

Abstract
Two new calcium-activated chloride channel (CLCA) family members, mCLCA5 and mCLCA6, have been cloned from mouse eye and intestine, respectively. mCLCA5 is highly homologous to hCLCA2, and mCLCA6 is highly homologous to hCLCA4. mCLCA5 is widely expressed with strong expression in eye and spleen, whereas mCLCA6 is primarily expressed in intestine and stomach. mCLCA6 is also expressed as a splice variant lacking exon 8 and part of exon 10 in intestine and stomach. Transfection of tsA201 cells with enhanced green fluorescent protein-tagged versions of the three cDNAs reveals protein products of 155 and 65 kDa for mCLCA5 and mCLCA6 and 145 and 65 kDa for the mCLCA6 splice variant. In vitro translation of mCLCA5 generates a 90-kDa protein that does not appear to be glycosylated. mCLCA6 also generates a 90-kDa protein that is glycosylated to a 110-kDa product, whereas the mCLCA6 splice variant generates an 80-kDa product that is 100 kDa after glycosylation. Treatment of enhanced green fluorescent protein-tagged mCLCA6 with PNGase F (peptide: N-glycosidase F) to remove N-linked glycosyl groups shows a reduction in size of the 65 kDa product to 60 kDa. Consistent with the hypothesis that mCLCA5, mCLCA6, and its splice variant encode calcium-activated chloride channels, in HEK293 cells expressing CLCAs ionomycin-evoked increases in intracellular calcium stimulated a current that reversed near Cl− equilibrium potential, ECl. Furthermore, these currents were inhibited by the chloride channel blocker niflumic acid. Given the prominent role of hCLCA2 in cancer cell adhesion and the unique high level of expression of hCLCA4 in brain, the identification of their murine counterparts presents the opportunity to clarify the role of CLCAs in disease and normal cell physiology.
Calcium-activated chloride currents have been characterized physiologically in many cell types including epithelium (1–4), smooth muscle (5–7), skeletal muscle (8), and neurons (9, 10). However, the molecular identities of channels responsible for calcium-activated chloride currents have not been determined. Proteins of the bestrophin and CLCA1 families have been proposed to function as calcium-activated chloride channels (11–19).
The CLCA gene family encodes proteins with four or five putative transmembrane domains, and many family members appear to function as calcium-activated chloride channels (bCLCA1, mCLCA1, mCLCA4, hCLCA1, and hCLCA2) (15–19). Other studies suggest that CLCAs may not be chloride channels themselves but may instead modulate chloride channels (20, 21)
In addition to their potential roles as ion channels or regulators of ion channels, some CLCA family members function as adhesion molecules. For example, bCLCA2 (Lu-ECAM-1), mCLCA1, and hCLCA2 all appear to be cell-cell adhesion molecules that help mediate the colonization of lung by lung metastatic murine B16-F10 melanoma cells (22–24). β4 integrin expressed on the surface of these melanoma cells can adhere to mCLCA1 in lung endothelium. This leads to focal adhesion kinase complexing, activation, and downstream signaling to extracellular signal-regulated kinase, which promotes intra-vascular metastatic growth (25). Human breast cancer cells can colonize the lung by expression of α6β4 integrin and adhesion to hCLCA2 (22). In addition, endothelial CLCA proteins (e.g. hCLCA2, mCLCA5, mCLCA1, bCLCA2) have β4 integrin binding domains in their 90- and 35-kDa fragments (26).
Some CLCA isoforms, both human and mouse, may have roles as tumor suppressors. In apoptosis-resistant tumor cell lines and in HC11 cells (mammary epithelial cell line) resistant to detachment-induced apoptosis, splicing of mCLCA2 is disrupted, and the mCLCA1 message is down-regulated. In addition, mCLCA2 promotes apoptosis in serum-starved mammary epithelial cells (27). hCLCA2 expression is lost in breast cancer and in tumorigenic breast cancer cell lines (28). Moreover, hCLCA1 and hCLCA4 transcription is down-regulated in 80% of colorectal carcinomas (29).
CLCA family members may also be involved in asthma and cystic fibrosis. Interleukin-13, a cytokine involved in the pathogenesis of asthma, enhances the calcium-activated chloride conductance in human bronchial epithelial cells (30), and hCLCA1 expression is likewise up-regulated in patients with bronchial asthma (31, 32). Overexpression of interleukin-9 in transgenic mice results in the induction of mCLCA3 in lung epithelium and an asthma phenotype (30, 33). Additionally, CLCA proteins may have a role in compensating for the pathology of cystic fibrosis given the up-regulation of a calcium-activated chloride conductance in cystic fibrosis transmembrane conductance regulator (−/−) mice with prolonged survival (34, 35).
Examination of the human and mouse CLCA loci indicates that mCLCA5 and mCLCA6 are the previously unidentified murine counterparts of hCLCA2 and hCLCA4 (36). The importance of cloning mCLCA5 and mCLCA6 is, thus, apparent in terms of the interesting roles of their human homologues. mCLCA5 is highly homologous to hCLCA2, a protein involved in the vascular arrest of lung metastatically competent cancer cells (22, 25). Both hCLCA2 and mCLCA5 have β4 integrin binding motifs (26). These motifs in hCLCA2 are directly involved in the adhesion of cancer cells (26). hCLCA2 has also been reported to mediate a calcium-activated chloride current when expressed in HEK293 cells (19). mCLCA6 is highly homologous to hCLCA4. The function of hCLCA4 (previously referred to as hCaCC-2) has not been reported, but it is expressed at high levels in brain, a unique property for a CLCA family member (37). In the context of determining the role and potential functional homology of mCLCA5 and mCLCA6 to other family members, we chose to focus on their tissue expression patterns, biochemical characteristics, and electrophysiological properties.
We report here the cloning of two new mouse CLCA family members: mCLCA5 and mCLCA6. mCLCA5 has ubiquitous expression with high levels in eye and spleen, whereas mCLCA6 is highly expressed in intestine and stomach with trace levels of expression in eye, liver, and spleen. mCLCA6 is also expressed as a splice variant in intestine and stomach. We transiently expressed the three proteins in tsA201 cells and were able to detect each by Western blot. Examination of the glycosylation states of mCLCA5, mCLCA6, and the mCLCA6 splice variant by in vitro translation in the presence of microsomal membranes indicates that mCLCA6 and its splice variant are glycosylated. We also show evidence that expression of all of these proteins results in calcium-activated chloride currents.
