

Abstract
Phosphoinositide 3-kinase (PI3K) plays a crucial role in triggering cell division. To initiate this process, PI3K induces two distinct routes, of which one promotes cell growth and the other regulates cyclin-dependent kinases. Fine-tuned PI3K regulation is also required for later cell cycle phases. Here, we review the multiple points at which PI3K controls cell division and discuss its impact on human cancer.
Keywords: cancer, cell cycle, p85 regulatory subunit, phosphatidylinositol 3-kinase
Introduction
Cell division is the process by which a cell replicates its DNA and increases its mass to generate two daughter cells. Cell division acts during embryonic development to generate tissues and organs, and in the adult for tissue repair and renewal. Initiation of cell division is a highly controlled process that depends on cell type, differentiation stage, cell context and growth factor receptor (GFR) activation. GFR frequently have intrinsic or associated Tyr kinase (Tyr K) activity; this allows Tyr-phosphorylated GFR to bind to SH2-domain containing proteins, which act as adaptors for a number of downstream signaling molecules such as phosphoinositide 3-kinase (PI3K), phospholipase C (PLC), Ras and microtubule-associated protein kinase (MAPK) (reviewed in Schlessinger, 2000; Cantley, 2002). In addition to classical GFR, other signals activate PI3K and influence cell division, including lysophosphatidic acid, adhesion molecules and nuclear receptors (Simoncini et al, 2000; Yart et al, 2002).
Class I PI3K family members
The PI3K are a family of enzymes that transfer phosphate to the D3 position of the inositol ring of membrane phosphoinositides (PI). Sequence comparison has allowed identification of several PI3K classes (reviewed in Fruman et al, 1998; Downes et al, 2005; Wymann and Marone, 2005). Here, we focus on the dual-specificity lipid and protein kinases of the class I family, the only class able to generate PI(3,4)P2 and PI(3,4,5)P3 (PIP2 and PIP3) in vivo. These lipids are at very low levels in quiescent cells, increase rapidly and transiently following GFR stimulation, and are crucial for control cell survival, migration and division.
Class I PI3K are composed of a regulatory and a catalytic (p110) subunit, of which there are four isoforms. Three of them, p110α, p110β and p110δ, associate p85-related regulatory subunits (class IA PI3K) and are activated by Tyr K. The fourth isoform, p110γ (class IB), has distinct regulatory counterparts (p101 or p84) and is activated by G-protein-coupled receptors (GPCR). p110α and p110β are expressed ubiquitously, whereas p110δ and p110γ expression is more abundant in hematopoietic cells (Figure 1).
Figure 1.
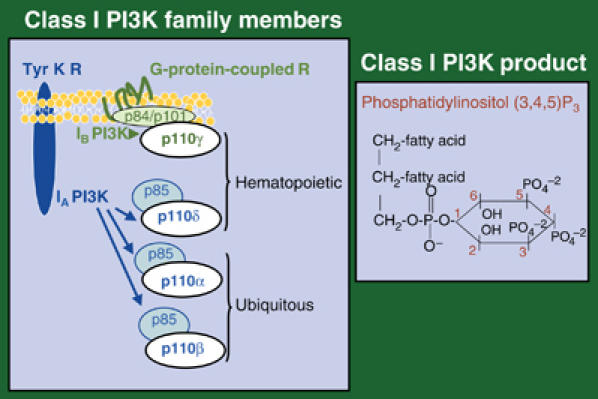
Class I PI3K. Scheme of heterodimeric class I PI3K family members and their classification as IA and IB. The figure illustrates the receptors that activate these enzymes, as well as PI3K tissue distribution. The PIP3 molecule is shown on the right. Tyr K R, Tyr kinase receptor.
GPCR activation of IB p110γ involves dissociation of G protein βγ subunits, which subsequently associate and activate p110γ (Stephens et al, 1994). IA PI3K are activated by Tyr K GFR, whose binding to p85-SH2 domains induce p85/p110 translocation to the plasma membrane. IA PI3K are also activated by cytosolic Tyr K, such as Src family members that phosphorylate the p85 subunit. Finally, class I PI3K are further activated by Ras (Jiménez et al, 2002). As exceptions to the class IA/Tyr K and class IB/GPCR activation rule, the IA isoform p110β can be activated by GPCR (Yart et al, 2002), whereas IB p110γ is involved in T-cell receptor-induced T-cell activation, a process dependent on Src kinase activation (Simoncini et al, 2000; Barber et al, 2005a).
Several downregulation mechanisms ensure that PIP3 formation is transient; these include the SH2-containing inositol D5-phosphatases (SHIP), which transform PIP3 into PIP2, and the D3-phosphatase PTEN (phosphatase and tensin homolog deleted in chromosome ten). PTEN reduces both PIP3 and PIP2 levels, although details as to how its action is regulated remain unclear (Cantley, 2002).
