

Abstract
Despite designating catalytic roles of Asp299 and Arg131 during the transfer of γ-phosphate from ATP to Ado (adenosine) [R. Datta, Das, Sen, Chakraborty, Adak, Mandal and A. K. Datta (2005) Biochem. J. 387, 591–600], the mechanisms that determine binding of substrate and cause product inhibition of adenosine kinase from Leishmania donovani remained unclear. In the present study, employing homology-model-guided site-specific protein mutagenesis, we show that Asp16 is indispensable, since its replacement with either valine or arginine resulted in a >200-fold increase in Km (Ado) with a 1000-fold decrease in kcat/Km, implying its critical importance in Ado binding. Even glutamate replacement was not tolerated, indicating the essentiality of Asp16 in the maintenance of steric complementarity of the binding pocket. Use of 2′or 3′-deoxygenated Ado as substrates indicated that, although both the hydroxy groups play important roles in the formation of the enzyme–Ado complex, the binding energy (ΔΔGB) contribution of the former was greater than the latter, suggesting possible formation of a bidentate hydrogen bond between Asp16 and the adenosyl ribose. Interestingly, AMP-inhibition and AMP-binding studies revealed that, unlike the R131A mutant, which showed abrogated AMP-binding and insensitivity towards AMP inhibition despite its unaltered Km (Ado), all the Asp16 mutants bound AMP efficiently and displayed AMP-sensitive catalytic activity, suggesting disparate mechanisms of binding of Ado and AMP. Molecular docking revealed that, although both Ado and AMP apparently occupied the same binding pocket, Ado binds in a manner that is subtly different from AMP binding, which relies heavily on hydrogen-bonding with Arg131 and thus creates an appropriate environment for competition with Ado. Hence, besides its role in catalysis, an additional novel function of the Arg131 residue as an effector of product-mediated enzyme regulation is proposed.
Keywords: adenosine kinase, AMP regulation, homology modelling, Leishmania donovani
Abbreviations: AdK, adenosine kinase; Ado, adenosine; LdAdK, Leishmania donovani AdK; RK, ribokinase
INTRODUCTION
AdK (adenosine kinase; EC 2.7.1.20), an important enzyme of the purine salvage pathway, catalyses the magnesium-dependent direct in-line transfer of the terminal phosphate from ATP to Ado (adenosine) [1–3]. The physiological function of AdK is associated with the regulation of extracellular Ado levels and maintenance of intracellular adenylate pools [4]. Apart from this, the inherent capability of this enzyme to phosphorylate a large number of therapeutically useful purine nucleoside analogues has provided impetus to a large number of workers in assessing its prospects as one of the drug targets in various systems [5].
In parasitic protozoa, this crucial purine salvage enzyme becomes additionally important, as these organisms lack the ability to synthesize purine nucleotides de novo and hence depend entirely on their host for the salvage of these metabolites [6,7]. In Leishmania donovani, a dimorphic parasitic protozoon that causes kala-azar in humans, this enzyme (LdAdK) is being studied extensively [8–14]. Studies showed that, unlike other higher eukaryotic AdKs, the parasitic enzyme possesses unique biochemical characteristics [8]. For instance, while the higher eukaryotic AdKs are prone to inhibition at high Ado concentrations, LdAdK is refractory to such inhibition. Moreover, in contrast with higher eukaryotic AdKs, LdAdK is much more sensitive to inhibition by ATP. Besides these, there are significant differences in catalytic properties between the host and parasitic AdK with respect to the Km values for substrates, sensitivity to chemical modifiers and their immunological properties [8,10,12,13].
More interestingly, the enzyme from all sources, unlike the other nucleoside and nucleotide kinases, belongs to the PfkB family of structurally related carbohydrate kinases that include RK (ribokinase) and phosphofructokinase. Members of this family are characterized by the presence of two common sequence motifs that includes a highly conserved di-glycine motif located near the N-terminal end and a DTXGAGD motif, positioned near the C-terminus [15,16]. From the available crystal structures of the enzyme form human and Toxoplasma gondii, it has now been established unequivocally that the enzyme, in general, consists of two domains: a large and a small domain between which the active site resides. The small domain, upon substrate binding, undergoes a conformational rotation and forms a lid-like structure to cover the active site [17,18]. The structures of human and T. gondii AdK further predicted the putative Ado- and ATP-binding sites at the cleft between the two domains. Although the ATP-binding site of the two enzymes are quite different, their Ado-binding pockets are structurally similar. Various relatively conserved hydrophobic residues have been shown to constitute the Ado-binding pocket. These residues possibly help in initial trapping of the substrate that is later stabilized by other enzyme–substrate hydrogen bonds. Since adenosyl adenine is bound in a solvent-accessible region, most of the contacts are water-mediated. In contrast, the adenosyl ribose points to the core of the protein and thus has been predicted to be engaged in multiple hydrogen bonds with different highly conserved amino acid side chains of the enzyme, e.g. Asp18, Gly64, Asn68 and Asp300 of human AdK (corresponding residues of T. gondii AdK are Asp24, Gly69, Asn73 and Asp318) [17,18].
In the absence of the X-ray structure of LdAdK, its modelled structure, based on the known co-ordinates of human and T. gondii AdK, has recently reaffirmed the existence of a similar, but not identical, structure of the parasite enzyme [19]. Furthermore, by analysis of several site-specific mutants, located in the neighbourhood of the Ado-binding site, and modelling of those mutant proteins, we confirmed the involvement of the di-glycine motif (Gly61-Gly62 for LdAdK) in Ado binding and substrate-induced domain rotation. A crucial catalytic role for Arg131 in the stabilization of the quinquivalent transition state formed during proton abstraction from the adenosyl O5′ moiety by Asp299, and subsequent nucleophilic attack on the acceptor molecule of the phosphoryl group, has also been proposed [19]. However, despite these studies, we still have very little information about the substrate-binding determinants and the mechanism by which the enzyme is regulated by its products. Moreover, why the R131A mutant, as shown in our earlier studies, although displaying a similar affinity for Ado as compared with the wild-type enzyme in the kinetic reaction, failed to bind to the AMP column remained unexplained [19].