EXPERIMENTAL PROCEDURES
Cloning of mCLCA5 and mCLCA6
RNA was extracted from B6SJL/J hybrid mouse eye and large intestine with a Tissue-Tearor™ homogenizer (Biospec Products, Inc., Bartlesville, OK) using Trizol (Invitrogen) with its standard protocol. 1 μg of RNA (eye for mCLCA5, intestine for mCLCA6) was used for RT-PCR. Gene-specific cDNA and PCR product were generated in the same tube (50 °C for 30 min; 94 °C for 2 min; 40 cycles at 94 °C for 15 s, 55 °C for 30 s, and 72 °C for 3 min 10 s; with a final extension at 72 °C for 10 min) with a Superscript One-Step RT-PCR for Long Templates kit (Invitrogen). Primers were designed based on predicted mRNAs for mouse homologues of hCLCA2 and hCLCA4 as reported in GenBank™. mCLCA5 primers are: forward, 5′-TCCCCGTGAATAAACCAGAGG-3′, and reverse, 5′-GTAAAGGTCATGTCCATCCTAC-3′. mCLCA6 primers are: forward, 5′-CTCTGGGTGACCAGGCTGGA-3′, and reverse, 5′-TTCATGATGACATGGGATATG-3′. RT-PCR products were subjected to a subsequent PCR (94 °C for 2 min; 40 cycles at 94 °C for 15 s, for 55 °C 30 s, 68 °C for 3 min; with a final extension at 68 °C 10 min) with a high fidelity enzyme, Platinum Pfx Polymerase (Invitrogen), to introduce restriction sites. mCLCA5 primers are: forward, 5′-GCTAAGATCTCATGACCCACAGGGACAGCAC-3′, and reverse, 5′-GCTAGTCGACGCTAGAAATTTTGTCTCATTCTCT-3′. mCLCA6 primers are: forward, 5′-AAAACTGCAGATGGCTTTCTCCAGAGGGCC-3′, and reverse, 5′-TGCTACCCCGGGAAAATTTTGTTATAGCTGCTGA-3′. Purified and restriction-digested products were then subcloned into pEGFP-N1 (BD Biosciences). mCLCA5 was inserted at BglII and SalI restriction sites, and mCLCA6 was inserted at PstI and SmaI sites. The pEGFP-N1 vector expresses proteins as EGFP fusions, with EGFP at the carboxyl terminus. Constructs were sequenced by the chain termination method with an ABI Prism 377 (Applied Biosystems, Foster City, CA). The mCLCA6 splice variant was identified by comparison of the sequence with the predicted exon/intron pattern in National Center for Biotechnology Information LocusLink and the presence of gt/ag splice sites.
Tissue Distribution of mCLCA5 and mCLCA6 by RT-PCR
RT-PCR was done as previously described with primers specific to each isoform and chosen to amplify across exon boundaries to rule out amplification from genomic DNA. In addition, mCLCA6 primers were chosen to flank exon 8, which is removed in the splice variant. Cycling conditions were (50 °C for 30 min; 94 °C for 2 min; 30 cycles for mCLCA5 or 35 cycles for mCLCA6 at 94 °C for 15 s, 58 °C for 30 s, 72 °C for 35 s; with a final extension at 72 °C 10 min). mCLCA5 primers are: forward, 5′-AAATCCGAGCCTCGCTGCA-3′, and reverse, 5′-CCAGTCTGCCACGTGACTAG-3′. mCLCA6 primers are: forward, 5′-TATGTGCCTAGTTCTTGATG-3′, and reverse, 5′-CTTCTGGGTGATATCAGCATTTTCTGAAGCCAG-3′. The predicted amplified fragments of mCLCA5, mCLCA6, and the mCLCA6 splice variants are ~550, 540, and 360 base pairs, respectively. Glyceraldehyde-3-phosphate dehydrogenase was amplified as a control for RNA quality and quantity. A negative control with water instead of RNA was also included. mCLCA5 and mCLCA6 products were sequenced to establish primer specificity. The smaller mCLCA6 splice variant band was verified by using splice variant specific primers (primers flanking the new exon boundaries) for RT-PCR.
Subcloning into pcDNA3.1 Expression Vector
The mCLCA5 and mCLCA6 open reading frames were subcloned directly from pRc/CMV (Invitrogen) with NotI and XbaI (Promega, Madison, WI) into pcDNA3.1 (Invitrogen). The 5′ and 3′ ends of the inserts were sequenced as previously described.
Expression of EGFP-tagged Protein Constructs in tsA201 Cells, Immunoprecipitation, and Immunoblotting
pEGFP-N1 constructs of mCLCA5, mCLCA6, and the mCLCA6 splice variant were used to transfect tsA201 cells by the calcium phosphate precipitate method. For immunoprecipitation, cells were harvested 48 h after transfection in the presence of complete, mini-protease inhibitor mixture (Roche Applied Science). Lysates were precleared with protein A-agarose (Santa Cruz Biotechnology, Santa Cruz, CA) at 4 °C for 30 min, then incubated for 4 h with protein A-agarose that had been incubated with a BD Living Colors™ full-length Aequorea victoria polyclonal GFP (BD Biosciences) antibody for 1 h. Immunoprecipitates were resolved by SDS-PAGE on a 5–15% gradient gel, transferred to PVDF, and analyzed by Western blotting with a BD Living Colors™ A. victoria monoclonal GFP antibody (BD Biosciences) as primary antibody and a horseradish peroxidase-conjugated goat anti-mouse antibody as secondary antibody (Promega). The blot was developed with Supersignal® West Pico chemiluminescence reagents (Pierce).
In Vitro Translation and N-Glycosidase F Assay
The TNT® Coupled Reticulocyte Lysate system (Promega) was used to transcribe and translate mCLCA5, mCLCA6, and the mCLCA6 splice variant from pcDNA3.1 in the presence of l-[35S]methionine (MP Biomedicals, Inc., Irvine, CA). Reactions were incubated at 30 °C for 1 h with or without canine pancreatic microsomal membranes (Promega). Samples were run on a 5–15% gradient SDS-polyacrylamide gel. The gel was then dried and exposed to a phosphorscreen for 3 days. A PGNase (New England Biolabs, Inc., Beverly, MA) digestion was performed on 200 μg of lysate from tsA201 cells transfected with mCLCA6 pEGFP-N1. The standard protocol for digestion was followed, and 60 μg of untreated or treated lysate were resolved on a 5–15% gradient SDS-polyacrylamide gel. Samples were then immunoblotted as described in the previous section.
Microscopy
Cells were transfected as above with pEGFP-N1 constructs of mCLCA5, mCLCA6, and the mCLCA6 splice variant. Cells were fixed with VECTASHIELD® Hard Set™ Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA) 48 h post-transfection. Images were obtained on a Nikon Microphot FX microscope equipped with an Optronics digital camera and Bioquant imaging software.
Electrophysiology
pEGFP-N1 constructs of mCLCA5, mCLCA6, and the mCLCA6 splice variant were used to transfect HEK293 cells plated on coverslips using GenePorter transfection reagent (Gene Therapy Systems, San Diego, CA). Mock-transfected control cells were transfected with an empty vector containing only pEGFP-N1. Successfully transfected cells were identified by EGFP fluorescence 24–48 h after transfection.