PI3K effectors
PIP3 and PIP2 act as docking sites for a subgroup of proteins bearing a pleckstrin homology (PH) domain (Downes et al, 2005; Wymann and Marone, 2005). Among these, there are several exchange factors for Rac GTPase, which mediates many PI3K effects on actin polymerization (Jiménez et al, 2000; Raftopoulou and Hall, 2004). Phosphoinositide dependent-kinase 1 (PDK1) also has these PH domains and regulates activation of enzymes such as p70 ribosomal S6 kinase (p70S6K), which controls protein synthesis (Kozma and Thomas 2002), serum-and-glucocorticoid kinase, and protein kinase B (PKB) (Figure 2). PKB activation requires not only PDK1-dependent phosphorylation, but also a second phosphorylation event mediated by a second kinase (PDK2), for which various candidates have been proposed (Wymann and Marone, 2005). PKB acts as a hub to control several critical enzymes for cell division, including the mammalian target of rapamycin (mTOR) and the FoxO members of the forkhead family of transcription factors (TF) (del Peso et al, 1999; Medema et al, 2000; Schmelzle and Hall, 2000). PKB also acts on a variety of apoptotic regulators (reviewed in Vivanco and Sawyers, 2002); for instance, PKB phosphorylation of MDM2 results in p53 inactivation (Mayo and Donner, 2002). Finally, PKB inhibits glycogen synthase kinase-3β (GSK-3β), an enzyme that triggers degradation of cell cycle regulators (Figure 2) (van Weeren et al, 1998).
Figure 2.
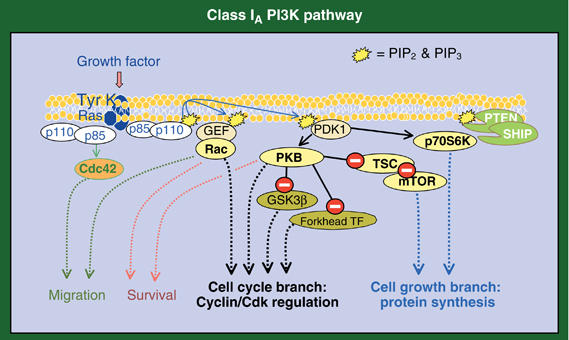
The two branches of the class I PI3K pathway. The scheme shows proteins regulated by class IA PI3K, including molecules whose activation is dependent on (yellow) or independent of (orange) PIP3 formation. Phosphatases that downregulate the pathway are shown in green. Inhibition is indicated by a white minus sign on red background. Some of these molecules promote cell growth (cell growth branch, blue dashed lines) and others cyclin/Cdk stability or activity (cell cycle branch, black dashed lines). GEF, GTPase exchange factor; GSK, glycogen-synthase kinase; mTOR, murine target of rapamycin; PDK, PI-dependent kinase; PKB, protein kinase B; PTEN, phosphatase/tensin homolog deleted in chromosome ten; SHIP, SH2 inositol-phosphatases; TSC, tuberous sclerosis complex.
A different set of PI3K effectors is activated independently of p110 lipid kinase activity. p110α phosphorylates translation initiation factor 4E binding protein-1 and Ras, potentially influencing their action (Foukas and Shepherd, 2004). p110γ acts as a scaffold for phosphodiesterase 3B; this association is required to maintain phosphodiesterase activity (Patrucco et al, 2004). Finally, p85 also binds and regulates other enzymes, such as Cdc42 and Jun N-terminal kinase, independently of p110 activity (Jiménez et al, 2000) (Figure 2).
PI3K actions
Expression of a gain-of-function IA PI3K mutant (p65PI3K) in T cells enhances cell survival, division and migration, inducing development of an invasive lymphoproliferative autoimmune disease (Jiménez et al, 1998; Rodríguez-Borlado et al, 2000). Reduced expression of the PI3K-negative regulator PTEN induces a similar phenotype (Di Cristofano et al, 1999). Apart from these effects on lymphocytes, PI3K regulates survival, migration and division in numerous cell types; indeed, PTEN deletion induces tumor development in tissues of diverse origins (Di Cristofano et al, 1998). PI3K also regulates glucose metabolism and oxidative stress (Cantley, 2002; Barthel and Klotz, 2005). PKB is the main effector of cell survival (Vivanco and Sawyers, 2002; Downward, 2004), whereas Rac is the main effector for cell migration (Raftopoulou and Hall, 2004). In cell division, PI3K has been implicated both in induction of cell growth and regulation of cyclin-dependent kinase (Cdk) activity. To trigger these processes, PI3K induces two distinct but interconnected routes; one branch promotes the cell growth and the other regulates cyclin/Cdk activation (Figure 2).
PI3K in the initiation of cell division: cell growth
Quiescent cells have a slow metabolism, with low transcription and translation activity and minimal energy consumption. Cells in this phase (G0) express high levels of cell cycle inhibitors and low levels of the G1 cyclins (Martínez-Gac et al, 2004). Division requires induction of protein synthesis (cell growth) and of DNA duplication, orchestrated by the cell cycle machinery (Figure 3). Cell growth is necessary to ensure that macromolecular composition and cell size are conserved in daughter cells. Cell division cannot occur without growth, although cells can grow in the absence of cell division (Tapon et al, 2001). Cell growth is therefore a prerequisite for cell cycle entry.
Figure 3.
