFM images and the apparent protofilament diameter in the earlier EM images may reflect an approximate 3:1 ratio of lateral dimensions (see Fig. 8), with fibrils adsorbing to mica on the 5 nm faces.
Figure 1.
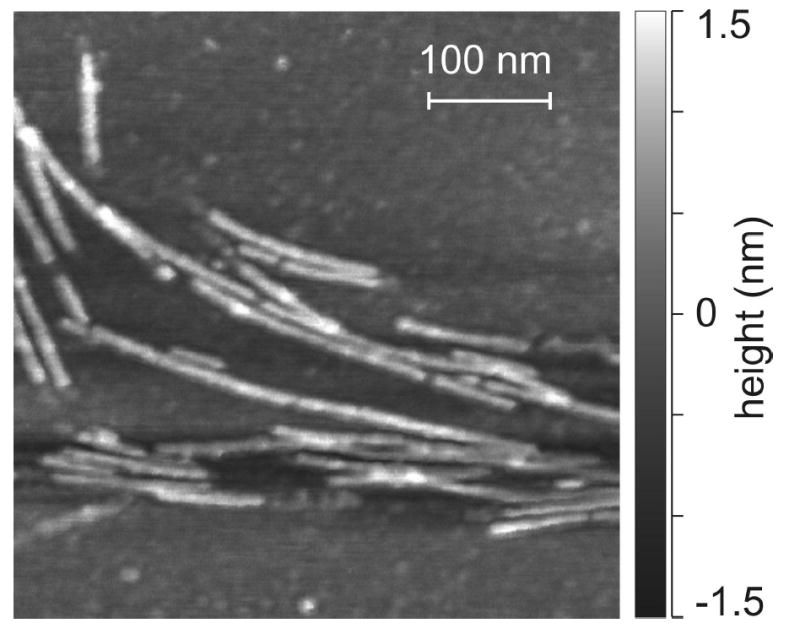
Atomic force microscope image of Ure2p10–39 fibrils, deposited on mica and recorded in air in tapping mode.
X-ray diffraction data on unaligned Ure2p10–39 fibrils obtained by Baxa et al. show the sharp 4.75 Å and broad 10 Å reflections characteristic of β-sheets in amyloid fibrils (68). Electron diffraction data on partially aligned Ure2p10–39 fibrils establish the cross-β orientation of the β-sheets relative to the long axis of the fibrils that is a defining feature of amyloid fibrils (68).
Solid state NMR spectra support an ordered β-sheet structure in Ure2p10–39 fibrils
Figs. 2a and 2b show solid state 13C and 15N NMR spectra of Ure2p10–39-AGF and Ure2p10–39-AQ fibrils in lyophilized form, as well as spectra of crude Ure2p10–39-AQ (ether precipitate after cleavage from the synthesis resin and drying, with no purification or fibrillization). The similarity of Ala15 methyl signals in 13C NMR spectra of Ure2p10–39-AGF and Ure2p10–39-AQ fibrils demonstrates the reproducibility of the fibril structures. Linewidths in 13C NMR spectra of fibrillized Ure2p10–39-AQ (2.0–2.9 ppm full width at half maximum) are less than in spectra of crude Ure2p10–39-AQ (3.0–5.1 ppm), indicating a higher degree of structural order in the fibrils. The development of resolved 13C and 15N NMR signals for the Gln18 sidechain carbonyl carbon and amide nitrogen upon fibril formation indicates that Gln18 sidechains are well ordered. In the fibrils, 13C chemical shifts for carbonyl sites of Gln18 and Phe37 (171.9 ppm and 171.6 ppm), the α-carbon of Gln18 (52.3 ppm), and the β-carbon of Ala15 (19.4 ppm) exhibit deviations from random coil values (174.3 ppm, 174.1 ppm, 54.0 ppm, and 17.4 ppm, respectively (81)) that are consistent in both sign and magnitude with β-strand backbone conformations at these residues (i.e., upfield shift for carbonyls and α-carbons, downfield shift for β-carbons (6,82–84)). The α-carbon signal for Gly22 is split into two components (45.2 ppm and 42.1 ppm, versus a random coil value of 43.4 ppm), suggesting the coexistence of two backbone conformations at Gly22 in Ure2p10–39 fibrils, at least one of which may be a non-β-strand conformation. Similar splittings of 13C NMR lines have been observed for certain residues in the non-β-strand segment of Aβ fibrils (30). The conformational interpretation of α-carbon chemical shifts for glycine residues is generally more ambiguous than for other residues, as the range of secondary shifts for glycines in proteins is relatively small and does not show a clear separation of values corresponding to helical and extended backbone conformations.
In crude Ure2p10–39-AQ, 13C chemical shifts for the carbonyl and α-carbon of Gln18 (176.4 ppm and 56.8 ppm) and the β-carbon of Ala15 (15.9 ppm) are consistent with α-helical conformations at these residues. Thus, the peptide conformations in ether-precipitated and fibrillized forms are substantially different, as previously observed for Aβ fibrils (7).
The assignment of 15N NMR lines to sidechain and backbone amide sites indicated in Fig. 2 was determined from 13C-detected frequency-selective REDOR measurements (73), in which selective excitation of the 15N NMR line at 111 ppm produced greater dephasing of the Gln18 δ-carbon and selective excitation of the 15N NMR line at 125 ppm produced greater dephasing of the Gln18 α-carbon. The measurements were performed with MAS at 9.00 kHz, Gaussian-shaped selective pulses with 1.111 ms durations, and 1.78 ms dephasing periods.
