

Abstract
MutS plays a critical role in DNA mismatch repair in Escherichia coli by binding to mismatches and initiating repair in an ATP-dependent manner. Mutational analysis of a highly conserved glutamate, Glu38, has revealed its role in mismatch recognition by enabling MutS to discriminate between homoduplex and mismatched DNA. Crystal structures of MutS have shown that Glu38 forms a hydrogen bond to one of the mismatched bases. In this study, we have analyzed the crystal structures, DNA binding and the response to ATP binding of three Glu38 mutants. While confirming the role of the negative charge in initial discrimination, we show that in vivo mismatch repair can proceed even when discrimination is low. We demonstrate that the formation of a hydrogen bond by residue 38 to the mismatched base authorizes repair by inducing intramolecular signaling, which results in the inhibition of rapid hydrolysis of distally bound ATP. This allows formation of the stable MutS–ATP–DNA clamp, a key intermediate in triggering downstream repair events.
Keywords: ATP binding, ATP hydrolysis, DNA binding, DNA mismatch repair, X-ray crystallography
Introduction
DNA mismatch repair plays a crucial role in ensuring genomic stability. In bacteria, absence of functional mismatch repair results in a mutator phenotype and removal of the interspecies barrier against recombination between slightly diverged sequences (reviewed in Schofield and Hsieh, 2003). Defects in the mismatch repair cascade in humans predispose to hereditary non-polyposis colorectal cancer and are associated with a variety of sporadic cancers (reviewed in Lynch and de la Chapelle, 1999; Peltomaki, 2003). DNA mismatch repair is initiated by the protein MutS (in Escherichia coli) or its MSH homologs (MSH2 and MSH6 that form the predominant MutSα heterodimer in humans). MutS recognizes and binds to mismatches or unpaired bases that have escaped the proofreading capacity of the DNA replication machinery or are present in the genome during recombinatorial events between non-fully complementary DNA strands. Mismatch binding triggers the uptake of ATP in the MutS nucleotide-binding domain and it is this mismatch-dependent ATP state that authorizes repair by recruitment of additional mismatch repair components (recently reviewed in Marti et al, 2002; Kunkel and Erie, 2005).
Molecular details of the initial recognition and binding of DNA containing different mismatched or unpaired bases have been visualized in several crystal structures of both E. coli and Thermus aquaticus MutS (Lamers et al, 2000; Obmolova et al, 2000; Natrajan et al, 2003). MutS is a homodimer consisting of two structurally different monomers, A and B (Figure 1A). The DNA is kinked by 60° at the mismatch-binding site (Figure 1B) and only the monomer A binds to the mismatch. In E. coli, this monomer also has an ADP bound to its ATPase domain. The mismatch itself is bound by two highly conserved residues. These are a phenylalanine (Phe 36 in E. coli), which stacks onto one of the mismatched bases, and a glutamate (Glu38 in E. coli), which forms a hydrogen bond to the same base (Figure 1C). Whereas mutating Phe 36 leads to elimination of both DNA binding and mismatch repair (Yamamoto et al, 2000; Drotschmann et al, 2001), the role of Glu38 has been less clear. It was proposed that the electrostatic repulsion between the negatively charged side chain of Glu38 and the DNA backbone facilitated the kinking of the DNA at the site of the mismatch (Lamers et al, 2000; Schofield et al, 2001). Electrophoretic mobility shift assays (‘band-shifts') using oligonucleotides containing base analogs have shown that the hydrogen bond only contributes marginally to mismatch binding (Schofield et al, 2001). Mutating Glu38 to an alanine or glutamine slightly increases the affinity of the enzyme for DNA carrying a mismatch, but even more so for homoduplex DNA (Schofield et al, 2001; Junop et al, 2003). MutS carrying these mutations was unable to complement mismatch repair in a MutS-deficient strain, presumably because the mutants lost their ability to discriminate between homoduplex and mismatched DNA.
Figure 1.

Crystal structures of wild-type MutS and E38 variants. (A) Schematic view of the E. coli MutS dimer, with the mismatch-binding monomer A shown in yellow, the supporting monomer B shown in orange and the mismatch containing DNA in blue. (B) Superposition of the mismatch-binding clamp domains and DNA of wild-type and mutant MutS obtained by superposing the Cα atoms of the mismatch-binding monomer A of all the structures. (C) G.T mismatch binding by the wild-type MutS. (D) G.T mismatch binding by the E38T mutant, with the bound water molecule in red, along with its mFo−DFc map contoured at 3σ (in green). (E) G.T mismatch binding by the E38Q mutant. (F) G.T mismatch binding by E38A, shown with the bound water molecule (in red) along with its mFo−DFc density map contoured at 3σ (in green). Dashed red lines indicate hydrogen bonds and residue 38 in panels C–F is shown in purple. All figures were made using PyMOL (Copyright© 2004 DeLano Scientific).
Mismatch binding results in a conformational change that is propagated toward the other end of the molecule, altering the nucleotide-binding capacity of the ATPase domains. The two ATPase domains are asymmetric in nucleotide binding and ATP hydrolysis (Bjornson et al, 2000; Lamers et al, 2003; Antony and Hingorani, 2004). In the absence of DNA, the rate-limiting step for ATPase activity is release of ADP, whereas binding of MutS to a DNA mismatch greatly enhances the rate of ADP–ATP exchange (Gradia et al, 1997; Blackwell et al, 2001; Acharya et al, 2003). After binding to perfectly paired DNA, this ATP is hydrolyzed quickly, but binding to mismatched DNA inhibits this fast hydrolysis. This would indicate the formation of an ATP-bound MutS state on mismatched DNA with a relatively long lifetime, a state that allows mismatch-dependent recruitment of MutL and initiation of repair (Bjornson et al, 2000; Antony and Hingorani, 2003, 2004). Meanwhile, ATP reduces affinity of MutS for the mismatch itself and induces conversion of the protein into a sliding clamp that can diffuse along the DNA helix (Gradia et al, 1999; Acharya et al, 2003). In contrast, ATP binding to MutS induces direct dissociation of the protein from homoduplex DNA (Iaccarino et al, 2000; Selmane et al, 2003). Specific inhibition of ATP hydrolysis by mismatched DNA binding, and the different modes of dissociation from homo- and heteroduplex DNA indicate that MutS uses ATP to verify mismatch binding and initiate repair, as proposed by Junop et al (2001). This may explain the high efficiency of the DNA mismatch repair process even though initial discrimination between homoduplex and mismatch containing DNA by E. coli MutS is only 8- to 20-fold (Schofield et al, 2001; Hays et al, 2005).
The nucleotide-dependent conformational changes and their effect on DNA binding indicate that residues that are in contact with the DNA are not only involved in initial recognition of mismatches, but also contribute to proper reciprocal signaling between DNA- and ATP-binding domains. To better understand this process, we investigated the role of glutamate 38 in more detail, using a combination of in vivo, crystallographic, biophysical and biochemical approaches. Our data demonstrate that the Glu38 side chain of MutS plays a dual role in initial mismatch discrimination, by its negative charge, as well as in transmitting conformational changes that induce formation of a stable MutS-ATP state and sliding clamp, by its ability to specifically form a hydrogen bond with the mismatched base.