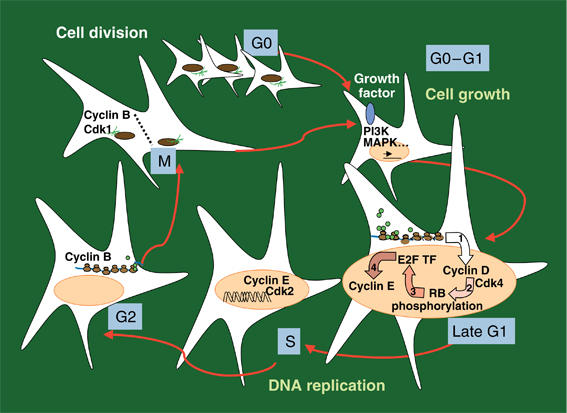
Steps in cell division. Quiescent cells require environmental signals (growth factors, GF) to initiate cell division. The signals generated following GF receptor activate PI3K, MAPK and other cascades, which trigger cell growth and cyclin/Cdk activation. In mid-G1, cyclin D/Cdk4 (or Cdk6) activation is required to induce cyclin E synthesis; cyclin E/Cdk2 regulates initiation of DNA replication. Once DNA is duplicated, cells enter G2 and synthesize proteins required for mitosis. Cyclin B/Cdk1 activation promotes entry into mitosis, in which the two DNA copies separate and cytosol divides. RB, retinoblastoma protein; E2F TF, E2F family transcription factors.
The signaling pathway that controls cell mass increase was recently elucidated, and includes two key enzymes, p70S6K and mTOR (Figure 2) (Tapon et al, 2001; Fingar et al, 2002; Saucedo and Edgar, 2002; Sarbassov et al, 2005). mTOR acts as a sensor of optimal growth conditions, as its activation requires appropriate oxygen, ATP and amino-acid levels, as well as the absence of free radicals. When energy and nutrient conditions are optimal, mitogens enhance mTOR activation. mTOR is activated within a multiprotein complex (reviewed in Schmelzle and Hall, 2000; Carrera, 2004), in which mTOR is negatively regulated by tumor-suppressor tuberous sclerosis protein-1 and 2 (TSC1 and 2). mTOR controls phosphorylation of translation initiation factor 4E-binding protein 1, and regulates p70S6K, which phosphorylates ribosomal S6 protein, facilitating translation of ribosomal components. p70S6K also regulates translation elongation (Kozma and Thomas, 2002). Interestingly, mTOR also forms part of another protein complex that regulates motility (Sarbassov et al, 2005).
Regulation of mTOR activity is one mechanism for PI3K control of cell growth. PKB phosphorylates and inactivates TSC2 that, bound to TSC1, inhibits mTOR (Inoki et al, 2002; Carrera, 2004). In addition, the p85 regulatory subunit associates with mTOR, allowing p70S6K activation (González-García et al, 2002). PI3K also regulates zeta-PKC, Cdc42 and Rac, all of which influence p70S6K activity (Romanelli et al, 2002). Finally, the PI3K pathway enhances translation of ribosomal components independently of p70S6K (Stolovich et al, 2002). mTOR also increases PKB activation, whereas it reduces PI3K activation by the insulin receptor (Sarbassov et al, 2005). p85/PKB/mTOR and p70S6K thus represent the cell growth branch of the PI3K pathway (Figure 2).
PI3K in Cdk regulation at G0/G1 and G1/S
Besides regulating cell growth, growth factors induce a remarkable increase in cyclin D levels, required to activate Cdk 4 or Cdk 6 in the G1 phase (Figure 3). These Cdk phosphorylate the retinoblastoma protein (RB), allowing release of associated E2F TF, which induces cyclin E synthesis (Figure 3) (Sherr and Roberts, 1999). Cyclin E/Cdk2 regulates prereplication complex assembly and augments RB phosphorylation and E2F liberation in a positive feedback loop. Initiation of DNA replication also requires Ask (activator of S phase kinase) and cyclin A synthesis, which are induced by E2F and/or Myc TF (Sanchez and Dynlacht, 2005). This hierarchical activation of Cdk required for G1 progression represents a simplified view, as G1 can also progress in the absence of some of these components (Ortega et al, 2002). Cdk activation requires Cdk-activating kinases as well as inactivation of Cdk inhibitors, which are grouped into INK and Cip/Kip families. Although INK expression is induced by aging, TGFβ or cell stress, Cip/Kip inhibitors (p27kip1 and p21cip1) are expressed in normal quiescent cells and inhibit Cdk2 (Sherr and Roberts, 1999).
Pharmacological inhibition of PI3K at the time of GFR stimulation blocks cell growth and cell cycle entry (Wymann and Pirola, 1998). In addition, enhanced PIP3 production following GFR stimulation in cells expressing PI3K mutants accelerates cell cycle entry, whereas PIP3 reduction diminishes this process (Álvarez et al, 2001). The mechanisms by which PI3K controls G0/G1 transition include regulation of cyclin D synthesis through the PI3K effectors Rac and Cdc42 (Olson et al, 1995). PKB also inhibits Myc and cyclin D degradation by inactivating GSK-3β (van Weeren et al, 1998; Ryves and Harwood, 2003). Finally, PKB phosphorylates FoxO TF, inducing its sequestration in the cytosol and reducing FoxO TF-mediated expression of Ccng2, a cell cycle inhibitor that assists in maintaining quiescence (Medema et al, 2000; Martínez-Gac et al, 2004).
