

Abstract
All G protein-coupled receptors (GPCRs) share a common seven TM helix architecture and the ability to activate heterotrimeric G proteins. Nevertheless, these receptors have widely divergent sequences with no significant homology. We present a detailed structure–function comparison of the very divergent Class A and D receptors to address whether there is a common activation mechanism across the GPCR superfamily. The Class A and D receptors are represented by the vertebrate visual pigment rhodopsin and the yeast α-factor pheromone receptor Ste2, respectively. Conserved amino acids within each specific receptor class and amino acids where mutation alters receptor function were located in the structures of rhodopsin and Ste2 to assess whether there are functionally equivalent positions or regions within these receptors. We find several general similarities that are quite striking. First, strongly polar amino acids mediate helix interactions. Their mutation generally leads to loss of function or constitutive activity. Second, small and weakly polar amino acids facilitate tight helix packing. Third, proline is essential at similar positions in transmembrane helices 6 and 7 of both receptors. Mapping the specific location of the conserved amino acids and sites of constitutively active mutations identified conserved microdomains on transmembrane helices H3, H6, and H7, suggesting that there are underlying similarities in the mechanism of the widely divergent Class A and Class D receptors.
G protein-coupled receptors (GPCRs)1 are the largest and most diverse superfamily of membrane receptors. GPCRs are characterized by their common 7 transmembrane (TM) helix architecture and their ability to activate heterotrimeric G proteins. Surprisingly, there is no significant sequence similarity across the GPCR superfamily. In fact, GPCRs have been grouped into at least five distinct classes (1) with no recognizable sequence similarity between the various classes (1, 2). Nevertheless, the working assumption has been that these receptors have a common fold and activation mechanism. For example, the amino acid sequences of the yeast pheromone GPCRs, such as the α-factor receptor, diverge considerably from those of the mammalian hormone GPCRs, such as the β-adrenergic receptor. Yet, in both of these receptors, ligand binding occurs within the core of the 7 TM helices and is, in part, mediated by aromatic amino acids on transmembrane helix H6 (3–5). In both receptors, the third intracellular loop has been implicated in G protein activation (6–8), and the cytoplasmic C-terminus, which is not required for signaling, instead acts as a negative regulatory domain that is a target for desensitization by phosphorylation and ligand-mediated down-regulation by receptor endocytosis (9, 10). There is direct evidence for a conserved mechanism in that the α-factor receptor can activate mammalian Gαolf subunits (11), and certain mammalian GPCRs can activate the pheromone-responsive G protein pathway in yeast (12, 13).
Despite their shared structural and functional properties, recent evidence has suggested that the mechanisms of activation may not be conserved across the GPCR superfamily. One line of evidence for this comes from the analysis of receptors with constitutively active mutations (CAMs). These receptors have been widely used to elucidate the mechanism of GPCR activation (14, 15). CAMs are thought to disrupt interactions that stabilize the inactive state of GPCRs or strengthen the interactions that stabilize the active state. The different ways CAMs activate GPCRs and their different interactions with agonists, antagonists, and inverse agonists have led to the conclusion that GPCRs have the potential to adopt multiple activated conformational states (14, 15). Each of these states may have different affinities for ligands, G-proteins, and the proteins that control receptor internalization (16–19). The existence of many active conformations has raised the question as to whether there is a common mechanism of activation for all GPCRs.
To search for common aspects of GPCR structure and function, we compare two well-studied prototypes of divergent classes: the prototypical Class A GPCR rhodopsin and the Class D α-factor receptor (Ste2) from Saccharomyces cerevisiae (Figure 1). In addition to rhodopsin, Class A includes many other medically important GPCRs such as the serotonin and adrenergic receptors. Rhodopsin was selected for this study because it is one of the most intensively studied GPCRs. The crystal structure of the dark (inactive) state of rhodopsin has been determined (20, 21) and provides a framework for understanding the wide range of biochemical and mutational data that has been amassed. Together, the structural and functional data on rhodopsin and other Class A receptors have provided the basic outlines of a common activation mechanism, at least for this receptor class. A central part of this mechanism involves the role of key “signature” amino acids that are highly conserved (>80%) and are mostly found in the TM helices. These amino acids mediate helix–helix interactions, which lock the receptor in an inactive conformation. Binding of ligand (or isomerization of the retinal chromophore in rhodopsin) is thought to alter the interactions between helices, allowing motion of TM helices H5, H6, and H7 (22, 23). This leads to a change in the structure or interactions in the third cytoplasmic loop which bridges H5 and H6 (24).
Figure 1.
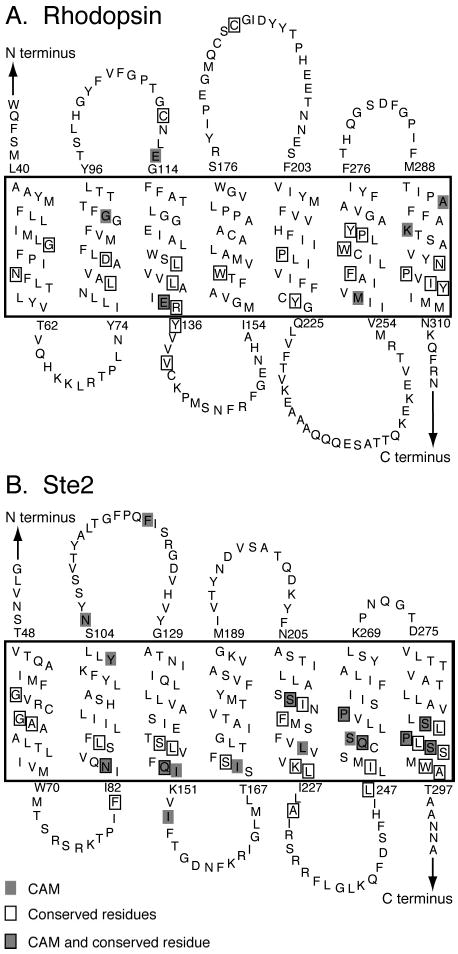
Secondary structural model of rhodopsin (A) and Ste2 (B) showing the position of conserved (boxed) and constitutively active (shaded) amino acids. The transmembrane regions are enclosed in the large boxes. The topology diagrams are oriented so that the extracellular regions are shown above and the intracellular regions are shown below.
Ste2 (or Ste2p) is a member of the Class D GPCRs (1, 25). Class D receptors have two major subfamilies: Ste2 and Ste3. Only the relatively newly discovered group of 7 TM receptors that includes the Drosophila Frizzled and Smoothened receptors are thought to be more divergent from rhodopsin (26). To emphasize the diversity of the Class D receptors, there is even no obvious sequence similarity between the Ste2 and Ste3 subfamilies (discussed below). The Ste3 receptors are present on cells of opposite mating type from those that produce Ste2, yet both types of pheromone receptors activate the same G protein signal pathway. The Class D receptors do not have many of the features characteristic of the consensus Class A GPCRs. In particular, they lack the ERY or DRY sequence on H3, the NPxxY sequence on H7, and the disulfide bridge between the extracellular end of H3 and extracellular loop 2 (EL2). Also, whereas intracellular loop 2 (IL2) is essential for G protein interaction in many GPCRs (27–29), the bulk of it can be removed in Ste2 (30). Ste2 also differs from rhodopsin in that its ligand is a small peptide, which binds to the extracellular loops and ends of the TM helices. However, as in the case of rhodopsin, a wealth of biochemical, genetic, and mutational data has been obtained for Ste2 (4, 31–36). Moreover, genome sequencing efforts have revealed over 20 related receptors that provide information on sequence conservation within the Ste2 subfamily.
In view of the lack of obvious sequence homology, several strategies were used to facilitate comparison of these well-characterized representatives of Class A and D GPCRs. First, the available structural and mutational data were used as a guide to align the Ste2 sequence with rhodopsin. Multiple sequence alignments with evolutionarily related receptors were constructed to identify conserved motifs that likely represent residues important in receptor structure and function. In these comparisons, special emphasis was given to amino acid subgroups with similar structural or functional roles in membrane proteins. For instance, strongly polar amino acids in the TM regions were highlighted since they generally mediate helix interactions rather than face the hydrophobic membrane lipids. Positions where there is conservation of residues with small and/or weakly polar side chains (i.e., Gly, Ala, Ser, Thr, and Cys) are highlighted. Although they do not show up with high identities in sequence comparisons, this group of residues permit close interactions that can stabilize packing between TM helices (37). Second, helical hydrophobic and packing moments were calculated for the predicted TM helices of Ste2 and compared with those of rhodopsin. The helical packing moment is a new method for predicting likely sites of TM helix interaction (38). Together, the sequence and packing moment analyses were used to generate a three-dimensional model of Ste2. The rhodopsin and Ste2 structures were evaluated using the mutational data for both receptors. Particularly important were positions where mutation results in loss of function or constitutive activity. Mapping the location of the conserved amino acids and the residues that exhibit constitutive activity when mutated suggests that there are conserved microdomains on transmembrane helices H3, H6, and H7 with similar functions in both receptor classes. These studies are the first step in a comprehensive analysis of the large GPCR superfamily.
MATERIALS AND METHODS
Multiple Sequence Alignments of Receptors
Receptor sequences were taken from the GPCR database (25) for the Class A family and the Ste3-subfamily or downloaded for the Ste2-subfamily from GenBank (39) as annotated sequences or, in the case of some pheromone receptor sequences, as open reading frames identified in the genome of partially sequenced organisms (40, 41). Initial alignments were made using CLUSTALW and then were transferred into a representation of the 7 TM topology of receptors to facilitate comparison (Supporting Information). Figure 2 shows the alignment of rhodopsin, Ste2, and Ste3 of S. cerevisiae together with the opsin, Class A, Ste2-subfamily, and Ste3-subfamily consensus sequences.
Figure 2.
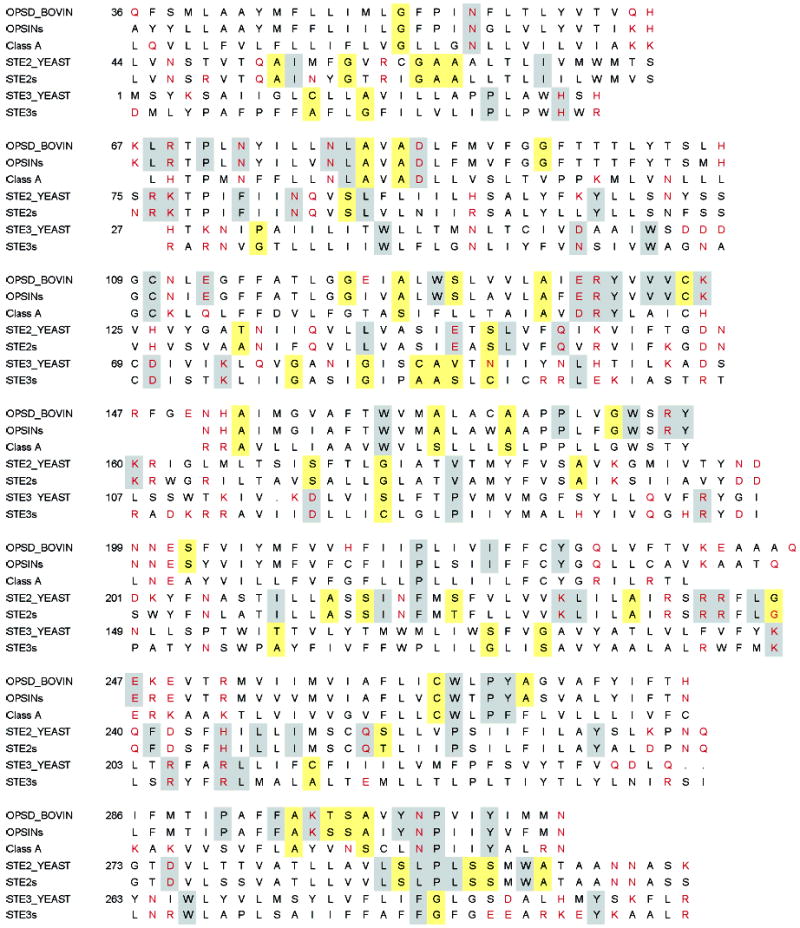
Sequence alignments of rhodopsin and the Ste2 and Ste3 receptors of S. cerevisiae. The consensus sequences of the opsin subfamily, Class A family, Ste2 subfamily, and Ste3 subfamily are shown for comparison. Strongly polar residues are in red. Gray boxes denote identity, and yellow boxes denote small and weakly polar group-conserved residues.