In a continuing effort to identify the amino acids that are responsible for Ado binding, in particular the adenosyl ribose-anchoring site, we have chosen Asp16 (homologous with Asp18 of human and Asp24 of T. gondii AdK) as the residue for mutational analysis. In the present study, by combining site-directed mutagenesis with molecular recognition using deoxygenated Ado analogues to study the energetics of the LdAdK–Ado transitionstate complex, we unambiguously established Asp16 as the vital Ado-binding residue.
AdK from various sources has been reported to bind self-generated AMP, resulting in reversible competitive inhibition with respect to Ado [9,20,21]. Yet, despite various studies, it is still not clear whether Ado and AMP occupy a common pocket involving the same amino acids for binding. In the present study, availability of the Asp16 mutants, defective with respect to Ado binding, has thus allowed us to investigate the mechanism of AMP inhibition. Our studies revealed that impaired Ado binding does not hamper binding of AMP, suggesting different binding modes. Simulated docking of AMP into the LdAdK active site demonstrated that, although Ado and AMP occupy a nearly overlapping position, resulting in apparent competition between the two, their mode of interaction with the enzyme are not exactly similar. Interestingly, Arg131, which was identified previously as one of the key catalytic residues [19], has been found to play an additional role in AMP binding, thereby acting as an effector for product-mediated enzyme regulation. Mutation of this residue to alanine, despite showing unaltered affinity for Ado, markedly reversed AMP inhibition because of significant loss of its AMP-binding affinity. The dual role of Arg131 (both in catalysis as well as in regulation) was supported further with the help of the AMP-docked structure of LdAdK, which for the first time provided structural insights into the differential mode of Ado and AMP binding to the enzyme. The possibility of exploiting the AMP-docked structure towards future designing of a product-based inhibitor of LdAdK is also discussed.
EXPERIMENTAL
Materials
Unless otherwise mentioned, all reagents were purchased from Sigma Chemicals. Oligonucleotides were obtained from Isogen. The QuikChange mutagenesis kit was the product of Stratagene. N-terminal His-tag expression vector (pQE30) and Ni-NTA (Ni2+-nitrilotriacetate)–agarose resin were purchased from Qiagen.
Site-directed mutagenesis
Oligonucleotide-directed in vitro mutagenesis was performed using the QuikChange mutagenesis kit following the manufacturer's protocol. For carrying out the desired mutations, the following sense primers (along with their antisense counterparts), with their substitution sites underlined were used: D16A, 5′-GTGCAACCCGCTCCTCGCCGTGTCTGCCCCTGTC-3′; D16V, 5′-GTGCAACCCGCTCCTCGTCGTGTCTGCCCCTGTC-3′; D16N, 5′-GTGCAACCCGCTCCTCAACGTGTCTGCCCCTGTC-3′; and D16E, 5′-GTGCAACCCGCTCCTCGAAGTGTCTGCCCCTGTC-3′. The primer sequence for carrying out the R131A mutation is described elsewhere [19]. The N-terminal His-tagged expression plasmid (pQE30) harbouring the wild-type AdK gene was used as the template. Mutations were verified by automated DNA sequencing in an ABI Prism™ DNA sequencer.
Heterologous expression and purification of LdAdKs
The wild-type and mutant LdAdKs were overexpressed in Escherichia coli M15 [pREP4] and were purified as described previously [19]. It should, however, be mentioned that the D16A mutant LdAdK could not be purified owing to the formation of inclusion body. Purity of other proteins was examined by SDS/13% PAGE, and protein concentrations were determined using the method of Bradford [22] using Bio-Rad reagent with BSA as the standard.
CD spectroscopy
CD spectra were measured in 20 mM phosphate buffer (pH 7.5) on a Jasco700 spectropolarimeter. The spectra were obtained at a protein concentration of 2.5 μM in a 2 mm pathlength cuvette. Five scans were made at 50 nm/min between 195 and 260 nm in 1.00 nm increments at room temperature (25 °C). After subtraction of buffer spectra, the data were converted into molecular ellipticity units.
AdK assay for determination of kinetic parameters and inhibition studies
To reach the steady-state turnover condition kinetically, the Asp16 mutants required concentrations of Ado in the milimolar range. However, in the radiometric assay, it was not technically possible to achieve concentrations of [3H]Ado beyond 300 μM with the necessary specific radioactivity. Therefore, to measure the kinetic parameters of the Asp16 mutants, a pyruvate kinase–lactate dehydrogenase-coupled spectrophotometric assay, as described in [8], was used. [In the pyruvate kinase–lactate dehydrogenase-coupled assay, ADP, a product inhibitor of AdK, is continuously depleted. Hence, the kinetic parameters determined by the coupled assay system differ from those obtained by radiometric assay [19] in which ADP is accumulated in the assay system.] The reaction was followed for 2–5 min for accurate measurements of the initial rates. For determination of initial velocities, the data were fitted into the Michaelis–Menten equation, i.e. rectangular hyperbola and double-reciprocal plots were obtained as described by Cleland [23]. Secondary plots were used to determine kinetic constants. For inhibition studies, assays were carried out radiochemically following a procedure described previously [9]. The linearity of all plots was checked graphically for all substrate concentrations.
Transition-state stabilization energy
The changes in transition-state binding energy, i.e. ΔΔGB, was calculated from the difference in the specificity constant (kcat/Km) between the wild-type and a mutant LdAdK or the parent substrate and an analogue using the equation:
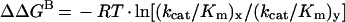 |
(1) |
where x represents either a mutant enzyme or a substrate analogue and y is the wild-type enzyme or parent substrate [24–26].
Comparison of AMP-binding affinity
AMP-binding abilities of the wild-type and mutant LdAdKs were compared by AMP-affinity chromatography as described previously [19]. Briefly, aliquots (500 μl) from the original stock (200 μg/ml) of each purified protein preparation (both wild-type and various mutants) were loaded on to identically sized preequilibrated AMP–agarose columns. In each case, the flow-through was recycled three times to ensure maximum binding. Columns were washed extensively, and protein remaining bound to the column was then eluted with 3 ml of buffer containing 5 mM Ado. The entire 3 ml eluate was then freeze-dried to a final volume of 500 μl. For direct comparison of binding to the AMP column, identical volumes (15 μl) of the original stock (200 μg/ml) and the freeze-dried eluate of each protein were subjected to SDS/13% PAGE analysis.