PI3K activity increases after GFR stimulation at the G0–G1 transition; a second activity peak is observed in mid-G1 (Figure 4A) (Jones and Kazlauskas, 2001; Álvarez et al, 2001). Pharmacological inhibition of PI3K in mid-G1 blocks DNA replication, whereas PIP3 addition at this point is sufficient to trigger DNA synthesis (Jones and Kazlauskas, 2001). Neither the signals that control this activity peak nor its specific role in DNA synthesis are known; nonetheless, as Ras is activated in mid-G1, it may regulate PI3K activity at this point (Jones and Kazlauskas, 2001).
Figure 4.
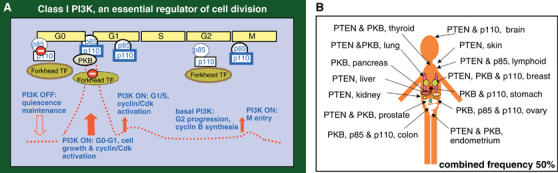
(A) PI3K activation throughout the cell cycle. (B) Deregulation of the PI3K pathway in human cancer. The figure shows mutations in p85, p110, PTEN and PKB in human cancer, and tissues/organs in which they are found.
PI3K might act in mid-G1 to control Cdk2, as expression of an active p110 mutant increases Cdk2 activity (Klippel et al, 1998). The PI3K pathway may regulate Cdk2 by stabilizing cyclin E, as PKB inactivates GSK-3β, an enzyme that targets cyclin E for degradation (van Weeren et al, 1998; Ryves and Harwood, 2003). Moreover, PKB regulates Cdk2 inhibitors in different ways. PKB phosphorylates p21cip, relocating it to the cytosol and inducing its release from proliferating cell nuclear antigen (PCNA), an event required for DNA polymerase activity (Vivanco and Sawyers, 2002). PKB also inhibits FoxO TF-mediated p27kip transcription (Medema et al, 2000) and phosphorylates p27kip, provoking its exit from the nucleus (Vivanco and Sawyers, 2002). These observations suggest that the PI3K effectors PKB, GSK-3β, Rac and Cdc42 regulate cyclin/Cdk activity and stability in early and mid-G1. These effectors thus represent the cell cycle regulatory branch of the PI3K pathway (Figure 2).
PI3K, a coordinator of cell growth and cell cycle entry
The independent processes of cell growth and cyclin/Cdk activation must be coordinated to ensure that DNA duplication is initiated only when the cell reaches appropriate size. In yeast, the connection between growth and cell cycle entry is mediated by cyclin D (cln3); cln3 mRNA has two in-frame ATG (initiation codon), and translates efficiently only when protein synthesis machinery is fully active (Tapon et al, 2001). In mammals, this link is still unknown, although PI3K appears to coordinate cell growth and cell cycle entry. Following GFR stimulation in cells expressing PI3K mutants, enhancement or reduction of transient PIP3 production accelerates or reduces protein synthesis levels. These variations in PIP3 production also affect cell cycle entry rates, triggering changes in the time of division, but not in cell size (Álvarez et al, 2003). The mechanism by which PI3K concerts regulation of cell growth and cell cycle progression is incompletely understood; however, as cyclin E has two in-frame ATG in its promoter, it is tempting to speculate that by regulating cell growth, PI3K determines the level of cyclin E translation and, in turn, the rate of S phase entry.
PI3K two-branch model for cell cycle entry
The observations described suggest that two routes downstream of PI3K regulate cell division, one involved in triggering cell cycle entry and the other, in promoting cell growth; PKB would be common to both branches (Figure 2). In fact, inhibition of PI3K or its effectors in the growth branch (p70S6K and mTOR) blocks cell growth (Fingar et al, 2002). Nonetheless, only activation of PI3K, but not of p70S6K and mTOR, promotes cell cycle entry (Álvarez et al, 2001; Jones and Kazlauskas, 2001). This shows that a selective set of PI3K substrates, different from mTOR and p70S6K, triggers PI3K-mediated cyclin/Cdk regulation.
Confirming this model, studies in Drosophila wing imaginal disc show that mutations in PI3K activity regulators (Inr, dp110, dPTEN and dRas) affect cell growth and cell cycle simultaneously, whereas mutations in the cell growth branch (dTSC, dTOR, d4EBP and dS6K) only affect cell size. In organs in which growth occurs without division, dp110 mutation enlarges cell size (Saucedo and Edgar, 2002). Similarly, in mammals, PI3K enhances cell growth in postmitotic cells of the heart, and increases cell division in mitotic tissues such as T cells or neurons of the developing retina (Rodríguez-Borlado et al, 2000; Shioi et al, 2000; Pimentel et al, 2002).
PI3K in G2 and M
Apart from controlling cell cycle entry, PI3K influences G2/M phases. During G2, cells prepare for mitosis by inducing centrosome maturation and synthesis of mitotic regulators such as cyclin B and polo-like kinase (Plk) (Figure 3). M phase begins when cyclin B/Cdk1 is activated and triggers nuclear division (DNA separation) and cytokinesis (cytosolic division).