Figs. 2c and 2d show solid state NMR spectra of polycrystalline 5%-U-13C, 15N-Gln. Linewidths in these spectra are less than 0.8 ppm, as expected for a polycrystalline material. Linewidths for Ure2p10–39 fibrils are typical of conformationally ordered peptides in rigid, noncrystalline environments (16,30,85–87).
Intermolecular 13C-13C dipole-dipole couplings indicate parallel β-sheets
Fig. 3 shows measurements of intermolecular 13C-13C nuclear magnetic dipole-dipole couplings for Ure2p10–39-AGF fibrils, using the fpRFDR-CT technique as in earlier studies of Aβ fibrils (13,30). The time scale for decay of 13C NMR signals in fpRFDR-CT measurements decreases with decreasing distance between 13C labels on neighboring peptide molecules, due to the 1/R3 dependence of dipole-dipole coupling strengths on internuclear distance R. Intermolecular distances for Ala15 methyl labels and Phe37 carbonyl labels were measured separately by positioning the rf carrier frequency in the fpRFDR-CT pulse sequence at either 32.2 ppm (Ala15 methyl data) or 162.1 ppm (Phe37 carbonyl data). The time scale for signal decay was approximately 30 ms in both measurements. Comparison with numerical simulations (Fig. 3b) indicates intermolecular distances of 5.0 ± 0.3 Å for both Ala15 methyl labels and Phe37 carbonyl labels. This result is consistent with an in-register parallel β-sheet structure in Ure2p10–39 fibrils, as previously observed for fibrils formed by the 40-residue and 42-residue variants of full-length Aβ and fibrils formed by residues 10–35 of Aβ (12,13,30). In an ideal in-register parallel β-sheet, the intermolecular distances would be 4.7–4.8 Å.
Computer modeling of parallel β-sheets with a one-residue shift from in-register alignment of neighboring peptide chains shows that nearest-neighbor intermolecular distances would be approximately 5.2 Å for backbone carbonyl labels and approximately 6.7 Å for alanine methyl labels. Thus, the data in Fig. 3 for Ala15 rule out a one-residue (or greater) shift. Data in Fig. 3 are also inconsistent with an antiparallel β-sheet structure, which could not produce the observed 5.0 ± 0.3 Å intermolecular distances for both Ala15 and Phe37 simultaneously.
Intermolecular 15N-13C dipole-dipole couplings indicate Gln18 sidechain interactions
Fig. 4b shows measurements of intermolecular 15N-13C nuclear magnetic dipole-dipole couplings among sidechain amide groups of Gln18 residues in Ure2p10–39-AQ fibrils, using the DSQ-REDOR technique shown in Fig. 4a. As explained above, the DSQ-REDOR technique was designed specifically to permit measurements of intermolecular 15N-13C couplings in the presence of the stronger intraresidue 15N-13C couplings in the Ure2p10–39-AQ sample. Intraresidue couplings are present because Gln18 is uniformly 15N and 13C labeled in this sample. Data points in Fig. 4b were obtained from the experimental 13C and 15N NMR spectra in Fig. 4c. Only signals from Gln18 sidechain carbonyl carbons contribute to the 13C NMR spectra in Fig. 4c because other carbon signals are removed by DSQ filtering (i.e., by the rf phase shifts ξ1, ξ2, ξ3, and ξ4 in Fig. 4a).
That the DSQ-REDOR signal decay for Ure2p10–39-AQ fibrils is primarily due to intermolecular couplings is supported by DSQ-REDOR data for polycrystalline 5%-U-13C, 15N-Gln, also shown in Fig. 4b, which decay significantly more slowly. Based on the reported crystal structure of L-glutamine (74), an isotopically labeled sidechain amide group in 5%-U-13C, 15N-Gln has a 26% probability of being within 4.5 Å of a 15N or 13C site on a neighboring molecule to which it will couple under the DSQ-REDOR pulse sequence, and the intramolecular distance between sidechain carbonyl 13C and amine 15N sites is 4.491 Å. The experimental DSQ-REDOR decay for 5%-U-13C, 15N-Gln in Fig. 4b is consistent with the crystal structure and isotopic dilution level.
The DSQ-REDOR data for Ure2p10–39-AQ fibrils are analyzed by comparison with numerical simulations. As depicted in Fig. 5a, these simulations assume a geometry for Gln18 sidechain amide groups in which the intraresidue 15N-13C chemical bond distance d1 is fixed at 1.338 Å (74), the intermolecular distance d2 is fixed at a value between 4.50 Å and 5.10 Å, and the angle θ between the chemical bonds and the intermolecular displacement is allowed to vary. Note that this assumed geometry applies to all possible Gln18 sidechain conformations, provided that the β-sheets are planar and the structure has translational symmetry. Deviations from planarity in the β-sheets of amyloid fibrils are expected to be small, corresponding to a twist by less than 10° per β-strand, based on the fact that fibril morphologies commonly exhibit twist periods greater than 20 nm (2,5,12,14,15,30). Approximate translational symmetry in Ure2p10–39 fibrils is supported by the relatively sharp solid state NMR lines for Gln18 in Fig. 2. Fig. 5b shows a contour plot of the χ2 deviation between experimental and simulated DSQ-REDOR data as a function of θ and d2. Simulated DSQ-REDOR data for d2 = 4.80 Å are compared with the experimental data in Fig. 5c. Given that d2 lies in the 4.65–4.85 Å range in β-sheets (based on examination of Cα-Cα distances across parallel β-sheets in high-resolution protein crystal structures, e.g., Protein Data Bank files 1R2R and 1J8Q), we conclude that θ = 25 ± 5°. Fig. 5d shows a molecular model for Gln18 sidechain interactions that is consistent with the best-fit geometry. This model appears compatible with the polar zipper hydrogen bonds proposed by Perutz (43,53,55).
