

Abstract
Vascular endothelial growth factor-A (VEGF-A) induces actin reorganization and migration of endothelial cells through a p38 mitogen-activated protein kinase (MAPK) pathway. LIM-kinase 1 (LIMK1) induces actin remodeling by phosphorylating and inactivating cofilin, an actin-depolymerizing factor. In this study, we demonstrate that activation of LIMK1 by MAPKAPK-2 (MK2; a downstream kinase of p38 MAPK) represents a novel signaling pathway in VEGF-A-induced cell migration. VEGF-A induced LIMK1 activation and cofilin phosphorylation, and this was inhibited by the p38 MAPK inhibitor SB203580. Although p38 phosphorylated LIMK1 at Ser-310, it failed to activate LIMK1 directly; however, MK2 activated LIMK1 by phosphorylation at Ser-323. Expression of a Ser-323-non-phosphorylatable mutant of LIMK1 suppressed VEGF-A-induced stress fiber formation and cell migration; however, expression of a Ser-323-phosphorylation-mimic mutant enhanced these processes. Knockdown of MK2 by siRNA suppressed VEGF-A-induced LIMK1 activation, stress fiber formation, and cell migration. Expression of kinase-dead LIMK1 suppressed VEGF-A-induced tubule formation. These findings suggest that MK2-mediated LIMK1 phosphorylation/activation plays an essential role in VEGF-A-induced actin reorganization, migration, and tubule formation of endothelial cells.
Keywords: endothelial cell migration, LIM-kinase, MAPKAPK-2, p38 MAPK, VEGF-A
Introduction
Angiogenesis is a process whereby new blood vessels are generated from pre-existing ones. It is essential for a number of physiological and pathological processes, including embryonic development, tissue regeneration, wound healing, diabetic retinopathy, and tumor growth and metastasis (Risau, 1997). Vascular endothelial growth factor-A (VEGF-A, hereafter simply referred to as VEGF) is a potent stimulator of angiogenesis, and promotes migration, proliferation, and tubule formation of endothelial cells (Ferrara, 2004). Although VEGF-induced endothelial cell migration is an essential step for angiogenesis, the underlying signaling mechanisms are not well understood (Rousseau et al, 2000).
VEGF exerts its angiogenic effects through the receptor tyrosine kinases, VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1), which are expressed on the surface of endothelial cells (Zachary and Gliki, 2001). Activation of VEGFR-2 by VEGF leads to stimulation of various intracellular signaling cascades, including activation of the extracellular signal-regulated kinase (Erk) and p38 mitogen-activated protein kinase (MAPK) pathways (Zachary and Gliki, 2001). As the p38 MAPK inhibitor SB203580 inhibits VEGF-induced stress fiber formation and cell migration, the p38 MAPK pathway is believed to play a crucial role in endothelial cell migration by regulating actin cytoskeletal organization (Rousseau et al, 1997). p38 MAPK is activated by the upstream MAPK kinases, MKK3/MKK6 (Ono and Han, 2000). Activation of p38 MAPK by VEGF leads to the activation of MAPK-activated protein kinase-2 (MK2), which in turn stimulates phosphorylation of heat-shock protein 27 (Hsp27) (Guay et al, 1997; Rousseau et al, 1997; Landry and Huot, 1999). Hsp27 behaves as an actin filament cap-binding protein and, in its non-phosphorylated form, inhibits actin polymerization. MK2-catalyzed phosphorylation of Hsp27 allows its release from capped actin filaments, thereby stimulating actin polymerization (Landry and Huot, 1999). Thus, p38/MK2-mediated phosphorylation of Hsp27 is believed to be involved in VEGF-induced actin filament assembly and stress fiber formation, both of which are involved in endothelial cell migration (Rousseau et al, 2000). However, other signaling mechanisms for VEGF-induced and p38/MK2-mediated actin reorganization and cell migration remain to be elucidated.
Cofilin plays an essential role in actin reorganization and cell migration by depolymerizing and severing actin filaments (Bamburg and Wiggan, 2002). The activity of cofilin is reversibly regulated by phosphorylation and dephosphorylation of Ser-3, with the phosphorylated form being inactive. LIM-kinase (LIMK) and TES-kinase are responsible for phosphorylation of this site, and thereby inactivate cofilin (Arber et al, 1998; Yang et al, 1998; Toshima et al, 2001). Cofilin phosphatases (termed Slingshot and chronophin) reactivate cofilin by dephosphorylating Ser-3 (Niwa et al, 2002; Nagata-Ohashi et al, 2004; Gohla et al, 2005). LIMK1 is activated through phosphorylation of Thr-508, in the kinase domain of LIMK1, by downstream kinases of the Rho family small GTPases, such as ROCK and PAK (Edwards et al, 1999; Maekawa et al, 1999; Ohashi et al, 2000). Thus, by phosphorylating cofilin, LIMK1 appears to play a critical role in stimulus-induced actin reorganization. LIMK1 is activated in response to various extracellular signals that stimulate cell migration, and stimulus-induced LIMK1 activation is required for chemokine-induced cell migration or LPA-induced tumor cell invasion (Nishita et al, 2002, 2005; Yoshioka et al, 2003). In contrast, overexpression of LIMK1 suppresses cell motility and polarized cell migration (Dawe et al, 2003; Endo et al, 2003). Thus, it seems likely that proper regulation of LIMK1 activity and cofilin phosphorylation is required for cell migration.
In this study, we examined the role of LIMK1 in VEGF-induced actin reorganization, migration, and tubule formation of vascular endothelial cells. We report here that LIMK1 is activated in VEGF-stimulated endothelial cells, by a novel signaling pathway, composed of p38 MAPK–MK2–LIMK1, and that MK2 activates LIMK1 by phosphorylating Ser-323 in the extracatalytic domain of LIMK1. We also provide evidence that MK2-mediated LIMK1 phosphorylation/activation is critical for VEGF-induced stress fiber formation, cell migration, and tubule formation.
Results
VEGF induces LIMK1 activation and cofilin phosphorylation
To examine whether LIMK1 is involved in VEGF-induced actin reorganization, we first analyzed changes in the kinase activity of LIMK1 and the level of Ser-3-phosphorylated cofilin (P-cofilin) in VEGF-stimulated human umbilical vein endothelial cells (HUVECs) and MSS31 endothelial cells. MSS31 cells have the properties of endothelial cells; they express PECAM-1, VE-cadherin, and VEGFR-1 and -2 (Supplementary Figure 1). We used 10 ng/ml VEGF for stimulation in this study. An in vitro kinase assay, using cofilin as a substrate, demonstrated that the kinase activity of LIMK1 increased 1.6- to 1.7-fold by 15 min after VEGF treatment, and then decreased by 30 min (Figure 1A). The 1.3-fold increase in LIMK1 activity was retained up to 6 h (Supplementary Figure 2). The level of P-cofilin (measured by immunoblots with anti-P-cofilin antibody) increased about two-fold by 15 min after VEGF treatment, and then decreased by 30 min (Figure 1B). Similar results were obtained by two-dimensional gel immunoblot analysis of P-cofilin levels, using an anti-cofilin antibody. P-cofilin represented 16% of cofilin in unstimulated cells. After VEGF stimulation, the relative abundance of P-cofilin reached a maximum (46%) by 15 min, and decreased to 24% by 30 min (Figure 1C). These results suggest that VEGF stimulates both LIMK1 activity and cofilin phosphorylation in a time-dependent manner, and that through cofilin phosphorylation, LIMK1 is involved in VEGF-induced actin reorganization.
Figure 1.
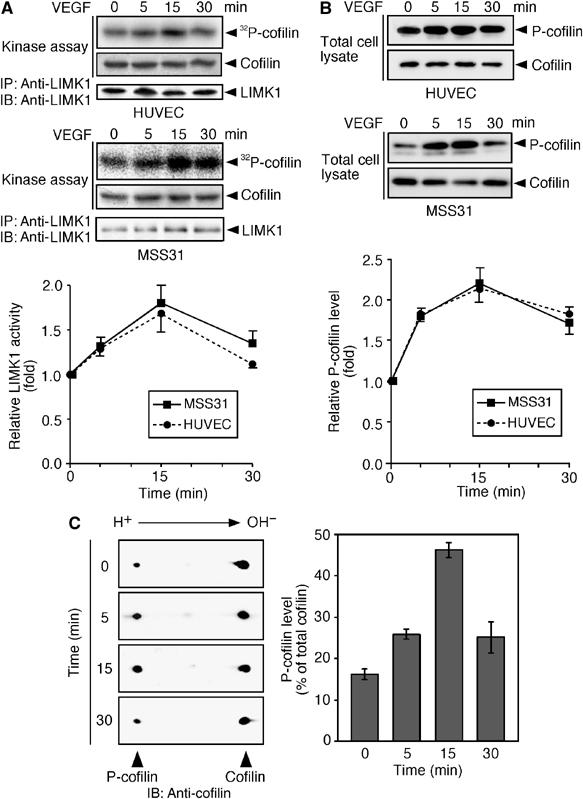
VEGF induces LIMK1 activation and cofilin phosphorylation. (A) LIMK1 activation. HUVEC and MSS31 cells were stimulated with VEGF. At the indicated time, cells were lysed and LIMK1 was immunoprecipitated (IP) with anti-LIMK1, and subjected to an in vitro kinase assay. Reaction mixtures were analyzed by autoradiography (32P-cofilin), Amido black staining (cofilin), and immunoblotting (IB) with anti-LIMK1 antibody. The bottom panel indicates the relative kinase activities of LIMK1, as measured by 32P incorporation into cofilin. Data are means±s.d. of five independent experiments. (B) Cofilin phosphorylation. HUVEC and MSS31 cells were stimulated with VEGF. Cell lysates were analyzed by immunoblotting with anti-P-cofilin and anti-cofilin antibodies. The bottom panel indicates the relative P-cofilin levels as means±s.d. of five independent experiments. (C) Two-dimensional gel analyses of P-cofilin levels. MSS31 cells were stimulated with VEGF, and cell lysates were analyzed by two-dimensional gel electrophoresis, followed by immunoblotting with anti-cofilin antibody. The right panel indicates the mean abundance of P-cofilin (means±s.d. of triplicate experiments), as the percentage of total cofilin.