Homology modelling and docking simulations
The substrate-bound three-dimensional model of LdAdK was constructed as described previously [19]. The interacting distances between the enzyme and Ado were determined using our in-house package MODELYN (a molecular modelling program; version PC-1.0; Indian Copyright no. 9/98, 1998, developed by C. Mandal). The structure of AMP was built using the program ‘Builder’ module of Insight II, followed by energy minimization and molecular dynamics with the ‘Discover’ module on a Silicon Graphics® OCTANE workstation. The initial docking of AMP into the active site was carried out by manually placing the molecule at different locations. Structural optimization involved energy minimization (100 steps each of steepest descent and conjugate gradient) with a convergence criterion of 0.001 kcal/mol (1 kcal/mol=4.184 kJ/mol) using cff91 forcefield followed by molecular dynamics simulations. A typical molecular dynamics run consisted of 1000 steps of 1 fs each after 100 steps of equilibr-ation with a conformational sampling of 1 in 10 steps at 300 K. At the end of the molecular dynamics simulation, the conformation having the lowest potential energy was picked for the next cycle of refinement. This combination of minimization and molecular dynamics was repeated until satisfactory conformational parameters were obtained. While carrying out energy minimization and molecular dynamics of the AMP-bound enzyme, positional constraints were applied to all residues that were more than 10 Å (1 Å=0.1 nm) away from the ligand. Multiple sequence alignment was performed using CLUSTAL W [28].
RESULTS
Structural analysis of the putative ribose-anchoring site of the LdAdK–Ado complex
Based on the homology model, we recently illustrated the structural features of LdAdK in its substrate-bound conformation [19]. Taking advantage of the spatial co-ordinates of the model, we searched for the amino acid residues that are within interacting distance (≤3.5 Å) of the adenosyl ribose. Table 1 depicts nine possible interactions between the three oxygen atoms of the ribose (O2′, O3′ and O5′) and the protein (Asp16, Gly62, Asn66 and Asp299). Furthermore, there are two additional interactions between the C5′ of ribose and the Asn295 of LdAdK. Except for Asn66 and Asn295, the other ribose interacting residues are absolutely invariant in all AdKs [19], indicating that these residues possibly play crucial role either in anchoring of Ado or catalysis. By site-directed mutagenesis coupled with comprehensive biochemical study, we recently established the functional roles of Gly62 (important for maintaining domain flexibility and substrate binding) and Asp299 (deprotonates the 5′ OH of Ado and promotes a nucleophilic attack on the phosphate donor) [19]. Our preliminary observation indicates that alanine replacement of Asn66 does not hamper the enzyme activity under standard assay conditions (R. Datta and A. K. Datta, unpublished work), suggesting non-involvement of this residue in proper functioning of the enzyme. However, despite these efforts, the exact function of Asp16 remains to be delineated. Comparison of the Ado-binding site of AdK and ribose binding site of RK revealed that this particular residue of LdAdK is not only conserved among AdKs from various sources (Figure 1A), but a similar aspartate residue (Asp16) is also involved in interaction with the ribose in RK [29], thus signifying a general mechanistic role of the residue in all the members of this carbohydrate kinase family. Crystal structures of human and T. gondii AdK indicated that the side chains of Asp18 and Asp24 (the sequence homologues of LdAdK Asp16 in the respective enzymes) formed hydrogen bonds with both the O2′ and O3′ ribose hydroxyls [17,18]. Our modelled structure of LdAdK shows that the carboxy group of its Asp16 also points towards the adenosyl ribose (Figure 1B) and is quite proximal to its O2′ and O3′ groups (2.67 Å and 3.86 Å respectively), suggesting a bidentate interacting mode. These findings suggest that Asp16 of LdAdK may have an indispensable role and thus encouraged us to conduct an in-depth study of its function.
Table 1. Hydrogen bonds and close contacts (≤3.5 Å) between the ribose subsite of Ado and amino acid residues of LdAdK in the modelled structure.
| Ribose subsite | Amino acid | Distance (Å) |
|---|---|---|
| O2′ | Asp16 OD1 | 2.67 |
| O2′ | Gly62 N | 3.24 |
| O3′ | Gly62 N | 2.96 |
| O3′ | Gly62 CA | 3.48 |
| O3′ | Gly62 C | 3.25 |
| O3′ | Asn66 ND2 | 2.94 |
| C5′ | Asn295 C | 3.31 |
| C5′ | Asn295 O | 3.22 |
| O5′ | Asp299 OD2 | 2.50 |
Figure 1. Sequence alignment and close-up view of the Ado-binding site of LdAdK.

(A) Alignment of the N-terminal region of LdAdK amino acid sequence with those from T. gondii (NCBI accession number AAF01261), Babesia canis (NCBI accession number CAA11263), human (NCBI accession number AAA97893) and rat (NCBI accession number AAH81712) showing the invariant aspartate residue. (B) Structural model illustrating the position of Asp16 relative to the bound Ado; the broken lines depict the possible interaction between the Asp16 carboxy group and ribose hydroxy groups of Ado.
Conformational property of the wild-type and mutant LdAdKs
The CD spectra of the purified mutant enzymes overlapped almost completely with that of the wild-type LdAdK in the entire wavelength region from 195 to 260 nm, indicating that none of the mutations caused significant disruption of secondary structure or protein folding (results not shown).