Effects of hydration on solid state NMR spectra argue against a water-filled structure
Perutz and coworkers also proposed that amyloid fibrils formed by glutamine- and asparagine-rich peptides might have water-filled, tubular structures (55). X-ray fiber diffraction data for Sup35 fibrils have been presented as support for this proposal (69). Fig. 6a shows solid state 13C NMR spectra of Ure2p10–39-AQ fibrils, examined as a centrifuged pellet of the initial fibrillized solution prior to lyophilization, as a dry, lyophilized powder, and as a lyophilized powder to which a small aliquot of 10 mM phosphate buffer (approximately 90 water molecules per peptide molecule) has been added. No differences in 13C NMR chemical shifts are observed in these three hydration states. The increased 13C NMR linewidths in the dry, lyophilized state (by 0.6 ppm, 0.4 ppm, 0.4 ppm, and 2.0 ppm for Gln18 sidechain carbonyl, Gln18 backbone carbonyl, Gln18 α-carbon, and Ala15 β-carbon lines, respectively) can be attributed to inhomogeneous broadening from local static structural disorder in these noncrystalline materials, e.g., minor variations in backbone and sidechain torsion angles from one Ure2p10–39 molecule to the next. In the hydrated state, librational motions and sidechain dynamics can average out some of the local structural disorder, producing sharper NMR lines.
Figure 6.
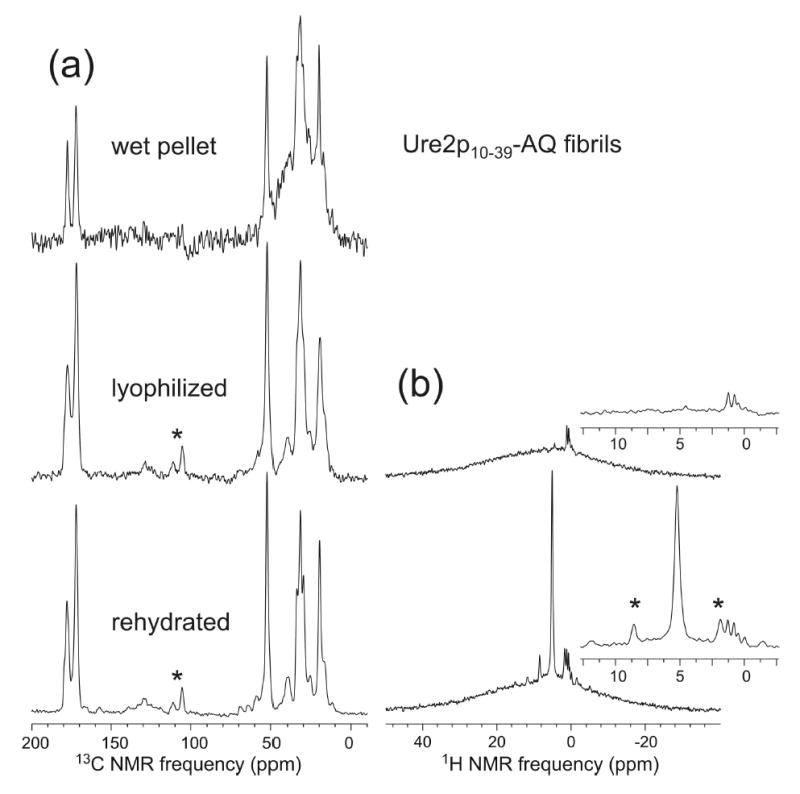
(a) 13C solid state NMR spectra of Ure2p10–39-AQ fibrils in a fully hydrated state after fibril formation (wet pellet), after lyophilization, and after rehydration. Minor changes in 13C NMR linewidths, but no changes in 13C NMR chemical shifts, are observed. Signal-to-noise for the wet pellet is lower and background signal (hump centered at 30 ppm) is higher because of the smaller sample quantity. (b) Proton NMR spectra of the lyophilized and rehydrated samples, indicating the absence of detectable mobile water (sharp peak at 5 ppm) in the lyophilized state. Asterisks indicate spinning sidebands.
1H NMR spectra in Fig. 6b show that the dry, lyophilized state indeed contains no detectable, mobile water (less than three mobile water molecules per peptide molecule, based on the signal-to-noise ratio for the water peak in Fig. 6b). Data in Fig. 6 are incompatible with the possibility that fully hydrated Ure2p10–39 fibrils have a water-filled tubular structure that undergoes a major change upon drying, such as collapse to a laminated β-sheet structure (69). Hydration apparently affects molecular motions, but not structure. These conclusions from solid state NMR spectra may be related to the work of Diaz-Avalos et al., in which diffraction measurements on a seven-residue, glutamine- and asparagine-rich peptide in an amyloid-like nanocrystalline form revealed a low density of water, attributed to sidechain hydrogen bonding (88).
Rehydration of the Ure2p10–39-AGF fibril sample also produced no changes in the 13C chemical shifts (less than 0.1 ppm change in peak positions). 13C NMR lines for the Ph37 carbonyl and Gly22 α-carbon sites were reduced by approximately 0.5 ppm by rehydration. Similarly, 15N NMR linewidths for Gln18 in Ure2p10–39-AQ fibrils were reduced from approximately 8 ppm to approximately 4 ppm by rehydration, but no changes in 15N chemical shifts were observed.
Discussion
Data presented above lead to the following conclusions: (1) Ure2p10–39 fibrils contain well ordered β-sheets with an in-register parallel structure; (2) Ala15, Gln18, and Phe37 are contained in β-strands that participate in the parallel β-sheets, but Gly22 may be contained in a non-β-strand segment with two alternative conformations; (3) the amide groups of Gln18 sidechains exhibit intermolecular 15N-13C dipole-dipole couplings that are consistent with polar zipper hydrogen bond interactions; (4) the molecular structure of Ure2p10–39 fibrils is not strongly dependent on hydration.