Kinase-negative LIMK1(D460A) suppresses VEGF-induced stress fiber formation
We next examined whether LIMK1 is involved in VEGF-induced stress fiber formation. MSS31 cells were transfected with cyan fluorescence protein (CFP)-tagged wild-type LIMK1 (LIMK1(WT)-CFP) or the kinase-dead form LIMK1(D460A)-CFP, in which the catalytic Asp-460 is replaced by alanine. Cells were either untreated or stimulated with VEGF, and stained with rhodamine-phalloidin to visualize F-actin (Figure 2A). In the control MSS31 cells (transfected with vector alone), thick and transcytoplasmic actin stress fibers were induced in response to VEGF treatment. Expression of LIMK1(WT)-CFP caused a marked increase in the induction of thick stress fibers, even in the absence of VEGF. Following VEGF treatment, induction of thicker stress fibers increased further, in comparison to the surrounding LIMK1-non-expressing cells, indicating that LIMK1 has the potential to stimulate stress fiber formation. In contrast, expression of LIMK1(D460A)-CFP significantly suppressed VEGF-induced stress fiber formation. Figure 2B shows the data of quantitative analysis of the ratio of the cells with thick stress fibers. These results suggest that LIMK1 plays a critical role in VEGF-induced stress fiber formation in endothelial cells.
Figure 2.
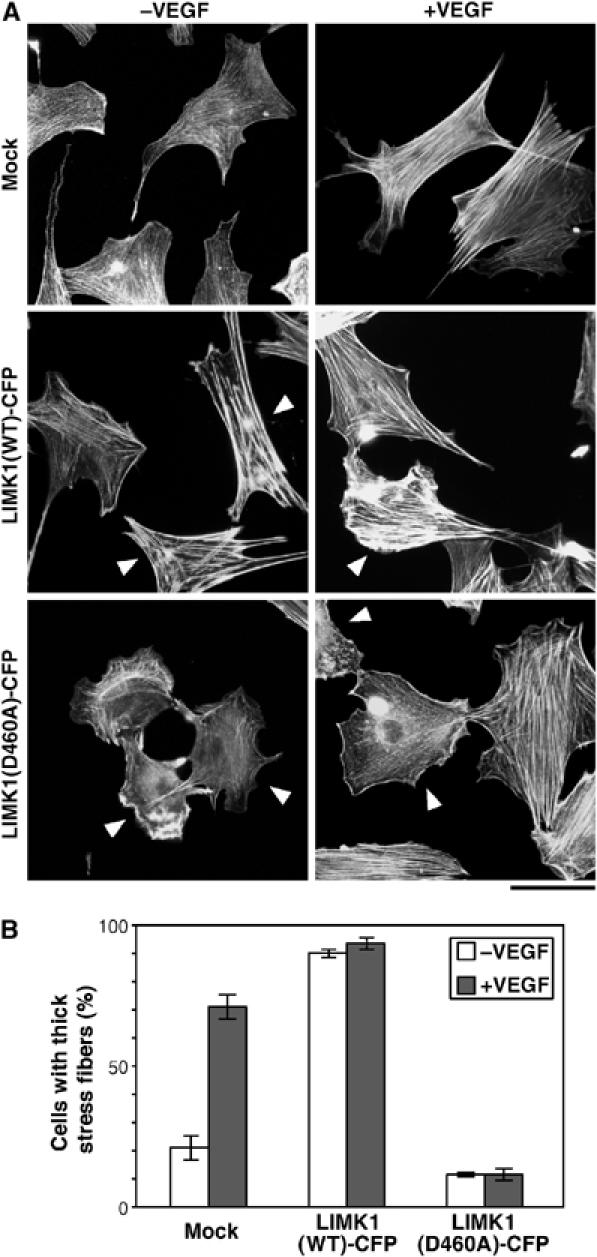
Kinase-negative LIMK1(D460A) suppresses VEGF-induced stress fiber formation. (A) MSS31 cells were transfected with control vector (mock) or plasmid encoding LIMK1(WT)-CFP or LIMK1(D460A)-CFP. They were unstimulated or stimulated with VEGF for 15 min and then stained with rhodamine-phalloidin. Arrowheads indicate CFP fluorescence-positive cells (see Supplementary Figure 3A). Bar, 50 μm. (B) Quantitative analysis of data shown in (A). The percentages of the cells with thick stress fibers in the total CFP-positive cells are shown as means±s.d. of triplicate experiments.
p38 MAPK mediates VEGF-induced LIMK1 activation, cofilin phosphorylation, and stress fiber formation
Because p38 MAPK has been shown to mediate VEGF-induced actin reorganization (Rousseau et al, 1997), we examined the effects of SB203580, a specific inhibitor for p38 MAPK, on VEGF-induced LIMK1 activation and cofilin phosphorylation in MSS31 cells. Almost complete inhibition of both VEGF-induced LIMK1 activation and cofilin phosphorylation was achieved by pretreatment with 1 μM SB203580 (Figure 3A and B). This indicates that LIMK1 activation and cofilin phosphorylation are stimulated downstream of p38 MAPK. We also examined the effects of SB203580 on VEGF-induced stress fiber formation (Figure 3C). Pretreatment with SB203580, but not with vehicle alone, suppressed VEGF-induced stress fiber formation in the control MSS31 cells expressing CFP. In contrast, stress fibers induced by LIMK1(WT)-CFP expression were not affected by SB203580 treatment, either before or after VEGF stimulation, in comparison to the surrounding LIMK1-non-expessing cells. This result is a further indication that LIMK1 exerts its actions downstream of p38. Thus, p38 MAPK appears to mediate VEGF-induced LIMK1 activation, cofilin phosphorylation, and stress fiber formation.
Figure 3.
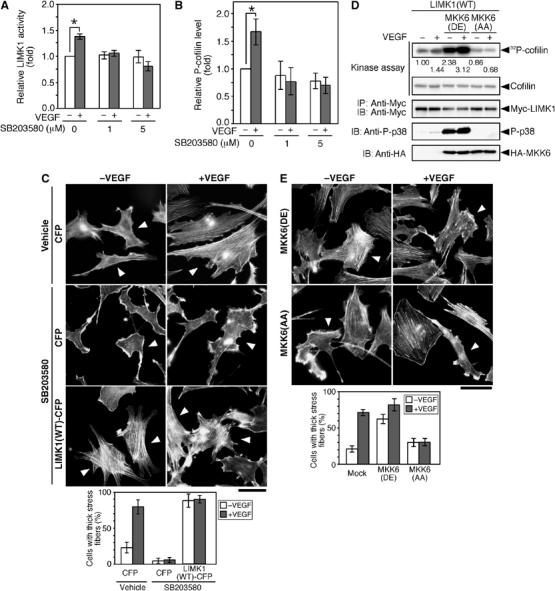
p38 MAPK mediates VEGF-induced LIMK1 activation, cofilin phosphorylation, and stress fiber formation. (A, B) SB203580 inhibits VEGF-induced LIMK1 activation and cofilin phosphorylation. MSS31 cells were pretreated with SB203580 (0, 1, and 5 μM) for 30 min and then stimulated with VEGF for 15 min. The levels of LIMK1 activity and P-cofilin were determined, as in Figure 1. Relative kinase activities of LIMK1 (A) and relative P-cofilin levels (B) are shown as means±s.d. of triplicate experiments. *P<0.001. (C) SB203580 inhibits stress fiber formation. MSS31 cells transfected with CFP or LIMK1(WT)-CFP were pretreated with 5 μM SB203580 or vehicle (DMSO) for 30 min and then stimulated with VEGF for 15 min. Cells were stained with rhodamine-phalloidin. Arrowheads indicate CFP-positive cells (see Supplementary Figure 3B). Bar, 50 μm. The bottom panel shows the data of quantitative analysis of triplicate experiments. (D) MKK6(DE) stimulates LIMK1 activity. MSS31 cells cotransfected with Myc-LIMK1, Flag-p38, and either HA-MKK6(DE) or HA-MKK6(AA) were treated with VEGF for 15 min, and Myc-LIMK1 was precipitated and subjected to an in vitro kinase assay. Kinase activities were quantified by autoradiography, and relative levels are indicated under the top panel. Expression of Myc-LIMK1 and HA-MKK6 mutants and activation of p38 (P-p38) were analyzed by immunoblotting, as indicated. (E) MKK6(AA) suppresses VEGF-induced stress fiber formation. MSS31 cells, transfected with HA-MKK6(DE) or HA-MKK6(AA), were stimulated with VEGF for 15 min and stained with rhodamine-phalloidin. Arrowheads indicate cells expressing MKK6(DE) or MKK6(AA), as measured by anti-HA staining (Supplementary Figure 3C). Bar, 50 μm. The bottom panel shows the data of quantitative analysis of triplicate experiments.