Kinetic parameters of the Asp16 mutants
All the Asp16 mutants showed significantly lower, but nonetheless detectable, activity when measured under the standard assay conditions. To evaluate the decreased activities of the Asp16 mutants, we performed the kinetic analyses under steady-state conditions (Table 2). Replacement of Asp16 with either valine or asparagine caused a drastic reduction in their affinities for Ado, as evident from 235- and 286-fold increases in Km (Ado) for the respective mutants. The effect of mutations on the catalytic activity (kcat), which exhibited a moderate 5-fold decrease in activity, was, however, not so pronounced, suggesting that this residue as such is not directly involved in catalysis. Nevertheless, the kcat/Km values for Ado of both D16V and D16N AdKs were lowered more than 1000-fold, indicating absolute essentiality of the Asp16 in maintaining the catalytic efficiency of the enzyme. However, none of the mutations significantly affected the apparent Km for ATP. To ascertain whether any acidic group at this position would function equally well, we made a conservative replacement of Asp16 with a glutamate residue. With increasing concentrations of Ado, the D16E mutant showed non-saturation kinetics up to a concentration of 2 mM, whereas the wild-type enzyme was saturated at much lower concentrations (Figure 2). The specific activity of the D16E mutant at the highest attainable Ado concentration was, however, only approx. 2.5-fold lower than that of the wild-type enzyme (Figure 2, inset), suggesting almost unaltered catalytic activity. The non-saturation kinetics exhibited by the D16E mutant are indicative of weaker binding, implying that not only the charge, but also the optimum length, of the side chain are also essential for making proper interaction with Ado. An increase in the length of the side chain probably resulted in some distortion of the pocket that is responsible for properly accommodating Ado. The results are consistent with our structural predictions and explain the strict conservation of this residue among all AdKs sequenced to date.
Table 2. Steady-state kinetic parameters for the wild-type and mutated LdAdKs.
Km and kcat values are the means±S.D. for at least three separate experiments. ND, values could not be determined owing to non-saturation kinetics.
| Km (μM) | ||||
|---|---|---|---|---|
| Enzyme | Ado | ATP | kcat (min−1) | kcat/Km (Ado) (μM−1·min−1) |
| Wild-type | 5.5±1.5 | 36±4 | 1449±130 | 263.4 |
| D16V | 1293±55 | 20±2 | 306±18 | 0.23 |
| D16N | 1575±50 | 17±2 | 313±22 | 0.20 |
| D16E | ND | ND | ND | ND |
| R131A | 5.9±1.3 | 50±5 | 44±2 | 7.4 |
Figure 2. Phosphate-transfer activities of the wild-type and D16E mutant LdAdK.

Under standard assay conditions, the rate of AMP formation by the wild-type (●, Wt) and D16E (○) enzymes were plotted against increasing concentrations of Ado (0.4 and 0.8 μg of enzyme was used respectively). Inset: specific activities of the respective enzymes, where the specific activity (Sp. Activity) of the D16E mutant was measured at 2 mM Ado. Results are means±S.D. for at least three experimental determinations.
Contributions of the 2′ and 3′ hydroxy groups of adenosyl ribose in the LdAdK–Ado interaction
We observed that, similar to AdK from higher eukaryotes [30], LdAdK is also capable of phosphorylating deoxyadenosine, although with a lower catalytic efficiency as compared with Ado (Table 3). Results show that deletion of either the 2′- or the 3′-OH group from the adenosyl ribose caused an increase in their Km values by more than 14-fold and 6-fold respectively, suggesting reduced affinity for the enzyme, which correlates well with the relative impotency of the 2′- and 3′-deoxyadenosine to inhibit the Ado-driven reaction, as evident from their high Ki values (15.75 and 3.3 mM respectively). It should be noted that other Ado analogues like 6-methylmercaptopurine and tubercidin, which retain the ribose hydroxy groups, are potent inhibitors of LdAdK, with Ki values in the micromolar range [9]. Since the formation of enzyme–substrate complex is known to be dependent upon the status of binding energy, to assess the relative contribution of each hydroxy group, we studied the energetics of binding involved during the interaction of different substrates with the wild-type and mutant enzymes. Because, neither of the hydroxy groups (i.e. 2′- and 3′-hydroxy) of Ado nor the Asp16 residue of LdAdK appear to be directly involved in the catalytic step, as evident from the determined kinetic constants (Tables 2 and 3), the incremental binding energy was calculated from eqn (1) [24–26]. Table 4 shows that substitution of either valine or asparagine for Asp16 is accompanied by significant destabilization of the transition-state complex by approx. 3.9 kcal/mol. On the other hand, the free energy change caused due to selective replacement of either the 2′- or the 3′-hydroxy group of adenosyl ribose was 2.25 kcal/mol and 1.71 kcal/mol respectively. Taken together, these results suggest that mutation of Asp16 weakens the enzyme–substrate complex substantially. It should, however, be noted that none of the Asp16 mutants displayed measurable activity with either of the Ado analogues, thereby restricting the determination of ΔΔGB in the D16mutant-deoxyadenosine system, suggesting that residues other than the Asp16, that interact via the 2′ and 3′-hydroxy groups of the adenosyl ribose, are apparently responsible for Ado binding. Gly62, which is close to both the hydroxy groups (Table 1) and its role in Ado binding having already been established [19], is a likely candidate for such interaction. Notwithstanding these constraints, taking the prediction from the modelled structure (Figure 1) into consideration and the contributions of the 2′- and 3′-hydroxy groups to the empirical binding energy, it seems likely that Asp16 of LdAdK forms a bidentate hydrogen bond simultaneously with the 2′- and 3′-hydroxy groups of the ribose of Ado. Under such circumstance, the contribution of the 2′-hydroxy group appears to be greater than that of the 3′-hydroxy group, and that loss of uncharged hydrogen bond in the modified substrate might account for the observed result of an altered binding affinity.
Table 3. Kinetic parameters for the wild-type LdAdK with Ado analogues.
Results are means±S.D.
| Wild-type LdAdK | ||||
|---|---|---|---|---|
| Substrate | Km (μM) | kcat (min−1) | kcat/Km (μM−1. min−1) | Ki (Ado) (mM) |
| Ado | 5.5±1.5 | 1449±130 | 263.4 | − |
| 2′-Deoxyadenosine | 77±5 | 315±8 | 4.09 | 15.75 |
| 3′-Deoxyadenosine | 36±3 | 403±15 | 11.19 | 3.3 |
Table 4. Binding energy contributions (ΔΔGB) of the key functional groups.