Results
Mutants of E38 in E. coli MutS
The conserved glutamate at position 38 (E38) in E. coli MutS forms a hydrogen bond to one of the mismatched bases and is thought to participate in the phenylalanine 36 (F36)-induced kinking of the DNA at the mismatch site, by causing electrostatic repulsion between its negatively charged side chain and the phosphates of the DNA (Lamers et al, 2000; Obmolova et al, 2000; Schofield et al, 2001; Natrajan et al, 2003). To address the individual roles of the negative charge of the carboxylate side chain and of hydrogen bonding to the mismatched base, we have replaced this glutamate with a glutamine, a threonine and an alanine residue. Mutations were introduced in the full-length MutS protein for genetic and biochemical analysis, as well as in the ΔC800 protein, which lacks the 53 C-terminal amino acids (Lamers et al, 2000), for crystallographic analysis. All mutant side chains lack the negative charge but they differ in size and hydrophilicity. The mutants were efficiently overexpressed and purified. All have the same molecular weight according to size-exclusion chromatography, indicating identical quaternary structure (data not shown).
Mismatch repair in vivo
To analyze the mismatch repair capabilities of the E38 mutants, we performed in vivo mismatch repair assays, based on complementation of repair in an E. coli strain lacking MutS (Wu and Marinus, 1994). These assays were performed at 37 and 22°C, the temperatures at which most subsequent in vitro assays were performed (Table I). At both temperatures, the E38A substitution is highly impaired in its ability to repair mismatches, as has been reported for this mutant in an earlier study (Schofield et al, 2001). The E38Q mutant has also been reported to be deficient in mismatch correction (Junop et al, 2003), and this correlates with its inability to complement MutS deficiency at 37°C. However, at room temperature, E38Q appears to be almost as efficient in mismatch repair as wild-type MutS. Unexpectedly, the threonine mutant is proficient in repair at both temperatures. Thus, mismatch repair can proceed in the absence of a negatively charged residue at MutS position 38.
Table 1.
In vivo mismatch repair
| 20°C |
37°C |
|||
|---|---|---|---|---|
| Mutation frequency (× 10−9) | Mutation rate (× 10−9) | Mutation frequency (× 10−9) | Mutation rate (× 10−9) | |
| WT MutS | 6±1 | 4.4 | 5±1 | 3.8 |
| E38A | 68±15 | 26.9 | 822±455 | 212 |
| E38T | 13±2 | 7.6 | 15±4 | 8.4 |
| E38Q | 12±1 | 7.2 | 445±255 | 126 |
| Control |
136±33 |
47.1 |
2058±80 |
469 |
| WT: wild type. | ||||
Crystal structures of E38 mutants bound to mismatched DNA
To investigate the molecular details of mismatch recognition by the MutS E38 mutants, we solved their crystal structures in complex with DNA containing a G.T mismatch (Table II). The overall structures of the mutants are very similar to that of the wild-type protein (Figure 1A and B), with an r.m.s.d. (Cα) of 0.27 Å between the wild type and the E38T, E38A mutants and 0.30 Å between the wild type and the E38Q mutant. The only differences are seen in the binding of the mismatch itself by the A subunit; no significant changes are seen in the other mutation site in the B subunit. In the mismatch-binding site, F36 from the A subunit stacks upon the thymidine in all of the structures. As in wild-type MutS, a hydrogen bond is formed by the thymidine to residue 38 of the A subunit in the E38T and the E38Q mutants, but in a different way in each mutant (Figure 1D and E). The E38T mutant has a water molecule bridging the OG of the threonine and the N3 of the mismatched thymidine base (Figure 1D). The glutamine in the E38Q mutant adopts an alternative side-chain conformation similar to the glutamate binding to an A.A mismatch (Natrajan et al, 2003), and now forms two hydrogen bonds to the thymidine, using its OE1 and NE2 (Figure 1E). In contrast, the E38A mutant cannot form a hydrogen bond to the thymidine (Figure 1F). There is weak electron density between the alanine side chain and the thymidine, suggesting only partial occupancy of this site by a water molecule. This water molecule is hydrogen-bonded to the N3 of the thymidine and is located at a distance of 3.5 Å from the alanine side chain, suggesting a Van der Waals contact between the two. This, therefore, is in contrast with the two other mutants and the wild-type protein, where at least one direct, or water-mediated, hydrogen bond is formed between the side chain of residue 38 and the thymidine. A single hydrophobic contact of glutamate 38 with methionine 33 is lost in all mutants and is therefore not relevant for functional mismatch repair. All other contacts to the DNA in the mutants are identical to those in the wild-type protein. The correlation between in vivo mismatch repair capabilities and hydrogen-bond formation suggests that this latter interaction may be critical for mismatch repair.
Table 2.
Crystallographic statistics for MutS E38 mutants bound to a G.T mismatched DNA
| E38T-G.T | E38A-G.T | E38Q-G.T | |
|---|---|---|---|
| Data collection | |||
| Resolution limits (Å) | 20–2.1 (2.15–2.10) | 20–2.5 (2.59–2.50) | 20–2.4 (2.49–2.40) |
| I/σ(I) | 14.46 (1.52) | 9.81 (1.47) | 11.90 (1.80) |
| Completeness (%) | 91.7 (47.0) | 99.3 (98.3) | 99.3 (92.9) |
| Rmerge (%) | 7.1 (48.8) | 11.4 (72.2) | 12.2 (69.3) |
| Cell parameters (Å) | a=89.61 | a=89.55 | a=89.62 |
| b=92.17 | b=92.49 | b=92.04 | |
| c=260.53 | c=261.66 | c=260.97 | |
| Refinement | |||
| Resolution (Å) | 20–2.1 | 20–2.5 | 20–2.4 |
| R-factor (%) | 18.8 | 21.9 | 21.2 |
| Rfree (%) | 23.3 | 26.9 | 25.8 |
| R.m.s.d. (bonds) (Å) | 0.014 | 0.011 | 0.014 |
| R.m.s.d. (angles) (deg) | 1.421 | 1.225 | 1.460 |
| No. of atoms | 13 862 | 13 190 | 13 160 |
| No. of waters |
900 |
240 |
250 |
| Values in parentheses refer to the highest resolution shell. | |||
Mismatch recognition by E38 mutants
To understand what causes the variation in mismatch repair capability by the different MutS mutants at amino acid 38, we first studied their mismatch recognition properties and the ability to discriminate against homoduplex DNA. We used isothermal titration calorimetry (ITC), native band-shifts and surface plasmon resonance (SPR) in a Biacore flow system. In all cases, data were fit to a single-site-binding model (see Discussion).
ITC measures the heat liberated owing to binding of the protein to the substrate in solution. We find that the E38A, E38T and E38Q mutants have very similar affinities for DNA containing a G.T mismatch as wild-type MutS (Figure 2A and Table III). We could not perform these experiments successfully using homoduplex DNA, owing to low signal to noise ratios.
Figure 2.

MutS–DNA binding assays using oligonucleotides with free ends. (A) Representative ITC binding curves for wild-type and mutant MutS (13–15 μM) at 37°C to a 21 bp DNA substrate containing a G.T mismatch. (B) Representative band-shifts showing DNA binding by increasing concentrations of wild-type MutS and E38 mutants. Reactions with homoduplex and heteroduplex DNA are shown in alternating lanes.