With the exception of studies of the designed peptide ccβ (23) and the HET-s prion protein (35), previous solid state NMR studies of amyloid fibril structures have focused on peptides with hydrophobic segments, at least five residues in length (1,3–8,12,13,16,18,22,24,25,30,89,90). Both parallel and antiparallel β-sheet structures have been observed in these studies, but in all cases the structures are such that hydrophobic contacts within a single β-sheet layer are maximized (3,5–8,12,13,16,30) or nearly maximized (1,25). Ure2p10–39 is a qualitatively different case because of its lack of hydrophobic segments. The observation of an in-register parallel β-sheet structure (Fig. 3) and the evidence for polar zipper interactions in Ure2p10–39 fibrils (Figs. 4 and 5) suggest that the parallel β-sheet structure is favored in amyloid fibrils by sidechain interactions among polar groups as well as hydrophobic groups within a single β-sheet layer. In particular, an in-register parallel β-sheet structure would be favored over out-of-register or antiparallel structures when the peptide sequence contains an asymmetric distribution of glutamine and asparagine residues, as depicted in Fig. 7. A sequence that is palindromic with respect to glutamine and asparagine residues could form either parallel or antiparallel β-sheets, with the choice being dictated by electrostatic or other interactions. We hypothesize that hydrogen bonds between a glutamine sidechain and an asparagine sidechain would not occur, because of the different sidechain lengths, so a peptide containing a mixture of glutamine and asparagine residues would adopt a β-sheet structure that aligns glutamines with glutamines and asparagines with asparagines.
Figure 7.
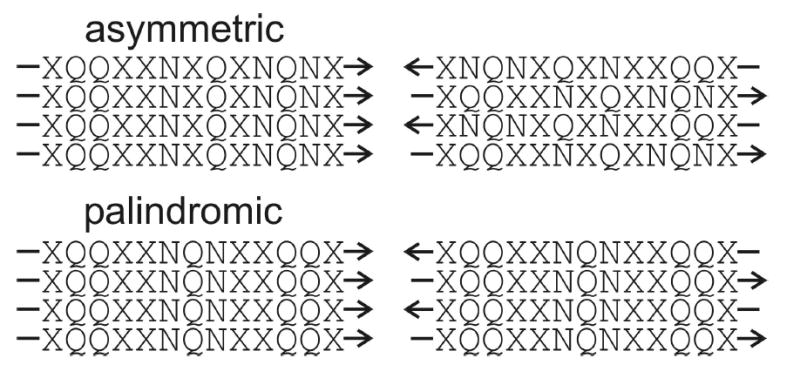
Proposal regarding the preference for in-register parallel (left) or antiparallel (right) β-sheets in amyloid fibrils formed by peptides with glutamine- and asparagine-rich sequences. Sequences with asymmetric distributions of glutamine and asparagine residues (such as Ure2p10–39) can maximize their polar zipper interactions only in an in-register parallel β-sheet, making this the preferred structure. Sequences with palindromic distributions of glutamine and asparagine residues can maximize their polar zipper interactions in either type of β-sheet, allowing electrostatic or other interactions to determine the preferred structure.
The recent observation by Ross et al. that scrambling of the Ure2p prion domain sequence does not suppress fibril formation (91) is consistent with an in-register parallel β-sheet structure stabilized by polar zipper interactions, since all polar zipper interactions could be present in such a structure with any ordering of the amino acids. In contrast, a rearrangement of the amino acid sequence of residues 10–35 of Aβ that eliminates the central hydrophobic segment of this peptide, but preserves the amino acid composition, has been shown to suppress both fibril formation and parallel β-sheet formation (12). Thus, amyloid fibril formation that is dependent on amino acid composition but independent of sequence order may be a unique property of glutamine- and asparagine-rich peptides.
Although both hydrophobic interactions and polar zipper interactions appear to favor an in-register parallel β-sheet structure for a generic amyloid-forming peptide with an asymmetric distribution of hydrophobic segments or glutamine/asparagine residues, such a structure could be destabilized by electrostatic repulsions between charged sidechains of neighboring peptide molecules if the charges were located in the low-dielectric core of the fibril. As exemplified by recent molecular structural models for full-length Aβ fibrils (16,30,31,92,93), electrostatic destabilization of an in-register parallel β-sheet structure can be avoided by adoption of a peptide conformation that places charges on the exterior of the fibril structure, where charged groups can be fully solvated, and by pairing of oppositely charged sidechains in the interior of the fibril structure. Fig. 8 shows two specific models for the structure of Ure2p10–39 fibrils, generated by restrained MD and energy minimization simulations (see Materials and Methods). These models are consistent with the solid state NMR data presented above and avoid electrostatic destabilization either by placing sidechains of Arg17, Arg24, Asp32, and Glu38 outside the fibril core (Fig. 8a) or by placing sidechains of Arg24 and Glu38 outside the fibril core and pairing sidechains of Arg17 with those of Asp31 (Fig. 8b). In these models, Ure2p10–39 adopts a strand-loop-strand conformation similar to the peptide conformation in the Aβ fibril model of Petkova et al. (16,30,31) The loop segment begins at Gly22, consistent with the solid state 13C NMR chemical shifts for this residue discussed above, and ends before the three threonines in the Ure2p10–39 sequence, consistent with the stabilization of β-strands in globular proteins by threonine residues (94,95). The approximate 1.5 nm fibril “height” in these models is consistent with our AFM data (Fig. 1) if the β-sheets are untwisted, while the approximate 5 nm fibril “width” may be consistent with the EM images reported by Kajava et al. (67) Interestingly, the model in Fig. 8a remains untwisted in the course of MD and energy minimization simulations, while the model in Fig. 8b develops a twist of roughly 8° per β-strand about the long axis of the fibril. Twisting of the model in Fig. 8b appears to result from strain due to the presence of the bulky Phe37 sidechain in the fibril core and the relatively tight loop conformation adopted by residues 22–26.