Fibroblasts expressing constitutive active PI3K/PKB forms show delayed G2/M progression and defective cell cycle exit (Álvarez et al, 2001). In normal G2 phase cells, FoxO TF relocates to the nucleus and regulates expression of cyclin B and Plk. In contrast, in fibroblasts expressing active PI3K/PKB, FoxO TF is cytosolic throughout the cell cycle, impairing cyclin B and Plk expression and mitotic progression (Álvarez et al, 2001). Further studies showed that FoxM, a forkhead TF whose expression increases in S to M phases and binds the same DNA motif as FoxO TF, has an essential role in G2 phase gene expression (Laoukili et al, 2005). As in yeast, forkhead TF thus appear to control expression of mitotic genes in mammals.
There is an additional minor PI3K activity peak at M phase entry. Release of epithelial cell lines from S phase arrest shows a first PI3K activity peak at S phase entry and another one simultaneous with M phase entry (Shtivelman et al, 2002). NIH3T3 cells show a strong activity peak at G0–G1, however, with two smaller peaks at mid-G1 (Álvarez et al, 2001) and M phase entry (our unpublished observations, Shtivelman et al, 2002). Inhibition of PI3K in late S phase blocks M phase entry in MDCK cells, whereas it delays entry into M phase in HeLa and NIH3T3 cells (Shtivelman et al, 2002). In HeLa cells, this delay correlates with postponed Cdc2 activation kinetics (Dangi et al, 2003). Reduction of basal PIP3 levels throughout S, G2 and M also enhances UV light-induced apoptosis (Dangi et al, 2003). These studies show that PI3K regulates mitosis entry, with a distinct relative contribution depending on cell type. Finally, it was recently shown that PIP3 localizes to cell poles and PTEN to the cleavage furrow in Dictyostelium discoideum, supporting the idea that a PI3K gradient regulates cytosolic division in this organism (Janetopoulos et al, 2005).
PI3K is an essential regulator for cell cycle progression
G0/G1 transition requires transient activation of PI3K and inactivation of FoxO TF. A second PI3K activity peak occurs in mid-G1 and is required for S phase entry. G2 phase progression involves forkhead TF activity and basal PIP3 levels, and M phase entry requires an additional PI3K activity peak. These observations support fine-tuned regulation of PI3K as an essential event for cell cycle progression (Figure 4A).
PI3K isoform-specific functions in cell division
Many of these studies were performed using general PI3K inhibitors or mutant forms of p110α and p85α. The phenotypes of mice deficient in each of the four PI3K catalytic subunits helped to define p110 isoform specificity. p110γ and p110δ are expressed more abundantly in hematopoietic tissues, and deletion of either gene thus impairs the immune response. p110γ regulates T-cell activation as well as neutrophil and macrophage migration, whereas p110δ controls T- and B-cell activation (Vanhaesebroeck et al, 2005). The ubiquitous expression of p110α and p110β anticipates a more generalized function for these isoforms. p110α-deficient mice die at embryonic day (E) E9.5–10.5; brain sections from these embryos show a DNA synthesis defect. p110α-deficient mice also show impaired angiogenesis (Lelievre et al, 2005).
The early embryonic lethality of p110β-deficient mice (lethal at E2.5) indicates that this isoform is essential in very early development (reviewed in Vanhaesebroeck et al, 2005), possibly at the embryonic stem (ES) cell phase. In vitro studies are controversial, as some authors found a PI3K activity requirement for ES cell division/survival, and others for ES cell self-renewal (Jirmanova et al, 2002; Paling et al, 2004).
Apart from the defects caused by p110α and p110β deficiency, excess PI3K pathway activation also impairs development (Di Cristofano et al, 1998). Further study is needed to define the specific p110α and p110β functions in cell division and to understand how distinct p110 subunits with identical lipid kinase activity have different functions. The p110α and p110β knockout mouse phenotypes nonetheless support regulation of cell division by these isoforms.
PI3K pathways and cancer
PI3K involvement in cell survival, migration and division explains the high frequency of mutations in this pathway in cancer. In fact, both enhanced PI3K activation (p65PI3K Tg mice) and heterozygous PTEN loss trigger a strikingly similar ‘polyclonal' invasive lymphoproliferative disorder and autoimmune disease. This phenotypic similarity shows that the tumor suppressor function of PTEN probably lies in its action on PIP3 rather than in its protein phosphatase activity. Clonal malignant tumors develop efficiently in PTEN+/− and in p65PI3K Tg/p53−/− mice, demonstrating the transforming potential of the PI3K pathway and suggesting that full transformation requires an additional mutation event(s) in individual clones (Di Cristofano et al, 1999; Rodríguez-Borlado et al, 2000).