To further examine the involvement of p38 MAPK in VEGF-induced LIMK1 activation, we tested the effects of expression of a constitutively active or a dominant-negative form of MKK6 in MSS31 cells. MKK6 is an MAPK kinase that phosphorylates and activates p38 MAPK. The constitutively active MKK6(DE) and dominant-negative MKK6(AA) mutants were constructed by replacing the two phosphorylation sites in MKK6 (Ser-207 and Thr-211) by Asp and Glu residues (DE), or two Ala residues (AA), respectively (Hanafusa et al, 1999). Coexpression of active MKK6(DE) significantly increased the kinase activity of LIMK1, in both unstimulated and VEGF-stimulated cells (Figure 3D). In contrast, coexpression of MKK6(AA) blocked VEGF-induced LIMK1 activation (Figure 3D). Immunoblot analysis with an anti-phospho-p38 (P-p38) antibody showed that p38 activity was enhanced by MKK6(DE) expression and/or VEGF treatment, but was suppressed by MKK6(AA) expression (Figure 3D). Expression of MKK6(DE) enhanced actin filament assembly in unstimulated cells, whereas expression of MKK6(AA) suppressed VEGF-induced stress fiber formation (Figure 3E). Taken together with the results of SB203580 treatment, these observations strongly suggest that an MKK6–p38 pathway mediates VEGF-induced LIMK1 activation, cofilin phosphorylation, and stress fiber formation.
Phosphorylation of Thr-508 is not required for VEGF-induced and p38-mediated LIMK1 activation
LIMK1 is activated by phosphorylation of Thr-508 by ROCK or PAK kinases. To examine whether Thr-508 phosphorylation is involved in VEGF-induced LIMK1 activation, we analyzed the VEGF-induced changes in the kinase activity of LIMK1(WT) and LIMK1(T508V), in which Thr-508 was replaced by a non-phosphorylatable valine. The basal kinase activity of LIMK1(T508V) is lower than that of LIMK1(WT) in unstimulated cells; however, VEGF stimulation resulted in an approximately 1.5-fold increase in the activity of both LIMK1(WT) and LIMK1(T508V) (Figure 4A). When either LIMK1(WT) or LIMK1(T508V) was coexpressed in 293T cells with active MKK6(DE) and p38, there was an approximately two-fold increase in activity (Figure 4B). These results suggest that both LIMK1(WT) and LIMK1(T508V) are activated by VEGF or active MKK6 to a similar extent, and that phosphorylation of Thr-508 is not required for VEGF-induced and p38-mediated LIMK1 activation. Coexpression with MKK6(DE) resulted in gel mobility shifts of LIMK1(WT) and LIMK1(T508V) (Figure 4B). These mobility shifts are the result of LIMK1 phosphorylation, as they were abolished by treatment with alkaline phosphatase (Figure 4C). Accordingly, LIMK1 is probably activated downstream of p38 MAPK, by phosphorylation of a site(s) other than Thr-508.
Figure 4.
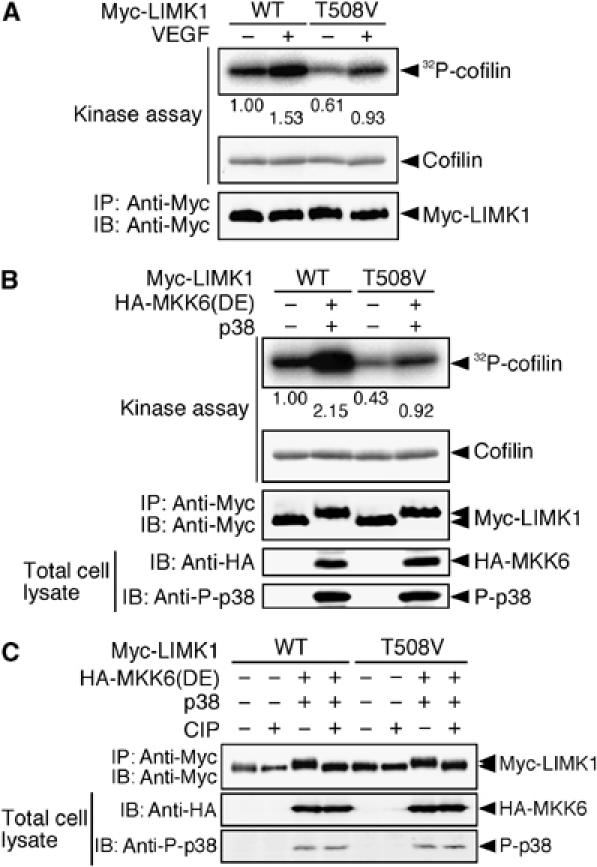
Phosphorylation of Thr-508 is not required for VEGF-induced LIMK1 activation. (A) LIMK1(T508V) is activated by VEGF. MSS31 cells transfected with Myc-LIMK1(WT) or Myc-LIMK1(T508V) were stimulated with VEGF for 15 min. Relative kinase activities of these proteins were analyzed as in Figure 3D and are indicated under the top panel. (B) LIMK1(T508V) is activated by MKK6(DE). 293T cells were cotransfected with HA-MKK6(DE), Flag-p38, and either Myc-LIMK1(WT) or Myc-LIMK1(T508V). Myc-LIMK1 proteins were immunoprecipitated and subjected to an in vitro kinase reaction. Expression of Myc-LIMK1 and HA-MKK6(DE) and activation of p38 were analyzed by immunoblotting, as indicated. (C) Effects of alkaline phosphatase treatment on a gel mobility shift of LIMK1. Myc-LIMK1(WT) or Myc-LIMK1(T508V) cotransfected with HA-MKK6(DE) and Flag-p38 in 293T cells was precipitated with anti-Myc antibody, incubated with calf intestinal alkaline phosphatase (CIP), and then analyzed by immunoblotting with anti-Myc antibody.
p38 MAPK phosphorylates, but fails to activate, LIMK1
To determine if LIMK1 is directly phosphorylated by p38, Myc-tagged LIMK1(T508V) and kinase-dead LIMK1(D460A) were incubated in vitro with [γ-32P]ATP in the absence or presence of recombinant active p38 (GST-p38) (Figure 5A). In the absence of p38, 32P was incorporated into LIMK1(T508V), but not LIMK1(D460A), indicating that LIMK1(T508V) underwent autophosphorylation. In the presence of active p38, 32P incorporation into LIMK1(D460A) and LIMK1(T508V) was increased, which suggests that p38 phosphorylates LIMK1 at a residue(s) other than Thr-508. However, there was no change in the kinase activity of LIMK1(WT) or LIMK1(T508V), after incubation with active p38 (Figure 5B). Thus, p38 has the potential to phosphorylate LIMK1, but is unable to activate LIMK1 by itself. A database search predicted that Ser-310 of LIMK1 was the most probable site for p38-catalyzed phosphorylation. In an in vitro kinase assay, active p38 enhanced phosphorylation of LIMK1(T508V), but not of LIMK1(S310A/T508V), in which Ser-310 and Thr-508 were replaced by alanine and valine, respectively (Figure 5C). These results indicate that Ser-310 of LIMK1 is the primary site of phosphorylation by p38.
Figure 5.
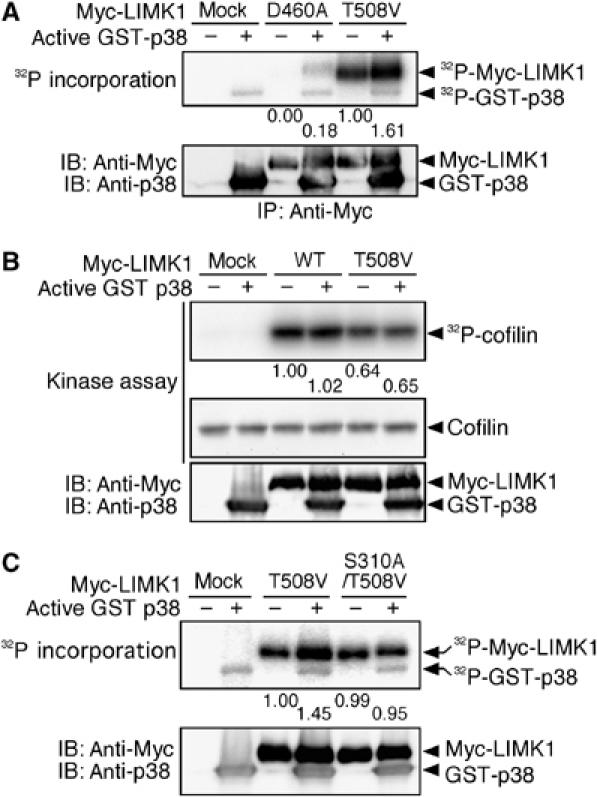
p38 MAPK phosphorylates, but does not activate, LIMK1. (A) p38 phosphorylates LIMK1. Myc-LIMK1(D460A) and Myc-LIMK1(T508V) expressed in 293T cells were precipitated with anti-Myc antibody and incubated with [γ-32P]ATP and active GST-p38. Reaction mixtures were separated by SDS–PAGE and analyzed by autoradiography. Relative values of 32P incorporation into Myc-LIMK1 mutants are indicated under the top panel. (B) LIMK1 is not activated by p38. Myc-LIMK1(WT) and Myc-LIMK1(T508V) expressed in 293T cells were immunoprecipitated, incubated with active GST-p38, and then subjected to an in vitro kinase assay. Relative kinase activities of LIMK1 are indicated under the top panel. (C) p38 phosphorylates Ser-310 of LIMK1. Myc-LIMK1(T508V) and Myc-LIMK1(S310A/T508V) expressed in 293T cells were precipitated with anti-Myc antibody and incubated with [γ-32P]ATP and active GST-p38. Reaction mixtures were separated by SDS–PAGE and analyzed by autoradiography. Relative values of 32P incorporation into Myc-LIMK1 mutants are indicated.