Whole cell recordings were obtained using patch electrodes pulled from borosilicate pipettes (1.2-mm outer diameter, 0.95-mm inner diameter, with internal filament) using a Narishige PP-830 vertical puller. The recording pipettes had tips of 1–1.5-μm outer diameter (R = 8–12 megaohms) and were filled with a solution containing 98 mm KCH3SO4, 44 mm KCl, 3 mm NaCl, 5 mm HEPES, 3 mm MgCl2, 1 mm CaCl2, 3 mm EGTA, 2 mm glucose, 1 mm MgATP, 1 mm GTP, 1 mm reduced glutathione (pH 7.8). Free Ca2+ in this solution was estimated to be 57 nm using MaxChelator. Cells were voltage-clamped at −50 mV using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Test pulses were applied, and currents were acquired using PClamp 8.2 with a Digidata 1200 interface (Axon Instruments). During recording, cells were perfused at room temperature using a single pass, gravity-feed perfusion system (1 ml/min) with an oxygenated medium containing 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, 10 mm glucose (pH 7.4). Ionomycin and niflumic acid were diluted into this solution from stock solutions prepared in dimethylsulfoxide. Experiments were conducted at room temperature. All chemicals were obtained from Sigma except KCH3SO4, which was obtained from Pfaltz and Bauer (Waterbury, CT).
Nucleotide Sequence Accession Numbers
The GenBank™ accession number for the mCLCA6 mRNA is AY560902 and for the mCLCA6 splice variant mRNA is AY560903.
RESULTS
Cloning and Sequence Analysis of mCLCA5, mCLCA6, and a Splice Variant of mCLCA6
To clone new mCLCA family members, GenBank™ was searched using BLAST (basic local alignment search tool) with sequences of known CLCAs. A predicted mRNA for a mouse homologue of hCLCA2 and of hCLCA4 was identified. RT-PCR with primers to amplify the complete open reading frame of each transcript was utilized. The mCLCA homologue of hCLCA2 (mCLCA5) was amplified from eye, whereas the mCLCA homologue of hCLCA4 (mCLCA6) was amplified from intestine. Direct sequencing of amplified products indicated that both transcripts agreed with the predicted mRNAs. The open reading frame of mCLCA5 is about 2.8 kilobases. mCLCA5 was also recently cloned by R. C. Elble and J. R. Beckley (GenBank™ accession number AY161007). The mCLCA6 open reading frame is also about 2.8 kilobases. In addition, a splice variant of mCLCA6 with an open reading frame of about 2.5 kilobases was identified.
Alignment of all known mCLCA isoforms indicates a high degree of homology between mCLCA5, mCLCA6, and other family members (Fig. 1). At the amino acid level, mCLCA5 is 69% identical to hCLCA2, and mCLCA6 is 69% identical to hCLCA4. Both proteins display conserved family features, including a symmetrical cysteine-rich region, a signal peptidase cleavage site, a site for monobasic proteolytic cleavage (38), sites for N-linked glycosylation,2 and consensus sites for phosphorylation by PKA, PKC, and Ca2+/calmodulin-dependent protein kinase II. The symmetrical cysteine-rich region for mCLCA5 is CX9CX4CX4X9C and for mCLCA6 is CX12CX4CX4CX12C. Assembly of a phylogenetic tree based on the complete sequences of all known CLCA proteins shows that mCLCA5 and mCLCA6 are highly homologous to hCLCA2 and hCLCA4, respectively (Fig. 2).
Fig. 1. Alignment of all mouse CLCA isoforms.
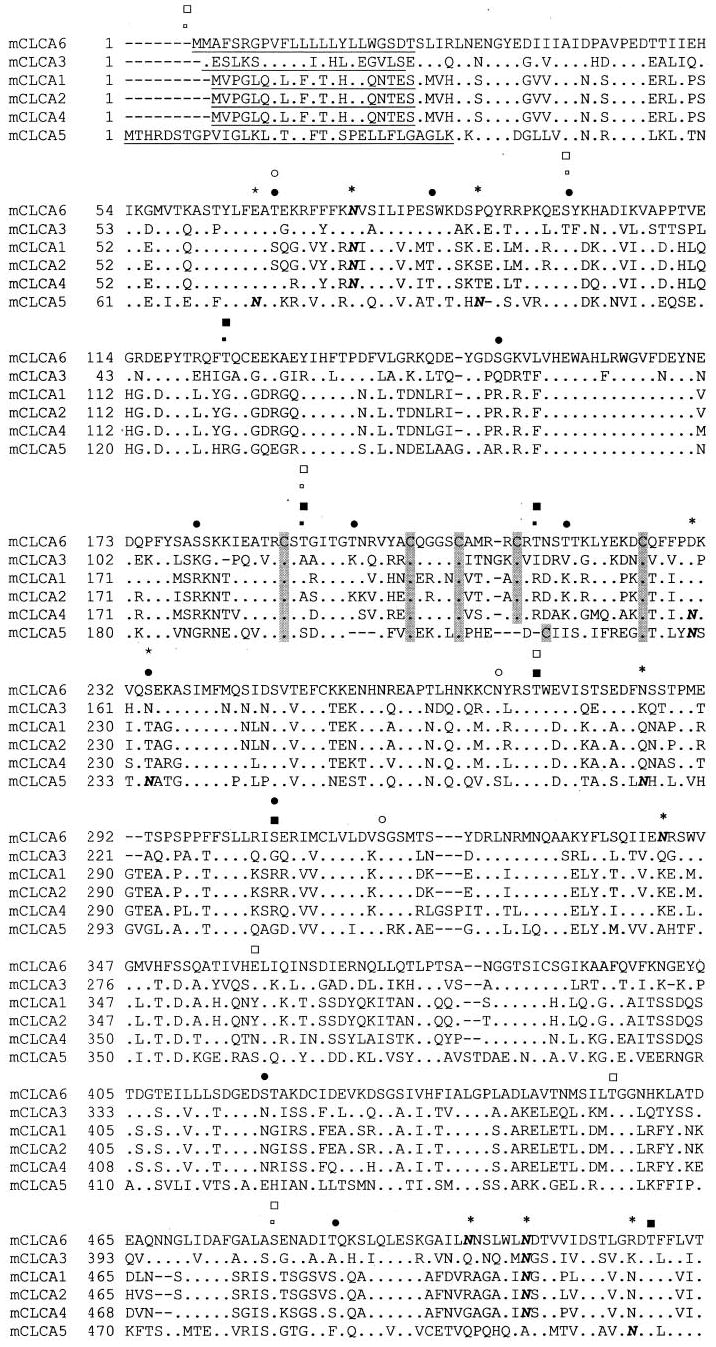

Consensus sites for phosphorylation by Ca2+/calmodulin-dependent protein kinase II, PKA, and PKC are marked for mCLCA5 and mCLCA6 (small open square, mCLCA5 Ca2+/calmodulin-dependent protein kinase II; large open square, PKA; open circle, PKC; small filled square, mCLCA6 Ca2+/calmodulin-dependent protein kinase II; large filled square, PKA; filled circle, PKC (PhosphoBase version 2.0, Center for Biological Sequence Analysis). Sites are marked for all mCLCAs for monobasic proteolytic cleavage (↓), signal sequence (underlined) (SignalPV1.1, Center for Biological Sequence Analysis), and multiple cysteine motif (C). Additionally, consensus sites for N-linked glycosylation are indicated (N with * overhead). The alignment was generated by the Clustal method.