In human cancer, PTEN phosphatase mutations appear with a combined frequency of 50% (Cantley and Neel, 1999). In addition, the PI3K/PKB pathway can be activated in human tumors by PTEN transcriptional silencing, expression of p65PI3K-like mutations and increased expression/mutation of p110α/β or PKB isoforms (Philp et al, 2001; Vivanco and Sawyers, 2002; Samuels et al, 2004; Stephens et al, 2005) (Figure 4B). Consistent with the two-branch PI3K pathway model, mutations in TSC1 and TSC2 genes in the PI3K cell growth branch give rise to benign tumors, whereas mutations that affect PI3K or PTEN, which induce cell growth and cell cycle entry, are found in malignant tumors (Inoki et al, 2002; Wymann and Marone, 2005).
The mutations reported in the PI3K pathway often allow transient PI3K activation. In fact, PTEN deletion only induces a moderate increase in basal PIP3, which is still upregulated transiently following GFR stimulation (Myers et al, 1998). Selection of mutations that permit transient PIP3 formation may reflect the deleterious consequences of sustained PI3K for cell growth and cell cycle exit (Álvarez et al, 2001, 2003).
Cell transformation is a stepwise process that involves acquisition of phenotypes including self-sufficient growth, insensitivity to growth inhibition, evasion of apoptosis, immortalization and the ability of transformed cells to trigger angiogenesis and invasion (Hanahan and Weinberg, 2000). The contribution of PI3K to cell division and survival suggests that the biological advantage of tumors with PI3K pathway mutations may be prolonged survival or the lower mitogenic threshold required for cell division. PI3K also regulates immortalization (Kang et al, 1999), angiogenesis and invasion (Lelievre et al, 2005); these events may also be affected by PI3K deregulation in human cancer.
Future studies will determine whether interference with specific PI3K catalytic and/or regulatory subunit isoforms impairs any of these cell responses involved in human cancer. Although p110α and p110β have essential functions during embryonic development, in the adult most cells are terminally differentiated and have low PIP3 levels. This suggests that inhibition of specific p110 isoforms may affect transformed cells selectively, without causing a generalized impairment of normal cell viability. A frequent side effect of chemotherapy is a reduction in the number of hematopoietic cells, because of their high mitotic index. The existence of hematopoietic-specific p110δ/γ PI3K isoforms could result in selective targeting of tumor cells using p110α- and β-specific inhibitors. Indeed, selective inhibition of the hematopoietic p110γ isoform ameliorates chronic inflammatory autoimmune disease, with no other side effects (Barber et al, 2005a, 2005b). As not all cells deregulate the pathway at the same level or show the same sensitivity to PI3K activity, individual phenotypic tumor analysis would be required for rational PI3K-based therapy. Preliminary studies in vitro or in xenografts using nonisoform specific PI3K inhibitors are promising, as PI3K inhibition reduces cell survival, invasion or proliferation in different tumor cell lines (Wymann and Marone, 2005). The mTOR inhibitor rapamycin also shows selective antitumor activity in cancers with PI3K/PTEN mutations (Vivanco and Sawyers, 2002).
These observations show that PI3K is needed for many events involved in cell division, such as regulation of cyclin/Cdk, translation initiation, TSC2 inactivation, etc. MAPK and other pathways also control many of these events in complementary ways. PI3K cooperates with these pathways to trigger cell division. However, in many cell types, PI3K activity is sufficient to support cell survival and is necessary for cell division. The hope for the future is that interfering with a necessary PI3K signal might be sufficient to impair altered survival and division of malignant cells.
Acknowledgments
We thank DF Barber, A González, M Serrano, M Torres and I Mérida for critical reading of the manuscript and C Mark for editorial assistance. Some of this work was supported by grants from the European Union (QLRT2001-02171) and the Spanish DGCyDT (SAF2004-05955). The Department of Immunology and Oncology was founded and is supported by the Spanish National Research Council (CSIC) and Pfizer.