The models in Fig. 8 differ from the “β-serpentine” model for Ure2p10–39 fibrils proposed recently by Kajava et al. (67) (which was based in part on the data in Fig. 3) in that the models in Fig. 8 contain two, rather than four, β-strand segments, place Gly22 in the loop, rather than in a β-strand, and (for the model in Fig. 8b) contain charged sidechains in the core. Additional experimental constraints are required to confirm any of these models or to provide directions for their refinement.
Finally, Thakur and Wetzel have shown that the effects on aggregation kinetics and fibril formation of the introduction of Pro-Gly pairs into long polyglutamine chains are sensitive to the spacing between Pro-Gly pairs, with strong effects when Pro-Gly pairs are separated by less than nine glutamine residues (60). This result suggests that the β-strands in polyglutamine fibrils are at least eight residues in length. Although Thakur and Wetzel interpret their data in terms of an antiparallel β-sheet structure (60), their data may also be consistent with structures analogous to the models in Fig. 8. For polyglutamine peptides of the type studied by Thakur and Wetzel, most polar zipper interactions could be present in either antiparallel or parallel β-sheet structures. Other interactions may then dictate the β-sheet structures that actually form in their experiments.
Acknowledgments
We thank Drs. R.B. Wickner and U. Baxa for useful discussions throughout the course of this work.
References
- 1.Lansbury PT, Costa PR, Griffiths JM, Simon EJ, Auger M, Halverson KJ, Kocisko DA, Hendsch ZS, Ashburn TT, Spencer RGS, Tidor B, Griffin RG. Structural model for the β-amyloid fibril based on interstrand alignment of an antiparallel-sheet comprising a C-terminal peptide. Nat Struct Biol. 1995;2:990–998. doi: 10.1038/nsb1195-990. [DOI] [PubMed] [Google Scholar]
- 2.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF. Common core structure of amyloid fibrils by synchrotron x-ray diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 3.Benzinger TLS, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Propagating structure of Alzheimer’s β-amyloid(10–35) is parallel β-sheet with residues in exact register. Proc Natl Acad Sci U S A. 1998;95:13407–13412. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gregory DM, Benzinger TLS, Burkoth TS, Miller-Auer H, Lynn DG, Meredith SC, Botto RE. Dipolar recoupling NMR of biomolecular self-assemblies: Determining inter- and intrastrand distances in fibrilized Alzheimer’s β-amyloid peptide. Solid State Nucl Magn Reson. 1998;13:149–166. doi: 10.1016/s0926-2040(98)00086-1. [DOI] [PubMed] [Google Scholar]
- 5.Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R. Multiple quantum solid state NMR indicates a parallel, not antiparallel, organization of β-sheets in Alzheimer’s β-amyloid fibrils. Proc Natl Acad Sci U S A. 2000;97:13045–13050. doi: 10.1073/pnas.230315097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW, Dyda F, Reed J, Tycko R. Amyloid fibril formation by Aβ16–22, a seven-residue fragment of the Alzheimer’s β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]
- 7.Benzinger TLS, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Two-dimensional structure of β-amyloid(10–35) fibrils. Biochemistry. 2000;39:3491–3499. doi: 10.1021/bi991527v. [DOI] [PubMed] [Google Scholar]
- 8.Burkoth TS, Benzinger TLS, Urban V, Morgan DM, Gregory DM, Thiyagarajan P, Botto RE, Meredith SC, Lynn DG. Structure of the β-amyloid(10–35) fibril. J Am Chem Soc. 2000;122:7883–7889. [Google Scholar]
- 9.Kheterpal I, Zhou S, Cook KD, Wetzel R. Aβ amyloid fibrils possess a core structure highly resistant to hydrogen exchange. Proc Natl Acad Sci U S A. 2000;97:13597–13601. doi: 10.1073/pnas.250288897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serpell LC, Smith JM. Direct visualisation of the β-sheet structure of synthetic Alzheimer’s amyloid. J Mol Biol. 2000;299:225–231. doi: 10.1006/jmbi.2000.3650. [DOI] [PubMed] [Google Scholar]
- 11.Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R. Structural features of the Aβ amyloid fibril elucidated by limited proteolysis. Biochemistry. 2001;40:11757–11767. doi: 10.1021/bi010805z. [DOI] [PubMed] [Google Scholar]
- 12.Antzutkin ON, Leapman RD, Balbach JJ, Tycko R. Supramolecular structural constraints on Alzheimer’s β-amyloid fibrils from electron microscopy and solid state nuclear magnetic resonance. Biochemistry. 2002;41:15436–15450. doi: 10.1021/bi0204185. [DOI] [PubMed] [Google Scholar]
- 13.Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, Tycko R. Supramolecular structure in full-length Alzheimer’s β-amyloid fibrils: Evidence for a parallel β-sheet organization from solid state nuclear magnetic resonance. Biophys J. 2002;83:1205–1216. doi: 10.1016/S0006-3495(02)75244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimenez JL, Nettleton EJ, Bouchard M, Robinson CV, Dobson CM, Saibil HR. The protofilament structure of insulin amyloid fibrils. Proc Natl Acad Sci U S A. 2002;99:9196–9201. doi: 10.1073/pnas.142459399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jimenez JL, Guijarro JL, Orlova E, Zurdo J, Dobson CM, Sunde M, Saibil HR. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. Embo J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torok M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG, Langen R. Structural and dynamic features of Alzheimer’s Aβ peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 18.Antzutkin ON, Balbach JJ, Tycko R. Site-specific identification of non-β-strand conformations in Alzheimer’s β-amyloid fibrils by solid state NMR. Biophys J. 2003;84:3326–3335. doi: 10.1016/S0006-3495(03)70057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Der-Sarkissian A, Jao CC, Chen J, Langen R. Structural organization of α-synuclein fibrils studied by site-directed spin labeling. J Biol Chem. 2003;278:37530–37535. doi: 10.1074/jbc.M305266200. [DOI] [PubMed] [Google Scholar]