The sequences for the Ste2 subfamily of pheromone receptors are identified by the initials of the genus and species of the corresponding organism as follows: Sc, S. cerevisiae; Sb, S. bayanus; Sca, S. castellii; Sk, S. kluyveri; Su, S. unisporus; Km, Kluyveromyces marxianus; Kl, K. lactis; Kw, K. waltii; Eg, Eremothecium gossypii; Cg, Candida glabrata; Yl, Yarrowia lipolytica; Pa, Pichia angusta; Dh, Debaryomyces hansenii; Zr, Zygosaccharomyces rouxii; Ca, C. albicans; Sp, Schizosaccharomyces pombe; Af, Aspergillus fumigatus; An, A. nidulans; Cp, Coccidioides posadasii; Mg, Magnaporthe grisea; Gz, Gibberella zeae; Fv, Fusarium virguliforme; Sm, Sordaria macrospora; Nc, Neurospora crassa.
Hydrophobic Moment and Helical Packing Moment Analysis
The hydrophobic moments were calculated according to Eisenberg et al. (42). The scale for calculating hydrophobic moments was taken from Engelman, Goldman, and Steitz (43).
The helix packing moment analysis was carried out as described previously (38). Briefly, packing moments provide a measure of the propensity for three specific amino acids to lie in a helical interface. Each moment corresponds to a 40° arc on a helical wheel diagram. The 40° range captures 3 amino acids separated by 1, 2, or 3 helical turns. In the helical wheel diagrams shown below, only the strongest helix packing moments are displayed with a cutoff of 0.452 (38). Using the sequence alignments for Class A and Class D receptors, we also calculated the average packing moments for the opsin, Ste2, and Ste3 subfamilies. The sequence alignments allowed us to identify the positions in the TM helices that are strictly conserved and group-conserved across GPCR families and subfamilies. For the analysis of group conservation, the amino acids with similar functional properties were categorized as follows: small and weakly polar (G, A, S, C, T), strongly polar (D, N, E, Q, H, K, R), aromatic (F, W, Y), or hydrophobic (I, L, M, F, V). On the basis of the helical hydrophobic and packing moments, as well as the position of the conserved and group-conserved amino acids, it is possible to predict how each face of a helical wheel diagram is oriented relative to the other TM helices. A similar type of analysis was previously used by Baldwin and co-workers (44, 45) to propose a model of Class A GPCRs before the high-resolution crystal structure was solved.
Structural Modeling of Ste2
A structural model of Ste2 was constructed by first manually aligning the ends of the transmembrane sequences of Ste2 with those of rhodopsin. The TM helix boundaries for rhodopsin were identified from the rhodopsin crystal structure (20) based on the position of the polar residues that bracket the hydrophobic core and from solvent accessibility data (24, 46–48). Boundaries for Ste2 were inferred from the solvent accessibility of residues near the plasma membrane interface (4, 36). The Rho-Ste2 alignment was refined on the basis of the helical wheel analysis described above.
On the basis of the Ste2-rhodopsin alignment, the program Modeler (49, 50) implemented in the Insight II package (Molecular Simulations, San Diego, CA) generated a homology model. The resulting structure was further refined with the use of the all-atom force-field ff94 (51) and Amber simulation package (Case, D. A., Darden, T. A., Cheatham, T. E., III, Simmerling, C. L., Wang, J., Duke, R. E., Luo, R., Merz, K. M., Wang, B., Pearlman, D. A., Crowley, M., Brozell, S., Tsui, V., Gohlke, H., Mongan, J., Hornak, V., Cui, G., Beroza, P., Schafmeister, C., Caldwell, J. W., Ross, W. S., and Kollman, P. A. (2004), AMBER 8, University of California, San Francisco). We used simulated annealing with restrained molecular dynamics in order to refine the position of the protein side chains. First, a short (100 ps) heating from 300 to 550 K was performed, followed by 50 ps of dynamics at 550 K. A set of six random structures was collected from the latter stage. These structures were slowly cooled to 100 K during much longer (10–50 ns) molecular dynamics runs. All simulations were run in a vacuum with a constant dielectric of 4 to simulate the nonpolar membrane environment. Because only the transmembrane region was modeled (with the loop regions being truncated), weak positional restraints (0.5 kcal/mol Å) were applied on the alpha carbons in the initial model.
RESULTS
Location and Role of Conserved Amino Acids in Rhodopsin
Evolutionarily conserved amino acids are predicted to play a key role in the structure and function of GPCRs. Figure 1A presents a topographical representation of the rhodopsin sequence that highlights the location of the highly conserved amino acids (boxed) and sites where mutation can lead to constitutive receptor activation (shaded). The conserved residues were identified in a multiple sequence alignment of all Class A GPCRs (25). Figure 2 presents the sequence for bovine rhodopsin along with the consensus sequences for the Class A receptors and opsin subfamily. This presentation can be used to distinguish residues that are conserved only within the visual pigment family. The putative function of these amino acids was assessed by analysis of the hydrophobic moments and the helical packing moments (38) of their respective helix. The helical wheel representation of rhodopsin, the opsin subfamily, and Class A family showing the orientation of the helical hydrophobic moment along with helical packing moments are shown in Figure 3A–C. For rhodopsin (A), occluded (solvent inaccessible) and exposed (solvent accessible) sites are indicated by open circles and closed circles, respectively, based on the high-resolution structure.
Figure 3.
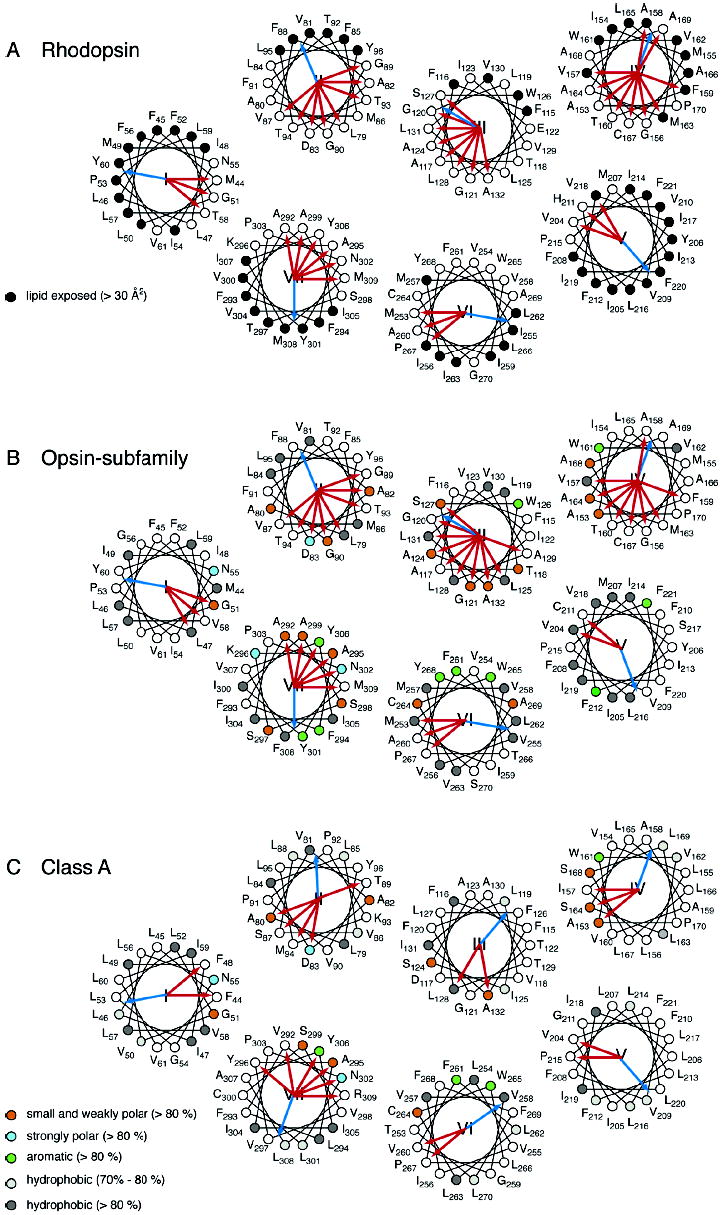
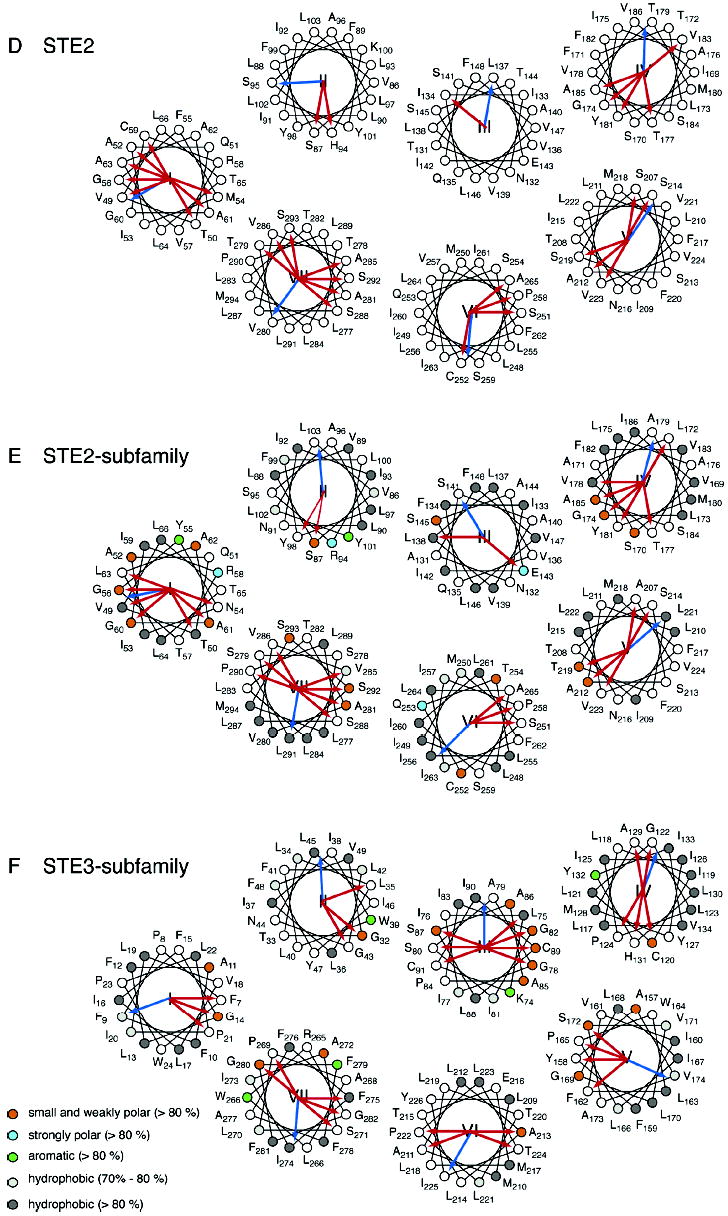
Helical wheel diagrams of G protein-coupled receptors. The helical wheel diagrams show the hydrophobic moment (blue arrow) (42) and helical packing moments (red arrow) (38) for rhodopsin (A), the opsin subfamily (B), the Class A family (C), the Ste2 receptor (D), the Ste2 subfamily (E), and the Ste3 subfamily (F). For panels A–C, the positions are numbered as in bovine rhodopsin; for panels D and E, the positions are numbered as in Ste2, and in panel F, the positions are numbered as in Ste3. The sites that are group-conserved (>80%) across a family or subfamily are colored as follows: small and weakly polar (orange), strongly polar (azure), aromatic (green), and hydrophobic (dark gray). The hydrophobic sites with group conservation between 70% and 80% are light gray. For rhodopsin (A), occluded (solvent inaccessible) and exposed (solvent accessible) residues are indicated by open circles and closed circles, respectively, based on the high-resolution structure. Note that H2 in the Ste2 subfamily (D) has no helical packing moment according to our definition (38). The two strongest moments are shown as thin red arrows.