References
- Álvarez B, Garrido E, Garcia-Sanz JA, Carrera AC (2003) PI3K activation regulates cell division time by coordinated control of cell mass and cell cycle progression rate. J Biol Chem 278: 26466–26473 [DOI] [PubMed] [Google Scholar]
- Álvarez B, Martínez AC, Burgering BM, Carrera AC (2001) Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature 413: 744–747 [DOI] [PubMed] [Google Scholar]
- Barber DF, Bartolomé A, Hernandez C, Flores JM, Fernandez-Arias C, Rodríguez-Borlado C, Hirsch E, Wymann M, Balomenos D, Carrera AC (2005a) Class IB-PI3K deficiency ameliorates IA-induced systemic lupus but not T cell invasion. J Immunol 176: 589–593 [DOI] [PubMed] [Google Scholar]
- Barber DF, Bartolome A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, Camps M, Ruckle T, Schwarz MK, Rodriguez S, Martinez-Alonso C, Balomenos D, Rommel C, Carrera AC (2005b) PI3Kγ inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med 11: 933–935 [DOI] [PubMed] [Google Scholar]
- Barthel A, Klotz LO (2005) Phosphoinositide 3-kinase signaling in the cellular response to oxidative stress. J Biol Chem 386: 207–216 [DOI] [PubMed] [Google Scholar]
- Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296: 1655–1657 [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the PI3K/AKT pathway. Proc Natl Acad Sci USA 96: 4240–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrera AC (2004) TOR signaling in mammals. J Cell Sci 117: 4615–4616 [DOI] [PubMed] [Google Scholar]
- Dangi S, Cha H, Shapiro P (2003) Requirement for PI3K activity during progression through S-phase and entry into mitosis. Cell Signal 15: 667–675 [DOI] [PubMed] [Google Scholar]
- del Peso L, Gonzalez VM, Hernandez R, Barr FG, Nuñez G (1999) Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene 18: 7328–7333 [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Kotsi P, Feng Peng Y, Cordon-Cardo C, Elkon KB, Pandolfi PP (1999) Impaired Fas response and autoimmunity in Pten−/− mice. Science 285: 2122–2125 [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP (1998) Pten is essential for embryonic development and tumor suppression. Nat Genet 19: 348–355 [DOI] [PubMed] [Google Scholar]
- Downes CP, Gray A, Lucocq JM (2005) Probing phosphoinositide functions in signaling and membrane trafficking. Trends Cell Biol 15: 259–268 [DOI] [PubMed] [Google Scholar]
- Downward J (2004) PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol 15: 177–182 [DOI] [PubMed] [Google Scholar]
- Fingar DC, Salama S, Tsou C, Harlow E, Blenis J (2002) Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev 16: 1472–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foukas LC, Shepherd PR (2004) eIF4E binding protein 1 and H-Ras are novel substrates for the protein kinase activity of class-I PI3K. Biochem Biophys Res Commun 319: 541–549 [DOI] [PubMed] [Google Scholar]
- Fruman DA, Meyers RE, Cantley LA (1998) Phosphoinositide kinases. Ann Rev Biochem 67: 481–507 [DOI] [PubMed] [Google Scholar]
- González-García A, Garrido E, Hernandez C, Alvarez B, Jiménez C, Cantrell DA, Pullen N, Carrera AC (2002) A new role for the p85-PI3K regulatory subunit linking FRAP to p70 S6 kinase activation. J Biol Chem 277: 1500–1508 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57–70 [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657 [DOI] [PubMed] [Google Scholar]
- Janetopoulos C, Borleis J, Vazquez F, Iijima M, Devreotes P (2005) Temporal and spatial regulation of phosphoinositide signaling mediates cytokinesis. Dev Cell 8: 467–477 [DOI] [PubMed] [Google Scholar]
- Jirmanova L, Afanassieff M, Gobert-Gosse S, Markossian S, Savatier P (2002) Differential contributions of ERK and PI3K to the regulation of cyclin D1 expression and to the control of the G1/S transition in mouse embryonic stem cells. Oncogene 21: 5515–5528 [DOI] [PubMed] [Google Scholar]
- Jiménez C, Hernandez C, Pimentel B, Carrera AC (2002) The p85 regulatory subunit controls sequential activation of PI3K by Tyr kinases and Ras. J Biol Chem 277: 41556–41562 [DOI] [PubMed] [Google Scholar]
- Jiménez C, Jones DR, Rodríguez-Viciana P, González-García A, Leonardo E, Wennstrom S, von Kobbe C, Toran JL, Rodríguez-Borlado L, Calvo V, Copín SG, Albar JP, Gaspar ML, Diez E, Marcos MA, Downward J, Martínez-Alonso C, Mérida I, Carrera AC (1998) Identification and characterization of a new oncogene derived from the regulatory subunit of PI3K. EMBO J 17: 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez C, Portela RA, Mellado M, Rodríguez-Frade JM, Collard J, Serrano A, Martínez-Alonso C, Avila J, Carrera AC (2000) Role of the PI3K regulatory subunit in the control of actin organization and cell migration. J Cell Biol 151: 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Kazlauskas A (2001) Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat Cell Biol 3: 165–172 [DOI] [PubMed] [Google Scholar]
- Kang SS, Kwon T, Kwon DY, Do SI (1999) Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem 274: 13085–13090 [DOI] [PubMed] [Google Scholar]
- Klippel A, Escobedo MA, Wachowicz MS, Apell G, Brown TW, Giedlin MA, Kavanaugh WM, Williams LT (1998) Activation of PI3K is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol Cell Biol 18: 5699–5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma SC, Thomas G (2002) Regulation of cell size in growth, development and human disease: PI3K, PKB and S6K. Bioessays 24: 65–71 [DOI] [PubMed] [Google Scholar]
- Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH (2005) FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 7: 126–136 [DOI] [PubMed] [Google Scholar]