MK2 activates LIMK1 by phosphorylation of Ser-323
MK2 was reported to mediate p38-induced stress fiber formation (Rousseau et al, 2000). As p38 did not directly activate LIMK1, we then asked whether MK2 is involved in p38-mediated LIMK1 phosphorylation and activation. When LIMK1(WT) and LIMK1(T508V) were incubated with [γ-32P]ATP and recombinant active MK2 (GST-MK2-Myc), 32P incorporation into LIMK1(WT) and LIMK1(T508V) was increased approximately two-fold (Figure 6A). The kinase activities of LIMK1(WT) and LIMK1(T508V) also increased to the same degree, after incubation with active MK2 (Figure 6A). These findings suggest that MK2 phosphorylates and activates LIMK1, and that phosphorylation of Thr-508 is not related to this reaction.
Figure 6.
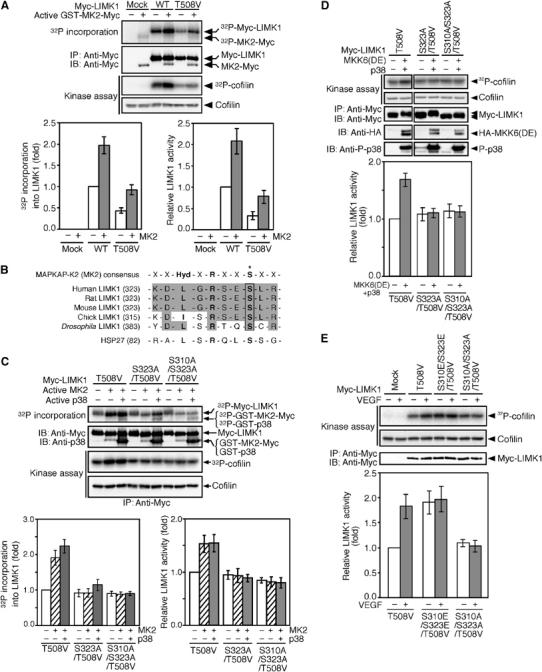
MK2 activates LIMK1 by phosphorylation of Ser-323. (A) MK2 phosphorylates and activates LIMK1. Myc-LIMK1(WT) and Myc-LIMK1(T508V) expressed in 293T cells were precipitated with anti-Myc antibody and incubated with [γ-32P]ATP and active GST-MK2-Myc. Reaction mixtures were separated by SDS–PAGE and analyzed by autoradiography. Relative values of 32P incorporation into Myc-LIMK1 are indicated in the bottom left panel. After incubation with active MK2, Myc-LIMK1 immunoprecipitates were subjected to an in vitro kinase assay. Relative kinase activities are indicated in the bottom right panel. Data are means±s.d. of three independent experiments. (B) Alignment of sequences of putative MK2 phosphorylation sites in LIMK1 and HSP27, and the consensus sequence for MK2 substrates. An asterisk indicates the phosphorylation site. (C) MK2 activates LIMK1 by Ser-323 phosphorylation. Myc-LIMK1 mutants were expressed in 293T cells, immunoprecipitated, and incubated with [γ-32P]ATP and active GST-MK2-Myc, with or without active GST-p38. Both 32P incorporation into Myc-LIMK1 and LIMK1 activity were analyzed as in (A). Relative values of 32P incorporation and relative kinase activities are indicated in the bottom panels. (D) Activation of LIMK1 by Ser-323 phosphorylation in cultured cells. Myc-LIMK1 mutants were coexpressed with HA-MKK6(DE) plus Flag-p38 in 293T cells, immunoprecipitated, and subjected to an in vitro kinase assay. Relative kinase activities are indicated in the bottom panel as means±s.d. of five independent experiments. (E) Kinase activities of LIMK1 mutants. MSS31 cells transfected with Myc-LIMK1 mutants were stimulated with VEGF for 15 min. Myc-LIMK1 mutants were immunoprecipitated and subjected to an in vitro kinase assay. Relative kinase activities are shown in the bottom panel as means±s.d. of five experiments.
MK2 phosphorylates several proteins, including HSP-27, at the serine residue in the consensus sequence motif, -Hyd-x-Arg-x-x-Ser-, where Hyd is a bulky hydrophobic residue (Stokoe et al, 1993). Human LIMK1 contains the corresponding sequence LGRSES323 in the region between a PDZ domain and a kinase domain, and the MK2 consensus motif is conserved between LIMK1 proteins from various species (Figure 6B). To determine if MK2 phosphorylates Ser-323 of LIMK1, and whether or not this phosphorylation is related to LIMK1 activation, we constructed the mutant LIMK1(S323A/T508V), in which Ser-323 and Thr-508 are replaced by alanine and valine, respectively. We also constructed the mutant LIMK1(S310A/S323A/T508V). After incubation with active MK2, we compared the phosphorylation level and kinase activity of these mutants to those of LIMK1(T508V). In cell-free assays, active MK2 phosphorylated LIMK1(T508V), but did not phosphorylate either (S323A/T508V) or (S310A/S323A/T508V) mutant (Figure 6C). Incubation with active MK2 together with active p38 resulted in a more highly phosphorylated LIMK1(T508V), a slightly phosphorylated LIMK1(S323A/T508V), and no increased phosphorylation of LIMK1(S310A/S323A/T508V) (Figure 6C). Similarly, active MK2 phosphorylated LIMK1(WT), but not LIMK1(S310A/S323A) (Supplementary Figure 4A). These results suggest that p38 and MK2 specifically phosphorylate LIMK1 at Ser-310 and Ser-323, respectively. Incubation with active MK2 increased the kinase activity of LIMK1(T508V), but did not alter the activity of either LIMK1(S323A/T508V) or LIMK1(S310A/S323A/T508V) (Figure 6C). Incubation with active MK2 and active p38 did not further increase the kinase activities of these mutants (Figure 6C). In a similar manner, active MK2 activated LIMK1(WT), but not LIMK1(S310A/S323A) (Supplementary Figure 4A). These findings suggest that in cell-free assays, MK2 stimulates the kinase activity of LIMK1 by phosphorylation of Ser-323. Although p38 phosphorylates LIMK1 at Ser-310, it fails to activate LIMK1.
To examine whether Ser-323 phosphorylation is involved in MKK6/p38-mediated LIMK1 activation in cultured cells, LIMK1 mutants were coexpressed with MKK6(DE) and p38 in 293T cells, and their kinase activities were measured (Figure 6D). By coexpression with active MKK6(DE) and p38, the kinase activity of LIMK1(T508V) increased 1.7-fold, but the activities of S323A/T508V and S310A/S323A/T508V mutants did not alter. Similarly, coexpression with active MKK6(DE) and p38 increased the kinase activity of LIMK1(WT), but not the activity of LIMK1(S310A/S323A) (Supplementary Figure 4B). Thus, these results suggest that phosphorylation of Ser-323 is essential for p38-mediated LIMK1 activation in cultured cells.
We also analyzed kinase activities of non-phosphorylatable LIMK1(S310A/S323A/T508V) and phosphorylation-mimic LIMK1(S310E/S323E/T508V) in MSS31 cells, before and after VEGF stimulation (Figure 6E). The activity of LIMK1(T508V) increased ∼1.8-fold, after stimulation with VEGF. In unstimulated cells, the kinase activity of LIMK1(S310A/S323A/T508V) was similar to that of LIMK1(T508V); however, unlike LIMK1(T508V), it was not activated by VEGF treatment. In unstimulated cells, LIMK1(S310E/S323E/T508V) showed an ∼2-fold increase in kinase activity, compared with LIMK1(T508V). After stimulation with VEGF, the kinase activity of LIMK1(S310E/S323E/T508V) did not increase further. In a similar experiment, VEGF stimulated the activity of LIMK1(WT), but not LIMK1(S310A/S323A) (Supplementary Figure 4C). These results support the suggestion that VEGF induces LIMK1 activation by phosphorylation of Ser-310 and Ser-323 residues.