Table 3.
MutS binding constants (μM) for unblocked mismatched (G.T) and homoduplex (A.T) oligonucleotide
| WT | E38A | E38T | E38Q | |
|---|---|---|---|---|
| ITC at 37°C | ||||
| G.T | 0.10±0.02 | 0.15±0.06 | 0.09±0.02 | 0.06±0.01 |
| Band-shift at 22°C | ||||
| G.T | 0.31±0.06 | 0.24±0.05 | 0.10±0.02 | 0.17±0.04 |
| A.T | 2.4±0.7 | 0.52±0.09 | 0.26±0.04 | 0.42±0.05 |
| G.T/A.T | 7.7±2.7 | 2.2±0.6 | 2.6±0.7 | 2.5±0.7 |
| Band-shift at 37°C | ||||
| G.T | 0.38±0.01 | 0.34±0.1 | 0.10±0.02 | 0.14±0.02 |
| A.T | 3.4±0.9 | 0.73±0.09 | 0.33±0.1 | 0.42±0.01 |
| G.T/A.T | 9.0±2.4 | 2.2±0.7 | 3.3±1.5 | 3.0±0.4 |
| SPR at 25°C | ||||
| G.T | 0.015±0.001 | 0.020±0.003 | 0.018±0.001 | 0.018±0.001 |
| A.T | 0.31±0.1 | 0.24±0.01 | 0.19±0.07 | 0.13±0.05 |
| G.T/A.T | 21±7.3 | 12±1.8 | 11±3.8 | 7.2±2.9 |
| SPR at 25°C, blocked DNA | ||||
| G.T | 0.015±0.001 | 0.048±0.003 | 0.013±0.002 | 0.013±0.001 |
| A.T | 0.21±0.07 | 0.13±0.02 | 0.067±0.01 | 0.070±0.01 |
| G.T/A.T |
14±5 |
2.6±0.4 |
5.2±1.2 |
5.3±1.1 |
| WT: wild type; ITC: isothermal titration calorimetry; SPR: surface plasmon resonance. | ||||
In band-shift studies, using native polyacrylamide gel electrophoresis with 32P end-labeled DNA substrates with free ends, we found that E38T and E38Q mutants bind marginally stronger to DNA containing a G.T mismatch than the wild-type protein, whereas the E38A protein has wild-type affinity (Figure 2B and Table III). This result indicates a marginal contribution of the hydrogen bond between MutS residue 38 and the mismatched base. A more pronounced difference between the wild-type and mutant proteins is seen in homoduplex binding. Wild-type MutS binds to homoduplex DNA with low affinity, whereas in all mutants this affinity is 5- to 10-fold higher.
The ability to discriminate between mismatched and paired DNA can be quantified using the ratio of the Kd values between these substrates (Table III, G.T/A.T). All mutants show a decrease in this ratio, demonstrating loss in discriminatory capacity as a consequence of loss of repulsion of the DNA backbone, by absence of the negative charge on residue 38. The general trend is similar at 22 and 37°C, and the values we find at 37°C for wild type and E38Q and E38A mutants are in good agreement with the results reported earlier at this temperature (Table III; Schofield et al, 2001; Junop et al, 2003).
Finally, we measured affinities in real time in solution using SPR in a Biacore system. The affinities measured in this assay (Table III) are considerably higher than those measured by band-shift analysis and ITC, which may relate to the different experimental setup of the continuous flow in the Biacore system. However, in the SPR experiments as well, we observe that the ability of the mutants to discriminate between mismatched and homoduplex DNA is clearly diminished, primarily as a consequence of an increase in homoduplex binding.
Mismatch recognition on DNA with blocked ends
A potential problem in MutS binding studies is the ability of MutS protein to bind to DNA ends (Acharya et al, 2003; Yang et al, 2005). Therefore, we have included binding studies to double end-blocked DNA, both in band-shift experiments (Figure 3) and using SPR (Table III). In the DNA band-shifts, the DNA was blocked on both ends with biotin–streptavidin, and in the SPR experiments, we used a fluorescein–antibody complex in addition to the biotin–streptavidin coupling of the DNA on the chip (Acharya et al, 2003).
Figure 3.

MutS–DNA binding assays using end-blocked oligonucleotides. Representative band-shifts showing wild-type and mutant MutS binding, in the absence of ATP, (A) to homoduplex, (B) to heteroduplex DNA, and in the presence of ATP, (C) to homoduplex and (D) to heteroduplex DNA.
In the band-shift experiments using double end-blocked DNA (Figure 3B), the E38T and E38Q mutants start binding to the G.T mismatch at lower concentrations (∼50 nM) than the E38A (∼150 nM), again supporting a small but reproducible contribution of the hydrogen bond to mismatch binding. It can also be seen that all mutants have a similar affinity to homoduplex DNA, which is considerably higher than that of wild-type MutS. This agrees with the loss of discrimination seen in the experiment using unblocked DNA.
In the SPR binding studies to double end-blocked DNA (Table III), we see that the mutants have retained their affinities to mismatched DNA, compared to unblocked DNA. Only in this experiment, the E38A mutant bound slightly weaker to the double end-blocked G.T substrate, although binding to unblocked DNA is similar to that of the other mutants and wild-type MutS. In agreement with the result obtained with unblocked DNA (Table III), all mutants display a three-fold-reduced discrimination compared to wild-type MutS.
The experiments described above confirm previously published conclusions, based on band-shifts with unblocked oligonucleotides, that removal of the carboxylate group from amino acid 38 of MutS does not strongly affect heteroduplex binding, but results in enhanced homoduplex binding and consequent reduction in the discriminatory capacity of the protein (Schofield et al, 2001; Junop et al, 2003). However, the reduced discrimination against homoduplex DNA, which is similar for the three E38 mutants, does not correlate with their significantly different capability to repair mismatches in vivo. These paradoxical results, therefore, suggest that initial discrimination conferred by the carboxylate group of MutS amino acid 38 is not the critical factor in DNA mismatch repair.
Mismatch-binding-induced signaling toward the nucleotide-binding domains
It has been well documented that after mismatch binding, the conformational change in the DNA-binding domains is propagated toward the distant nucleotide-binding sites (Gradia et al, 1997; Acharya et al, 2003). Because initial mismatch recognition and discrimination did not correlate with in vivo repair capabilities, we investigated the activity of the ATPase sites and the ability of the mutants to signal toward these domains. Similar to wild-type MutS, all mutants retain a single ADP molecule per MutS dimer during purification, indicating retention of the high-affinity site for nucleotide-diphosphate (data not shown). In addition, the high-affinity site for the nucleotide-triphosphate is not affected by the mutation, as binding of ATPγS is in the low micromolar range for all MutS variants, in agreement with published values (Table IV; Bjornson and Modrich, 2003b; Lamers et al, 2004). The rate of ADP–ATP exchange, which is slow in the absence of mismatched DNA, was equally unaffected by the mutations (Table IV). Furthermore, we found very similar kinetic constants for steady-state ATP hydrolysis for wild-type MutS and E38 mutants (Table IV). Taken together, these observations indicate that the intrinsic properties of the nucleotide-binding sites are not affected by the mutations at position 38 in the DNA-binding domains.
Table 4.