Fig. 2. Phylogenetic tree showing the relation of all CLCAs.
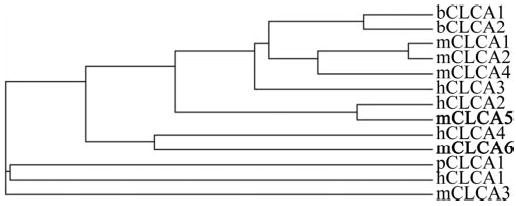
The dendogram was generated by Clustal method alignment on the European Bioinformatics Institute website with complete amino acid sequences for all CLCAs.
Analysis of the mCLCA6 splice variant sequence indicates a loss of exon 8 and a portion of exon 10. The removal of exon 8 causes a shift in the reading frame. This generates a new sequence for exon 9 that we call exon 9a. The reading frame then reverts back to the original frame in the remaining portion of exon 10 (Fig. 3).
Fig. 3. Diagram of the mCLCA6 splice variant.
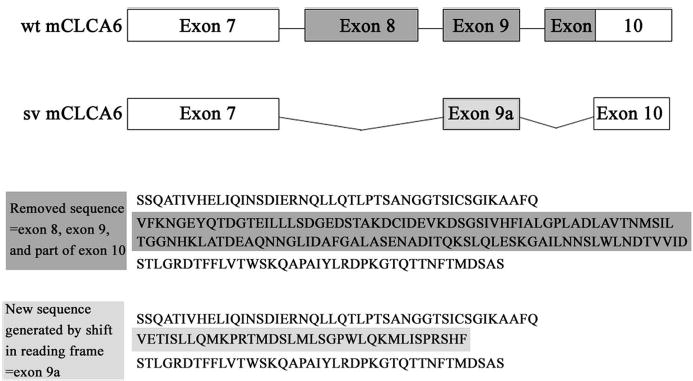
Exon 8 and a portion of exon 10 are removed in the mCLCA6 splice variant (mCLCA6sv). The removal of exon 8 (starts at amino acid 397) causes a shift in the reading frame, thus changing the sequence of exon 9 (which we call exon 9a). The removal of a portion of exon 10 causes a reversion to the normal reading frame in the latter portion of exon 10. Shown below the splice variant schematic is a diagram of the amino acid sequence removed from the splice variant (sv) (as compared with wild type (wt, dark gray)) and the new sequence generated in exon 9a (light gray).
Tissue Expression of mCLCA5 and mCLCA6
RT-PCR was performed on multiple tissues to determine the tissue expression pattern of mCLCA5, mCLCA6, and the mCLCA6 splice variant (Fig. 4). mCLCA5 showed the highest levels of expression in eye and spleen, whereas mCLCA6 showed the highest levels in intestine and stomach. Low levels of mCLCA6 are also present in eye, liver, and spleen. The mCLCA6 splice variant, the smaller band on the RT-PCR gel, was detected in both intestine and stomach, although at lower levels than wild type.
Fig. 4. Tissue expression of mCLCA5 and mCLCA6 by RT-PCR.
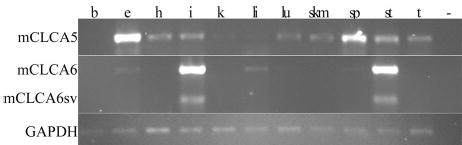
mCLCA5 shows the highest levels of expression in eye and spleen, whereas mCLCA6 is expressed predominantly in intestine and stomach. The lower band in the mCLCA6 intestine and stomach lanes represents the splice variant (mCLCA6sv). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) verifies RNA integrity and loading. b, brain; e, eye; h, heart; i, intestine; k, kidney; li, liver, lu, lung, skm, skeletal muscle; sp, spleen; st, stomach; t, testes; (−), negative control.
Biochemical Characterization of mCLCA5, mCLCA6, and mCLCA6 Splice Variant Proteins
The predicted full-length size for untagged mCLCA5 is 103.6 kDa, for mCLCA6 is 101.9 kDa, and for the mCLCA6 splice variant is 93.7 kDa. Lysates of tsA201 cells transiently transfected with EGFP-tagged versions of either mCLCA5, mCLCA6, or the mCLCA5 splice variant were immunoblotted for the corresponding protein. The EGFP tag is at the carboxyl terminus of all three proteins and adds 30 kDa to the size. Proteins of the CLCA family are generally processed from a ~125-kDa precursor to a ~90-kDa amino-terminal fragment and a ~35-kDa carboxyl-terminal fragment (15, 16, 18, 19, 40, 41). This cleavage likely occurs at the conserved monobasic proteolytic cleavage site, as indicated in Fig. 1. Only the cleaved carboxyl terminus of mCLCA6 was detected (~65 kDa; data not shown). Immunoprecipitation with a polyclonal GFP antibody was used to concentrate CLCAs for detection. A 155- and 65-kDa product for mCLCA5 and mCLCA6 and 145- and 65-kDa product for the mCLCA6 splice variant were detected by immunoblotting (Fig. 5A). The 165 kDa (145 for the splice variant) product corresponds to full-length EGFP-tagged protein and the 65 kDa to the cleaved carboxyl terminus. The additional increase in size as compared with the predicted size is likely due to glycosylation. These results are consistent with the proteolytic processing that occurs with other CLCA family members (15, 16, 18, 19, 40, 41).
Fig. 5. Biochemical characterization of mCLCA5, mCLCA6, and mCLCA6 splice variant proteins.