Phosphorylation of Ser-323 of LIMK1 is required for VEGF-induced cell migration and stress fiber formation
It was reported that a p38–MK2 pathway plays a crucial role in the VEGF-induced endothelial cell migration and stress fiber formation (Rousseau et al, 2000). The finding that VEGF induced LIMK1 activation through the MK2-mediated phosphorylation of Ser-323 prompted us to investigate whether phosphorylation of Ser-323 is required for VEGF-induced cell migration. We examined the effects of expression of various LIMK1 mutants on VEGF-induced endothelial cell migration. MSS31 cells were cotransfected with LIMK1 mutants and YFP and subjected to a migration assay, using Transwell chambers. The number of YFP-positive cells that migrated across the membrane was determined 18 h after the addition of VEGF (Figure 7A). For control MSS31 cells expressing YFP alone, VEGF increased the number of migrating cells about two-fold. Expression of LIMK1(WT) increased the number of migrating cells in both unstimulated and VEGF-stimulated conditions. In contrast, expression of LIMK1(D460A) suppressed VEGF-induced migration. These results indicate that LIMK1 activity is crucial for VEGF-induced cell migration. Additionally, expression of LIMK1(T508V) increased cell migration in both unstimulated and VEGF-stimulated conditions, although the levels of enhancement were lower than those for the LIMK1(WT)-expressing cells. In contrast, for cells expressing S323A/T508V, S310A/S323A/T508V, or S310A/S323A mutant, VEGF-induced cell migration was suppressed. Expression of LIMK1(S310E/S323E/T508V) markedly increased the number of migrating cells in the absence of VEGF, but no further increase was observed with stimulation with VEGF. Taken together, these results suggest that LIMK1 activation, through MK2-catalyzed phosphorylation of Ser-323, plays an essential role in VEGF-induced endothelial cell migration. Immunoblot analysis demonstrated that there were similar levels of expression of the different Myc-LIMK1 mutants (about five- to seven-fold greater than the amount of endogenous LIMK1) in these experiments (Figure 7B).
Figure 7.
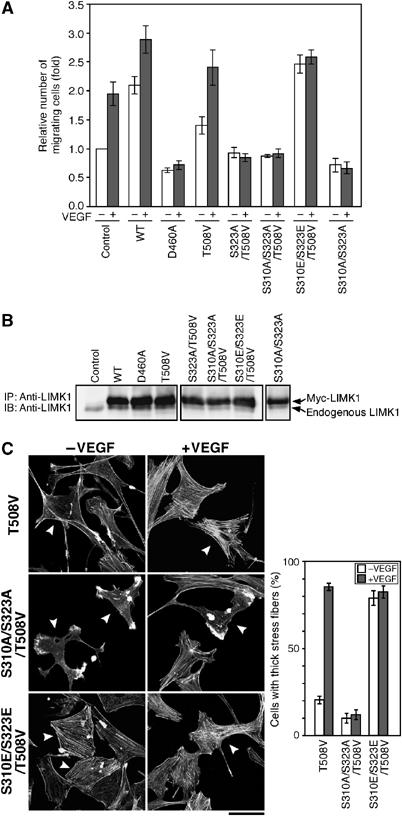
Effects of expression of LIMK1 mutants on VEGF-induced cell migration and stress fiber formation. (A) Effects on cell migration. Migration of MSS31 cells expressing YFP alone (control) or YFP plus Myc-LIMK1 mutant was analyzed by Transwell assays. Relative numbers of migrating cells are shown as means±s.d. of four experiments. (B) Expression levels of LIMK1 mutants. MSS31 cells were transfected as above, and cell lysates were immunoprecipitated and immunoblotted with anti-LIMK1 antibody. (C) Effects on stress fiber formation. MSS31 cells transfected with Myc-LIMK1 mutants were stimulated with or without VEGF for 15 min. The cells were costained with rhodamine-phalloidin and anti-Myc antibody. Arrowheads indicate the cells expressing Myc-LIMK1 mutant (see Supplementary Figure 3D). Bar, 50 μm. The right panel shows the data of quantitative analysis of triplicate experiments.
We also examined the effects of expression of LIMK1 mutants on VEGF-induced stress fiber formation (Figure 7C). Expression of LIMK1(T508V) had no apparent effect on the actin cytoskeleton, when compared with the surrounding non-expressing cells. In contrast, expression of LIMK1(S310A/S323A/T508V) caused a significant suppression of VEGF-induced stress fiber formation, and expression of LIMK1(S310E/S323E/T508V) enhanced stress fiber formation in unstimulated cells. These findings suggest that phosphorylation of Ser-310/Ser-323 in LIMK1 is crucial to VEGF-induced stress fiber formation.
Knockdown of MK2 suppresses VEGF-induced LIMK1 activation, stress fiber formation, and cell migration
We analyzed the effect of knockdown of MK2 expression on VEGF- or MKK6(DE)-induced LIMK1 activation. MSS31 cells were transfected with either an siRNA plasmid targeting MK2, or the mutated siRNA plasmid. Immunoblot analysis revealed that transfection with the MK2 siRNA plasmid efficiently suppressed the expression of endogenous MK2 protein, but transfection with the mutated siRNA plasmid did not alter expression (Figure 8A). In cells transfected with siRNA vector plasmid or mutated siRNA plasmid, LIMK1 activity increased after VEGF treatment or expression of MKK6(DE) (Figure 8B). In contrast, VEGF- or MKK6(DE)-induced LIMK1 activation was abolished in cells transfected with the MK2 siRNA plasmid (Figure 8B). This suggests that MK2 plays an essential role in VEGF- and MKK6(DE)-induced LIMK1 activation. Similar results were obtained using two other MK2 siRNA plasmids that have distinct MK2 target sequences (data not shown).
Figure 8.
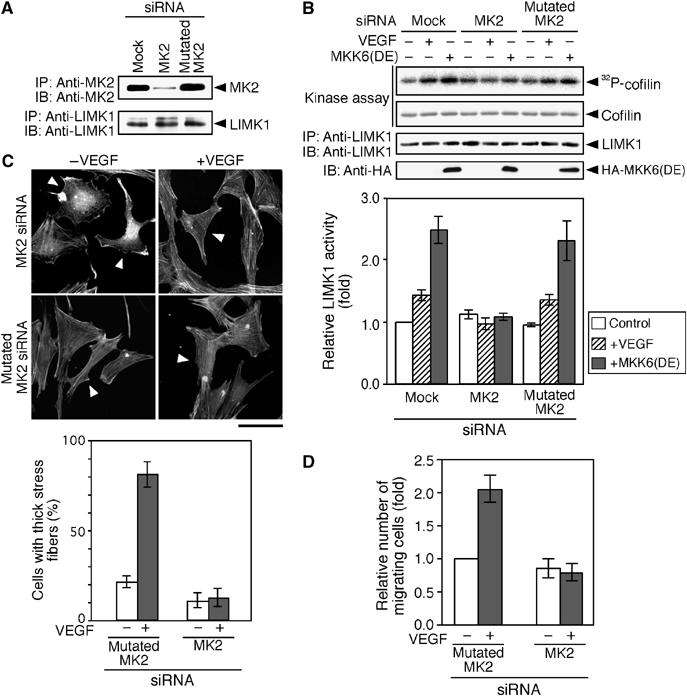
Knockdown of MK2 suppresses VEGF-induced LIMK1 activation, stress fiber formation, and cell migration. (A) Knockdown of MK2 expression. MSS31 cells were transfected with the siRNA vector (mock), MK2 siRNA plasmid, or mutated MK2 siRNA plasmid. Expression of endogenous MK2 and LIMK1 was analyzed by immunoprecipitation and immunoblotting. (B) Suppression of LIMK1 activation. MSS31 cells transfected with siRNA plasmids were stimulated with VEGF for 15 min or cotransfected with HA-MKK6(DE). Endogenous LIMK1 was precipitated and subjected to an in vitro kinase assay. Relative kinase activities are shown in the bottom panel as means±s.d. of five experiments. (C) Suppression of stress fiber formation. MSS31 cells were cotransfected with YFP and siRNA plasmids (with a molar ratio of 1:9), stimulated with VEGF for 15 min, and stained with rhodamine-phalloidin. Arrowheads indicate YFP-positive cells (see Supplementary Figure 3E). Bar, 50 μm. The bottom panel shows the data of quantitative analysis of triplicate experiments. (D) Suppression of cell migration. MSS31 cells transfected with YFP and siRNA plasmids were subjected to cell migration assay in the absence or presence of VEGF. Data are shown as means±s.d. of four experiments.
We also examined the effect of MK2 siRNA on VEGF-induced stress fiber formation and migration of MSS31 cells. Transfection with MK2 siRNA plasmid suppressed VEGF-induced stress fiber formation significantly; however, the mutated MK2 siRNA had no apparent effect, when compared with the surrounding siRNA-non-transfected cells (Figure 8C). Additionally, VEGF-induced MSS31 cell migration was inhibited by transfection with MK2 siRNA, but not with the mutated MK2 siRNA (Figure 8D). These results suggest that MK2 is required for VEGF-induced stress fiber formation and migration of endothelial cells.