- 20.Kheterpal I, Lashuel HA, Hartley DM, Wlaz T, Lansbury PT, Wetzel R. Aβ protofibrils possess a stable core structure resistant to hydrogen exchange. Biochemistry. 2003;42:14092–14098. doi: 10.1021/bi0357816. [DOI] [PubMed] [Google Scholar]
- 21.Gordon DJ, Balbach JJ, Tycko R, Meredith SC. Increasing the amphiphilicity of an amyloidogenic peptide changes the β-sheet structure in the fibrils from antiparallel to parallel. Biophys J. 2004;86:428–434. doi: 10.1016/S0006-3495(04)74119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A. 2004;101:711–716. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kammerer RA, Kostrewa D, Zurdo J, Detken A, Garcia-Echeverria C, Green JD, Muller SA, Meier BH, Winkler FK, Dobson CM, Steinmetz MO. Exploring amyloid formation by a de novo design. Proc Natl Acad Sci U S A. 2004;101:4435–4440. doi: 10.1073/pnas.0306786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oyler NA, Tycko R. Absolute structural constraints on amyloid fibrils from solid state NMR spectroscopy of partially oriented samples. J Am Chem Soc. 2004;126:4478–4479. doi: 10.1021/ja031719k. [DOI] [PubMed] [Google Scholar]
- 25.Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. Solid state NMR reveals a pH-dependent antiparallel β-sheet registry in fibrils formed by a β-amyloid peptide. J Mol Biol. 2004;335:247–260. doi: 10.1016/j.jmb.2003.10.044. [DOI] [PubMed] [Google Scholar]
- 26.Shivaprasad S, Wetzel R. An intersheet packing interaction in Aβ fibrils mapped by disulfide cross-linking. Biochemistry. 2004;43:15310–15317. doi: 10.1021/bi048019s. [DOI] [PubMed] [Google Scholar]
- 27.Tycko R. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr Opin Struct Biol. 2004;14:96–103. doi: 10.1016/j.sbi.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Makin OS, Atkins E, Sikorski P, Johansson J, Serpell LC. Molecular basis for amyloid fibril formation and stability. Proc Natl Acad Sci U S A. 2005;102:315–320. doi: 10.1073/pnas.0406847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petkova AT, Leapman RD, Guo ZH, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 31.Tycko R. Insights into the amyloid folding problem from solid state NMR. Biochemistry. 2003;42:3151–3159. doi: 10.1021/bi027378p. [DOI] [PubMed] [Google Scholar]
- 32.Naito A, Kamihira M, Inoue R, Saito H. Structural diversity of amyloid fibril formed in human calcitonin as revealed by site-directed 13C solid state NMR spectroscopy. Magn Reson Chem. 2004;42:247–257. doi: 10.1002/mrc.1323. [DOI] [PubMed] [Google Scholar]
- 33.Kamihira M, Oshiro Y, Tuzi S, Nosaka AY, Saito H, Naito A. Effect of electrostatic interaction on fibril formation of human calcitonin as studied by high resolution solid state 13C NMR. J Biol Chem. 2003;278:2859–2865. doi: 10.1074/jbc.M205285200. [DOI] [PubMed] [Google Scholar]
- 34.Kamihira M, Naito A, Tuzi S, Nosaka AY, Saito H. Conformational transitions and fibrillation mechanism of human calcitonin as studied by high-resolution solid state 13C NMR. Protein Sci. 2000;9:867–877. doi: 10.1110/ps.9.5.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siemer AB, Ritter C, Ernst M, Riek R, Meier BH. High-resolution solid state NMR spectroscopy of the prion protein HET-s in its amyloid conformation. Angew Chem Int Ed. 2005;44:2441–2444. doi: 10.1002/anie.200462952. [DOI] [PubMed] [Google Scholar]
- 36.Booth DR, Sunde M, Bellotti V, Robinson CV, Hutchinson WL, Fraser PE, Hawkins PN, Dobson CM, Radford SE, Blake CCF, Pepys MB. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature. 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 37.Guijarro JI, Sunde M, Jones JA, Campbell ID, Dobson CM. Amyloid fibril formation by an SH3 domain. Proc Natl Acad Sci U S A. 1998;95:4224–4228. doi: 10.1073/pnas.95.8.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson CM. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci U S A. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pertinhez TA, Bouchard ML, Tomlinson EJ, Wain R, Ferguson SJ, Dobson CM, Smith LJ. Amyloid fibril formation by a helical cytochrome. FEBS Lett. 2001;495:184–186. doi: 10.1016/s0014-5793(01)02384-5. [DOI] [PubMed] [Google Scholar]
- 40.Fandrich M, Forge V, Buder K, Kittler M, Dobson CM, Diekmann S. Myoglobin forms amyloid fibrils by association of unfolded polypeptide segments. Proc Natl Acad Sci U S A. 2003;100:15463–15468. doi: 10.1073/pnas.0303758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 42.Jayasinghe SA, Langen R. Identifying structural features of fibrillar islet amyloid polypeptide using site-directed spin labeling. J Biol Chem. 2004;279:48420–48425. doi: 10.1074/jbc.M406853200. [DOI] [PubMed] [Google Scholar]