In addition to the highly conserved “signature” residues highlighted in Figures 1 and 2, we also identified for the opsin subfamily (B) and Class A family (C) the positions that are group-conserved as small and weakly polar (G, A, S, C, T), strongly polar (D, N, E, Q, H, K, R), aromatic (F, W, Y), or hydrophobic (I, L, M, F. V). Of particular interest are the group-conserved amino acids that are small and weakly polar (Gly, Ala, Ser, Thr, and Cys). These residues have low individual sequence identities, but are highly conserved when considered as a group. The small and weakly polar amino acids have high propensities for mediating helix–helix interactions (37). The key amino acids that show group-conservation across >80% of the Class A GPCR family are Gly51, Ala80, Ala82, Ala124, Ala132, Ala153, Ala164, Ala168, Cys264, Ala295, and Ala299 (Figures 2 and 3C). Consideration of the group-conserved amino acids has led to the proposal that helices H1 through H4 are locked in a stable structure that does not change upon receptor activation. H5, H6, and H7 have multiple contacts each with the H1–H4 core of the protein which lock the receptor in the inactive state (38). Retinal isomerization and translation contribute to disrupting these interactions (23).
For each of the seven TM helices in rhodopsin, we briefly discuss the location and potential role of the conserved and the group-conserved amino acids. For those amino acids where mutation can lead to constitutive activity, we describe how specific mutations might destabilize the inactive state of the receptor or stabilize the active state.
H1
Asn55 points to the center of the helical bundle and is roughly opposite the hydrophobic moment of H1 (Figure 3A–C). It is strictly conserved across the Class A GPCRs. Asn55 hydrogen-bonds to the backbone carbonyl of Ala299 on H7 and to the side chain of Asp83 on H2. The small side chains of Gly51 and Ala299, which are both group-conserved and part of the 40° arc of at least one packing moment (Figure 3A–C), allow close packing of H1 and H7 and facilitate this hydrogen-bonding interaction. In rhodopsin, mutation of Gly51 leads to retinitis pigmentosa (52), a disease associated with misfolding of the protein (53).
H2
Asp83 on H2 is highly conserved and part of the hydrogen-bonding network involving Asn55 on H1 and the backbone carbonyl of A299 on H7. The H1–H2 contact is facilitated by a group-conserved small amino acid, Ala80 (Figure 3C).
A second hydrogen-bonding network mediates interactions between H2, H3, and H4. The side chain of Asn78 (same position as Y96 in the helical wheel of Figure 3A–C) on H2 forms an interhelical hydrogen bond with the indole nitrogen of Trp161 on H4 (Figure 4A,C). Trp161 is conserved in 97% of Class A receptors. A polar amino acid at position 78 (Asn 54%, Ser 31%, His 7%) is also highly conserved. The bulky Trp side chain lies between H2 and H4 and effectively locks H3 into the helical bundle. There are several group-conserved amino acids that allow close helix packing of H2, H3, and H4 and, consequently, facilitate this interhelical hydrogen bond. These include Ala82 on H2 and Ala153, Ala164, and Ala168 on H4 (Figure 3A–C). These amino acids contribute to the packing moments observed in Figure 3A–C.
Figure 4.
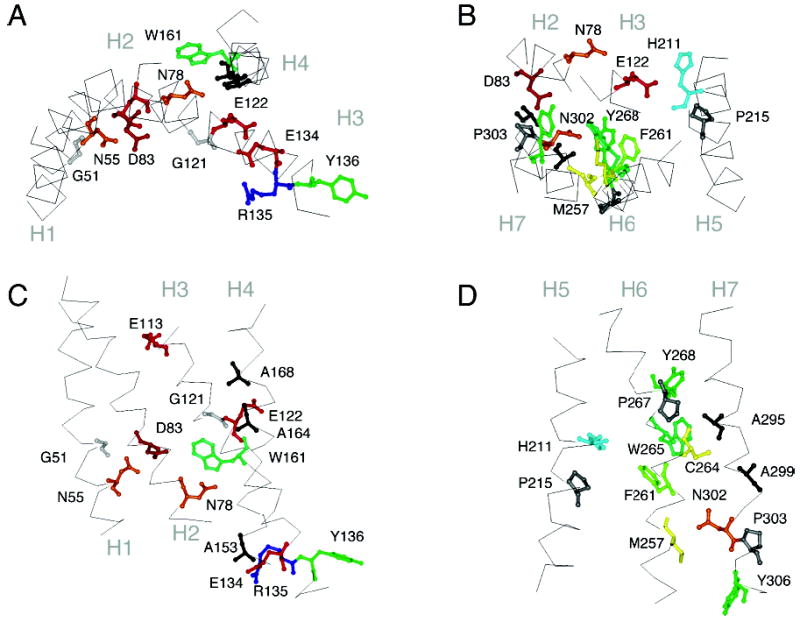
Crystal structure of rhodopsin. Panels A and C highlight the position of several of the signature and group-conserved amino acids in helices H1 to H4 in rhodopsin. Panels B and D highlight the position of several of the signature and group-conserved amino acids in helices H5, H6, and H7.
H3
H3 is the most buried TM helix in rhodopsin (Figure 3A) and has direct or water-mediated contacts with each of the other six TM helices. It has a high number of both conserved amino acids and sites where mutation leads to constitutive activity (Figure 1A). In the Class A–C receptors, there is a conserved Cys near the extracellular border of H3 that forms a disulfide link with the second extracellular loop (54, 55). These highly conserved cysteines (~92%) are not found in the Class D receptors. Also, at the extracellular end of H3, are residues that are known to interact with the retinal chromophore in rhodopsin (Glu113) (56) or the ligand in the biogenic amine receptors (Asp113) (55).
H3 in rhodopsin is also unusual in having a glutamic acid in the middle of the TM helix. Glu122 is conserved pairwise with His211 on H5. The Glu–His pair (Figure 4B) is responsible for the high sensitivity of the rhodopsin-like receptors and is not found in the cone pigments (57).
At the intracellular end of H3 is a conserved ERY or DRY sequence which is involved in interhelical contacts that are thought to lock the receptor off in the dark (27, 58–62) (Figure 4A,C). Mutation of Glu134 to Gln results in constitutive activation of the receptor (63, 64). Glu134 interacts electrostatically with Arg135, which in turn hydrogen-bonds with Glu247 and Thr251 (20). The hydrophobic sequence (VVV) immediately following the ERY sequence is also highly conserved. Ballesteros et al. (60) introduced the concept of a structural motif as a functional microdomain in describing these hydrophobic amino acids as a cage which limits the accessibility of Arg135 to bulk solvent.
H4
H4 is predicted to be part of the stable core of the Class A GPCRs (38). It has one highly conserved amino acid, Trp161, which hydrogen-bonds with Asn78 on H2, and effectively locks H3 within the helix bundle. While H4 has several strong helical packing moments that are oriented toward helix interfaces, there are two packing moments that align with the helical hydrophobic moment (Figure 3A). These packing moments are consistent with the intradimeric contact found by atomic force microscopy of native rod outer segment membranes (65). However, these packing moments are not conserved across the Class A family (Figure 3C), indicating that dimerization motifs are subfamily-specific and may not be present in all Class A receptors.
H5
H5 has been shown to be involved in receptor activation in a number of the Class A GPCRs. In rhodopsin, Glu122 on H3 is hydrogen-bonded to the backbone carbonyl of His211 on H5. Receptor activation causes a rearrangement of His211 hydrogen bonding (66). In the β-adrenergic receptors, there are two serines that interact with hydroxyl groups on the amine ligand and are critical to receptor activation (67). One serine is at the position of the His211 in rhodopsin, and the other serine is one helical turn away. H5 has few conserved amino acids other than a proline (Pro215) in the middle of the helix and a tyrosine (Tyr223) at the intracellular end of the helix.
H6
Our helix packing moment analysis suggests that H6 in Class A GPCRs is not designed to interact with H3 and H5 other than at the cytoplasmic boundary of the helices (Figure 3A–C). The strongest interhelical interactions are with H7 and involve Met257, Cys264, and Trp265. Met257 lies in the H6–H7 interface at the level of the conserved NPxxY sequence in H7 (see below). Mutation of Met257 in rhodopsin results in constitutive activity (68).
EPR studies have shown that H6 undergoes the largest displacement of the seven TM helices upon light-activation in rhodopsin (22). H6 has several conserved aromatic amino acids (Phe261, Trp265, and Tyr268) that interact with the retinal chromophore in rhodopsin and ligands in other Class A GPCRs (69, 70). Pro at position 267 in rhodopsin is one of the most highly conserved amino acids in GPCRs and is thought to facilitate rigid body motion of H6 (71).
H7
The key role of H7 in the activation mechanism of Class A GPCRs is reflected in the number of conserved amino acids and positions where mutation leads to constitutive activity. Furthermore, it has the most helix packing moments in the Class A receptors (Figure 3C). In the rhodopsin crystal structure, H7 has two distinct regions separated near a kink at Pro303. The sequence from Pro290 to Tyr301 appears to be ligand-specific and is only conserved within subfamilies of Class A GPCRs (72). In contrast, the sequence from Asn302 to Tyr306 is highly conserved across the Class A receptors, constituting the signature NPxxY motif.
Location and Role of Conserved Residues in Ste2
An overview of the positions of highly conserved amino acids and sites where mutation leads to constitutive receptor activity is shown on the model of Ste2 in Figure 1B. The sites of mutations that cause constitutive receptor activity, particularly those with strong phenotypes, are clustered on H3, H6, and H7 (Figure 1B and Supporting Information). The most highly conserved residues are clustered on H5 and H7. The conserved residues were identified by comparison of over 20 Ste2 sequences from different fungi as shown in the multiple sequence alignment in Figure 1 of the Supporting Information. There are several strictly conserved and many group-conserved amino acids. However, comparison of the rhodopsin (Class A) and Ste2 (Class D) sequences in Figure 2 shows that there are no significant identities.
To compare the position of the critical residues in Ste2 with rhodopsin, we assessed the likely orientation of the TM helices by analysis of the hydrophobic moments and the helical packing moments (38). Helical wheel representations of the Ste2 receptor along with the Ste2 and Ste3 subfamilies are presented in Figure 3D–F and show the orientation of the helical hydrophobic moments along with helical packing moments. As for the opsin subfamily and Class A family, the multiple sequence alignments for the Ste2 and Ste3 subfamilies allowed us to identify the conserved and group-conserved positions in the TM helices. These are represented on the helical wheel diagrams. A three-dimensional model of Ste2 developed on the basis of the available structural and mutational data provides a tool for discussing the strictly conserved and group-conserved amino acids in the TM helices (see also Tables 1 and 2 in the Supporting Information).
H1
There are no conserved Asn residues on H1 of Ste2 that correspond to Asn55 in rhodopsin. However, Arg58 in Ste2 is striking because it is unusual to find a charged or strongly polar amino acid in the middle of a transmembrane helix. In the Ste2 helical wheel presentation (Figure 3D) and model (Figure 5A,C), Arg58 is oriented inward. Interestingly, although Arg58 is not highly conserved in the Ste2 family, there is a pairwise conservation of polar residues at position 58 on H1 and position 94 on H2. In most of the receptors, an Arg or Lys at position 58 correlates with a His at position 94, whereas Gln at position 58 correlates with Arg at position 94 (Supporting Information). We propose that the position of the positively charged Arg functional group is likely to be in the same spatial position in the structure of Ste2 whether it occurs at position 58 or 94.
Figure 5.