Mechanisms of VEGF-induced MK2 activation
To examine whether MK2 is activated by VEGF treatment, the level of MK2 activity was analyzed by immunoblotting with the antibody specific to Thr-334-phosphorylated MK2 (P-MK2). After VEGF stimulation, the level of P-MK2 increased about two-fold by 15 min, then decreased gradually, and the 1.4-fold increased activity was retained up to 6 h (Supplementary Figure 2). Time course of the change in the level of P-MK2 was almost parallel to that of the kinase activity of LIMK1 (Supplementary Figure 2), which further supports the correlation between MK2 and LIMK1 activation.
We next asked which type of VEGFRs is involved in VEGF-induced MK2 and LIMK1 activation. Pretreatment of MSS31 cells with the neutralizing antibody against VEGFR2 almost completely inhibited VEGF-induced MK2 phosphorylation and LIMK1 activation, whereas the anti-VEGFR1 neutralizing antibody partially suppressed these processes (Supplementary Figure 5). This indicates that VEGFR2 is essential for VEGF-induced MK2 and LIMK1 activation, whereas VEGFR1 is only partially involved in the activation of MK2–LIMK1 signaling pathway.
ROCK and PAK are known to activate LIMK1 by Thr-508 phosphorylation. We analyzed the effects of suppression of ROCK and PAK activity on VEGF-induced MK2 and LIMK1 activation, by expression of a dominant-negative form of ROCK (ROCK(KD-IA)) or an autoinhibitory domain of PAK3 (PAK(AI)). Both ROCK(KD-IA) and PAK(AI) inhibited VEGF-induced MK2 phosphorylation and LIMK1 activation (Supplementary Figure 6). Taken together with the findings that Thr-508 phosphorylation is not required for VEGF-induced LIMK1 activation (Figure 4A), these results suggest that ROCK and PAK are involved in LIMK1 activation through MK2 activation.
LIMK1 activation is required for VEGF-induced tubule formation
VEGF stimulates the tubule formation of endothelial cells in Matrigel. To examine the role of LIMK1 in VEGF-induced tubule formation, MSS31 cells were infected with adenoviruses expressing YFP or YFP-tagged LIMK1(WT) or LIMK1(D460A) and subjected to the in vitro tubule formation assay in Matrigel (Figure 9A). The total tube length was measured by tracing the tube-like structures. For control cells expressing YFP, VEGF increased the total tube length about two-fold. Expression of LIMK1(WT) increased the total tube length in both unstimulated and VEGF-stimulated cells. In contrast, expression of kinase-dead LIMK1(D460A) suppressed VEGF-induced tubule formation. These results suggest that LIMK1 activity is required for VEGF-induced tubule formation. Additionally, for MSS31 cells expressing LIMK1(T508V), VEGF increased the total tube length about two-fold, but expression of LIMK1(S310A/S323A/T508V) suppressed VEGF-induced tubule formation. Thus, the p38/MK2-mediated Ser-310/Ser-323 phosphorylation of LIMK1 appears to be critical for the in vitro tubule formation of endothelial cells.
Figure 9.
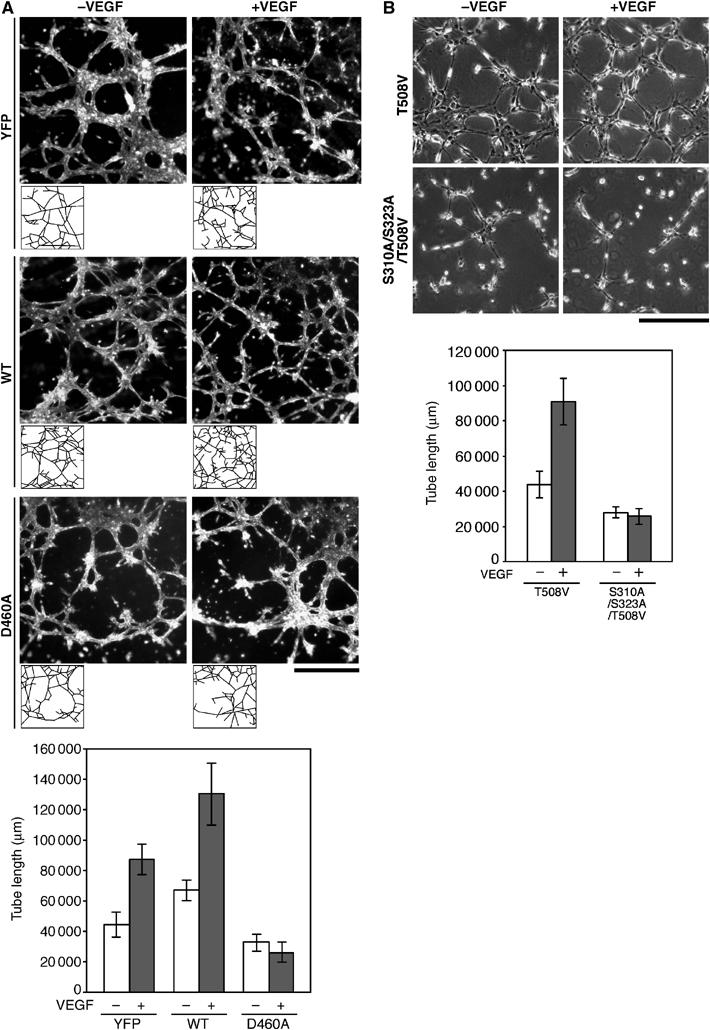
Effects of expression of LIMK1 mutants on VEGF-induced tubule formation. (A) Bright-field micrographs showing the tubule formation of MSS31 cells expressing YFP, YFP-LIMK1(WT), or YFP-LIMK1(D460A) in Matrigel, in the presence or absence of VEGF. Bar, 1 mm. The tube-like structures were traced, as shown at the bottom of each micrograph, and the total tube length was quantified using the image software. The results are shown in the bottom panel, as means±s.d. of four experiments. (B) Phase contrast micrographs showing the tubule formation of MSS31 cells expressing LIMK1(T508V) or LIMK1(S310A/S323A/T508V) in Matrigel. Bar, 0.5 mm. The quantitative data of the total tube length are shown as means±s.d. of four experiments.
Hyperosmotic shock induces LIMK1 activation through p38 MAPK
Hyperosmotic shock activates the p38 MAPK signaling pathway and induces actin reorganization (Ono and Han, 2000). To examine whether the osmotic shock induces LIMK1 activation through p38, we analyzed the changes in the kinase activity of LIMK1 and p38 in MSS31 cells after treatment with 0.3 M NaCl. Both the kinase activity of LIMK1 and the level of phospho-p38 increased at 5 and 15 min and then decreased 30 min after hyperosmotic treatment (Supplementary Figure 7A). Pretreatment of the cells with SB203580 abolished osmotic shock-induced LIMK1 activation and p38 phosphorylation (Supplementary Figure 7B), indicating that p38 mediates osmotic stress-induced LIMK1 activation. When expressed in MSS31 cells, LIMK1(T508V) was activated by hyperosmotic shock, but LIMK1(S323A/T508V) was not (Supplementary Figure 7C). These results suggest that MK2-mediated Ser-323 phosphorylation of LIMK1 is critical for osmotic stress-induced LIMK1 activation, and that there is a common signaling mechanism of p38/MK2-mediated LIMK1 activation in VEGF stimulation and hyperosmotic stress.
Discussion
In this study, we have identified a novel signaling pathway that is crucial for VEGF-induced stress fiber formation and migration of endothelial cells (Figure 10). We have demonstrated VEGF-induced LIMK1 activation and cofilin phosphorylation. In endothelial cells, expression of a kinase-inactive LIMK1 suppressed VEGF-induced stress fiber formation, cell migration, and tubule formation, implicating LIMK1 in these processes. Inhibition of p38 activation by SB203580 or expression of a dominant-negative mutant MKK6(AA) blocked VEGF-induced LIMK1 activation and stress fiber formation, and the expression of active MKK6(DE) augmented LIMK1 activity and stress fiber formation, indicating that the MKK6–p38 pathway mediates VEGF-induced LIMK1 activation and stress fiber formation. Although previous studies showed that ROCK and PAK activate LIMK1 by phosphorylating Thr-508 in the kinase domain of LIMK1 (Edwards et al, 1999; Ohashi et al, 2000), a non-phosphorylatable T508V mutant of LIMK1, similar to wild-type LIMK1, was activated by both VEGF treatment and active MKK6 expression. This indicates that VEGF-induced and MKK6/p38-mediated LIMK1 activation occurs independently of Thr-508 phosphorylation. In cell-free assays, p38 phosphorylated LIMK1 at Ser-310, but failed to activate LIMK1. In contrast, MK2, a major downstream kinase of p38, directly activated LIMK1 by phosphorylating Ser-323 in the consensus sequence motif for MK2 substrates. In cultured cells, an LIMK1 S323A mutant was not activated by VEGF treatment or active MKK6(DE) expression, indicating that MK2-catalyzed phosphorylation of Ser-323 is critical for VEGF-induced and p38-mediated LIMK1 activation. As p38 did not increase MK2-catalyzed LIMK1 activation further (Figure 6C), p38-catalyzed phosphorylation of Ser-310 does not appear to be involved in LIMK1 activation, to any great extent. As expression of either the S323A or the S310A/S323A mutant of LIMK1 inhibited VEGF-induced stress fiber formation, cell migration, and tubule formation, MK2-mediated phosphorylation of Ser-323 is essential for these processes. Finally, we showed that siRNA knockdown of MK2 inhibited VEGF-induced LIMK1 activation, stress fiber formation, and cell migration. This further supports the notion that MK2 is crucial to LIMK1 activation, actin remodeling, and cell migration, in response to VEGF. Thus, we propose a novel signaling cascade, comprising p38–MK2–LIMK1–cofilin, that plays an essential role in VEGF-induced actin reorganization, cell migration, and tube formation (Figure 10). Further studies are required to understand the role of this pathway in in vivo angiogenesis. We also provide evidence that hyperosmotic shock stimulates LIMK1 activation through the p38/MK2 signaling pathway. This suggests that p38 activation, in response to other stimuli such as cytokines, chemokines, and oxygen radical stresses (Ono and Han, 2000), may also induce actin reorganization through p38/MK2-mediated LIMK1 activation.