- 43.Perutz MF. Glutamine repeats and inherited neurodegenerative diseases: Molecular aspects. Curr Opin Struct Biol. 1996;6:848–858. doi: 10.1016/s0959-440x(96)80016-9. [DOI] [PubMed] [Google Scholar]
- 44.Macdonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, Macfarlane H, Jenkins B, Anderson MA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sedlacek Z, Poustka A, Frischauf AM, Lehrach H, Buckler AJ, Church D, Doucettestamm L, Odonovan MC, Ribaramirez L, Shah M, Stanton VP, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 45.Bevivino AE, Loll PJ. An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates parallel β-fibrils. Proc Natl Acad Sci U S A. 2001;98:11955–11960. doi: 10.1073/pnas.211305198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DePace AH, Santoso A, Hillner P, Weissman JS. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell. 1998;93:1241–1252. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 47.Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p prion-containing cells. Science. 1995;270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- 48.Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell. 2000;100:277–288. doi: 10.1016/s0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 49.Edskes HK, Wickner RB. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full-length protein. Proc Natl Acad Sci U S A. 2002;99:16384–16391. doi: 10.1073/pnas.162349599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baxa U, Taylor KL, Wall JS, Simon MN, Cheng NQ, Wickner RB, Steven AC. Architecture of Ure2p prion filaments: The N-terminal domains form a central core fiber. J Biol Chem. 2003;278:43717–43727. doi: 10.1074/jbc.M306004200. [DOI] [PubMed] [Google Scholar]
- 51.Urry DW. The change in Gibbs free energy for hydrophobic association: Derivation and evaluation by means of inverse temperature transitions. Chem Phys Lett. 2004;399:177–183. [Google Scholar]
- 52.Sandberg L, Edholm O. Calculated solvation free energies of amino acids in a dipolar approximation. J Phys Chem B. 2001;105:273–281. [Google Scholar]
- 53.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perutz MF, Staden R, Moens L, Debaere I. Polar zippers. Curr Biol. 1993;3:249–253. doi: 10.1016/0960-9822(93)90174-m. [DOI] [PubMed] [Google Scholar]
- 55.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci U S A. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perutz MF, Pope BJ, Owen D, Wanker EE, Scherzinger E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid β-peptide of amyloid plaques. Proc Natl Acad Sci U S A. 2002;99:5596–5600. doi: 10.1073/pnas.042681599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu YS, Gotte G, Libonati M, Eisenberg D. A domain-swapped RNAse A dimer with implications for amyloid formation. Nat Struct Biol. 2001;8:211–214. doi: 10.1038/84941. [DOI] [PubMed] [Google Scholar]
- 58.Yoder MD, Lietzke SE, Jurnak F. Unusual structural features in the parallel β-helix in pectate lyases. Structure. 1993;1:241–251. doi: 10.1016/0969-2126(93)90013-7. [DOI] [PubMed] [Google Scholar]
- 59.Jenkins J, Shevchik VE, Hugouvieux-Cotte-Pattat N, Pickersgill RW. The crystal structure of pectate lyase Pel9a from Erwinia chrysanthemi. J Biol Chem. 2004;279:9139–9145. doi: 10.1074/jbc.M311390200. [DOI] [PubMed] [Google Scholar]
- 60.Thakur AK, Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci U S A. 2002;99:17014–17019. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wickner RB. URE3 as an altered Ure2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 62.Edskes HK, Gray VT, Wickner RB. The URE3 prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci U S A. 1999;96:1498–1503. doi: 10.1073/pnas.96.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lacroute F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J Bacteriol. 1971;106:519–522. doi: 10.1128/jb.106.2.519-522.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Umland TC, Taylor KL, Rhee S, Wickner RB, Davies DR. The crystal structure of the nitrogen regulation fragment of the yeast prion protein Ure2p. Proc Natl Acad Sci U S A. 2001;98:1459–1464. doi: 10.1073/pnas.041607898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taylor KL, Cheng NQ, Williams RW, Steven AC, Wickner RB. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science. 1999;283:1339–1343. doi: 10.1126/science.283.5406.1339. [DOI] [PubMed] [Google Scholar]
- 66.Baxa U, Speransky V, Steven AC, Wickner RB. Mechanism of inactivation on prion conversion of the Saccharomyces cerevisiae Ure2 protein. Proc Natl Acad Sci U S A. 2002;99:5253–5260. doi: 10.1073/pnas.082097899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kajava AV, Baxa U, Wickner RB, Steven AC. A model for Ure2p prion filaments and other amyloids: The parallel superpleated β-structure. Proc Natl Acad Sci U S A. 2004;101:7885–7890. doi: 10.1073/pnas.0402427101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baxa, U., Cheng, N., Winkler, D.C., Chiu, T., Davies, D.R., Sharma, D., Inouye, H., Kirschner, D.A., Wickner, R.B., and Steven, A.C. (2005) Filaments of the Ure2p prion protein have a cross-β core structure, J. Struct. Biol. in press [DOI] [PubMed] [Google Scholar]