Affinity for ATP and kinetic constants for DNA-induced nucleotide exchange, steady-state ATP hydrolysis and its stimulation by DNA for WT MutS and E38 mutants
| ATPγS binding | ADP–ATP exchange |
ATPase |
Steady-state ATPase stimulation by DNA |
||||
|---|---|---|---|---|---|---|---|
| KD(ATPγS) (μM) |
koff (ADP) (s−1) |
KM (μM) | kcat (min−1) |
n-fold |
|||
| −DNA | +DNA | Homoduplex | Heteroduplex | ||||
| WT | 2.3 | 0.030 | 0.39 | 7.8±0.4 | 5.4±0.1 | 3.7±0.4 | 3.2±0.4 |
| E38A | 2.1 | 0.029 | 0.38 | 7.4±0.7 | 4.7±0.7 | 4.7±0.4 | 3.6±0.4 |
| E38T | 3.0 | 0.028 | 0.36 | 11.0±0.4 | 4.1±0.1 | 5.3±0.3 | 4.1±0.4 |
| E38Q |
2.4 |
0.031 |
0.38 |
9.0±2.2 |
5.4±0.1 |
4.8±0.1 |
3.6±0.5 |
| WT: wild type. | |||||||
We next investigated whether mutation of glutamate 38 affects the signaling from the mismatch-binding domains toward the nucleotide-binding sites by measuring the rate of ADP to ATP exchange in the presence of mismatched DNA. As reported before (Gradia et al, 1997; Acharya et al, 2003), this exchange rate, which is the rate-limiting step in steady-state ATP hydrolysis, is highly accelerated by mismatched DNA and we find that this is conserved in all mutants (Table IV). In addition, this increased release of ADP is reflected by the ability of DNA to stimulate the steady-state ATPase activity of wild-type MutS and of the amino acid 38 mutants (Table IV). These results indicate that, following DNA binding, signaling from the DNA-binding domains toward the nucleotide-binding domains depends neither on the presence of a negative charge nor on the formation of a hydrogen bond by the residue at position 38.
Formation of a stable MutS–DNA–ATP complex
ATP binding to MutS induces a rearrangement of the nucleotide-binding domains toward each other (Lamers et al, 2004) and these conformational changes are propagated toward the mismatch-binding and/or clamp domains, resulting in release of the mismatch and formation of a sliding clamp. We analyzed whether the mutants were affected in clamp formation by investigating their behavior upon ATP addition to prebound, end-blocked DNA–MutS complexes (see Materials and methods). The band-shifts show that the wild-type protein binds to G.T-mismatched DNA at a much lower concentration (∼20 nM) compared to binding without subsequent challenge with ATP (Figure 3B and D). This indicates that this ATP-bound state of MutS, presumably the sliding clamp, is more stably associated with DNA than the initial interaction between MutS and the mismatch itself. The mutants bind in varying degrees, with E38Q starting to bind at lower concentrations than the others and the E38A mutant lagging behind the other MutS mutants. Thus, glutamate 38 appears to be important for this increased ATP-mediated heteroduplex affinity. The reproducible appearance of double bands on heteroduplex DNA at 300 nM protein concentrations in all MutS proteins except E38A is unexpected. We speculate that the faster migrating band may represent a form of the ATP-bound sliding clamp that is induced at higher protein concentrations. Aberrant forms of MutS–DNA band-shift complexes have been reported in several other studies (Gradia et al, 1997; Acharya et al, 2003; Antony and Hingorani, 2003).
ATP has a remarkable effect on the binding of the MutS mutants to homoduplex DNA (Figure 3A and C). Even at the highest protein concentration, there is no binding by any of the mutants, although in the absence of ATP all mutants bind well to homoduplex DNA at this concentration. Because both ends of the homoduplex DNA were blocked by biotin–streptavidin, this ATP-induced release of the homoduplex must have occurred via direct dissociation of the protein (Iaccarino et al, 2000; Selmane et al, 2003). These results indicate qualitatively that the binding of ATP to the protein, after DNA binding, brings about a secondary discrimination step, or verification. This verification results, on the one hand, in stable clamp formation on mismatched DNA and, on the other hand, in direct dissociation after homoduplex binding. The ATP-mediated dissociation from homoduplex is operational in all mutants, even in the near absence of initial discrimination, and therefore does not involve residue 38.
MutL is recruited by the stable sliding clamp form of the MutS–ATP state on heteroduplex DNA (Acharya et al, 2003). We analyzed whether MutL binding was affected in the mutants by including MutL in the end-blocked band-shifts described above. At high protein concentrations, at which all mutants are forced into a stable ATP-bound complex with DNA (Figure 4, left panel), we see a supershifted band for all the mutants upon addition of MutL (Figure 4, right panel). At low protein concentrations, the ability to interact with MutL correlates with the amount of stable clamp formation (data not shown). Together, these data show that none of the mutants have lost the ability to interact with MutL and therefore indicate that MutS residue 38 is not directly involved in the interaction of MutS with MutL.
Figure 4.
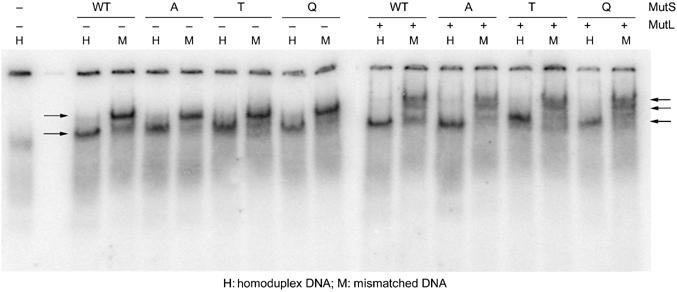
MutL binding to MutS–DNA complex, with G.T mismatched double end-blocked oligonucleotides. Wild-type and mutant MutS (400 nM) bound to mismatched DNA and supershifted by binding to MutL (1.6 μM), in the presence of 1 mM ATP during electrophoresis. Single lane on the left: DNA with biotin–streptavidin blocks on both ends. Left panel: mismatch-dependent trapping of wild-type and mutant MutS sliding clamps. Free DNA and sliding clamp complexes are indicated by arrows. Right panel: mismatch-dependent MutL complex formation by wild-type and mutant MutS sliding clamps. Lower arrow indicates unbound end-blocked DNA, arrow in the middle indicates MutS-associated DNA and upper arrow points at MutS–MutL supershifted DNA complexes.
In pre-steady-state ATP hydrolysis experiments, a burst of ADP formation has been observed, indicating that the first ATP molecule that is bound after ADP release is hydrolyzed rapidly. This occurs both in the absence of any DNA substrate or in the presence of homoduplex DNA (Bjornson et al, 2000; Antony and Hingorani, 2004). This rapid ADP formation is followed by a slower steady-state hydrolysis rate, consistent with the release of ADP being the rate-limiting process for steady-state ATP turnover. The presence of a mismatch results in almost complete suppression of rapid ADP formation, demonstrating that binding of a mismatch inhibits initial ATP hydrolysis. We determined the rate of ATP hydrolysis in the absence of DNA during the first few catalytic turnovers and obtained a reproducible burst amplitude for wild type and all three MutS mutants, corresponding to 0.6 ADP molecules produced per MutS monomer (Figure 5A and Supplementary data). This indicates rapid hydrolysis of one ATP molecule in only one subunit of the functional MutS dimer, in agreement with its expected asymmetry (Bjornson and Modrich, 2003b; Lamers et al, 2003; Antony and Hingorani, 2004). As expected, binding to mismatched DNA causes the loss of this rapid hydrolysis step in the wild type and E38Q and E38T mutants. However, in the E38A mutant, the ATP hydrolysis remains fast also after mismatch binding (Figure 5A). Apparently, in this mutant, mismatch binding can no longer induce inhibition of rapid initial ATP hydrolysis. This suggests that E38A is not able to remain in the relatively long-lived MutS-mismatched DNA–ATP state that has been proposed to play an important role in initiating downstream events (Bjornson et al, 2000; Antony and Hingorani, 2003, 2004).