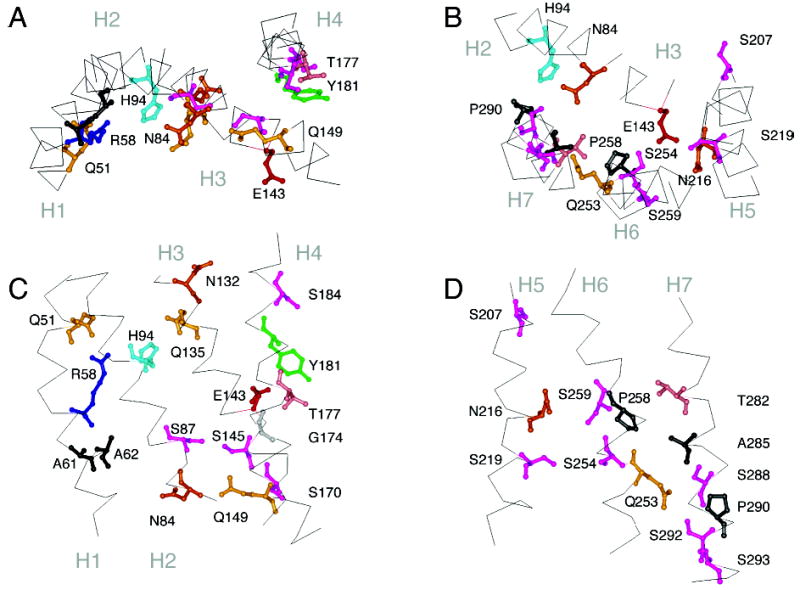
Molecular model of the Ste2 receptor. Panels A and C highlight the position of several of the signature and group-conserved amino acids in helices H1 to H4. Panels B and D highlight the position of several of the signature and group-conserved amino acids in helices H5, H6, and H7. The orientations shown are the same as those in Figure 4 of rhodopsin.
An important question is how Arg58 is accommodated in the interior of the Ste2 helix bundle. The only relatively well-conserved negatively charged amino acid in the TM region of Ste2 is Glu143. However, Glu143 is not conserved pairwise with Arg58, and mutational studies also indicate that they are not likely to form a salt-bridge (76). There is some precedent for Arg in the middle of TM helices without a counterion. These include Arg82 in bacteriorhodopsin (pdb access code 1C3W) and Arg265 in the ABC bacterial transporter for vitamin B12 (BtuCD; pdb access code 1L7V). In bacteriorhodopsin, Arg82 is hydrogen-bonded with three water molecules (77). In the BtuCD transporter, Arg265 is hydrogen-bonded with His262 and Gln78 (78). This is similar to the hydrogen-bond interactions between Arg58, His94, and Gln51 in the structural model of Ste2 (Figure 5).
Analysis of the helical packing moments of H1 identified two conserved moments that are oriented toward the H7 interface (Figure 3D,E). These packing moments are associated with Ala61 and Thr50. Position 61 is an Ala in all of the Ste2 sequences, except for the Kw (Thr), Ca (Ser), and Sp (Leu) homologues of Ste2. Thr50 is not as well-conserved. However, it is often replaced with amino acids that have higher packing values and propensities to occur in helix interfaces (37). For instance, there are several pheromone receptors that have an Asn at this position. The highly polar Asn would not be predicted to have a lipid facing orientation. There are no conserved helix packing moments oriented toward H2, in part because of the occurrence of Arg58, which has a low intrinsic packing value (37). Several relatively well-conserved residues are predicted to be in the H1–H2 interface, including Ala62.
Finally, two conserved glycines (Gly56 and Gly60) and a conserved Ala (Ala52) on H1 are predicted to be oriented outward, away from the interior of the helical bundle. The GxxxG sequence has been identified as a dimerization motif in proteins with single TM helices (79, 80) and has been proposed to mediate dimerization of Ste2 (81). This potential dimerization site is absent in the Ste3 subfamily (Figure 3F).
H2
H2 is predicted to interact with H1, H3, and H7. The most striking feature of H2 is the presence of a large bulky hydrophobic face from Ile83 to Leu93. This face is predicted to be oriented toward the surrounding lipid. The hydrophobic face is opposite to a hydrophilic face which contains several conserved amino acids: Phe81, Asn84, His94, and Tyr98.
His94 may correspond functionally to Asp83 in rhodopsin in mediating an interaction between H2 and H1. As indicated above, the amino acid at position 94 or 58 is typically an Arg with the other position being histidine or glutamine. This suggests that the residues at 58 and 94 are oriented inward and face the same position. A small residue at position 95 (Ser95) in the H1–H2 interface allows His to interact with Arg58 in the structural model of Ste2. Other H1–H2 interactions include two relatively well-conserved large, hydrophobic residues in the H2–H1 interface: Leu88 and Ile92. They bracket conserved Ala62 on H1 as part of a potential leucine zipper interaction.
Of the two strong packing moments on H2, one is associated with Ser87 (Figure 3D). In the structural model of Ste2, Ser87 is located in the H2–H3 interface and hydrogen-bonds to Ser145 on H3. Also in the structural model of Ste2, Asn84 hydrogen-bonds to a highly conserved Ser293 on H7. This interaction is similar to the hydrogen-bonding interaction between Asn73 and Tyr306 in rhodopsin. Mutational studies, however, suggest that Asn84 interacts functionally with Gln149 on H3 (35). Asn84, Ser293, and Gln149 may form a hydrogen-bonding network preventing G protein activation.
The two tyrosines (Tyr98 and Tyr101) on the extracellular end of H2 are relatively well-conserved. Tyr98 is positioned one helical turn from and on the same face as His94. In our model, the side chain of Tyr98 is hydrogen-bonded to the side chain of Gln135 on H3. Mutation of Tyr98 to His results in constitutive activity (35). Tyr101 and Lys100 are facing out toward the lipids and are predicted to be at the level of the polar lipid headgroups.
H3
H3 differs from the other helices in Ste2 in that it contains several highly polar residues. At the intracellular end of H3 is a cluster of residues where mutation leads to constitutive activity (see Figure 1, (35)). Although Ste2 lacks the E/DRY motif at the cytoplasmic end of H3, one of the highly conserved features of Class A GPCRs, H3 is a hotspot for CAMs in both Ste2 and rhodopsin. Gln149 on the cytoplasmic end of H3 in Ste2 may correspond to Glu134 in rhodopsin, which is part of the ERY sequence. Gln149 is adjacent to two conserved Ile residues on H3 similar to hydrophobic patch near the ERY sequence in rhodopsin.
Glu143 is in the middle of H3, in a position similar to Glu122 in rhodopsin and is relatively well-conserved across the Ste2 family. Glu143 interacts with Ser219, Asn216, and Ser254 in a hydrogen-bonding network (Figure 5B).
Gln135 and Asn132 are two strongly polar residues at the extracellular end of H3. In the Ste2 model, Gln135 interacts with His94 and Tyr98. Asn132 is not hydrogen-bonded. Certain mutations affecting Gln135 and Asn132 can cause a dominant-negative phenotype, which indicates that mutation of these sites can interfere with ligand binding or receptor activation (33, 82). There is a parallel role for Glu113 at the extracellular end of H3 in rhodopsin.
H3 has two strong helix packing moments in the Ste2 subfamily (Figure 3E). One is associated with Ser145 and is oriented toward the H2–H3 interface. As mentioned above, Ser145 hydrogen-bonds with Ser87 in the Ste2 model, and mutations affecting either site can cause constitutive signaling (31, 83). The second helix packing moment is associated with Glu143 and is oriented toward the H3–H5 interface.
H4
H4 in Ste2 is similar to rhodopsin in having no strongly polar or charged residues. The sequence alignment with rhodopsin orients positions Tyr181, Ser184, and Gly188, where there are polar residues in related receptors, toward the helix bundle. For instance, arginine and lysine occur at position 181 in the Af, An, Cp, Mg, and Gz homologues of Ste2 and would not be expected to face the lipids. The alignment with rhodopsin also places Ser170, Gly174, and Val178 in the H3–H4 interface. Gly174, a conserved small residue, is predicted to lie at the H3–H4 crossing point opposite Thr144. Glycines typically have a high occurrence at helix crossing points (84). Conserved Ser170 packs against Phe148 (next to Gln149) but does not appear to form an interhelical hydrogen bond. Ser184, a conserved polar residue at the extracellular end of H4, may form an interhelical hydrogen bond with Ser207 on H5. Thr177 and Tyr181 are sites where mutation leads to loss of function (85). Both amino acids are oriented toward the center of the helical bundle between H3 and H5. The side-chain hydroxyl of Tyr181 is hydrogen-bonded to the backbone of Ser214 on H5.
This alignment orients Ser168, Thr172, Ala176, and Thr179 toward the lipids. These residues are associated with helix packing moments (Figure 3D,E) and may represent a second receptor dimerization face.
H5
Asn216 in the middle of H5 defines the rotational orientation of the helix. In the energy minimized structure of Ste2, Asn216 is predicted to hydrogen-bond with Glu143 on H3 and with Ser254 on H6. Substitution of Asn216 and Leu226 with Asp and Trp, respectively, results in constitutive activity (83, 86). Val223 on H5 and Leu247 on H6, when changed to Cys, are able to form a disulfide link (34). Ser214 and Ser219 are both highly conserved and are both predicted to be involved in hydrogen-bond interactions as described above. Mutation of both results in constitutive activity (34).
H6
TM helix H6 is perhaps the most important helix for coupling ligand binding on the extracellular side of GPCRs to a conformational change in the cytoplasmic loops. The proline in the middle of H6 (Pro258) is highly conserved across the GPCR superfamily. Substitution of Pro258 with a variety of different amino acids all cause strong constitutive activity (87, 88). In fact, mutations affecting position 258 are the strongest CAMs that have been identified to date. Substitution of the adjacent Ser259 by leucine can enhance the constitutive activity of Pro258 mutations (87), and a single substitution of Ser259 by a Pro causes constitutive activity (83). Ser259 is in close proximity to Asn216 and may be part of a hydrogen-bonding cluster with Glu143 and Ser254 (Figure 5B). Ser259 and Asn216 show partial pairwise conservation in Ste2 homologues with one or two exceptions (Supporting Information).
The rotational orientations of H6 and H7 in Ste2 are constrained by the interaction of a highly conserved residue, Gln253, with two conserved serines on H7, Ser288 and Ser292. Mutations at positions 253, 288, or 292 that disrupt this interaction result in constitutive receptor activity (89). Gln253, Ser288, and Ser292 are all highly conserved except for the Mg, Gz, Sm, and Nc homologues of Ste2 in which all three positions are no longer conserved.
The orientation of H6 places Ser254 against Ile150 and Ile153 on H3. Substitutions at each of these sites lead to constitutive activity (35). Interestingly, substitution of Ser254 with larger amino acids causes high levels of constitutive activity (89). In a set of roughly comparable studies on rhodopsin, it was found that substitution of Gly121 to larger residues led to activation of rhodopsin in the dark (90). Gly121 is in the H3–H6 interface, and the substitution of larger residues is consistent with an outward motion of H6 during receptor activation.
Finally, Ala265 in the H5–H6 interface of the Ste2 receptor is often a polar residue in other members of the Ste2 family. For instance, Ala265 is substituted with Asn, Gln, His, Asp, or Glu in the Yl, Af, Ca, Cp, Sp, Gz, Sm, or Nc homologues of Ste2. Moreover, there are corresponding polar residues on H5 in these receptors to accommodate hydrogen bonding or ionic interactions with H6. The adjacent residue Tyr266 plays an important role in promoting the activated state of the receptor in response to ligand binding (4, 33, 36, 91).
H7
H7 has a stretch of eight highly conserved residues, SLPLSSxWA, that may be functionally equivalent to the conserved NPxxY motif in the Class A receptors and mediate both H7–H6 and H7–H2 interactions. Pro290 generates a non-hydrogen-bonded carbonyl at Val286 that is oriented toward H2. Ser288 and Ser292 are thought to interact with Gln253 on H6 based on mutational data (89). Ile249, a conserved residue on H6, is in a pocket formed by Trp295 and Ala296, two highly conserved members of the SLPLSSx-WA motif. The structural model of Ste2 places the third conserved serine (Ser293) in this stretch in a position where it can hydrogen-bond to the conserved residue Asn84 on H2.