Figure 10.
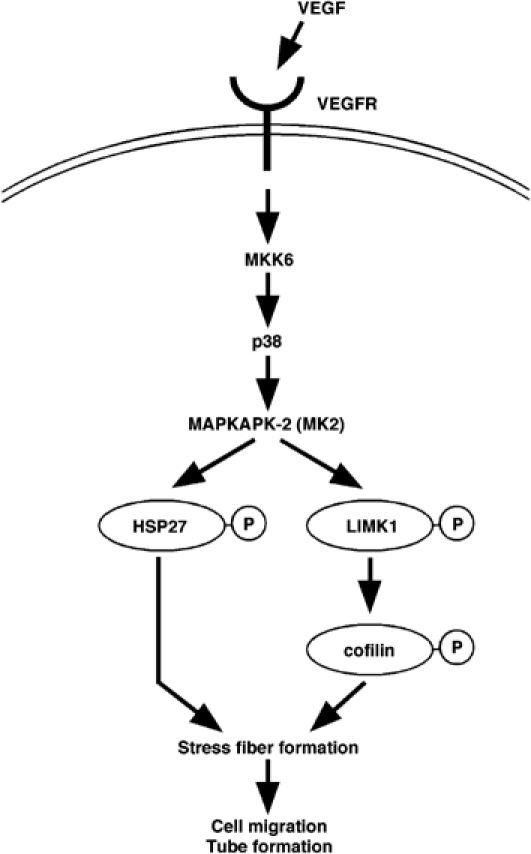
A proposed signaling pathway for VEGF-induced LIMK1 activation and cofilin phosphorylation. VEGF induces activation of MK2 via the MKK6 and p38 MAPK signaling cascade. MK2 activates LIMK1 by phosphorylation of Ser-323, which in turn stimulates phosphorylation of cofilin. Together with MK2-mediated Hsp27 phosphorylation, cofilin phosphorylation stimulates stress fiber formation, cell migration, and tube formation.
Previous studies have shown that a p38/MK2 signaling pathway is involved in VEGF-induced actin reorganization and cell migration through phosphorylation of Hsp27 (Rousseau et al, 2000). Phosphorylation of Hsp27 by MK2 probably suppresses its actin filament capping activity, thereby generating free barbed ends for actin polymerization. Thus, MK2 can stimulate actin filament assembly by two pathways: decapping the plus end of actin filaments through Hsp27 phosphorylation; and inhibiting actin-disassembling activity of cofilin through LIMK1 activation and cofilin phosphorylation. LIMK1 is activated by various extracellular cues that stimulate cell migration (Nishita et al, 2002). Inhibition of LIMK1 activity suppressed chemokine-induced T-cell migration, suggesting a critical role for LIMK1 in cell migration (Nishita et al, 2005). However, overexpression of LIMK1 is known to suppress cell migration and neurite extension (Dawe et al, 2003; Endo et al, 2003). Therefore, it is likely that the precise and balanced regulation of cofilin phosphorylation is required for cell migration.
LIMK1 structure comprises two LIM domains and a PDZ domain at the N-terminus and a kinase domain at the C-terminus (Okano et al, 1995). Here, we have demonstrated that LIMK1 is activated by MK2-catalyzed phosphorylation of Ser-323, which locates in the region connecting the PDZ domain and the kinase domain. In a previous study, we demonstrated that deletion of the N-terminal LIM and PDZ domains increased the kinase activity of LIMK1, and that excess N-terminal LIM fragments suppressed the kinase activity of the C-terminal kinase domain. This suggested that the N-terminal region had an autoinhibitory role in the regulation of the kinase activity of LIMK1 (Nagata et al, 1999). MK2-catalyzed phosphorylation of Ser-323 may increase the kinase activity of LIMK1 by inducing a conformational change, thus releasing the autoinhibitory effect of the N-terminal region.
Rho family small GTPases, including Rho, Rac, and Cdc42, play key roles in actin cytoskeletal reorganization and cell migration. VEGF stimulates Rho, Rac, and Cdc42 activation in endothelial cells, and they are implicated in VEGF-induced cell migration and tubule formation (Soga et al, 2001; Zeng et al, 2002; Lamalice et al, 2004). Whereas LIMK1 is known to be activated by ROCK and PAK by phosphorylation of Thr-508, our present study indicates that LIMK1 is activated through a p38–MK2 pathway, independent of Thr-508 phosphorylation, in endothelial cells. On the other hand, we showed that both ROCK(KD-IA) and PAK(AI) suppressed VEGF-induced MK2 activation and LIMK1 activation. Thus, ROCK and PAK seem to be involved in LIMK1 activation through MK2 activation, but not through direct phosphorylation of Thr-508, in endothelial cells. ROCK- and PAK-catalyzed phosphorylation of LIMK1 may be protected by spatially separating these proteins in these cells. As p38 MAPK was reported to be activated downstream of ROCK and PAK in other cell types (Zhang et al, 1995; Ashida et al, 2001), ROCK/PAK activation probably contributes to the VEGF-induced LIMK1 activation through the activation of the p38–MK2 pathway. In contrast to our results, using the antibody to phospho-Thr-508 LIMK1, Gong et al (2004) reported that VEGF induced Thr-508 phosphorylation of LIMK1 in a manner dependent on ROCK activity. However, they did not show the changes in the kinase activity of LIMK1, or the ratio of the Thr-508-phosphorylated form, relative to the total abundance of LIMK1. In this study, we provided evidence that VEGF increases the kinase activity of a LIMK1(T508V) mutant, to an extent similar to that of wild-type LIMK1 and that Thr-508 phosphorylation is not required for VEGF-induced LIMK1 activation, stress fiber formation, and cell migration. Our results strongly suggest that VEGF-induced LIMK1 activation is caused primarily by a p38–MK2 pathway, via phosphorylation of Ser-323, although this does not completely exclude the possibility that ROCK-catalyzed LIMK1 phosphorylation partially contributes to the process.
Our results indicate that LIMK1 is activated by multiple signaling pathways. Further study will be required to determine how the cells differently use these pathways and whether or not these pathways work separately or synergistically in the complex spatiotemporal regulation of actin cytoskeletons.
Materials and methods
Reagents and antibodies
VEGF-A (VEGF165) and SB203580 were purchased from PeproTech (London) and Calbiochem, respectively. Antibodies to P-cofilin, cofilin, and LIMK1 were prepared as described previously (Okano et al, 1995; Toshima et al, 2001). Antibodies against Myc (9E10; Roche), HA (3F10; Roche), FLAG (M2; Sigma), and Thr-180/Tyr-182-phosphorylated p38 MAPK (P-p38) (Cell Signaling) were purchased commercially.
Plasmids
Plasmids encoding Myc-LIMK1 and its D460A mutant were constructed as described previously (Ohashi et al, 2000). Plasmids for the point mutants of Myc-LIMK1 were constructed by using a site-directed mutagenesis kit (Stratagene). Plasmids for HA-MKK6(DE) and HA-MKK6(AA) were provided by E Nishida (Kyoto University). A plasmid for FLAG-p38α was constructed by subcloning p38α cDNA into the pCMV5-FLAG vector. A plasmid encoding Myc-MK2 was provided by J Han (Scripps Research Institute). The pSUPER vector for siRNA was provided by R Agami (Netherlands Cancer Institute). The MK2 siRNA plasmid that targets the mouse MK2 mRNA sequence (5′-GACGCATCCAACCCTCTGC-3′) and the plasmid for the mutated MK2 siRNA (5′-GACGAATCCAACCCGCTGC-3′) were constructed as described previously (Brummelkamp et al, 2002).
Cells, transfection, and siRNA
HUVECs were obtained from the Riken Cell Bank (Tsukuba, Japan) and cultured as described (Rousseau et al, 1997). MSS31 cells, a mouse spleen endothelial cell line (Yanai et al, 1991), were provided by N Yanai (Miyagi Gakuin University), and the cells were grown in α-MEM with 10% FCS. 293T cells (Cell Resource Center, Tohoku University) were cultured in DMEM containing 10% FCS. Cells were transfected with plasmids using FuGENE6 Transfection Reagent (Roche). Transfection efficiency of MSS31 cells was 50–60%, judging from CFP fluorescence.