Figure 5.
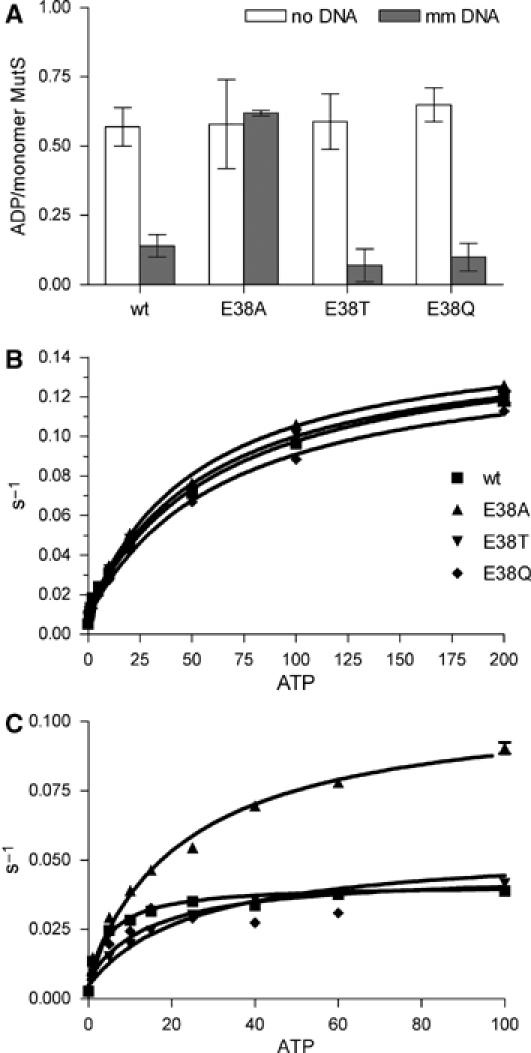
Kinetic response to ATP binding of wild-type MutS and E38 mutants. (A) Magnitude of burst amplitude of ADP formation during the first hydrolytic turnover in a rapid quench ATPase assay in the absence and presence of DNA containing a G.T mismatch. (B) Kinetics of ATP-induced MutS release from mismatched (mm) DNA with a free end and (C) from double end-blocked mismatched DNA determined by SPR.
To examine the kinetic effect of the E38 mutation on the stable ATP-bound state of MutS in more detail, we used SPR to determine the kinetics of ATP-induced release of MutS prebound to DNA with a free end or to DNA with both ends blocked (Figure 5B and C). The kinetics of dissociation from homoduplex DNA, either blocked on one end or double end-blocked, is too fast for quantification in this assay system. When one end of the DNA containing a G.T mismatch is kept unblocked, the rates of release of all the mutants are similar to that of the wild-type MutS (kmax of 0.13–0.14 s–1; Figure 5B and Supplementary data). This mainly reflects the rapid dissociation of wild-type and mutant proteins from the free end of the DNA. However, when both DNA ends are blocked, a much slower release is seen (Figure 5C). As MutS is trapped on double end-blocked ends in other systems (e.g. band-shifts above), this slow release must reflect the dissociation of ATP-bound MutS due to the extensive flow that is inherent to the SPR experimental setup. Strikingly, in this experiment, the E38A releases much faster (kmax of 0.098 s−1) than the wild-type, E38T and E38Q proteins (kmax∼0.036–0.049 s−1) (Figure 5C; Supplementary data). We conclude that, in the E38A mutant, the absence of a hydrogen bond with the mismatched base results in rapid hydrolysis of bound ATP and concomitant fast release of MutS from the mismatched DNA. This is in contrast to the mismatch repair-proficient variants in which the hydrogen bond-mediated interaction between amino acid 38 and the mismatched base inhibits ATP hydrolysis and retains the protein on the DNA.
Discussion
MutS glutamate 38 has been reported to allow discrimination between perfectly paired DNA and DNA containing a mismatch or unpaired base (Junop et al, 2001; Schofield et al, 2001). However, from the analysis of a series of MutS mutants at amino acid 38, we could not satisfactorily correlate the loss of discriminatory capacity in the mutants with their ability to repair mismatches in an in vivo complementation assay. We have therefore extended the analysis of these mutants in their down-stream signaling ability.
In these studies of the DNA–MutS interaction, it became obvious that the single-site-binding model that is traditionally used (Blackwell et al, 2001; Brown et al, 2001; Schofield et al, 2001) is insufficient to explain the data completely. This is particularly clear in the ITC data (Figure 2), but was also apparent from the statistical analysis of the SPR data (not shown). However, it is difficult to describe the process properly: the binding of MutS to a mismatch in DNA is associated with binding and release of nucleotide and conformational changes in the protein and DNA. Furthermore, monomer–dimer and dimer–tetramer transitions should be taken into account (Bjornson et al, 2003a; Lamers et al, 2004). In the absence of a better model, the one-site binding model is the best approximation, as it leads to acceptable fits with reliable statistics. Further studies will be required to establish a proper binding model for the DNA–MutS interaction.
Our studies into the effect of the E38 mutations were also complicated by the variable effect of temperature on the different mutants in the in vivo repair assay, in particular for E38Q, which repairs at 22°C but not at 37°C (Table I). As we did not find a temperature dependence in either DNA binding or mismatch discrimination for this mutant, it seems likely that the temperature-dependent in vivo mismatch repair ability of the E38Q mutant is due to kinetic differences. Our experimental setup does not allow a complete analysis of the effect of temperature on the kinetics of the reaction, but their importance fits very well with our overall conclusion that kinetic differences are responsible for the defects observed in the E38 mutants (see below).
A final complicating factor in our studies is the possibility of DNA end-binding. In two recent reports on DNA binding by E. coli and T. aquaticus MutS, it was found that especially E. coli MutS has strong affinity for free DNA ends (Acharya et al, 2003; Yang et al, 2005). We considered whether this could explain the discrepancy between low discriminatory capacity and high repair efficiency of E. coli MutS. We can assess end-binding directly by comparison of affinity constants obtained by SPR on DNA with one free end and both ends blocked. If significant end-binding to the free DNA ends occurs, affinity for homoduplex with a free end would be higher than for double end-blocked homoduplex DNA. However, the observed affinities for homoduplex are similar, or even lower, on an oligonucleotide with free ends, strongly indicating that end-binding does not occur in this continuous flow system (Table III). Meanwhile, the calculated specificities for mismatched versus homoduplex DNA are at most two-fold higher than specificities we find in band-shifts using DNA with free ends, and are similar to reported values (Schofield et al, 2001; Selmane et al, 2003). We therefore cannot conclude from our experiments that the specificity for a mismatch will be significantly better in the absence of end-binding.