DISCUSSION
GPCRs transduce signals for a wide variety of ligands ranging from molecules as small as Ca2+ to large glycoprotein hormones. These receptors share a common function in their ability to activate heterotrimeric G proteins, and they share a common overall structure of seven TM helices. It is therefore surprising that, despite these similarities, there is no significant sequence identity across the GPCR family to suggest that they have a common mechanism of action. The lack of sequence identity in the extracellular domains is expected since these regions mediate the binding of such diverse ligands. However, the lack of sequence similarity in the TM regions and intracellular sequences is more unexpected for proteins that carry out a common function. As a result of this sequence diversity, previous studies have largely focused on obviously related subgroups of receptors.
Here, we have carried out a detailed comparison of two very divergent receptors, which are the best characterized of the Class A and Class D GPCR families. Rhodopsin was selected to represent the Class A receptors because it is the only GPCR whose structure has been determined to high resolution by protein crystallography. Ste2, the yeast α-factor mating pheromone receptor, was selected for comparison because the experimental accessibility of yeast has generated a large body of genetic and biochemical data that have allowed us to extensively annotate the sequence in terms of function. In comparing rhodopsin and Ste2, we have primarily focused on the TM helices. The TM helices are responsible for transducing the ligand binding signal from the extracellular side of the receptor to the intracellular loops involved in G protein activation.
General Features of Receptor Structure Common to Ste2 and Rhodopsin
The comparison of Ste2 and rhodopsin was facilitated by a model of Ste2 that was generated using the crystal structure of rhodopsin as a template for the helix positions. We have taken advantage of recent cysteine accessibility data (4, 36) to define the extracellular ends of the Ste2 TM helices. This represents an improvement over our previous models which made use of computational algorithms alone to predict the TM boundaries (35, 36). Moreover, the rotational orientation for each transmembrane segment was based on the hydrophobic moments and helix packing moments, as well as on the positions of conserved and group conserved amino acids.
When comparing the structures of Ste2 and rhodopsin, particular emphasis was given to amino acids that were structurally and functionally conserved within the Class A and Class D receptors (Figure 6). Since there is no significant sequence identity between these two classes of receptors, the comparison of the rhodopsin and Ste2 families is based on a comparison of amino acid subgroups that have similar structural and functional roles in membrane proteins. Substitution databases derived from membrane protein sequences (92–94) and analyses of membrane proteins of known structure (95–97) have suggested that the following subgroups have similar roles in membrane proteins: the strongly polar amino acids (E, D, R, K, H, Q, N), the small and weakly polar amino acids (G, A, S, T, C), the large hydrophobic amino acids including phenylalanine (L, V, I, M, F), the aromatic amino acids capable of hydrogen-bonding (W, Y), and proline. The similarities and differences involving these different subgroups are discussed below.
Figure 6.
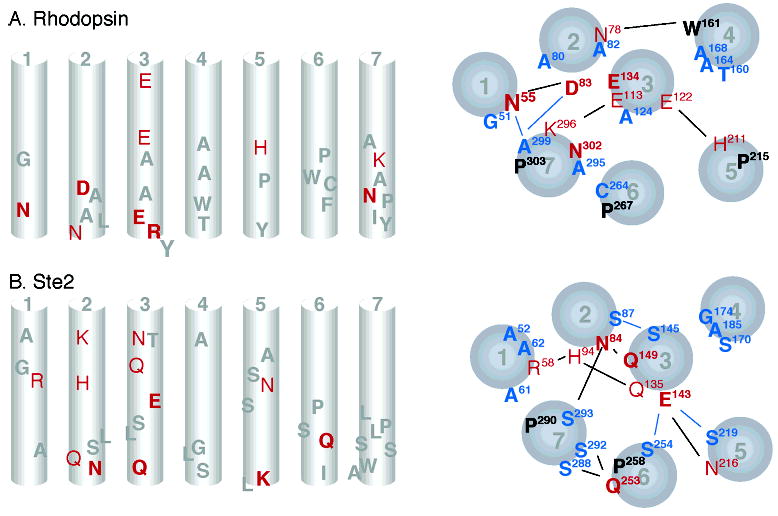
Schematic representations comparing the location of conserved and polar residues in rhodopsin (A) and the Ste2 receptor (B). The left-hand panels show a side view of the TM helices (in the plane of the membrane) with conserved residues in gray, strongly polar residues in red, and conserved strongly polar residues in bold red. The right-hand panels show the TM helices as viewed from the intracellular side. Conserved residues are shown in black, group-conserved small and weakly polar residues in blue, strongly polar residues in red, and conserved strongly polar residues in bold red. Lines indicate potential interhelical interactions.
Strongly Polar Amino Acids
Strongly polar amino acids (E, D, R, K, H, Q, N) that occur in the hydrophobic interior of GPCRs are generally thought to play key roles in receptor structure and function by mediating interhelical interactions. These amino acids can form single helix–helix contacts or hydrogen-bonding networks. We use the position of the polar residues and their pattern of conservation to identify functional equivalents in rhodopsin and Ste2 and to address the question of whether these receptors share a common mechanism (Figure 6).
A highly conserved pair of polar amino acids in rhodopsin, Asn55 on H1 and Asp83 on H2 (Figure 4A,C), is critical for function. They are strictly conserved across the Class A GPCRs, and mutation leads to either loss of function (98, 99) or constitutive activity (100). The polar side chains of Asp55 and Asp83 hydrogen-bond with each other and with the backbone carbonyl of Ala299 on H7. This hydrogen-bonding network stabilizes H7 in an inactive orientation in dark rhodopsin and is likely broken in light-activated rhodopsin. Importantly, the backbone carbonyl of Ala299 is free to hydrogen-bond because of a conserved proline (Pro303) on H7.
In Ste2, there are two strongly polar amino acids (Arg58 and His94; Figure 5A,C) that are moderately well-conserved and located at positions on H1 and H2 similar to the conserved Asn55 and Asp83 in rhodopsin. However, mutations affecting Arg58 do not have the strong functional phenotypes as in rhodopsin arguing that the corresponding residues are not functional equivalents. The Ste2 model indicates that Arg58 and H94 would serve to stabilize the H1–H2 interaction of the Ste2 but are not predicted to have hydrogen-bonding contacts with H7 (Figure 6).
There is a striking similarity between rhodopsin and Ste2 in the location and functional role of strongly polar amino acids on H3. In rhodopsin, there are three glutamic acids on H3: Glu113, Glu122, and Glu134. In Ste2, there are two glutamines and a glutamate on H3: Gln135, Glu143, and Gln149. Glu113 and Gln135 are both located at the extra-cellular end of H3 and show only partial conservation, suggesting that they have parallel functions in ligand activation. Glu113 on H3 in rhodopsin is only conserved in the opsin subfamily of Class A GPCRs. Glu113 interacts electrostatically with the protonated retinal Schiff’s base and stabilizes the inactive state. Mutating Glu113 to Gln results in constitutive receptor activity (73, 74). In a parallel fashion, mutational analyses indicate that Gln135 in Ste2 can influence α-factor activation (33). The similar location and the mutational sensitivity of Glu113 and Gln135 suggest that these residues may be functionally equivalent.
Glu122 in rhodopsin and Glu143 in Ste2 are both located in the middle of H3, and form hydrogen bonds with highly polar amino acids on H5. In rhodopsin, Glu122 hydrogen-bonds with the backbone carbonyl of His211 on H5, while Glu143 in Ste2 hydrogen-bonds with Asn216 on H5 and Ser254 on H6. The backbone carbonyl of His211 in rhodopsin is free to hydrogen-bond as a result of a highly conserved proline at position 215. This hydrogen bond is broken in the activated receptor (66) and is thought to reflect rotation of H5. In a parallel fashion, the highly conserved proline at position 258 in H6 of Ste2 results in a free carbonyl at Ser254. Both Ser254 and Pro258 are important in the activation of Ste2, and are involved in a key hydrogen-bonding cluster along with Glu143 and Asn216. The important point to note here is that in rhodopsin there are functionally important hydrogen-bonding networks centered on the backbone carbonyls at His211 (H5) and Ala299 (H7) due to the conserved Pro215 on H5 and Pro303 on H7. In contrast, Ste2 does not have a proline on H5, and the proline on H7 does not appear to be involved in a hydrogen-bonding cluster. The important hydrogen-bonding cluster in Ste2 is centered on H6.
At the cytoplasmic end of H3, Glu134 in rhodopsin and Gln149 in Ste2 are both strictly conserved and key to receptor function. Glu134 is part of the signature ERY sequence that stabilizes the inactive state of Class A GPCRs. Mutation of Glu134 results in constitutive activity (63, 64). Although the ERY sequence is not present in the Class D pheromone receptors, the highly conserved Gln149 may play an analogous role since mutation of Gln149 also results in constitutive receptor activation (35).
Finally, Asn302 is part of the conserved NPxxY sequence on H7 in rhodopsin. There is no strongly polar residue in the conserved SLPLSSxWA motif on H7 in Ste2. However, Ser288 and Ser292 of this conserved motif are predicted to interact with strongly polar Gln253 on H6 (89). As a result, Asn302 and Gln253 may have similar functions in mediating interhelical interactions involving the intracellular half of H7.
Small and Weakly Polar Amino Acids
Interactions between TM helices have long been recognized as important for stabilizing GPCRs in an inactive conformation in the absence of ligand. Until recently, the focus has been on highly conserved or strongly polar amino acids. However, an important role for the small and weakly polar amino acids in mediating helix interactions in membrane proteins was identified when it was recognized that these amino acids are highly conserved when considered as a group at sites of helix–helix contact in the Class A receptors (38).
A parallel analysis of the Class D Ste2 subfamily of receptors (Figure 3E) reveals the same high conservation of the small and weakly polar group of amino acids at specific sites in the TM helices (Figure 6). These include positions 52, 56, 60, 61, 62 on H1, 87 on H2, 145 on H3, 170, 174 on H4, 212, 214, 219 on H5, and 292, 293, 296 on H7 (see Table 2 in the Supporting Information). Importantly, these group-conserved residues were generally found in the Ste2 model at sites of interhelical contact. Furthermore, mutations affecting these sites commonly cause constitutive receptor activation (see Supporting Information).
A general similarity between rhodopsin and Ste2 is the location of the group-conserved residues on H2, H3, and H4. In rhodopsin, the small and weakly polar amino acids stabilize H2, H3, and H4 (Figure 6). In Ste2, the conserved serines at positions 87 and 145 are predicted to hydrogen-bond and mediate a strong H2–H3 contact, while the conserved SxxxG motif on H4 involving positions 170 and 174 is positioned to mediate close packing of H3 and H4. Interestingly, in the Ste3 subfamily, H3 has the largest number of strong packing moments, consistent with it being the central helix in the 7-TM bundle (Figure 3F).
Another important similarity is that both rhodopsin and Ste2 largely lack conserved small and weakly polar amino acids on H6. In rhodopsin, the lack of group-conserved amino acids on H6 suggests that this helix is designed to not interact strongly with adjacent helices. A similar lack of the group-conserved amino acids on H6 of Ste2 suggests that in both rhodopsin and Ste2 H6 undergoes rigid body motion upon activation.
Ste2 also has a conserved GxxxG motif on H1. Gly56 and Gly60 in Ste2 appear to face out toward a predicted dimer interface (Figure 3D,E and ref 81). A similar sequence (S38xxxA42) is present in rhodopsin but absent in the Ste3 subfamily (Figure 3F). Dimerization (or oligomerization) of GPCRs has been described for transport to the plasma membrane and signaling (for a recent review, see ref 101). Our helix packing moment analysis (Figure 3) shows that there is no common dimerization motif in the Class A or Class D families. All the subfamilies analyzed in this study have potential dimerization motifs on H4: A166xxA169 in the opsin subfamily, A176xxA179 in the Ste2 subfamily. The Ste3 subfamily has two helix packing moments facing the presumed lipid face.
Proline in GPCR Structure and Function
Proline plays a unique role in membrane protein structure and function. As mentioned above, the lack of an amide NH in proline results in a non-hydrogen-bonded carbonyl at the i-4 position in the helix. This carbonyl in turn is free to form interhelical hydrogen bonds. The lack of hydrogen bonding along the helix backbone in many cases introduces a flexible hinge for rigid body rotation or motion of the TM helix segments.