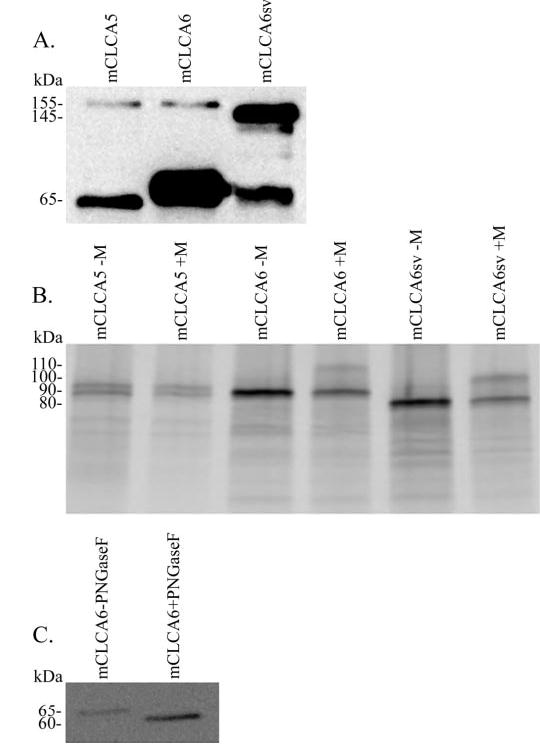
A, immunoblotting analysis of immunoprecipitated EGFP-tagged CLCAs from transfected tsA201 cells. mCLCA5 and mCLCA6 are detected as 65- and 155-kDa bands, whereas the mCLCA6 splice variant is detected as 65 and 145 kDa. The 65-kDa band corresponds to the cleaved carboxyl terminus, and the 155 (or 145)-kDa band corresponds to full-length protein. B, in vitro translation without (−M) and with (+M) microsomal membranes. l-[35S]Methionine-labeled proteins were resolved by SDS-PAGE and detected by phosphorscreen. mCLCA6 and the mCLCA6 splice variant (mCLCA6sv) show an increase in size due to glycosylation in the presence of microsomal membranes from 90 to 110 kDa for mCLCA6 or 80 to 100 kDa for the mCLCA6 splice variant. C, immunoblot analysis of a PGNase digestion of lysate from tsA201 cells transfected with EGFP-tagged mCLCA6. The mCLCA6 carboxyl terminus shows a decrease in size from 65 to 60 kDa due to the removal of N-linked glycosyl groups by PGNase.
To study the glycosylation states of all three CLCA proteins, cDNAs were translated in vitro and resolved by SDS-PAGE. mCLCA5 and mCLCA6 both generated products of 90 kDa, whereas the mCLCA6 splice variant produced a 80-kDa fragment (Fig. 5B). In vitro translation in the presence of canine pancreatic microsomal membranes led to a shift in size due to glycosylation for mCLCA6 to 110 kDa and for the mCLCA6 splice variant to 100 kDa, whereas mCLCA5 showed no change in size. mCLCA5 produced a doublet at 90 kDa in the presence and absence of microsomal membranes.
To further examine the glycosylation state of these proteins, we performed a PNGase digestion on lysate from tsA201 cells transfected with EGFP-tagged mCLCA6. For this experiment, we only analyzed mCLCA6, as it is the only protein of the three that can be detected in total lysate. PNGase specifically removes N-linked glycosyl groups. Treatment with PNGase caused a reduction in size of the EGFP-tagged mCLCA6 carboxyl terminus from 65 to 60 kDa, indicating removal of glycosyl groups (Fig. 5C).
Microscopy and Electrophysiological Characteristics
The three EGFP-tagged mCLCA proteins were expressed in tsA201 cells by transient transfection. After 48 h, microscopy revealed membrane expression of EGFP-tagged proteins (Fig. 6). Expression levels of mCLCA6 appeared highest of the three subtypes. The mCLCA6 splice variant does not appear to be as sharply localized to the membrane as mCLCA5 or mCLCA6. Similar expression patterns were observed after expression in HEK293 cells (not shown).
Fig. 6. tsA201 cells expressing EGFP-tagged mCLCA5 (A), mCLCA6 (B), and mCLCA6 splice variant (mCLCA6sv) (C).
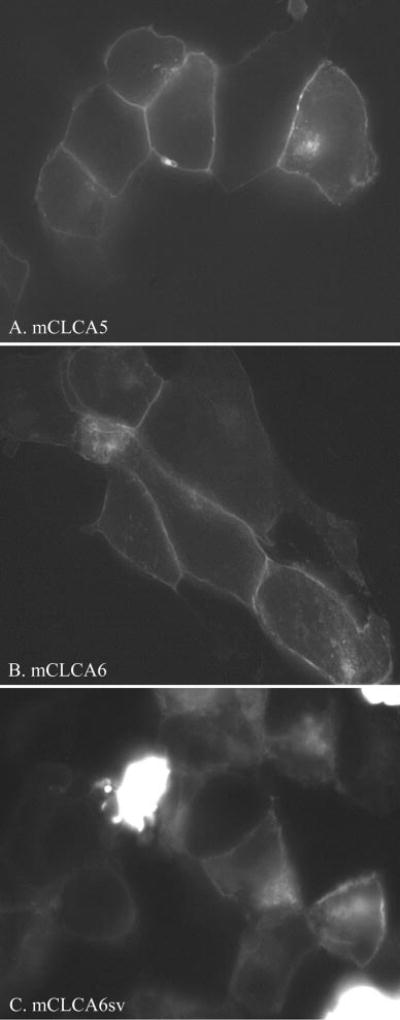
Note the apparent membrane localization of all three proteins. mCLCA5 and mCLCA6 show the sharpest membrane staining and are present at cell-cell junctions.
Whole cell recordings were obtained from HEK293 CLCA-expressing cells. A calcium ionophore, ionomycin (10 μm), was superfused to stimulate an increase in intracellular Ca2+. Fig. 7A shows whole cell currents evoked by a series of voltage steps (150 ms, −70 to +70 mV) applied from a holding potential of −50 mV in a cell expressing mCLCA6 before application of ionomycin. As shown in Fig. 7B, application of ionomycin evoked a conductance increase and an inward current at the holding potential of −50 mV. Inward currents measured 2–3 min after ionomycin application averaged −66 ± 38 pA, −110 ± 41 pA, and −60 ± 32 pA in cells transfected with mCLCA5, mCLCA6, and the mCLCA6 splice variant, respectively. To obtain the current/voltage relationship for the inward current stimulated by ionomycin, we subtracted the control currents from those obtained in the presence of ionomycin. Fig. 7C shows the steady state current/voltage relationship for the resulting difference current in this cell. The predicted Cl− equilibrium potential (ECl) under these experimental conditions was −23.7 mV, close to the mean of the reversal potentials for the ionomycin-evoked currents in cells expressing mCLCA5 (Erev = −21.9 ± 2.9 mV), mCLCA6 (−21.3 ± 2.3 mV), and the splice variant of mCLCA6 (−19.1 ± 2.4 mV; Fig. 7D). The slight differences in reversal potentials among the three CLCA subtypes were not significant (p = 0.8, one-way analysis of variance). In untransfected control cells, ionomycin evoked an inward current that required 6 min to attain its peak and reversed at a substantially more positive value of +24 mV. This slowly developing current in control cells may reflect currents arising from insertion of the calcium ionophore into the membrane. Inward currents exhibiting positive reversal potential were also observed in mock-transfected control cells (data not shown). As further evidence that mCLCA5, mCLCA6, and the mCLCA6 splice variant encode calcium-activated chloride channels, inward currents evoked by ionomycin in CLCA-expressing but not mock-transfected or untransfected control cells were strongly inhibited by the calcium-activated chloride channel blocker, niflumic acid (0.1 mm) (Fig. 8).