We have used a number of different techniques to study DNA recognition by MutS wild type and mutants of amino acid 38. Despite their intrinsic technical differences, all binding studies confirm that the three mutants show loss of discrimination between mismatched and homoduplex DNA. This was expected, as they lack the negatively charged carboxylate group (Schofield et al, 2001). The E38A mutant appears to have a slightly weaker ability to discriminate than the other mutants, which may be related to the absence of the hydrogen bond. However, the differences with the other mutants are minor and do not correlate with their large differences in repair capability. On the other hand, we have shown that efficient repair is possible even if the initial low discrimination capacity of wild-type MutS is decreased further by mutation of Glu38. This strongly indicates that MutS relies on an additional factor to verify binding to mismatched DNA. It was previously suggested that the increased rate of ADP–ATP exchange, induced by mismatch binding, plays a role in determining specificity of mismatch repair (Acharya et al, 2003). Furthermore, ADP to ATP exchange in MutS after binding to DNA might confer a second round of mismatch discrimination by inducing different modes of dissociation from bound hetero- and homoduplex DNA (Iaccarino et al, 2000; Junop et al, 2001; Selmane et al, 2003). From the band-shift experiments on DNA with double-blocked ends (Figure 3), it is clear that, in the presence of ATP, wild-type MutS has a higher affinity for mismatched DNA than in the absence of the nucleotide-triphosphate. In addition, exploiting the high affinity of the mutants for homoduplex DNA, which is a result of the loss of the negative charge, we were able to demonstrate that ATP binding causes direct dissociation of MutS from inadvertently bound homoduplex. As this phenotype was found in all mutants, this process requires neither the carboxylate side chain nor the hydrogen bond of E38. In conclusion, ATP binding induces enhanced heteroduplex binding as well as increased release from homoduplex DNA and this secondary discrimination, or verification, by MutS presumably allows E38T and E38Q to be active in vivo.
To understand the varying repair capabilities of the mutants, and especially the severe defect of E38A, we studied the signaling toward the ATPase domains, and conformational responses induced by ATP binding. In all mutants, the binding of a mismatch results in uptake of ATP, excluding involvement of E38 in this signaling process. In wild-type MutS, hydrolysis of this ATP is inhibited and a conformational change results in the formation of the ATP-bound sliding clamp. Here, we found a significant difference in the kinetic behavior of the E38A mutant. It releases double end-blocked heteroduplex DNA much faster than any of the other proteins after ATP binding. Concomitantly, E38A has lost the inhibition of rapid ATP hydrolysis that is induced by mismatch binding. Thus, despite reasonable initial mismatch binding, this mutant is defective in subsequent clamp formation. The isomorphic crystal structures of wild-type and mutant MutS reveal that the overall protein conformation and contacts to the DNA, apart from the direct interaction between residue 38 and the DNA mismatch, are not significantly different. The E38Q and E38T mutant side chains still form a hydrogen bond to the mismatched thymidine, whereas no such interaction is present in the E38A mutant. As MutS E38Q and E38T are repair proficient, we conclude that the hydrogen bond to the mismatched base, and not the negative charge on the glutamate, is critical for repair.
Based on these data, we propose the following model for glutamate 38-mediated authorization of mismatch repair: MutS scans the DNA for mismatches, attempting to kink the DNA. After mismatch binding, or after fortuitous homoduplex binding, MutS kinks the DNA and exchanges ADP for ATP in the distant nucleotide-binding domains. In addition, uniquely after mismatch binding, a hydrogen bond is formed between the mismatched base and glutamate 38 (or the threonine or glutamine residue in the respective mutants). The establishment of this hydrogen bond initiates intramolecular signaling, resulting in the inhibition of hydrolysis of bound ATP. This stable ATP binding results in tightening of the MutS dimer (Lamers et al, 2004), and propagation of this conformational change toward the DNA-binding domains initiates formation of the sliding clamp. The ability of MutS to form a stable sliding clamp after releasing the mismatch implies that the hydrogen bond is not involved in stabilizing the clamp while it is sliding, but solely in its induction. This stable clamp subsequently initiates repair by formation of a complex with MutL and recruitment of additional downstream factors. The hydrogen bond between the residue at position 38 and the mismatch cannot be formed in the E38A mutant, or when (wild type, or E38T or E38Q) MutS is inadvertently bound to homoduplex DNA. In these cases, there is no intramolecular signal generated to inhibit ATP hydrolysis. We hypothesize that this results in rapid hydrolysis of ATP and concomitant widening of the MutS dimer at the intertwined ATPase domains, leading to conformational changes at the DNA-binding domains that prevent stable clamp formation and result in release of the DNA by MutS. In conclusion, the hydrogen bond between MutS amino acid 38 and the mismatch validates mismatch binding and authorizes repair.
Materials and methods
Site-directed mutagenesis
MutS mutants E38A, E38T and E38Q were constructed by converting the Glu GAA codon of wild-type MutS plasmid pMQ372 (full length) or pM800 (ΔC800) into an Ala GCG, Thr ACC or Gln CAG codon, using QuickChange (Stratagene). Mutations were confirmed by DNA sequencing.
DNA substrates
We used double-stranded DNA oligonucleotides containing a single G.T mismatch or a perfectly paired, otherwise identical, oligonucleotide. A 30 bp oligonucleotide (Lamers et al, 2000) was used for crystallization, band-shifts with free DNA ends and DNA-dependent ATPase assays. A shortened version (21 bp) was used for the ITC (upper strand 5′ AGC TGC CAG GCA CCA GTG TCA, bottom strand 5′ TGA CAC TGG TGC TTG GCA GCT, mismatched nucleotides in bold). A 41 bp biotinylated oligonucleotide was used for band-shifts with DNA with blocked ends (upper strand 5′ ATC GAA TTA GAA GCT GCC AGG CAC CAG TGT CAG CGT CCT AT-biotin, bottom strand 5′ ATA GGA CGC TGA CAC TGG TGC TTG GCA GCT TCT AAT TCG AT-biotin).
In vivo mismatch repair
Mismatch repair complementation assays were performed as described (Wu and Marinus, 1994; Lamers et al, 2003). A MutS-deficient strain RK1517 was transformed with wild-type MutS or E38A, E38T, E38Q mutants or empty vector. Samples of nine overnight cultures for each strain, grown in LB at 37 or 22°C, were plated on LB with 100 μg/ml rifampicin to select for mutant colonies, and titers were determined on LB plates. Mutation frequency was calculated as the median value of nine mutation frequencies determined. Mutation rates were determined using the Luria and Delbrück equation 0.6RifR/C=Np log(CNp), where RifR is the number of rifampicin-resistant cells in a culture expanded to N cells, C is the number of different cultures and p is the number of new Rifampicin-resistant cells per cell division. Each experiment was performed three times.