Three prolines in the Class A GPCRs are highly conserved: Pro215 on H5, Pro267 on H6, and Pro303 on H7. In Ste2, there are no prolines on H5, but there are conserved prolines on H6 (Pro258) and H7 (Pro290). H5 has four serines, two of them in a row (Ser213 and Ser214). Ballesteros and co-workers have shown that Ser can introduce bends into transmembrane helices and therefore might replace Pro to maintain a similar structure (102, 103).
Our current model of the structure of Class A GPCRs is that H1, H2, H3, and H4 form a tightly packed core and H5, H6, and H7 move relative to that core upon activation (23). The conserved prolines appear to be essential elements in this movement. In both rhodopsin and Ste2, we envision that there is a significant reorientation of H6 which is coupled to motion of H5 and H7. In this regard, analysis of the Ste3 subfamily shows that H6 also has a conserved proline (67%).
Large Hydrophobic Amino Acids
Phenylalanine is often associated with the large nonaromatic hydrophobic (Leu, Val, Ile, Met) amino acids in substitution databases (92–94). Tryptophan and tyrosine with more polar side chains are associated as a distinct subgroup. The large hydrophobic amino acids (including Phe) have high occurrences in the transmembrane regions of membrane proteins. Tyrosine and tryptophan are generally found at the ends of the TM helices.
Although the large hydrophobic residues have the highest occurrence on the lipid-facing surface of the TM helices of rhodopsin and Ste2, there are sites that are conserved and appear to be essential for receptor structure and function. One example of conserved hydrophobic residues in rhodopsin is the three consecutive valines at the intracellular boundary of H3. These valines have been described as a hydrophobic lid over the conserved ERY sequence (60, 104). In a parallel fashion, in Ste2 there are conserved large hydrophobic residues at the cytoplasmic boundary of H3. Positions Ile150 and Ile153 are conserved as large hydrophobic amino acids in Ste2, and mutation can lead to constitutive receptor activation (35). Strikingly, Ile150 and Ile153 in Ste2 would be predicted to form a hydrophobic lid on Gln149, which is thought to be functionally equivalent to the ERY sequence in rhodopsin.
Tyrosine and Tryptophan
In both rhodopsin and Ste2, aromatic amino acids on H6 are important in forming the ligand-binding site. The side chains of tyrosine and tryptophan are largely hydrophobic but have polar groups capable of hydrogen-bonding and aromatic rings capable of cationic-π interactions. In rhodopsin, Trp265 and Tyr268 bracket the retinal chromophore and are involved in channeling the structural changes of the retinal upon light absorption into changes in the orientation of H6. In Ste2, Tyr266 is thought to interact with the α-factor ligand and plays a key role in promoting receptor activation (33, 36). Both receptor classes also have aromatic amino acids in the conserved motifs on H7: tyrosine in the NPxxY motif in rhodopsin and tryptophan in the SLPLSSxWA motif in Ste2.
Conserved Functional Microdomains and Implications for Mechanism of Activation
The discussion above highlights the similarities and differences between the Class A and Class D receptors based on the chemical nature of the amino acids at conserved and group-conserved positions in the TM helices. Studies of CAMs suggest that there is no simple common mechanism (14, 15). However, analyses of the role of helices H3 and H6 suggest a common mechanism by the similar function carried out by receptor microdomains in divergent receptors (22, 60, 105). In this final section, we address the question as to whether there are similar functional microdomains in these two classes of receptors.
There are several regions in the Class A and D families that appear to be conserved functional microdomains (Figure 6). On H3, there is a conserved strongly polar amino acid (Glu or Gln) at the cytoplasmic end of the helix that is capped by several large hydrophobic amino acids. In rhodopsin, Glu134 is part of the conserved ERY sequence, which is capped by three consecutive valines. In the α1b-adrenergic receptor, mutation of Val147 to alanine leads to constitutive activity (106). In Ste2, Gln149 is conserved and mutation results in constitutive activity. Gln149 is capped by two isoleucines (Ile150 and Ile153) that are oriented toward the center of the helical bundle and also result in constitutive activity when mutated (35). These data support the proposal of Weinstein, Sealfon, and colleagues that the hydrophilic residues at the end of helix 3 are “caged” in a microdomain to allow proper function of the receptor (60).
Interestingly, in rhodopsin, there is a strongly polar group at the extracellular end of H3 (Glu113) that mediates interaction with the retinal and is involved in locking the receptor off in the absence of retinal. A similar function may be ascribed to Gln135 in Ste2. It is difficult to assess whether sites on the extracellular side of these receptors function as microdomains since they tend to be receptor- or ligand-specific.
On H4, both rhodopsin and Ste2 have several small and weakly polar amino acids that mediate packing of H4 on H3 (Figure 6). In both rhodopsin and Ste2, H5 has a strongly polar site (His211 in rhodopsin, Asn216 in Ste2) involved in hydrogen bonding with a glutamate in the middle of H3 (Glu122 in rhodopsin, Glu143 in Ste2). In Ste2, mutation of Asn216 to Asp results in constitutive activity (86).
Conserved prolines are found on H6 and H7 in rhodopsin and Ste2. Conserved prolines in GPCRs generally might be considered microdomains. Prolines provide a point of flexibility and expose a free carbonyl that is usually positioned to mediate interhelical interactions. On H6, the conserved proline is associated with aromatic amino acids in both the Class A and Class D GPCRs. In rhodopsin, Trp265 on H6 appears to be responsible for channeling the energy involved in retinal isomerization into rotation of H6 (23). In Ste2, Tyr266 on H6 interacts with the α-factor ligand (36) and may play an analogous role as Trp265 in rhodopsin in changing the orientation of H6.
On H7, the conserved proline is part of a conserved motif: the NPxxY motif in Class A receptors and the SLPLSSxWA motif in Class D receptors. The role of these motifs in receptor activation is still poorly understood. However, a common element of both motifs is that they mediate H6–H7 interactions. In rhodopsin, Met257 on H6 interacts with Asn302 on H7. Mutation of Met257 results in constitutive activity (68). In Ste2, Gln253 on H6 interacts with Ser288 and Ser292. Mutation of Gln253, Ser288, or Ser292 results in constitutive activity (89). The last polar residue of the conserved motif (Tyr306 in rhodopsin and Ser293 in Ste2) is hydrogen-bonded to an asparagine at the cytoplasmic end of H2.
On the basis of our analysis of the Class A and Class D GPCRs, we can draw two general conclusions. First, the detailed mechanisms are not conserved. The intricate arrangement of hydrogen bonding and van der Waals interactions are different in the Class A and Class D receptors. As a result, the CAMs that are often used to support specific activation mechanisms are different between different families and subfamilies of GPCRs. Our analysis of the Class A and Class D GPCRs leads to the conclusion that there are multiple contacts that stabilize both inactive and active conformations. These not only involve hydrogen-bonding interactions of the strongly polar amino acids but also van der Waals interactions involving all of the other amino acid subgroups (20, 38). The presence of multiple contacts underlies suggestions that there are multiple activation mechanisms (16, 17).
The second general conclusion is that there are similar microdomains in the Class A and Class D receptors that mediate receptor activation. In fact, these microdomains are consistent with a general model for activation of GPCRs that not only involves TM helix motion (22) but specifically involves H5, H6, and H7 moving relative to a tightly packed core of helices formed by H1, H2, H3, and H4 (23, 38). Importantly, the comparison of the Class A and Class D GPCRs does not focus on the conserved disulfide bond (for example, see refs 54, 55). We found no evidence of an analogous functional domain in the Class D receptors. The focus is rather on the ERY, Pro-aromatic, and NPxxY regions on H3, H6, and H7 of Class A GPCRs that have conserved analogues in the Class D receptors.
Supplementary Material
Acknowledgments
We thank Thomas Wolff for discussions and the initial MD simulations on the Ste2 structure.
Footnotes
This work was supported by National Institutes of Health Grant GM55107 awarded to J.B.K. and Grant GM-41412 awarded to S.O.S.
Abbreviations: CAM, constitutively active mutation; GPCR, G protein-coupled receptor; H, helix; TM, transmembrane.
SUPPORTING INFORMATION AVAILABLE The sequence alignments of selected Class A GPCRs and Class D GPCRs of the Ste2- and Ste3 subfamily. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kolakowski LF. Gcrdb–a G-protein-coupled receptor database. Recept Channels. 1994;2:1–7. [PubMed] [Google Scholar]
- 2.Probst WC, Snyder LA, Schuster DI, Brosius J, Sealfon SC. Sequence alignment of the G-protein coupled receptor superfamily. DNA Cell Biol. 1992;11:1–20. doi: 10.1089/dna.1992.11.1. [DOI] [PubMed] [Google Scholar]
- 3.Henry LK, Khare S, Son C, Babu VV, Naider F, Becker JM. Identification of a contact region between the tridecapeptide α-factor mating pheromone of Saccharomyces cerevisiae and its G protein-coupled receptor by photoaffinity labeling. Biochemistry. 2002;41:6128–6139. doi: 10.1021/bi015863z. [DOI] [PubMed] [Google Scholar]
- 4.Lin JC, Duell K, Konopka JB. A microdomain formed by the extracellular ends of the transmembrane domains promotes activation of the G protein-coupled α-factor receptor. Mol Cell Biol. 2004;24:2041–2051. doi: 10.1128/MCB.24.5.2041-2051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen SH, Lin F, Xu M, Riek RP, Novotny J, Graham RM. Mutation of a single TMVI residue, Phe(282), in the β2-adrenergic receptor results in structurally distinct activated receptor conformations. Biochemistry. 2002;41:6045–6053. doi: 10.1021/bi012189c. [DOI] [PubMed] [Google Scholar]
- 6.O’Dowd BF, Hnatowich M, Regan JW, Leader WM, Caron MG, Lefkowitz RJ. Site-directed mutagenesis of the cytoplasmic domains of the human β2-adrenergic receptor. Localization of regions involved in G protein-receptor coupling. J Biol Chem. 1988;263:15985–15992. [PubMed] [Google Scholar]
- 7.Schandel KA, Jenness DD. Direct evidence for ligand-induced internalization of the yeast α-factor pheromone receptor. Mol Cell Biol. 1994;14:7245–7255. doi: 10.1128/mcb.14.11.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stefan CJ, Blumer KJ. The 3rd cytoplasmic loop of a yeast G-protein-coupled receptor controls pathway activation, ligand discrimination, and receptor internalization. Mol Cell Biol. 1994;14:3339–3349. doi: 10.1128/mcb.14.5.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen QJ, Konopka JB. Regulation of the G-protein-coupled α-factor pheromone receptor by phosphorylation. Mol Cell Biol. 1996;16:247–257. doi: 10.1128/mcb.16.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hicke L, Riezman H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 1996;84:277–287. doi: 10.1016/s0092-8674(00)80982-4. [DOI] [PubMed] [Google Scholar]