In vitro kinase assay
HUVEC and MSS31 cells were serum-starved for 4 h and stimulated with 10 ng/ml VEGF. Cells were lysed and LIMK1 was immunoprecipitated with anti-LIMK1 or anti-Myc antibody and then subjected to an in vitro kinase reaction, using His6-cofilin as a substrate, as described previously (Ohashi et al, 2000). p38- or MK2-catalyzed LIMK1 phosphorylation was analyzed as follows: Myc-LIMK1 mutants were expressed in 293T cells, immunoprecipitated with anti-Myc antibody, and incubated in 20 μl lysis/kinase buffer containing 50 μM ATP and 185 kBq [γ-32P]ATP (110 TBq/mmol; Amersham Pharmacia Biotech) with 100 ng recombinant active GST-p38 (Upstate) or active GST-MK2-Myc (Upstate) at 30°C for 20 min. The reaction mixture was separated by SDS–PAGE and analyzed by autoradiography to measure 32P-labeled LIMK1.
Cell staining
MSS31 cells were cultured on a 32-mm glass-bottom dish coated with 0.1 mg/ml type I-C collagen. After 24 h of culture (48 h for cells transfected with siRNA plasmids) in serum-containing α-MEM, cells were serum-starved for 4 h and then stimulated with 10 ng/ml VEGF for 15 min. Cells were fixed and stained with anti-HA or anti-Myc, as described (Nagata et al, 1999). To visualize F-actin, cells were stained with rhodamine-phalloidin (Molecular Probes). Fluorescent images were obtained using a confocal microscope (model LSM510, Carl Zeiss).
Cell migration assay
MSS31 cells were cotransfected with the plasmid for YFP and the plasmid for Myc-LIMK1 mutants or the siRNA plasmid (with a molar ratio of 1:9), and cultured for 18 h (48 h for cells transfected with siRNA plasmids). Cells were trypsinized, and 5 × 104 cell aliquots were loaded onto the upper well of a gelatin-coated Transwell chemotaxis chamber (8 μm pore size; Corning). The lower well was filled with α-MEM, with or without 10 ng/ml VEGF. After incubation for 18 h at 37°C, the membrane was fixed in 4% formaldehyde. After the non-migrating cells on the top of the membrane were removed by wiping and rinsing, the number of YFP-positive migrating cells on the lower face of the membrane was counted. Relative numbers of migrating cells were calculated, with the mean number of migrating cells (5201) for the control cells expressing YFP alone taken as 1.0.
Tubule formation assay
MSS31 cells were infected with adenoviruses expressing YFP, YFP-LIMK1(WT), or YFP-LIMK1(D460A), which were constructed using pAxCAwt vectors (Takara Biomedicals). At a multiplicity of infection of 100, almost all cells were positive for YFP. For expression of Myc-LIMK1(T508V) or Myc-LIMK1(S310A/S323A/T508V), MSS31 cells were transfected with pPURO (Clontech) and plasmids encoding these proteins, and selected with 1 μg/ml puromycin. After 24 h of incubation, cells were trypsinized and 1 × 104 cells were plated on Matrigel in a 7 mm well. After 4 h of incubation with or without 10 ng/ml VEGF, cells were fixed in 4% formaldehyde. The tube-like structures were traced in all viewing field in each well and the total tube length was integrated using IPLab image analysis software (Scanalytics). All tests were performed in duplicate and repeated four times.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Acknowledgments
We thank Dr E Nishida (Kyoto University) for providing plasmids for MKK6, Dr J Han (Scripps Research Institute) for plasmids for MK2, Dr N Yanai (Miyagi Gakuin University) for MSS31 cells, and Dr R Agami (Netherlands Cancer Institute) for pSUPER vector. We also thank Dr Y Sato (Tohoku University) for advice. This work was supported by a grant for Creative Scientific Research from the Japan Society of the Promotion of Science (KM).
References
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stayon CA, Bernard O, Caroni P (1998) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393: 805–809 [DOI] [PubMed] [Google Scholar]
- Ashida N, Arai H, Yamasaki M, Kita T (2001) Distinct signaling pathways for MCP-1-dependent integrin activation and chemotaxis. J Biol Chem 276: 16555–16560 [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Wiggan OP (2002) ADF/cofilin and actin dynamics in disease. Trends Cell Biol 12: 598–605 [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 [DOI] [PubMed] [Google Scholar]
- Dawe HR, Minamide LS, Bamburg JR, Cramer LP (2003) ADF/cofilin controls cell polarity during fibroblast migration. Curr Biol 13: 252–257 [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN (1999) Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol 1: 253–259 [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, Mizuno K (2003) Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci 23: 2527–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 25: 581–611 [DOI] [PubMed] [Google Scholar]
- Gohla A, Birkenfeld J, Bokoch GM (2005) Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat Cell Biol 7: 21–29 [DOI] [PubMed] [Google Scholar]
- Gong C, Stoletov KV, Terman BI (2004) VEGF treatment induces signaling pathways that regulate both actin polymerization and depolymerization. Angiogenesis 7: 313–321 [DOI] [PubMed] [Google Scholar]
- Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J (1997) Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci 110: 357–368 [DOI] [PubMed] [Google Scholar]
- Hanafusa H, Ninomiya-Tsuji J, Masuyama N, Nishita M, Fujisawa J, Shibuya H, Matsumoto K, Nishida E (1999) Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-b-induced gene expression. J Biol Chem 274: 27161–27167 [DOI] [PubMed] [Google Scholar]
- Lamalice L, Houle F, Jourdan G, Huot J (2004) Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene 23: 434–445 [DOI] [PubMed] [Google Scholar]
- Landry J, Huot J (1999) Regulation of actin dynamics by stress-activated protein kinase 2 (SAPK2)-dependent phosphorylation of heat-shock protein of 27 kDa (Hsp27). Biochem Soc Symp 64: 79–89 [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285: 895–898 [DOI] [PubMed] [Google Scholar]
- Nagata K, Ohashi K, Yang N, Mizuno K (1999) The N-terminal LIM domain negatively regulates the kinase activity of LIM-kinase 1. Biochem J 343: 99–105 [PMC free article] [PubMed] [Google Scholar]
- Nagata-Ohashi K, Ohta Y, Goto K, Chiba S, Mori R, Nishita M, Ohashi K, Kousaka K, Iwamatsu A, Niwa R, Uemura T, Mizuno K (2004) A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J Cell Biol 165: 465–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishita M, Aizawa H, Mizuno K (2002) Stromal cell-derived factor 1α activates LIM kinase 1 and induces cofilin phosphorylation for T-cell chemotaxis. Mol Cell Biol 22: 774–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishita M, Tomizawa C, Yamamoto M, Horita Y, Ohashi K, Mizuno K (2005) Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J Cell Biol 171: 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T (2002) Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108: 233–246 [DOI] [PubMed] [Google Scholar]
- Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K (2000) Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem 275: 3577–3582 [DOI] [PubMed] [Google Scholar]
- Okano I, Hiraoka J, Otera H, Nunoue K, Ohashi K, Iwashita S, Hirai M, Mizuno K (1995) Identification and characterization of a novel family of serine/threonine kinases containing two N-terminal LIM motifs. J Biol Chem 270: 31321–31330 [DOI] [PubMed] [Google Scholar]
- Ono K, Han J (2000) The p38 signal transduction pathway: activation and function. Cell Signal 12: 1–13 [DOI] [PubMed] [Google Scholar]
- Risau W (1997) Mechanisms of angiogenesis. Nature 386: 671–674 [DOI] [PubMed] [Google Scholar]
- Rousseau S, Houle F, Huot J (2000) Integrating the VEGF signals leading to actin-based motility in vascular endothelial cells. Trend Cardiovasc Med 10: 321–327 [DOI] [PubMed] [Google Scholar]
- Rousseau S, Houle F, Landry J, Huot J (1997) p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 15: 2169–2177 [DOI] [PubMed] [Google Scholar]
- Soga N, Namba N, McAllister S, Cornelius L, Teitelbaum SL, Dowdy SF, Kawamura J, Hruska KA (2001) Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp Cell Res 269: 73–87 [DOI] [PubMed] [Google Scholar]
- Stokoe D, Caudwell B, Cohen PT, Cohen P (1993) The substrate specificity and structure of mitogen-activated protein (MAP) kinase-activated protein kinase-2. Biochem J 296: 843–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima J, Toshima JY, Amano T, Yang N, Narumiya S, Mizuno K (2001) Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol Biol Cell 12: 1131–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai N, Satoh T, Obinata M (1991) Endothelial cells create a hematopoietic inductive microenvironment preferential to erythropoiesis in the mouse spleen. Cell Struct Funct 16: 87–93 [DOI] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K (1998) Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393: 809–812 [DOI] [PubMed] [Google Scholar]
- Yoshioka K, Foletta V, Bernard O, Itoh K (2003) A role for LIM kinase in cancer invasion. Proc Natl Acad Sci USA 100: 7247–7252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary I, Gliki G (2001) Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res 49: 568–581 [DOI] [PubMed] [Google Scholar]
- Zeng H, Zhao D, Mukhopadhyay D (2002) KDR stimulates endothelial cell migration through heterotrimeric G protein Gq/11-mediated activation of a small GTPase RhoA. J Biol Chem 277: 46791–46798 [DOI] [PubMed] [Google Scholar]
- Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, Bokoch GM (1995) Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem 270: 23934–23936 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7