- Lelievre E, Bourbon PM, Duan LJ, Nussbaum RL, Fong GH (2005) Deficiency in the p110alpha subunit of PI3K results in diminished Tie2 expression and Tie2−/− like vascular defects in mice. Blood 105: 3935–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Gac L, Marqués M, García Z, Campanero M, Carrera AC (2004) Control of cyclin G2 mRNA expression by Forkhead transcription factors: a novel mechanism for cell cycle control by PI3K and Forkhead. Mol Cell Biol 24: 2181–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo LD, Donner DB (2002) The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci 27: 462–467 [DOI] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM (2000) AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404: 782–787 [DOI] [PubMed] [Google Scholar]
- Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK (1998) The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci USA 95: 13513–13518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, Hall A (1995) An essential role for Rho, Rac and Cdc42 GTPases in cell cycle progression through G1. Science 269: 1270–1272 [DOI] [PubMed] [Google Scholar]
- Ortega S, Malumbres M, Barbacid M (2002) Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta 1602: 73–87 [DOI] [PubMed] [Google Scholar]
- Paling NR, Wheadon H, Bone HK, Welham MJ (2004) Regulation of embryonic stem cell self-renewal by PI3K-dependent signaling. J Biol Chem 279: 48063–48070 [DOI] [PubMed] [Google Scholar]
- Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E (2004) PI3Kg modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 118: 375–387 [DOI] [PubMed] [Google Scholar]
- Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJ, Phillips WA (2001) The PI3K p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res 61: 7426–7429 [PubMed] [Google Scholar]
- Pimentel B, Rodríguez-Borlado L, Hernandez C, Carrera AC (2002) The p85 regulatory subunit controls sequential activation of PI3K by Tyr kinases and Ras. Dev Biol 247: 295–306 [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A (2004) Cell migration: Rho GTPases lead the way. Dev Biol 265: 23–32 [DOI] [PubMed] [Google Scholar]
- Rodríguez-Borlado L, Redondo C, Álvarez B, Jiménez C, Criado LM, Flores J, Rodríguez-Marcos MA, Martínez-Alonso C, Balomenos D, Carrera AC (2000) Increased PI3K activity induces a lymphoproliferative disorder and contributes to tumor generation in vivo. FASEB J 14: 895–903 [DOI] [PubMed] [Google Scholar]
- Romanelli A, Dreisbach VC, Blenis J (2002) Characterization of PI3K-dependent phosphorylation of the hydrophobic motif site Thr(389) in p70 S6 kinase 1. J Biol Chem 277: 40281–40289 [DOI] [PubMed] [Google Scholar]
- Ryves WJ, Harwood AJ (2003) The interaction of glycogen synthase kinase-3 (GSK-3) with the cell cycle. Prog Cell Cycle Res 5: 489–495 [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304: 554. [DOI] [PubMed] [Google Scholar]
- Sanchez I, Dynlacht BD (2005) New insights into cyclins, CDKs and cell cycle control. Semin Cell Dev Biol 16: 311–321 [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17: 596–603 [DOI] [PubMed] [Google Scholar]
- Saucedo LJ, Edgar BA (2002) Why size matters: altering cell size. Curr Opin Genet Dev 12: 565–571 [DOI] [PubMed] [Google Scholar]
- Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103: 211–225 [DOI] [PubMed] [Google Scholar]
- Schmelzle T, Hall MN (2000) TOR, a central controller of cell growth. Cell 103: 253–262 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13: 1501–1512 [DOI] [PubMed] [Google Scholar]
- Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J, Cantley LC, Izumo S (2000) The conserved PI3K pathway determines heart size in mice. EMBO J 19: 2537–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtivelman E, Sussman J, Stokoe D (2002) A role for PI3K and PKB activity in the G2/M phase of the cell cycle. Curr Biol 12: 919–924 [DOI] [PubMed] [Google Scholar]
- Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK (2000) Interaction of oestrogen receptor with the regulatory subunit of PI3K. Nature 407: 538–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT (1994) A novel PI3K activity in myeloid-derived cells is activated by G protein beta gamma subunits. Cell 77: 83–93 [DOI] [PubMed] [Google Scholar]
- Stephens L, Williams R, Hawkins P (2005) Phosphoinositide 3-kinases as drug targets in cancer. Curr Opin Pharmacol 5: 357–365 [DOI] [PubMed] [Google Scholar]
- Stolovich M, Tang H, Hornstein E, Levy G, Cohen R, Bae SS, Birnbaum MJ, Meyuhas O (2002) Transduction of growth or mitogenic signals into translational activation of TOP mRNAs is fully reliant on the PI3K-mediated pathway but requires neither S6K1 nor rpS6 phosphorylation. Mol Cell Biol 22: 8101–8113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Moberg KH, Hariharan IK (2001) The coupling of cell growth to the cell cycle. Curr Opin Cell Biol 13: 731–737 [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC (2005) Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci 3: 194–204 [DOI] [PubMed] [Google Scholar]
- van Weeren PC, de Bruyn KM, de Vries-Smits AM, van Lint J, Burgering BM (1998) Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB. J Biol Chem 273: 13150–13156 [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL (2002) The PI3K AKT pathway in human cancer. Nat Rev Cancer 2: 489–501 [DOI] [PubMed] [Google Scholar]
- Wymann MP, Marone R (2005) Phosphoinositide 3-kinases in disease: timing, location and scaffolding. Curr Opin Cell Biol 17: 141–149 [DOI] [PubMed] [Google Scholar]
- Wymann MP, Pirola L (1998) Structure and function of phosphoinositide 3-kinases. Biochem Biophys Acta 1436: 127–150 [DOI] [PubMed] [Google Scholar]
- Yart A, Chap H, Raynal P (2002) PI3K in lysophosphatidic acid signaling: regulation and cross-talk with the Ras/mitogen-activated protein kinase pathway. Biochim Biophys Acta 1582: 107–111 [DOI] [PubMed] [Google Scholar]