- 11.Crowe ML, Perry BN, Connerton IF. Golf complements a GPA1 null mutation in Saccharomyces cerevisiae and functionally couples to the STE2 pheromone receptor. J Recept Signal Transduction Res. 2000;20:61–73. doi: 10.3109/10799890009150037. [DOI] [PubMed] [Google Scholar]
- 12.Broach JR, Thorner J. High-throughput screening for drug discovery. Nature. 1996;384:14–16. [PubMed] [Google Scholar]
- 13.Pausch MH. G-protein-coupled receptors in Saccharomyces cerevisiae: high-throughput screening assays for drug discovery. Trends Biotechnol. 1997;15:487–494. doi: 10.1016/S0167-7799(97)01119-0. [DOI] [PubMed] [Google Scholar]
- 14.Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 15.Parnot C, Miserey-Lenkei S, Bardin S, Corvol P, Clauser E. Lessons from constitutively active mutants of G protein-coupled receptors. Trends Endocrinol Metab. 2002;13:336–343. doi: 10.1016/s1043-2760(02)00628-8. [DOI] [PubMed] [Google Scholar]
- 16.Prossnitz ER, Gilbert TL, Chiang S, Campbell JJ, Qin S, Newman W, Sklar LA, Ye RD. Multiple activation steps of the N-formyl peptide receptor. Biochemistry. 1999;38:2240–2247. doi: 10.1021/bi982274t. [DOI] [PubMed] [Google Scholar]
- 17.Thomas WG, Qian H, Chang CS, Karnik S. Agonist-induced phosphorylation of the angiotensin II (AT1A) receptor requires generation of a conformation that is distinct from the inositol phosphate-signaling state. J Biol Chem. 2000;275:2893–2900. doi: 10.1074/jbc.275.4.2893. [DOI] [PubMed] [Google Scholar]
- 18.Carter PH, Petroni BD, Gensure RC, Schipani E, Potts JT, Gardella TJ. Selective and nonselective inverse agonists for constitutively active type-1 parathyroid hormone receptors: evidence for altered receptor conformations. Endocrinology. 2001;142:1534–1545. doi: 10.1210/endo.142.4.8103. [DOI] [PubMed] [Google Scholar]
- 19.Whistler JL, Gerber BO, Meng EC, Baranski TJ, von Zastrow M, Bourne HR. Constitutive activation and endocytosis of the complement factor 5a receptor: evidence for multiple activated conformations of a G protein-coupled receptor. Traffic. 2002;3:866–877. doi: 10.1034/j.1600-0854.2002.31203.x. [DOI] [PubMed] [Google Scholar]
- 20.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 21.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl Acad Sci USA. 2002;99:5982–5987. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 23.Patel AB, Crocker E, Eilers M, Hirshfeld A, Sheves M, Smith SO. Coupling of retinal isomerization to the activation of rhodopsin. Proc Natl Acad Sci USA. 2004;101:10048–10053. doi: 10.1073/pnas.0402848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altenbach C, Yang K, Farrens DL, Farahbakhsh ZT, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the cytoplasmic interhelical E–F loop region of rhodopsin: a site-directed spin-labeling study. Biochemistry. 1996;35:12470–12478. doi: 10.1021/bi960849l. [DOI] [PubMed] [Google Scholar]
- 25.Horn F, Weare J, Beukers MW, Horsch S, Bairoch A, Chen W, Edvardsen O, Campagne F, Vriend G. GPCRDB: an information system for G protein-coupled receptors. Nucleic Acids Res. 1998;26:275–279. doi: 10.1093/nar/26.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bockaert J, Pin JP. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acharya S, Saad Y, Karnik SS. Transducin-α C-terminal peptide binding site consists of C–D and E–F loops of rhodopsin. J Biol Chem. 1997;272:6519–6524. doi: 10.1074/jbc.272.10.6519. [DOI] [PubMed] [Google Scholar]
- 28.Neumann S, Krause G, Claus M, Paschke R. Structural determinants for G protein activation and selectivity in the second intracellular loop of the thyrotropin receptor. Endocrinology. 2005;146:477–485. doi: 10.1210/en.2004-1045. [DOI] [PubMed] [Google Scholar]
- 29.Havlickova M, Blahos J, Brabet I, Liu J, Hruskova B, Prezeau L, Pin JP. The second intracellular loop of metabotropic glutamate receptors recognizes C termini of G-protein α-subunits. J Biol Chem. 2003;278:35063–35070. doi: 10.1074/jbc.M306555200. [DOI] [PubMed] [Google Scholar]
- 30.Martin NP, Leavitt LM, Sommers CM, Dumont ME. Assembly of G protein-coupled receptors from fragments: identification of functional receptors with discontinuities in each of the loops connecting transmembrane segments. Biochemistry. 1999;38:682–695. doi: 10.1021/bi982062w. [DOI] [PubMed] [Google Scholar]
- 31.Martin NP, Celic A, Dumont ME. Mutagenic mapping of helical structures in the transmembrane segments of the yeast α-factor receptor. J Mol Biol. 2002;317:765–788. doi: 10.1006/jmbi.2002.5444. [DOI] [PubMed] [Google Scholar]
- 32.Arevalo E, Estephan R, Madeo J, Arshava B, Dumont M, Becker JM, Naider F. Biosynthesis and biophysical analysis of domains of a yeast G protein-coupled receptor. Biopolymers. 2003;71:516–531. doi: 10.1002/bip.10491. [DOI] [PubMed] [Google Scholar]
- 33.Dosil M, Giot L, Davis C, Konopka JB. Dominant-negative mutations in the G-protein-coupled α-factor receptor map to the extracellular ends of the transmembrane segments. Mol Cell Biol. 1998;18:5981–5991. doi: 10.1128/mcb.18.10.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dube P, DeCostanzo A, Konopka JB. Interaction between transmembrane domains five and six of the α-factor receptor. J Biol Chem. 2000;275:26492–26499. doi: 10.1074/jbc.M002767200. [DOI] [PubMed] [Google Scholar]
- 35.Parrish W, Eilers M, Ying WW, Konopka JB. The cytoplasmic end of transmembrane domain 3 regulates the activity of the Saccharomyces cerevisiae G-protein-coupled α-factor receptor. Genetics. 2002;160:429–443. doi: 10.1093/genetics/160.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin JC, Parrish W, Eilers M, Smith SO, Konopka JB. Aromatic residues at the extracellular ends of transmembrane domains 5 and 6 promote ligand activation of the G protein-coupled α-factor receptor. Biochemistry. 2003;42:293–301. doi: 10.1021/bi026766o. [DOI] [PubMed] [Google Scholar]
- 37.Eilers M, Patel AB, Liu W, Smith SO. Comparison of helix interactions in membrane and soluble α-bundle proteins. Biophys J. 2002;82:2720–2736. doi: 10.1016/S0006-3495(02)75613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu W, Eilers M, Patel AB, Smith SO. Helix packing moments reveal diversity and conservation in membrane protein structure. J Mol Biol. 2004;337:713–729. doi: 10.1016/j.jmb.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank: update. Nucleic Acids Res. 2004;32:D23–D26. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, de Montigny J, Marck C, Neuveglise C, Talla E, Goffard N, Frangeul L, Aigle M, Anthouard V, Babour A, Barbe V, Barnay S, Blanchin S, Beckerich JM, Beyne E, Bleykasten C, Boisrame A, Boyer J, Cattolico L, Confanioleri F, de Daruvar A, Despons L, Fabre E, Fairhead C, Ferry-Dumazet H, Groppi A, Hantraye F, Hennequin C, Jauniaux N, Joyet P, Kachouri R, Kerrest A, Koszul R, Lemaire M, Lesur I, Ma L, Muller H, Nicaud JM, Nikolski M, Oztas S, Ozier-Kalogeropoulos O, Pellenz S, Potier S, Richard GF, Straub ML, Suleau A, Swennen D, Tekaia F, Wesolowski-Louvel M, Westhof E, Wirth B, Zeniou-Meyer M, Zivanovic I, Bolotin-Fukuhara M, Thierry A, Bouchier C, Caudron B, Scarpelli C, Gaillardin C, Weissenbach J, Wincker P, Souciet JL. Genome evolution in yeasts. Nature. 2004;430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- 41.Dietrich FS, Voegeli S, Brachat S, Lerch A, Gates K, Steiner S, Mohr C, Pohlmann R, Luedi P, Choi SD, Wing RA, Flavier A, Gaffney TD, Phillippsen P. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science. 2004;304:304–307. doi: 10.1126/science.1095781. [DOI] [PubMed] [Google Scholar]
- 42.Eisenberg D, Weiss RM, Terwilliger TC. The helical hydrophobic moment–a measure of the amphiphilicity of a helix. Nature. 1982;299:371–374. doi: 10.1038/299371a0. [DOI] [PubMed] [Google Scholar]
- 43.Engelman DM, Steitz TA, Goldman A. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu Rev Biophys Biophys Chem. 1986;15:321–353. doi: 10.1146/annurev.bb.15.060186.001541. [DOI] [PubMed] [Google Scholar]
- 44.Baldwin JM. The probable arrangement of the helices in G protein-coupled receptors. EMBO J. 1993;12:1693–1703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldwin JM, Schertler GFX, Unger VM. An α-carbon template for the transmembrane helices in the rhodopsin family of G-protein-coupled receptors. J Mol Biol. 1997;272:144–164. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- 46.Farahbakhsh ZT, Ridge KD, Khorana HG, Hubbell WL. Mapping light-dependent structural changes in the cytoplasmic loop connecting helices C and D in rhodopsin: a site-directed spin labeling study. Biochemistry. 1995;34:8812–8819. doi: 10.1021/bi00027a033. [DOI] [PubMed] [Google Scholar]
- 47.Altenbach C, Cai K, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the sequence 306–322 extending from helix VII to the palmitoylation sites in rhodopsin: a site-directed spin-labeling study. Biochemistry. 1999;38:7931–7937. doi: 10.1021/bi9900121. [DOI] [PubMed] [Google Scholar]
- 48.Altenbach C, Klein-Seetharaman J, Hwa J, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the sequence 59–75 connecting helices I and II in rhodopsin: a site-directed spin-labeling study. Biochemistry. 1999;38:7945–7949. doi: 10.1021/bi990014l. [DOI] [PubMed] [Google Scholar]
- 49.Sali A, Blundell TL. Definition of general topological equivalence in protein structures. A procedure involving comparison of properties and relationships through simulated annealing and dynamic programming. J Mol Biol. 1990;212:403–428. doi: 10.1016/0022-2836(90)90134-8. [DOI] [PubMed] [Google Scholar]
- 50.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 51.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 52.Sung CH, Davenport CM, Nathans J. Rhodopsin mutations responsible for autosomal-dominant retinitis-pigmentosa – clustering of functional classes along the polypeptide-chain. J Biol Chem. 1993;268:26645–26649. [PubMed] [Google Scholar]
- 53.Hwa J, Garriga P, Liu X, Khorana HG. Structure and function in rhodopsin–packing of the helices in the transmembrane domain and folding to a tertiary structure in the intradiscal domain are coupled. Proc Natl Acad Sci USA. 1997;94:10571–10576. doi: 10.1073/pnas.94.20.10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karnik SS, Sakmar TP, Chen HB, Khorana HG. Cysteine residues 110 and 187 are essential for the formation of correct structure in bovine rhodopsin. Proc Natl Acad Sci USA. 1988;85:8459–8463. doi: 10.1073/pnas.85.22.8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strader CD, Fong TM, Tota MR, Underwood D, Dixon RA. Structure and function of G protein-coupled receptors. Annu Rev Biochem. 1994;63:101–132. doi: 10.1146/annurev.bi.63.070194.000533. [DOI] [PubMed] [Google Scholar]
- 56.Sakmar TP, Franke RR, Khorana HG. Glutamic acid-113 serves as the retinylidene Schiff base counterion in bovine rhodopsin. Proc Natl Acad Sci USA. 1989;86:8309–8313. doi: 10.1073/pnas.86.21.8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Imai H, Kojima D, Oura T, Tachibanaki S, Terakita A, Shichida Y. Single amino acid residue as a functional determinant of rod and cone visual pigments. Proc Natl Acad Sci USA. 1997;94:2322–2326. doi: 10.1073/pnas.94.6.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Acharya S, Karnik SS. Modulation of GDP release from transducin by the conserved Glu134-Arg135 sequence in rhodopsin. J Biol Chem. 1996;271:25406–25411. doi: 10.1074/jbc.271.41.25406. [DOI] [PubMed] [Google Scholar]