Structure determination
MutS lacking the C-terminal 53 amino acids (ΔC800) and its mutants were purified and crystallized as described (Natrajan et al, 2003). Crystals grew in space group P212121 and data were collected at the ID 14 beamlines, located at the ESRF Grenoble, at a wavelength of 0.91 Å. Processing was carried out using the HKL 2000 (Otwinowski and Minor, 1997) package (Table II). The structures of the mutants were solved by rigid-body refinement using the MutS structure as model (Lamers et al, 2000). The residues from 30 to 45 in both monomers and the mismatched bases were removed before the first restrained refinement and rebuilt into the difference density map. All refinements were performed using REFMAC5 (Murshudov et al, 1997) and TLS refinement (Winn et al, 2001), in the CCP4 (CCP4, 1994) program suite. The structures were checked using the WHATCHECK (Hooft et al, 1996) server.
Isothermal titration calorimetry
ITC experiments were performed using a MicroCal VP-ITC calorimeter at 37°C. Both protein and DNA were dialyzed overnight in the ITC buffer (25 mM Hepes–NaOH pH 7.5, 125 mM NaCl, 10 mM MgCl2, 10 mM β-mercaptoethanol, 10% glycerol, 0.5 μM ADP). DNA was injected into the protein solution (13–15 μM) and the increase in ligand concentration was ∼0.36 μM per injection. A long time interval of 300 s was given between injections and only the peak was selected for integration using a constant peak width to avoid a large scatter in the data points obtained. Although a baseline of DNA into buffer was essentially flat, the intrinsic scatter was very large, probably owing to dilution effects of the DNA. Therefore, a flat baseline taken from the end of each titration (Figure 2A–D) was used for peak integration. The data were analyzed using the Origin software (MicroCal) and the single-site-binding model was fitted to it. Standard errors were determined after repeating each experiment two to four times.
DNA band-shifts
For band-shifts using DNA with free ends, MutS (30 nM–20 μM) was bound to 1 nM 32P end-labeled 30 bp DNA substrate (see above) in binding buffer (25 mM Hepes–NaOH pH 7.5, 100 mM NaCl, 50 mM KCl, 10 mM MgCl2, 10 mM β-mercaptoethanol, 1% glycerol, 25 μg/ml BSA) in 10 μl total volume. After incubation of ∼10 min at 20 or 37°C, 3 μl of 50% sucrose was added to the samples, which were then loaded under voltage onto 4% native polyacrylamide gels (29:1 acrylamide:bisacrylamide) and run in TAE buffer+5 mM MgAc at 22 or 37°C. The gels were dried, exposed to phosphorimager and quantified using ImageQuant software. Data were analyzed using Microsoft Excel with the Solver add-in software. The single-site-binding model [DNA bound]=Bmax[protein]/(Kd+[protein]) was fitted to the data. Standard errors were determined from three independent experiments.
For band-shifts with blocked DNA ends, 1 nM 32P-labeled 41 bp DNA substrate (see above) with biotin at both 3′ ends was preincubated with streptavidin (10 ng/μl) for ∼10 min before binding to MutS (50–500 nM). For sliding clamp formation, after ∼10 min, the reaction with MutS was split into two reactions, of which one was further incubated with ATP (10 mM) and the other with ATP buffer for ∼10 min. Reactions were performed at 22 or 4°C in binding buffer. Data are reproducible, but quantification was not possible owing to smearing of the double end-blocked DNA. For MutL binding, after ∼10 min, 10 mM ATP was added to the reaction with MutS (400 nM). After ∼10 min, one-half of the reaction was further incubated with MutL (1600 nM) and the other with MutL buffer. To 10 μl volume of final reactions, 3 μl of 50% sucrose was added. Samples were run on 4 % native polyacrylamide gels (29:1 acrylamide:bisacrylamide) in TAE buffer+5 mM MgAc and 1 mM ATP both in the gel and running buffer.
Nucleotide binding and exchange
Binding of the poorly hydrolyzable analog ATPγS to the high-affinity site of MutS was assessed by filter binding (Schleicher & Schuell Minifold II slot-blot). 35S-labeled ATPγS was incubated with 0.1–10 μM MutS in reaction buffer (25 mM Hepes–NaOH, 150 mM NaCl, 10 mM MgCl2) and incubated on ice for several minutes. Samples were spotted in triplicate onto 0.45 μM nitrocellulose filters that were prewashed with reaction buffer. Filters were analyzed as described before (Lamers et al, 2003). Nucleotide exchange was monitored on a FLUOstar Optima spectrophotometer using the increase in fluorescence emission intensity of the ADP derivative MANT-ADP (Molecular Probes) upon binding to MutS. We used excitation and emission filters of 355 and 405 nm, respectively. Reactions contained 5 μM MutS and 10 μM MANT-ADP with or without 5 μM mismatched DNA. ADP exchange was initiated by fast titration of 1 volume of either reaction buffer or reaction buffer containing 100 μM ATP, and loss of fluorescence emission intensity was followed in time. After correction for buffer dilution effects, a function describing linear exponential decay was fitted to the data using nonlinear regression.
ATP hydrolysis
Kinetics for steady-state ATP hydrolysis were determined using an ATP-regenerating spectrophotometric assay as described (Lamers et al, 2003). Stimulation of the steady-state ATP hydrolysis rate by DNA was determined as described (Lamers et al, 2004). For determination of the magnitude of the pre-steady-state burst amplitude, 20 μl reactions contained 10 μM MutS in 25 mM Hepes–NaOH pH 7.5, 150 mM NaCl, 5 mM β-mercaptoethanol, 10 mM MgCl2 and 50 μM ATP containing α-32P-labeled ATP, with or without 10 μM mismatched DNA. Reactions without ATP were assembled at 0°C and hydrolysis was initiated by the addition of ATP. Samples (1 μl) were removed, quenched in 40 μl 0.2% SDS and 10 mM EDTA and spotted onto PEI cellulose plates (Merck). Plates were developed in 1 M orthophosphoric acid (pH 3.8) and analyzed by phosphorimaging.
Surface plasmon resonance
SPR spectroscopy was performed at 25°C on a BIAcore 3000. Streptavidin SA sensor chips were derivatized with ∼150 resonance units of biotin/fluorescein-derivatized 41 bp heteroduplex and homoduplex. In measurements using double end-blocked DNA, the 3′-fluorescein end was blocked using anti-fluorescein rabbit IgG Fab fragment (Molecular Probes, Invitrogen). Anti-fluorescein was flown across the chip at 5 μl/min before each MutS injection. MutS (50–300 nM) or MutS E38 mutants in running buffer (RB; 25 mM Hepes–NaOH, pH 7.5, 10 mM dithiothreitol, 150 mM NaCl, 5 mM MgCl2) were injected across the SA chip at 30 μl/min. In measurements of ATP-dependent MutS dissociation, the RB containing 0–200 μM ATP was injected immediately after MutS (300 nM). Chips were regenerated by a 20 μl injection of 0.05% SDS. The dissociation constant (Kd) for MutS–DNA complexes was determined by titration with increasing concentrations of MutS. Saturation binding values were fit according to a one-to-one binding model. ATP-dependent MutS dissociation was fit to one-phase exponential decay. The rate constants (k) from these fittings were plotted as a function of the ATP concentration and the hyperbolic function k=k0+kmax[ATP]/(Km+[ATP]) was fitted to the data.
Coordinates
Coordinates have been deposited in the PDB: 1WB9 (MutS-E38T), 1WBB (MutS-E38A) and 1WBD (MutS-E38Q).