For the wild-type column, the ratios were determined using the parent and modified substrates as indicated; for the mutant enzyme columns, the ratio was between the mutant and wild-type enzymes using the indicated substrates. For details refer to the text. ND, values could not be determined because assay sensitivities were too low to allow accurate determination of the kinetic parameters.
| ΔΔGB (kcal/mol) | |||
|---|---|---|---|
| Substrate | Wild-type | D16V | D16N |
| Ado | − | 3.82 | 3.89 |
| 2′-Deoxyadenosine | 2.25 | ND | ND |
| 3′-Deoxyadenosine | 1.71 | ND | ND |
AdK binds Ado and AMP by different mechanisms
Because AMP is the universally known competitive inhibitor of AdK with respect to Ado, it is generally agreed that AMP, at high concentration, occupies the Ado-binding site and causes the inhibition [9,20,21]. To verify whether AMP and Ado binding really correlate, the specific property of the wild-type and the mutant enzymes to bind to an AMP-affinity column and their relative sensitivities towards AMP inhibition in the AdK reaction were studied in order to probe the differential product binding properties of various mutants, if any, with respect to wild-type enzyme.
Contrasting observations obtained from the kinetic analysis (Table 2) of different mutants provided us with the impetus to design such an experiment. From the kinetic constants, it became apparent that, whereas all of the Asp16 mutants, because of drastic reduction in their affinity towards Ado, failed to catalyse Ado phosphorylation, the R131A mutant lost its catalytic efficiency, despite showing very little change in its Km value for Ado. Enzymes from Asp16 and R131A mutants were therefore subjected to inhibition as a function of AMP concentration. Interestingly, AMP, while it inhibited both D16V and D16N mutant enzymes to similar extents to that observed with the wild-type enzyme, displayed a much reduced ability in inhibiting R131A mutant enzyme (Figure 3A). Further support for these results was obtained when it was observed that, as opposed to R131A mutant enzyme, all the Asp16 mutant enzymes could efficiently bind to the AMP column (Figure 3B). Taken together, the results indicate that impairment of Ado binding does not necessarily prevent AMP binding, suggesting disparate binding mechanisms. Furthermore, the involvement of Arg131 in AMP binding (but not Ado binding) has also been established unambiguously for the first time.
Figure 3. AMP inhibition and AMP binding affinities of the wild-type and mutant LdAdK.

(A) Wild-type LdAdK (○) and D16V (■), D16N (△) and R131A (●) LdAdK mutants were assayed by direct radiometric methods in the presence of increasing concentrations of AMP. Results are means±S.D. for at least three experimental determinations and are expressed relative to the percentage of activity obtained without the exogenously added AMP (considered as 100% activity for the respective enzymes). Under standard assay conditions, the activity of the D16E mutant was too low for such an inhibition study. (B) AMP-binding affinity of wild-type and the mutant enzymes were compared by passing each of them through an AMP-agarose affinity column. Lane 1 represents the amount of protein loaded on to the column and lane 2 shows the amount that remained bound to the column after extensive washing.
Docking of AMP into the LdAdK active site
Owing to lack of availability of a product-bound AdK crystal from any source, an effort to reassess the above findings was made by carrying out computer-assisted docking of AMP into the LdAdK active site. Different binding locations were tested, and, based on the least-energy conformational requirement, AMP was placed into a site that, upon superposition, largely overlapped with the Ado-binding site (Figure 4A). In the modelled structure, although the purine subsites of Ado and AMP aligned almost completely, their ribose conformations were fairly different, suggesting that structural differences in the sugar puckers between Ado and AMP might be responsible for the different recognition modes of the two ligands. Nevertheless, the overlapping Ado–AMP structure is indicative of the fact that binding of the substrate and the product were mutually exclusive, a conclusion in agreement with the biochemical observation based on the competitive inhibition studies [9,20,21]. Further investigation showed that, among the AMP-binding residues, Arg131 is the most extensively hydrogen-bonded to the bound AMP (Figure 4B). Its guanidine group is engaged in the formation of two hydrogen bonds with the phosphate moiety of AMP and one additional bond with the O3′ of the AMP ribose. This finding is consistent with our mutagenesis results, thus indicating the essentiality of Arg131 in the formation of enzyme–AMP complex. Based on the mutational analysis, a function of Arg131 in LdAdK, as a bidentate electrophile in stabilizing the negative charge on the leaving group during the phosphate-transfer step, had recently been proposed [19]. The present studies therefore reveal an additional regulatory role of Arg131, acting as an effector of product binding. Consequently, this residue, by virtue of its dual role, appears to be a master player in the LdAdK reaction mechanism. The fact that this arginine residue is strictly conserved in AdK from all sources provides support for this interpretation.
Figure 4. Molecular docking of AMP into the LdAdK active site.

Computer-assisted docking of AMP into the LdAdK active site was carried out as described in the Experimental section. (A) Comparison of the AMP- and Ado-binding sites. The Figure was generated by superimposing the spatial locations of AMP (red) and Ado (yellow) in the catalytic site of LdAdK. (B) Hydrogen-bond networks (along with the inter-atomic distances) between the docked AMP and the side chain of Arg131 shown by the broken line.
DISCUSSION
Combined use of site-directed mutagenesis, molecular recognition studies with modified adenosine analogues and homology modelling guided us to identify Asp16 as the key molecular determinant for substrate binding and also established the novel AMP-anchoring property of Arg131 of LdAdK. Differential hydrogen bonding of adenosine and AMP with distinct interacting active-site residues to occupy a nearly overlapping position of the enzyme has provided important mechanistic insights into its catalytic process and regulation.
The in silico model of LdAdK positioned Asp16 in the active-site cleft of the enzyme. Its position relative to the bound Ado predicted hydrogen-bond formation between the ribose subsite of Ado and the carboxy group of Asp16. The prediction was confirmed by creating various point mutants and the results of the present study undoubtedly established that Asp16 is indeed a crucial Ado-binding residue. Replacement of this residue by either valine or asparagine caused a significant increase in the Km (Ado) value, thereby inducing considerable destabilization of the substrate in the active site. This is reflected in the transition-state binding energy differences for each of the mutants (3.82 and 3.89 kcal/mol respectively). These values fall within the range of energy value (0.5–5 kcal/mol) typical of an enzyme–substrate hydrogen bond [31]. Moreover, this particular aspartate residue of LdAdK is so intolerant to substitution that its replacement, even with a glutamate residue also known to possess the hydrogen-bonding functionality, led to weak substrate binding. This observation allowed us to propose that not only the negative charge of the carboxy group, but also the steric complementarity around this residue, is obligatory for proper recognition of the substrate, thus providing an explanation for the strict conservation of the residue among AdK from all sources sequenced to date.