Fig. 7. Cl− currents were evoked in CLCA-expressing cells by ionomycin-stimulated calcium influx.
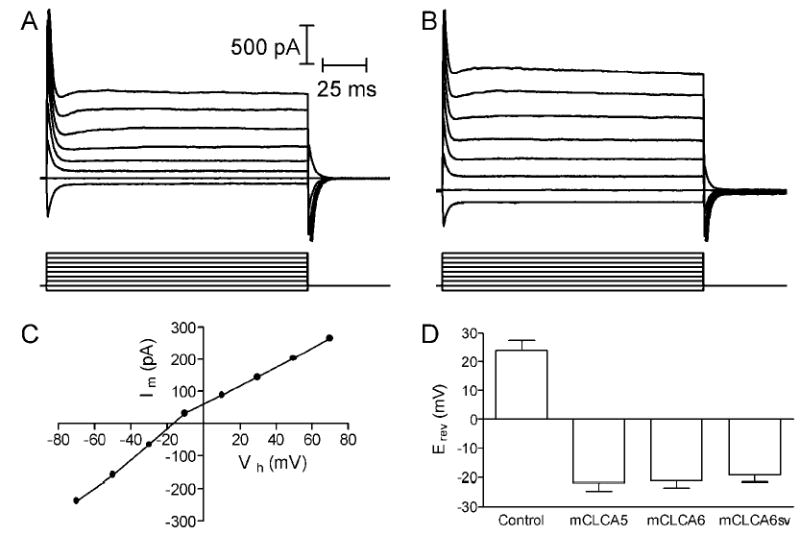
A, currents evoked in mCLCA6-expressing HEK293 cells by a series of voltage steps (−70 to +70 mV, 150 ms). B, currents evoked in the same cell by voltage steps provided after ionomycin (10 μm) application. Ionomycin stimulated an inward current at the holding potential of −50 mV and increased the amplitude of currents evoked by the various voltage steps. C, current/voltage relationship for the steady state difference current. The difference current was obtained by subtracting step-evoked currents obtained before the application of ionomycin from the currents obtained after ionomycin application. The difference current shows a positive slope conductance and reversed around −17 mV. D, mean reversal potentials for currents evoked by ionomycin in untransfected control cells (n = 10) and cells expressing mCLCA5 (n = 7), mCLCA6 (n = 7), and mCLCA6 splice variant (mCLCA6sv) (n = 7). Current/voltage relationships used to calculate reversal potentials were measured at the peak of the ionomycin-evoked current, which was attained 3 min after ionomycin application in transfected cells and 6 min after ionomycin application in untransfected control cells.
Fig. 8. Niflumic acid inhibits ionomycin-evoked currents in CLCA-expressing cells.
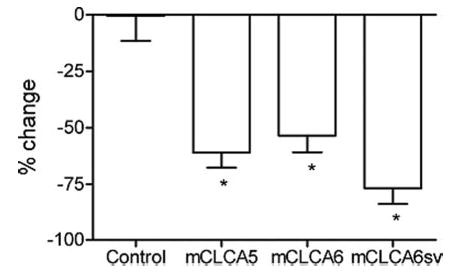
Ionomycin-evoked currents (Vh = − 50 mV) were significantly inhibited by niflumic acid (0.1 mm) in HEK293 cells expressing mCLCA5 (n = 7, p = 0.0004, t test), mCLCA6 (n = 7, p < 0.0001), and the mCLCA6 splice variant (mCLCA6sv) (n = 7, p < 0.0001) but not in untransfected (n = 10, p = 0.97) or mock-transfected (n = 4, p = 0.47, not shown) control cells.
DISCUSSION
The CLCA family of calcium-activated chloride channel proteins continues to grow. Here we describe the cloning and initial characterization of two new mCLCAs, mCLCA5 and mCLCA6. mCLCA5 is highly homologous to hCLCA2, and mCLCA6 is highly homologous to hCLCA4. We have also described the expression of the first functional splice variant in the family, a splice variant of mCLCA6. These new family members share features with other CLCA proteins and include a symmetrical cysteine-rich region that may be important for protein folding or metal ion incorporation. In addition, there is a signal peptidase cleavage site, a site for monobasic proteolytic cleavage, and consensus sites for N-linked glycosylation and phosphorylation by PKA, PKC, and Ca2+/calmodulin-dependent protein kinase II.
The monobasic proteolytic cleavage site is necessary for the processing of all family members to an ~90-kDa amino-terminal fragment and a 35-kDa carboxyl-terminal fragment. The size of these fragments varies slightly among family members and is often altered by glycosylation. This same proteolytic processing occurs with mCLCA5, mCLCA6, and the mCLCA6 splice variant. The functional importance of this processing has not yet been determined, although it seems for some other family members that both fragments are present on the cell surface (18, 40). Extracellular biotinylation of bovine aortic endothelial cells expressing bCLCA2 (Lu-ECAM-1) indicates that both the 90- and 35-kDa fragments are on the cell surface (40). Cell surface labeling of HEK293 cells expressing hCLCA1 with c-Myc tags in both the 90- and 35-kDa fragments also shows that both are on the cell surface (18).
Although this processing is conserved for all known CLCA family members, the structure of these proteins is unclear. hCLCA1 has been proposed to have 4 transmembrane domains in the 90kDa portion, with the 35-kDa fragment extracellularly associated with it (18). On the other hand, hCLCA2 has been proposed to have five transmembrane domains, with 3 in the 90-kDa fragment and 2 in the 35-kDa fragment (19). Interestingly, it has been shown that bCLCA1 still functions the same as wild type channels when the amino and carboxyl termini are truncated, leaving only the transmembrane domains (42). It may be that the termini are important for modulation of protein function or play a role in cell-cell adhesion. Whether these proteins form multimers or associate with other subunit proteins also remains to be determined. Co-expression of a large conductance K+ channel (BK channel) β-subunit with mCLCA1 in HEK293 cells alters the kinetics and Ca2+ sensitivity of the channel, and the two proteins interact in a mammalian two-hybrid system. This is the only published study that has examined potential accessory subunit interactions with a CLCA family member (43).
Glycosylation is another important modification for CLCAs. So far bCLCA1, bCLCA2 (Lu-ECAM-1), mCLCA1, mCLCA3, hCLCA1, hCLCA2, hCLCA3, and in this study mCLCA5 and mCLCA6 are glycosylated in an in vitro translation system in the presence of microsomal membranes or when expressed in cells (15, 16, 18, 19, 40, 41, 44). This conserved post-translational modification indicates that these proteins are likely cell surface molecules.