Supplementary Material
Supplementary Table I
Supplementary Table II
Acknowledgments
We thank the beamline staff at the European Synchrotron Radiation Facility, Grenoble, France, Dr A Perrakis for discussion and assistance during crystallographic data collection and M Lamers for useful discussions. The MutS-deficient strain RK1517 was kindly provided by Dr I Matic. We thank Professor AM Deelder, Department of Parasitology, Leiden University Medical Center, for the use of their Biacore facility and A van Remoortere for discussion and technical assistance. Dr D Georgijevic is gratefully acknowledged for his help in solving the Luria–Delbrück equation. Funding from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek-Chemische Wetenschappen (Jonge Chemici 99548 and a VENI fellowship to JHGL), Koningin Wilhelmina Fonds (project no. 04-3084), EU-Structural Proteomics in Europe (QLRT-2001-0988) and Association for International Cancer Research (Grant Ref. 99-142) is gratefully acknowledged.
References
- Acharya S, Foster PL, Brooks P, Fishel R (2003) The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol Cell 12: 233–246 [DOI] [PubMed] [Google Scholar]
- Antony E, Hingorani MM (2003) Mismatch recognition-coupled stabilization of Msh2–Msh6 in an ATP-bound state at the initiation of DNA repair. Biochemistry 42: 7682–7693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony E, Hingorani MM (2004) Asymmetric ATP binding and hydrolysis activity of the Thermus aquaticus MutS dimer is key to modulation of its interactions with mismatched DNA. Biochemistry 43: 13115–13128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornson KP, Allen DJ, Modrich P (2000) Modulation of MutS ATP hydrolysis by DNA cofactors. Biochemistry 39: 3176–3183 [DOI] [PubMed] [Google Scholar]
- Bjornson KP, Blackwell LJ, Sage H, Baitinger C, Allen D, Modrich P (2003a) Assembly and molecular activities of the MutS tetramer. J Biol Chem 278: 34667–34673 [DOI] [PubMed] [Google Scholar]
- Bjornson KP, Modrich P (2003b) Differential and simultaneous adenosine di- and triphosphate binding by MutS. J Biol Chem 278: 18557–18562 [DOI] [PubMed] [Google Scholar]
- Blackwell LJ, Bjornson KP, Allen DJ, Modrich P (2001) Distinct MutS DNA-binding modes that are differentially modulated by ATP binding and hydrolysis. J Biol Chem 276: 34339–34347 [DOI] [PubMed] [Google Scholar]
- Brown J, Brown T, Fox KR (2001) Affinity of mismatch-binding protein MutS for heteroduplexes containing different mismatches. Biochem J 354: 627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Drotschmann K, Yang W, Brownewell FE, Kool ET, Kunkel TA (2001) Asymmetric recognition of DNA local distortion. Structure-based functional studies of eukaryotic Msh2–Msh6. J Biol Chem 276: 46225–46229 [DOI] [PubMed] [Google Scholar]
- Gradia S, Acharya S, Fishel R (1997) The human mismatch recognition complex hMSH2–hMSH6 functions as a novel molecular switch. Cell 91: 995–1005 [DOI] [PubMed] [Google Scholar]
- Gradia S, Subramanian D, Wilson T, Acharya S, Makhov A, Griffith J, Fishel R (1999) hMSH2–hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell 3: 255–261 [DOI] [PubMed] [Google Scholar]
- Hays JB, Hoffman PD, Wang H (2005) Discrimination and versatility in mismatch repair. DNA Repair 4: 1463–1474 [DOI] [PubMed] [Google Scholar]
- Hooft RW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381: 272. [DOI] [PubMed] [Google Scholar]
- Iaccarino I, Marra G, Dufner P, Jiricny J (2000) Mutation in the magnesium binding site of hMSH6 disables the hMutSalpha sliding clamp from translocating along DNA. J Biol Chem 275: 2080–2086 [DOI] [PubMed] [Google Scholar]
- Junop MS, Obmolova G, Rausch K, Hsieh P, Yang W (2001) Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol Cell 7: 1–12 [DOI] [PubMed] [Google Scholar]
- Junop MS, Yang W, Funchain P, Clendenin W, Miller JH (2003) In vitro and in vivo studies of MutS, MutL and MutH mutants: correlation of mismatch repair and DNA recombination. DNA Repair (Amst) 2: 387–405 [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA (2005) DNA mismatch repair. Annu Rev Biochem 74: 681–710 [DOI] [PubMed] [Google Scholar]
- Lamers MH, Georgijevic D, Lebbink JH, Winterwerp HH, Agianian B, de Wind N, Sixma TK (2004) ATP increases the affinity between MutS ATPase domains. Implications for ATP hydrolysis and conformational changes. J Biol Chem 279: 43879–43885 [DOI] [PubMed] [Google Scholar]
- Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, de Wind N, Sixma TK (2000) The crystal structure of DNA mismatch repair protein MutS binding to a G × T mismatch. Nature 407: 711–717 [DOI] [PubMed] [Google Scholar]
- Lamers MH, Winterwerp HH, Sixma TK (2003) The alternating ATPase domains of MutS control DNA mismatch repair. EMBO J 22: 746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, de la Chapelle A (1999) Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet 36: 801–818 [PMC free article] [PubMed] [Google Scholar]
- Marti TM, Kunz C, Fleck O (2002) DNA mismatch repair and mutation avoidance pathways. J Cell Physiol 191: 28–41 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Natrajan G, Lamers MH, Enzlin JH, Winterwerp HH, Perrakis A, Sixma TK (2003) Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res 31: 4814–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obmolova G, Ban C, Hsieh P, Yang W (2000) Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 407: 703–710 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (eds) (1997) Processing of X-ray Data Collected in Oscillation Mode. New York: Academic Press [DOI] [PubMed] [Google Scholar]
- Peltomaki P (2003) Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol 21: 1174–1179 [DOI] [PubMed] [Google Scholar]
- Schofield MJ, Brownewell FE, Nayak S, Du C, Kool ET, Hsieh P (2001) The Phe-X-Glu DNA binding motif of MutS. The role of hydrogen-bonding in mismatch recognition. J Biol Chem 276: 45505–45508 [DOI] [PubMed] [Google Scholar]
- Schofield MJ, Hsieh P (2003) DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol 57: 579–608 [DOI] [PubMed] [Google Scholar]
- Selmane T, Schofield MJ, Nayak S, Du C, Hsieh P (2003) Formation of a DNA mismatch repair complex mediated by ATP. J Mol Biol 334: 949–965 [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D 57: 122–133 [DOI] [PubMed] [Google Scholar]
- Wu TH, Marinus MG (1994) Dominant negative mutator mutations in the mutS gene of Escherichia coli. J Bacteriol 176: 5393–5400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Schofield MJ, Biswas I, Hsieh P (2000) Requirement for Phe36 for DNA binding and mismatch repair by Escherichia coli MutS protein. Nucleic Acids Res 28: 3564–3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Sass LE, Du C, Hsieh P, Erie DA (2005) Determination of protein–DNA binding constants and specificities from statistical analyses of single molecules: MutS–DNA interactions. Nucleic Acids Res 33: 4322–4334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table I
Supplementary Table II