Additional kinetic studies employing deoxygenated adenosine analogues successfully identified 2′- and 3′-hydroxy groups of the adenosyl ribose as the key functional groups that are responsible for formation of the LdAdK–Ado adduct. Replacement of either of the hydroxy groups caused a significant decrease in the affinity of the adenosine analogues for the enzyme as revealed by: (i) their elevated Km values, (ii) very high inhibition constants for the 2′- and 3′-deoxyadenosine as compared with the other adenosine analogues, which retain the hydroxy groups, e.g. 6-methylmercaptopurine and tubercidin [9], and (iii) 2.25 kcal/mol and 1.71 kcal/mol increase in transition-state stabilization energies respectively upon removal of the 2′- and 3′-hydroxy groups. The greater importance of the 2′-hydroxy group relative to the 3′-hydroxy group reflects the differences in hydrogen-bond strengths between the respective hydroxy groups and Asp16, possibly due to varying inter-atomic distances. Differences in the interacting distances of the hydroxy groups with other active-site residues might also be partially responsible for this differential contribution. Our findings thus amply corroborate an earlier study with T. gondii AdK, which also concluded that the hydroxy groups at the 2′ and 3′ positions of adenosine are crucial for the enzyme–substrate interaction [32]. They showed that substitution of either hydroxy group with a sterically misfitted methoxy group abolished the binding completely [32], validating our observation that the longer side-chain glutamate residue, possibly owing to a steric clash, could not functionally replace Asp16 in this structurally defined binding pocket. Thus, when considered in combination with the information obtained from the structural studies on LdAdK and AdKs from other sources [17,18], the kinetic data of the present study suggested that the bidentate hydrogen-bond formation between the carboxy group of Asp16 and the 2′- and 3′-hydroxy groups of the ribose subsite of adenosine is one of the key determining factors for the enzyme–Ado interaction.
Kinetic studies from our laboratory [9], as well as many others [20,21], showed that AMP is a competitive product inhibitor of AdK with respect to Ado, having Ki values in the range of its physiological concentration. It has been suggested that AMP elicits its inhibitory effect by competitively occupying the Ado-binding site. This is precisely what one would expect of a product inhibitor in an ordered Bi–Bi reaction mechanism. However, the observation that all of the Asp16 mutants, despite drastic loss of their catalytic efficiency resulting from inefficient Ado binding, did bind AMP efficiently and were equally susceptible to AMP inhibition in comparison with the wild-type enzyme has been difficult to reconcile with this mechanism. Two plausible explanations could account for this paradoxical result: (i) AMP binds at a site distinct from the Ado-binding site, but indirectly perturbs the conformation of the Ado-binding pocket in a manner that leads to lowered affinity of the site for Ado, thus showing as an apparent competition between Ado and AMP, or (ii) although both AMP and Ado binds LdAdK at an overlapping site, their targets for interactions with the enzyme are different, i.e. different interacting amino acid residues might be involved in recognizing AMP and Ado. However, the AMP-docked structure of LdAdK in the present paper rules out the possibility of AMP binding to a distinct regulatory site; rather, it makes a strong case for the presence of a common binding pocket where AMP relies on the interaction of its phosphate moiety with the guanidinium group of Arg131 of the enzyme.
Further experimental support of this model-based prediction was obtained when it was observed that (i) the R131A mutant enzyme displayed resistance to AMP inhibition, and (ii) mutation of Arg131 to neutral alanine resulted in non-absorption of the enzyme on to the AMP column. The product-binding property of Arg131 documented in the present study is hitherto unknown and thus provided an attractive point of regulation of LdAdK activity. This particular arginine residue, along with Asp299, had previously been proposed to constitute the catalytic backbone of LdAdK. However, upon alanine substitution, the D299A mutant was affected more drastically (catalytic activity completely lost) as compared with the R131A mutant that showed significantly lowered, but detectable, activity [19]. The present study provides a reasonable explanation for this observation, suggesting that reduced AMP-binding ability for the R131A mutant probably resulted in unregulated functioning of the mutant enzyme, thereby partially compensating for its loss of catalytic function. Since Arg131 is involved both in catalysis and regulation, it is also understandable as to why replacement of Arg131 with alanine failed to enhance the catalytic activity, as has been observed on several occasions where deregulation of an enzyme by a point mutation that destroys the effector binding site has been associated with an increased enzymatic activity [33,34]. Since AdK is a potential controller of both extracellular Ado concentration and intracellular adenine nucleotide levels [4], its phosphorylating ability strictly depends on the intracellular ATP levels [9]. The enzyme thus, by assigning a dual responsibility to the particular arginine residue, has evolved a ‘fail-safe’ mechanism to prevent unregulated purine metabolism under unfavourable conditions. As a result, the loss of regulation is associated with reduced catalytic activity, thereby allowing the system to switch to a bypass pathway in order to adapt itself to the changing environment. This hypothesis seems relevant in the context of Leishmania physiology, where many such bypass pathways for salvaging the purines are operational [35].