Each new isoform has a unique tissue expression pattern, but there are some similarities with the localization of other family members. Importantly, both mCLCA5 and mCLCA6 are expressed in intestine as are most other CLCA isoforms. The high level of mCLCA5 expression in eye could parallel the role of hCLCA2 in cornea. Both mCLCA5 and hCLCA2 have two β4 integrin binding domains (26), and hCLCA2 is localized to the basal epithelium of the cornea, where it may interact with β4 integrin and help with basal cell-basement membrane adhesion (39). Additionally, mCLCA5 has a high level of expression in spleen and is also expressed at lower levels in a number of other tissues. mCLCA6 is highly expressed in intestine and stomach, and the mCLCA6 splice variant is also present in both of these tissues. mCLCA6 is also weakly expressed in eye, liver, and spleen. Although hCLCA4 is expressed in the brain, mCLCA6 is not. Analysis of whole brain by Northern blot and of various regions of the brain by RNA dot blot indicates that hCLCA4 is expressed in many parts of the brain (37). Our RT-PCR analysis of whole brain did not detect mCLCA6. The two proteins may not be functional homologues or this difference may be due to differences in experimental methodology. It is difficult to determine the significance of the expression patterns for each family member until more functional studies are done with CLCAs.
With our description of the first functionally expressed CLCA splice variant, it is clear that CLCAs undergo splicing. The mCLCA6 splice variant loses exon 8 and a portion of exon 10. The loss of exon 8 causes a reading frameshift that generates a new amino acid sequence, exon 9a. The reading frame then reverts to the original frame in the latter portion of exon 10. The new exon that is generated, exon 9a, does not seem to have any unique features, and no obviously important consensus sites are removed with the removal of exon 8 and part of 10. The mCLCA6 splice variant may have subtle differences in function from wild type.
Functionally, our data indicate a role for mCLCA5, mCLCA6, and the mCLCA6 splice variant in the production of calcium-activated chloride currents in HEK293 cells. The addition of ionomycin to cells expressing each of the three proteins induced a current that was different from that produced in untransfected or mock-transfected cells. Furthermore, the reversal potential for these ionomycin-evoked currents correlates with the predicted Cl− equilibrium potential. Niflumic acid, a chloride channel blocker, strongly inhibited the ionomycin-stimulated current. These three pieces of data indicate the presence of a calcium-activated chloride current in CLCA-expressing cells. The mCLCA6 splice variant does not behave differently from wild type mCLCA6 in the context of these experiments. Further studies will be necessary to determine how each of these proteins function and how the splice variant differs from wild type protein.
These electrophysiological studies correlate well with data reported for other family members. hCLCA1-expressing HEK293 cells produced ionomycin-evoked currents that could be inhibited by niflumic acid (18). The addition of ionomycin to hCLCA2-expressing HEK293 cells induces a current that is absent in untransfected or mock-transfected cells. A current could also be induced in these cells by the presence of 2 mm Ca2+ in the pipette and the bath that could be inhibited by niflumic acid (19). Similar results were obtained with mCLCA1-expressing HEK293 cells (16). Interestingly, bCLCA1 produced similar calcium-activated currents in COS-7 cells and Xenopus oocytes (15). This indicates that CLCA proteins produce these currents in multiple cell types and supports the idea that they are conducting the ions themselves and not modulating some other chloride-conducting protein.
The data we report here expands the CLCA family of proteins. Identification of mCLCA5 and mCLCA6 completes the identification of mCLCAs at the mouse CLCA locus (36). The functions of this large and diverse family are not yet fully understood, although it is clear that they have roles in the production of calcium-activated chloride currents and cell adhesion. The splice variant described here provides a way to further diversify the family, perhaps to produce subtle differences in function. The cloning of these mouse homologues presents the opportunity to learn more about the CLCA family and the role of these proteins in cancer and normal cell physiology.
Acknowledgments
We thank Nancy Philp for assistance and advice on imaging. We also thank Peying Fong, Achim Gruber, and Dick Horn for useful and helpful discussions.
Footnotes
The abbreviations used are: CLCA, calcium-activated chloride channel; HEK293, human embryonic kidney cell line 293; tsA201, transformed HEK293 cell line stably expressing an SV40 temperature-sensitive T antigen; GFP, green fluorescent protein; EGFP, enhanced GFP; RT, reverse transcription; PKA, cAMP-dependent protein kinase; PKC, protein kinase C; b, bovine; m, murine; h, human; p, porcine; PGNase, peptide: N-glycosidase F.
R. Gupta, E. Jung, and S. Brunak, manuscript in preparation.
References
- 1.Huang SJ, Fu WO, Chung YW, Zhou TS, Wong PY. Am J Physiol. 1993;264:C794–C802. doi: 10.1152/ajpcell.1993.264.4.C794. [DOI] [PubMed] [Google Scholar]
- 2.Evans MG, Marty A. J Physiol (Lond) 1986;378:437–460. doi: 10.1113/jphysiol.1986.sp016229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arreola J, Park K, Melvin JE, Begenisich T. J Physiol (Lond) 1996;490:351–362. doi: 10.1113/jphysiol.1996.sp021149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cliff WH, Frizzell RA. Proc Natl Acad Sci U S A. 1990;87:4956–4960. doi: 10.1073/pnas.87.13.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amedee T, Large WA, Wang Q. J Physiol (Lond) 1990;428:501–516. doi: 10.1113/jphysiol.1990.sp018224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clapp LH, Turner JL, Kozlowski RZ. Am J Physiol (Lond) 1996;270:H1577–H1584. doi: 10.1152/ajpheart.1996.270.5.H1577. [DOI] [PubMed] [Google Scholar]