- 69.Kishimoto A, Hasegawa K, Suzuki H, Taguchi H, Namba K, Yoshida M. β-helix is a likely core structure of yeast prion Sup35 amyloid fibers. Biochem Biophys Res Commun. 2004;315:739–745. doi: 10.1016/j.bbrc.2004.01.117. [DOI] [PubMed] [Google Scholar]
- 70.Sieber P, Riniker B. Protection of carboxamide functions by the trityl residue: Aapplication to peptide-synthesis. Tetrahedron Lett. 1991;32:739–742. [Google Scholar]
- 71.Ishii Y, Balbach JJ, Tycko R. Measurement of dipole-coupled lineshapes in a many-spin system by constant-time two-dimensional solid state NMR with high-speed magic-angle spinning. Chem Phys. 2001;266:231–236. [Google Scholar]
- 72.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J Magn Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 73.Jaroniec CP, Tounge BA, Herzfeld J, Griffin RG. Frequency selective heteronuclear dipolar recoupling in rotating solids: Accurate 13C-15N distance measurements in uniformly 13C, 15N-labeled peptides. J Am Chem Soc. 2001;123:3507–3519. doi: 10.1021/ja003266e. [DOI] [PubMed] [Google Scholar]
- 74.Wagner A, Luger P. Charge density and topological analysis of L-glutamine. J Molec Struct. 2001;595:39–46. [Google Scholar]
- 75.Sorensen OW, Eich GW, Levitt MH, Bodenhausen G, Ernst RR. Product operator formalism for the description of NMR pulse experiments. Prog Nucl Magn Reson Spectrosc. 1983;16:163–192. [Google Scholar]
- 76.Gullion T, Baker DB, Conradi MS. New, compensated Carr-Purcell sequences. J Magn Reson. 1990;89:479–484. [Google Scholar]
- 77.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–6958. [Google Scholar]
- 78.Petkova AT, Tycko R. Sensitivity enhancement in structural measurements by solid state NMR through pulsed spin locking. J Magn Reson. 2002;155:293–299. doi: 10.1006/jmre.2002.2519. [DOI] [PubMed] [Google Scholar]
- 79.Oas TG, Hartzell CJ, McMahon TJ, Drobny GP, Dahlquist FW. The carbonyl 13C chemical-shift tensors of five peptides determined from 15N dipole-coupled chemical shift powder patterns. J Am Chem Soc. 1987;109:5956–5962. [Google Scholar]
- 80.Koradi R, Billeter M, Wuthrich K. Molmol: A program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 81.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C, and 15N random coil NMR chemical shifts of the common amino acids. 1 Investigations of nearest-neighbor effects. J Biomol NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 82.Saito H. Conformation-dependent 13C chemical shifts: A new means of conformational characterization as obtained by high-resolution solid state 13C NMR. Magn Reson Chem. 1986;24:835–852. [Google Scholar]
- 83.Spera S, Bax A. Empirical correlation between protein backbone conformation and Cα and Cβ 13C nuclear magnetic resonance chemical shifts. J Am Chem Soc. 1991;113:5490–5492. [Google Scholar]
- 84.Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 85.Long HW, Tycko R. Biopolymer conformational distributions from solid state NMR: α-helix and 310-helix contents of a helical peptide. J Am Chem Soc. 1998;120:7039–7048. [Google Scholar]
- 86.Weliky DP, Bennett AE, Zvi A, Anglister J, Steinbach PJ, Tycko R. Solid state NMR evidence for an antibody-dependent conformation of the V3 loop of HIV-1 gp120. Nat Struct Biol. 1999;6:141–145. doi: 10.1038/5827. [DOI] [PubMed] [Google Scholar]
- 87.Sharpe S, Kessler N, Anglister JA, Yau WM, Tycko R. Solid state NMR yields structural constraints on the V3 loop from HIV-1 gp120 bound to the 447–52d antibody Fv fragment. J Am Chem Soc. 2004;126:4979–4990. doi: 10.1021/ja0392162. [DOI] [PubMed] [Google Scholar]
- 88.Diaz-Avalos R, Long C, Fontano E, Balbirnie M, Grothe R, Eisenberg D, Caspar DLD. Cross-β order and diversity in nanocrystals of an amyloid-forming peptide. J Mol Biol. 2003;330:1165–1175. doi: 10.1016/s0022-2836(03)00659-4. [DOI] [PubMed] [Google Scholar]
- 89.Tycko R, Ishii Y. Constraints on supramolecular structure in amyloid fibrils from two-dimensional solid state NMR spectroscopy with uniform isotopic labeling. J Am Chem Soc. 2003;125:6606–6607. doi: 10.1021/ja0342042. [DOI] [PubMed] [Google Scholar]
- 90.Jaroniec CP, MacPhee CE, Astrof NS, Dobson CM, Griffin RG. Molecular conformation of a peptide fragment of transthyretin in an amyloid fibril. Proc Natl Acad Sci U S A. 2002;99:16748–16753. doi: 10.1073/pnas.252625999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ross ED, Baxa U, Wickner RB. Scrambled prion domains form prions and amyloid. Mol Cell Biol. 2004;24:7206–7213. doi: 10.1128/MCB.24.16.7206-7213.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guo JT, Wetzel R, Ying X. Molecular modeling of the core of Aβ amyloid fibrils. Proteins. 2004;57:357–364. doi: 10.1002/prot.20222. [DOI] [PubMed] [Google Scholar]
- 93.Ma BY, Nussinov R. Stabilities and conformations of Alzheimer’s β-amyloid peptide oligomers (Aβ16–22, Aβ16–35, and Aβ10–35): Sequence effects. Proc Natl Acad Sci USA. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smith CK, Withka JM, Regan L. A thermodynamic scale for the β-sheet forming tendencies of the amino acids. Biochemistry. 1994;33:5510–5517. doi: 10.1021/bi00184a020. [DOI] [PubMed] [Google Scholar]
- 95.Minor DL, Kim PS. Measurement of the β-sheet-forming propensities of amino acids. Nature. 1994;367:660–663. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]