Sequence of events in the LdAdK reaction: a composite proposal
The differential mode of interaction of LdAdK with adenosine and AMP, together with the previously reported kinetic and crystallographic studies on AdKs, provided a basis for analysing the ordered reaction pathway of the enzyme. Based on this evidence [8,17–19], we propose that Ado is the first substrate to bind in an open active site of the enzyme, inducing a conformational change leading to a relatively closed positioning of the lid, which places the second substrate (i.e. ATP) in a catalytically competent position. The finding of a relatively uncommon ribose sugar pucker in the O4′-endo conformation of Ado bound to human or T. gondii AdK crystals [17,18] suggests that the binding conformation of the substrate possibly plays an important role in the AdK reaction mechanism. It is likely that Ado, in a more common puckered form of the ribose (i.e. C2′ or C3′-endo), probably binds to the active site at the initial stage and subsequently gets distorted to an energetically strained O4′-endo conformation which is stabilized by favourable hydrogen-bond interactions, particularly between the carboxy group of Asp16 and O2′ and O3′ of adenosyl ribose. A similar type of induced-fit binding of substrate has been proposed for RK [36]. At the time of progression through the phosphate-transfer pathway, the ribose contact with the Asp16 probably undergoes a remodelling in synchrony with the substrate-induced conformational switch of the enzyme, which ultimately results in disruption of the contact of Asp16 with the ribose hydroxy group in the product, AMP. Concomitantly, Arg131, present in the smaller domain, swings into the active site, where, apart from its catalytic function through charge-stabilizing contacts with the ATP [19], it provides crucial hydrogen-bond interactions with the phosphate moiety of AMP, thus functioning as an effector of product-mediated enzyme inhibition. This type of mechanism is consistent with a growing body of evidence demonstrating ligand-altered protein dynamics [37,38]. The reaction scheme of the present study assumes that Ado and AMP, by binding independently at two contiguous overlapping sites, sterically interfere with the binding of each other. Although the proposed model adequately explains the competition between the substrate and the product, it failed to clarify how the product is released. The possibility that the enzyme may release AMP once it encounters an additional dose of substrate or, alternatively, the enzyme might undergo yet another conformational change after the catalysis step, causing domain re-opening and subsequent expulsion of the product from the active site, cannot be ruled out. Direct X-ray crystallographic work on LdAdK complexed with substrate analogues or the products, combined with hinge engineering experiments, may provide definitive mechanistic clues regarding the product clearance and other events associated with the enzyme turnover.
Conclusion
In conclusion, our work identified key molecular determinants (Asp16 and Arg131 respectively) for substrate binding and product inhibition in LdAdK. Although they may not be the only factors, their crucial roles in the differential recognition of Ado and AMP binding provide important insights into the phosphorylation mechanism of LdAdK. In particular, the product-binding property of Arg131 has not been envisaged previously and thus represents a unique regulatory feature among this class of enzymes. An additional point of significance of these findings is that, by understanding the structural requirements of product binding, one can certainly conceive of strategies to design inhibitors that are capable of interacting with the product-binding site. Therefore the analysis of the AMP-docked model of LdAdK using mutagenesis experiments could be used as the lead towards designing selective product-based inhibitors.
Acknowledgments
The work was supported by Grant SP/SO/D-38/2000 from the Department of Science and Technology, Government of India. Additional financial support was obtained from CSIR network projects (CMM0017 and SMM003), Government of India. R. D. and B. S. were each supported by individual fellowships from the Council of Scientific & Industrial Research, Government of India. Authors wish to thank Mr Jaganmoy Guin of Bose Institute, Kolkata for his technical support with CD experiments.
References
- 1.Caputto R. The enzymatic synthesis of adenylic acid: adenosine kinase. J. Biol. Chem. 1951;189:801–814. [PubMed] [Google Scholar]
- 2.Kornberg A., Pricer W. E., Jr Enzymatic phosphorylation of adenosine and 2,6-diaminopurine riboside. J. Biol. Chem. 1951;193:481–495. [PubMed] [Google Scholar]
- 3.Richard J. P., Carr M. C., Ives D. H., Frey P. A. The stereochemical course of thiophosphoryl group transfer catalyzed by adenosine kinase. Biochem. Biophys. Res. Commun. 1980;94:1052–1056. doi: 10.1016/0006-291x(80)90525-2. [DOI] [PubMed] [Google Scholar]
- 4.Decking U. K., Schlieper G., Kroll K., Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ. Res. 1997;2:154–164. doi: 10.1161/01.res.81.2.154. [DOI] [PubMed] [Google Scholar]
- 5.Miller R. L., Adamczyk D. L., Miller W. H., Koszalka G. W., Rideout J. L., Beacham L. M., 3rd, Chao E. Y., Haggerty J. J., Krenitsky T. A., Elion G. B. Adenosine kinase from rabbit liver. II. Substrate and inhibitor specificity. J. Biol. Chem. 1979;254:2346–2352. [PubMed] [Google Scholar]
- 6.Looker D. L., Berens R. L., Marr J. J. Purine metabolism in Leishmania donovani amastigotes and promastigotes. Mol. Biochem. Parasitol. 1983;9:15–28. doi: 10.1016/0166-6851(83)90053-1. [DOI] [PubMed] [Google Scholar]
- 7.Gottlieb M. The surface membrane 3′-nucleotidase/nuclease of trypanosomatid protozoa. Parasitol. Today. 1989;5:257–260. doi: 10.1016/0169-4758(89)90259-7. [DOI] [PubMed] [Google Scholar]
- 8.Datta A. K., Bhaumik D., Chatterjee R. Isolation and characterization of adenosine kinase from Leishmania donovani. J. Biol. Chem. 1987;262:5515–5521. [PubMed] [Google Scholar]
- 9.Bhaumik D., Datta A. K. Reaction kinetics and inhibition of adenosine kinase from Leishmania donovani. Mol. Biochem. Parasitol. 1988;28:181–187. doi: 10.1016/0166-6851(88)90002-3. [DOI] [PubMed] [Google Scholar]