- 59.Scheer A, Fanelli F, Costa T, De Benedetti PG, Cotecchia S. The activation process of the α1B-adrenergic receptor: potential role of protonation and hydrophobicity of a highly conserved aspartate. Proc Natl Acad Sci USA. 1997;94:808–813. doi: 10.1073/pnas.94.3.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ballesteros J, Kitanovic S, Guarnieri F, Davies P, Fromme BJ, Konvicka K, Chi L, Millar RP, Davidson JS, Weinstein H, Sealfon SC. Functional microdomains in G-protein-coupled receptors–the conserved arginine-cage motif in the gonadotropin-releasing hormone receptor. J Biol Chem. 1998;273:10445–10453. doi: 10.1074/jbc.273.17.10445. [DOI] [PubMed] [Google Scholar]
- 61.Scheer A, Costa T, Fanelli F, De Benedetti PG, Mhaouty-Kodja S, Abuin L, Nenniger-Tosato M, Cotecchia S. Mutational analysis of the highly conserved arginine within the Glu/Asp-Arg-Tyr motif of the α1b-adrenergic receptor: effects on receptor isomerization and activation. Mol Pharmacol. 2000;57:219–231. [PubMed] [Google Scholar]
- 62.Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SGF, Shi L, Gether U, Javitch JA. Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 63.Cohen GB, Yang T, Robinson PR, Oprian DD. Constitutive activation of opsin: influence of charge at position 134 and size at position 296. Biochemistry. 1993;32:6111–6115. doi: 10.1021/bi00074a024. [DOI] [PubMed] [Google Scholar]
- 64.Kim JM, Altenbach C, Thurmond RL, Khorana HG, Hubbell WL. Structure and function in rhodopsin–rhodopsin mutants with a neutral amino acid at E134 have a partially activated conformation in the dark state. Proc Natl Acad Sci USA. 1997;94:14273–14278. doi: 10.1073/pnas.94.26.14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J Biol Chem. 2003;278:21655–21662. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patel AB, Crocker E, Reeves PJ, Getmanova EV, Eilers M, Khorana HG, Smith SO. Changes in interhelical hydrogen bonding upon rhodopsin activation. J Mol Biol. 2005;347:803–812. doi: 10.1016/j.jmb.2005.01.069. [DOI] [PubMed] [Google Scholar]
- 67.Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RA. Identification of two serine residues involved in agonist activation of the β-adrenergic receptor. J Biol Chem. 1989;264:13572–13578. [PubMed] [Google Scholar]
- 68.Han M, Smith SO, Sakmar TP. Constitutive activation of opsin by mutation of methionine 257 on transmembrane helix 6. Biochemistry. 1998;37:8253–8261. doi: 10.1021/bi980147r. [DOI] [PubMed] [Google Scholar]
- 69.Javitch JA, Ballesteros JA, Weinstein H, Chen JY. A cluster of aromatic residues in the sixth membrane-spanning segment of the dopamine D2 receptor is accessible in the binding-site crevice. Biochemistry. 1998;37:998–1006. doi: 10.1021/bi972241y. [DOI] [PubMed] [Google Scholar]
- 70.Hovelmann S, Hoffmann SH, Kuhne R, ter Laak T, Reilander H, Beckers T. Impact of aromatic residues within transmembrane helix 6 of the human gonadotropin-releasing hormone receptor upon agonist and antagonist binding. Biochemistry. 2002;41:1129–1136. doi: 10.1021/bi0113162. [DOI] [PubMed] [Google Scholar]
- 71.Luo X, Zhang D, Weinstein H. Ligand-induced domain motion in the activation mechanism of a G-protein-coupled receptor. Protein Eng. 1994;7:1441–1448. doi: 10.1093/protein/7.12.1441. [DOI] [PubMed] [Google Scholar]
- 72.Madabushi S, Gross AK, Philippi A, Meng EC, Wensel TG, Lichtarge O. Evolutionary trace of G protein-coupled receptors reveals clusters of residues that determine global and class-specific functions. J Biol Chem. 2004;279:8126–8132. doi: 10.1074/jbc.M312671200. [DOI] [PubMed] [Google Scholar]
- 73.Robinson PR, Cohen GB, Zhukovsky EA, Oprian DD. Constitutively active mutants of rhodopsin. Neuron. 1992;9:719–725. doi: 10.1016/0896-6273(92)90034-b. [DOI] [PubMed] [Google Scholar]
- 74.Cohen GB, Oprian DD, Robinson PR. Mechanism of activation and inactivation of opsin: role of Glu113 and Lys296. Biochemistry. 1992;31:12592–12601. doi: 10.1021/bi00165a008. [DOI] [PubMed] [Google Scholar]
- 75.Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, Ernst OP. Role of the conserved NPxxY(x)(5,6)F motif in the rhodopsin ground state and during activation. Proc Natl Acad Sci USA. 2003;100:2290–2295. doi: 10.1073/pnas.0435715100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sommers CM, Dumont ME. Genetic interactions among the transmembrane segments of the G protein coupled receptor encoded by the yeast STE2 gene. J Mol Biol. 1997;266:559–575. doi: 10.1006/jmbi.1996.0816. [DOI] [PubMed] [Google Scholar]
- 77.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 angstrom resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 78.Locher KP, Lee AT, Rees DC. The E-coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 79.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane α-helices. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 80.Smith SO, Song D, Shekar S, Groesbeek M, Ziliox M, Aimoto S. Structure of the transmembrane dimer interface of glycophorin A in membrane bilayers. Biochemistry. 2001;40:6553–6558. doi: 10.1021/bi010357v. [DOI] [PubMed] [Google Scholar]
- 81.Overton MC, Chinault SL, Blumer KJ. Oligomerization, biogenesis, and signaling is promoted by a glycophorin A-like dimerization motif in transmembrane domain 1 of a yeast G protein-coupled receptor. J Biol Chem. 2003;278:49369–49377. doi: 10.1074/jbc.M308654200. [DOI] [PubMed] [Google Scholar]
- 82.Dosil M, Schandel KA, Gupta E, Jenness DD, Konopka JB. The C terminus of the Saccharomyces cerevisiae α-factor receptor contributes to the formation of preactivation complexes with its cognate G protein. Mol Cell Biol. 2000;20:5321–5329. doi: 10.1128/mcb.20.14.5321-5329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sommers CM, Martin NP, Akal-Strader A, Becker JM, Naider F, Dumont ME. A limited spectrum of mutations causes constitutive activation of the yeast α-factor receptor. Biochemistry. 2000;39:6898–6909. doi: 10.1021/bi992616a. [DOI] [PubMed] [Google Scholar]
- 84.Javadpour MM, Eilers M, Groesbeek M, Smith SO. Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys J. 1999;77:1609–1618. doi: 10.1016/S0006-3495(99)77009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leavitt LM, Macaluso CR, Kim KS, Martin NP, Dumont ME. Dominant negative mutations in the α-factor receptor, a G protein-coupled receptor encoded by the STE2 gene of the yeast Saccharomyces cerevisiae. Mol Gen Genet. 1999;261:917–932. doi: 10.1007/s004380051039. [DOI] [PubMed] [Google Scholar]
- 86.Parrish, W. R. (2001) The cytoplasmic end of transmembrane domain three regulates the activity of the G protein-coupled α-factor receptor, Ph.D. Thesis, Department of Molecular Microbiology, State University of New York at Stony Brook, Stony Brook, NY.
- 87.Konopka JB, Margarit SM, Dube P. Mutation of pro-258 in transmembrane domain 6 constitutively activates the G protein-coupled α-factor receptor. Proc Natl Acad Sci USA. 1996;93:6764–6769. doi: 10.1073/pnas.93.13.6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stefan CJ, Overton MC, Blumer KJ. Mechanisms governing the activation and trafficking of yeast G protein-coupled receptors. Mol Biol Cell. 1998;9:885–899. doi: 10.1091/mbc.9.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dube P, Konopka JB. Identification of a polar region in transmembrane domain 6 that regulates the function of the G protein-coupled α-factor receptor. Mol Cell Biol. 1998;18:7205–7215. doi: 10.1128/mcb.18.12.7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Han M, Lin SW, Smith SO, Sakmar TP. The effects of amino acid replacements of glycine 121 on transmembrane helix 3 of rhodopsin. J Biol Chem. 1996;271:32330–32336. doi: 10.1074/jbc.271.50.32330. [DOI] [PubMed] [Google Scholar]
- 91.Lee BK, Lee YH, Hauser M, Son CD, Khare S, Naider F, Becker JM. Tyr266 in the sixth transmembrane domain of the yeast α-factor receptor plays key roles in receptor activation and ligand specificity. Biochemistry. 2002;41:13681–13689. doi: 10.1021/bi026100u. [DOI] [PubMed] [Google Scholar]
- 92.George DG, Barker WC, Hunt LT. Mutation data matrix and its uses. Methods Enzymol. 1990;183:333–351. doi: 10.1016/0076-6879(90)83022-2. [DOI] [PubMed] [Google Scholar]
- 93.Jones DT, Taylor WR, Thornton JM. A mutation data matrix for transmembrane proteins. FEBS Lett. 1994;339:269–275. doi: 10.1016/0014-5793(94)80429-x. [DOI] [PubMed] [Google Scholar]
- 94.Ng PC, Henikoff JG, Henikoff S. PHAT: a transmembrane-specific substitution matrix. Bioinformatics. 2000;16:760–766. doi: 10.1093/bioinformatics/16.9.760. [DOI] [PubMed] [Google Scholar]
- 95.Wallin E, Tsukihara T, Yoshikawa S, Von Heijne G, Elofsson A. Architecture of helix bundle membrane proteins: an analysis of cytochrome c oxidase from bovine mitochondria. Protein Sci. 1997;6:808–815. doi: 10.1002/pro.5560060407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eilers M, Shekar SC, Shieh T, Smith SO, Fleming PJ. Internal packing of helical membrane proteins. Proc Natl Acad Sci USA. 2000;97:5796–5801. doi: 10.1073/pnas.97.11.5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ulmschneider MB, Sansom MSP. Amino acid distributions in integral membrane protein structures. Biochim Biophys Acta. 2001;1512:1–14. doi: 10.1016/s0005-2736(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 98.Sealfon SC, Chi L, Ebersole BJ, Rodic V, Zhang D, Ballesteros JA, Weinstein H. Related contribution of specific helix 2 and 7 residues to conformational activation of the serotonin 5-HT2A receptor. J Biol Chem. 1995;270:16683–16688. doi: 10.1074/jbc.270.28.16683. [DOI] [PubMed] [Google Scholar]
- 99.Parent JL, Le Gouill C, Rola-Pleszczynski M, Stankova J. Mutation of an aspartate at position 63 in the human platelet-activating factor receptor augments binding affinity but abolishes G-protein-coupling and inositol phosphate production. Biochem Biophys Res Commun. 1996;219:968–975. doi: 10.1006/bbrc.1996.0341. [DOI] [PubMed] [Google Scholar]
- 100.Scheer A, Fanelli F, Costa T, De Benedetti PG, Cotecchia S. Constitutively active mutants of the α1B-adrenergic receptor: role of highly conserved polar amino acids in receptor activation. EMBO J. 1996;15:3566–3578. [PMC free article] [PubMed] [Google Scholar]
- 101.Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci. 2005;26:131–137. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 102.Ballesteros JA, Deupi X, Olivella M, Haaksma EEJ, Pardo L. Serine and threonine residues bend α-helices in the χ1 = g− conformation. Biophys J. 2000;79:2754–2760. doi: 10.1016/S0006-3495(00)76514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure–function analysis of rhodopsin-like receptors. Mol Pharmacol. 2001;60:1–19. [PubMed] [Google Scholar]
- 104.Menon ST, Han M, Sakmar TP. Rhodopsin: structural basis of molecular physiology. Physiol Rev. 2001;81:1659–1688. doi: 10.1152/physrev.2001.81.4.1659. [DOI] [PubMed] [Google Scholar]
- 105.Sheikh SP, Vilardarga JP, Baranski TJ, Lichtarge O, Iiri T, Meng EC, Nissenson RA, Bourne HR. Similar structures and shared switch mechanisms of the β2-adrenoceptor and the parathyroid hormone receptor–Zn(II) bridges between helices III and VI block activation. J Biol Chem. 1999;274:17033–17041. doi: 10.1074/jbc.274.24.17033. [DOI] [PubMed] [Google Scholar]
- 106.Greasley PJ, Fanelli F, Scheer A, Abuin L, Nenniger-Tosato M, DeBenedetti PG, Cotecchia S. Mutational and computational analysis of the α1b-adrenergic receptor. Involvement of basic and hydrophobic residues in receptor activation and G protein coupling. J Biol Chem. 2001;276:46485–46494. doi: 10.1074/jbc.M105791200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.