- 7.Wang Q, Hogg RC, Large WA. J Physiol (Lond) 1992;451:525–537. doi: 10.1113/jphysiol.1992.sp019177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hume RI, Thomas SA. J Physiol (Lond) 1989;417:241–261. doi: 10.1113/jphysiol.1989.sp017799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes S, Hille B. J Gen Physiol. 1989;94:719–743. doi: 10.1085/jgp.94.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallani M, Lynch JW, Barry PH. J Membr Biol. 1998;161:163–171. doi: 10.1007/s002329900323. [DOI] [PubMed] [Google Scholar]
- 11.Sun H, Tsunenari T, Yau KW, Nathans J. Proc Natl Acad Sci U S A. 2002;99:4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qu Z, Wei RW, Mann W, Hartzell HC. J Biol Chem. 2003;278:49563–49572. doi: 10.1074/jbc.M308414200. [DOI] [PubMed] [Google Scholar]
- 13.Tsunenari T, Sun H, Williams J, Cahill H, Smallwood P, Yau KW, Nathans J. J Biol Chem. 2003;278:41114–41125. doi: 10.1074/jbc.M306150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qu Z, Fischmeister R, Hartzell C. J Gen Physiol. 2004;123:327–340. doi: 10.1085/jgp.200409031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham SA, Awayda MS, Bubien JK, Ismailov II, Arrate MP, Berdiev BK, Benos DJ, Fuller CM. J Biol Chem. 1995;270:31016–31026. doi: 10.1074/jbc.270.52.31016. [DOI] [PubMed] [Google Scholar]
- 16.Gandhi R, Elble RC, Gruber AD, Schreur KD, Ji HL, Fuller CM, Pauli BU. J Biol Chem. 1998;273:32096–32101. doi: 10.1074/jbc.273.48.32096. [DOI] [PubMed] [Google Scholar]
- 17.Elble RC, Ji G, Nehrke K, DeBiasio J, Kingsley PD, Kotlikoff MI, Pauli BU. J Biol Chem. 2002;277:18586–18591. doi: 10.1074/jbc.M200829200. [DOI] [PubMed] [Google Scholar]
- 18.Gruber AD, Elble RC, Ji HL, Schreur KD, Fuller CM, Pauli BU. Genomics. 1998;54:200–214. doi: 10.1006/geno.1998.5562. [DOI] [PubMed] [Google Scholar]
- 19.Gruber AD, Schreur KD, Ji HL, Fuller CM, Pauli BU. Am J Physiol. 1999;276:C1261–C1270. doi: 10.1152/ajpcell.1999.276.6.C1261. [DOI] [PubMed] [Google Scholar]
- 20.Loewen ME, Bekar LK, Gabriel SE, Walz W, Forsyth GW. Biochem Biophys Res Commun. 2002;298:531–536. doi: 10.1016/s0006-291x(02)02498-1. [DOI] [PubMed] [Google Scholar]
- 21.Loewen ME, Smith NK, Hamilton DL, Grahn BH, Forsyth GW. Am J Physiol Cell Physiol. 2003;285:C1314–C1321. doi: 10.1152/ajpcell.00210.2003. [DOI] [PubMed] [Google Scholar]
- 22.Abdel-Ghany M, Cheng HC, Elble RC, Pauli BU. J Biol Chem. 2001;276:25438–25446. doi: 10.1074/jbc.M100478200. [DOI] [PubMed] [Google Scholar]
- 23.Pauli, B. U., Goodwin, A. E., and Abdel-Ghany, M. (1995) in Adhesion Molecules and the Lung (Ward, P. A., and Fantone, J. C., eds) pp. 211–241, Marcel Dekker, Inc., New York
- 24.Pauli, B. U., and Lin, H. (1997) in Encyclopedia of Cancer (Bertino, J. R., ed) Academic Press Inc., San Diego, CA
- 25.Abdel-Ghany M, Cheng HC, Elble RC, Pauli BU. J Biol Chem. 2002;277:34391–34400. doi: 10.1074/jbc.M205307200. [DOI] [PubMed] [Google Scholar]
- 26.Abdel-Ghany M, Cheng HC, Elble RC, Lin H, DiBiasio J, Pauli BU. J Biol Chem. 2003;278:49406–49416. doi: 10.1074/jbc.M309086200. [DOI] [PubMed] [Google Scholar]
- 27.Elble RC, Pauli BU. J Biol Chem. 2001;276:40510–40517. doi: 10.1074/jbc.M104821200. [DOI] [PubMed] [Google Scholar]
- 28.Gruber AD, Pauli BU. Cancer Res. 1999;59:5488–5491. [PubMed] [Google Scholar]
- 29.Bustin SA, Li SR, Dorudi S. DNA Cell Biol. 2001;20:331–338. doi: 10.1089/10445490152122442. [DOI] [PubMed] [Google Scholar]
- 30.Atherton H, Mesher J, Poll CT, Danahay H. Naunyn-Schmiedeberg’s. Arch Pharmacol. 2003;367:214–217. doi: 10.1007/s00210-002-0668-1. [DOI] [PubMed] [Google Scholar]
- 31.Toda M, Tulic MK, Levitt RC, Hamid Q. J Allergy Clin Immunol. 2002;109:246–250. doi: 10.1067/mai.2002.121555. [DOI] [PubMed] [Google Scholar]
- 32.Hoshino M, Morita S, Iwashita H, Sagiya Y, Nagi T, Nakanishi A, Ashida Y, Nishimura O, Fujisawa Y, Fujino M. Am J Respir Crit Care Med. 2002;165:1132–1136. doi: 10.1164/ajrccm.165.8.2107068. [DOI] [PubMed] [Google Scholar]
- 33.Zhou Y, Dong Q, Louahed J, Dragwa C, Savio D, Huang M, Weiss C, Tomer Y, McLane MP, Nicolaides NC, Levitt RC. Am J Respir Cell Mol Biol. 2001;25:486–491. doi: 10.1165/ajrcmb.25.4.4578. [DOI] [PubMed] [Google Scholar]
- 34.Rozmahel R, Wilschanski M, Matin A, Plyte S, Oliver M, Auerbach W, Moore A, Forstner J, Durie P, Nadeau J, Bear C, Tsui LC. Nat Genet. 1996;12:280–287. doi: 10.1038/ng0396-280. [DOI] [PubMed] [Google Scholar]
- 35.Clarke LL, Grubb BR, Yankaskas JR, Cotton CU, McKenzie A, Boucher RC. Proc Natl Acad Sci U S A. 1994;91:464–476. doi: 10.1073/pnas.91.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ritzka M, Weinel C, Stanke F, Tummler B. Genome Lett. 2003;2:149–154. [Google Scholar]
- 37.Agnel M, Vermat T, Culouscou JM. FEBS Lett. 1999;455:295–301. doi: 10.1016/s0014-5793(99)00891-1. [DOI] [PubMed] [Google Scholar]
- 38.Devi L. FEBS Lett. 1991;280:189–194. doi: 10.1016/0014-5793(91)80290-j. [DOI] [PubMed] [Google Scholar]
- 39.Connon CJ, Yamasaki K, Kawasaki S, Quantock AJ, Koizumi N, Kinoshita S. J Histochem Cytochem. 2004;52:415–418. doi: 10.1177/002215540405200313. [DOI] [PubMed] [Google Scholar]
- 40.Elble RC, Widom J, Gruber AD, Abdel-Ghany M, Levine R, Goodwin A, Cheng HC, Pauli BU. J Biol Chem. 1997;272:27853–27861. doi: 10.1074/jbc.272.44.27853. [DOI] [PubMed] [Google Scholar]
- 41.Leverkoehne I, Gruber AD. J Histochem Cytochem. 2002;50:829–838. doi: 10.1177/002215540205000609. [DOI] [PubMed] [Google Scholar]
- 42.Ji HL, DuVall MD, Patton HK, Satterfield CL, Fuller CM, Benos DJ. Am J Physiol. 1998;274:C455–C464. doi: 10.1152/ajpcell.1998.274.2.C455. [DOI] [PubMed] [Google Scholar]
- 43.Greenwood IA, Miller LJ, Ohya S, Horowitz B. J Biol Chem. 2002;277:22119–22122. doi: 10.1074/jbc.C200215200. [DOI] [PubMed] [Google Scholar]
- 44.Gruber AD, Pauli BU. Biochim Biophys Acta. 1999;1444:418–423. doi: 10.1016/s0167-4781(99)00008-1. [DOI] [PubMed] [Google Scholar]