- 10.Bhaumik D., Datta A. K. Immunochemical and catalytic characteristics of adenosine kinase from Leishmania donovani. J. Biol. Chem. 1989;264:4356–4361. [PubMed] [Google Scholar]
- 11.Bhaumik D., Datta A. K. Active site thiol(s) in Leishmania donovani adenosine kinase: comparison with hamster enzyme and evidence for the absence of regulatory adenosine binding site. Mol. Biochem. Parasitol. 1992;52:29–38. doi: 10.1016/0166-6851(92)90033-g. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh M., Datta A. K. Probing the function(s) of active-site arginine residue in Leishmania donovani adenosine kinase. Biochem. J. 1994;298:295–301. doi: 10.1042/bj2980295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagui T. K., Ghosh M., Datta A. K. Two conformationally vicinal thiols at the active site of Leishmania donovani adenosine kinase. Biochem. J. 1996;316:439–445. doi: 10.1042/bj3160439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinha K. M., Ghosh M., Das I., Datta A. K. Molecular cloning and expression of adenosine kinase from Leishmania donovani: identification of unconventional P-loop motif. Biochem. J. 1999;339:667–673. [PMC free article] [PubMed] [Google Scholar]
- 15.Wu L. F., Reizer A., Reizer J., Cai B., Tomich J. M., Saier M. H., Jr Nucleotide sequence of the Rhodobacter capsulatus fruK gene, which encodes fructose-1-phosphate kinase: evidence for a kinase superfamily including both phosphofructokinases of Escherichia coli. J. Bacteriol. 1991;173:3117–3127. doi: 10.1128/jb.173.10.3117-3127.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bork P., Sander C., Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993;2:31–40. doi: 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathews I. I., Erion M. D., Ealick S. E. Structure of human adenosine kinase at 1.5 Å resolution. Biochemistry. 1998;37:15607–15620. doi: 10.1021/bi9815445. [DOI] [PubMed] [Google Scholar]
- 18.Schumacher M. A., Scott D. M., Mathews I. I., Ealick S. E., Roos D. S., Ullman B., Brennan R. G. Crystal structures of Toxoplasma gondii adenosine kinase reveal a novel catalytic mechanism and prodrug binding. J. Mol. Biol. 2000;298:875–893. doi: 10.1006/jmbi.2000.3753. [DOI] [PubMed] [Google Scholar]
- 19.Datta R., Das I., Sen B., Chakraborty A., Adak S., Mandal C., Datta A. K. Mutational analysis of the active-site residues crucial for catalytic activity of adenosine kinase from Leishmania donovani. Biochem. J. 2005;387:591–600. doi: 10.1042/BJ20041733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palella T. D., Andres C. M., Fox I. H. Human placental adenosine kinase: kinetic mechanism and inhibition. J. Biol. Chem. 1980;255:5264–5269. [PubMed] [Google Scholar]
- 21.Hawkins C. F., Bagnara A. S. Adenosine kinase from human erythrocytes: kinetic studies and characterization of adenosine binding sites. Biochemistry. 1987;26:1982–1987. doi: 10.1021/bi00381a030. [DOI] [PubMed] [Google Scholar]
- 22.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 23.Cleland W. W. The Enzymes, vol. 2. In: Boyer P. D., editor. 3rd edn. New York: Academic Press; 1970. pp. 1–65. [Google Scholar]
- 24.Wilkinson A. J., Fersht A. R., Blow D. M., Winter G. Site-directed mutagenesis as a probe of enzyme structure and catalysis: tyrosyl-tRNA synthetase cysteine-35 to glycine-35 mutation. Biochemistry. 1983;22:3581–3586. doi: 10.1021/bi00284a007. [DOI] [PubMed] [Google Scholar]
- 25.Street I. P., Armstrong C. R., Withers S. G. Hydrogen bonding and specificity: fluorodeoxy sugars as probes of hydrogen bonding in the glycogen phosphorylase–glucose complex. Biochemistry. 1986;25:6021–6027. doi: 10.1021/bi00368a028. [DOI] [PubMed] [Google Scholar]
- 26.Sierks M. R., Svensson B. Kinetic identification of a hydrogen bonding pair in the glucoamylase–maltose transition state complex. Protein Eng. 1992;5:185–188. doi: 10.1093/protein/5.2.185. [DOI] [PubMed] [Google Scholar]
- 27. Reference deleted.
- 28.Thompson J. D., Higgins D. G., Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sigrell J. A., Cameron A. D., Jones T. A., Mowbray S. L. Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 Å resolution: insights into a new family of kinase structures. Structure. 1998;6:183–193. doi: 10.1016/s0969-2126(98)00020-3. [DOI] [PubMed] [Google Scholar]
- 30.Hurley M. C., Lin B., Fox I. H. Regulation of deoxyadenosine and nucleoside analog phosphorylation by human placental adenosine kinase. J. Biol. Chem. 1985;260:15675–15681. [PubMed] [Google Scholar]
- 31.Fersht A. R., Shi J. P., Knill-Jones J., Lowe D. M., Wilkinson A. J., Blow D. M., Brick P., Carter P., Waye M. M., Winter G. Hydrogen bonding and biological specificity analysed by protein engineering. Nature (London) 1985;314:235–238. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 32.Iltzsch M. H., Uber S. S., Tankersley K. O., el Kouni M. H. Structure-activity relationship for the binding of nucleoside ligands to adenosine kinase from Toxoplasma gondii. Biochem. Pharmacol. 1995;49:1501–1512. doi: 10.1016/0006-2952(95)00029-y. [DOI] [PubMed] [Google Scholar]
- 33.Rothman S. C., Voorhies M., Kirsch J. F. Directed evolution relieves product inhibition and confers in vivo function to a rationally designed tyrosine aminotransferase. Protein Sci. 2004;3:763–772. doi: 10.1110/ps.03117204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adak S., Crooks C., Wang Q., Crane B. R., Tainer J. A., Getzoff E. D., Stuehr D. J. Tryptophan 409 controls the activity of neuronal nitric-oxide synthase by regulating nitric oxide feedback inhibition. J. Biol. Chem. 1999;274:26907–26911. doi: 10.1074/jbc.274.38.26907. [DOI] [PubMed] [Google Scholar]
- 35.Hassan H. F., Coombs G. H. Leishmania mexicana: purine-metabolizing enzymes of amastigotes and promastigotes. Exp. Parasitol. 1985;59:139–150. doi: 10.1016/0014-4894(85)90066-9. [DOI] [PubMed] [Google Scholar]
- 36.Sigrell J. A., Cameron A. D., Mowbray S. L. Induced fit on sugar binding activates ribokinase. J. Mol. Biol. 1999;290:1009–1018. doi: 10.1006/jmbi.1999.2938. [DOI] [PubMed] [Google Scholar]
- 37.Volkman B. F., Lipson D., Wemmer D. E., Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 38.Revington M., Holder T. M., Zuiderweg E. R. NMR study of nucleotide-induced changes in the nucleotide binding domain of Thermus thermophilus Hsp70 chaperone DnaK: implications for the allosteric mechanism. J. Biol. Chem. 2004;279:33958–33967. doi: 10.1074/jbc.M313967200. [DOI] [PubMed] [Google Scholar]