

Abstract
Uric acid (UA) is a purine metabolite that selectively inhibits peroxynitrite-mediated reactions implicated in the pathogenesis of multiple sclerosis (MS) and other neurodegenerative diseases. Serum UA levels are inversely associated with the incidence of MS in humans because MS patients have low serum UA levels and individuals with hyperuricemia (gout) rarely develop the disease. Moreover, the administration of UA is therapeutic in experimental allergic encephalomyelitis (EAE), an animal model of MS. Thus, raising serum UA levels in MS patients, by oral administration of a UA precursor such as inosine, may have therapeutic value. We have assessed the effects of inosine, as well as inosinic acid, on parameters relevant to the chemical reactivity of peroxynitrite and the pathogenesis of EAE. Both had no effect on chemical reactions associated with peroxynitrite, such as tyrosine nitration, or on the activation of inflammatory cells in vitro. Moreover, when mice treated with the urate oxidase inhibitor potassium oxonate were fed inosine or inosinic acid, serum UA levels were elevated markedly for a period of hours, whereas only a minor, transient increase in serum inosine was detected. Administration of inosinic acid suppressed the appearance of clinical signs of EAE and promoted recovery from ongoing disease. The therapeutic effect on animals with active EAE was associated with increased UA, but not inosine, levels in CNS tissue. We, therefore, conclude that the mode of action of inosine and inosinic acid in EAE is via their metabolism to UA.
The prospect that the manipulation of serum urate (UA) levels may provide a natural approach to therapeutic intervention in multiple sclerosis (MS) and perhaps other neurodegenerative processes is supported by several lines of evidence, including (i) the specificity of UA for the inhibition of certain peroxynitrite-mediated chemical reactions, such as tyrosine nitration (1–5), (ii) evidence that these reactions may contribute to the pathogenesis of MS and other neurodegenerative diseases (6–9), (iii) the demonstration that UA is an effective treatment in several animal models of CNS inflammation (8, 10–13), and (iv) observations of a reciprocal relationship between the incidence of MS and serum UA levels (10, 14–17). A common feature of the neuroinflammatory conditions that are influenced by UA is the presence in CNS tissues of cells expressing inducible nitric oxide (iNOS), an enzyme that produces high levels of NO (11). When NO and superoxide are produced in close proximity, for example, by activated monocytes (18), they combine to form peroxynitrite, which rapidly decomposes in a biological milieu to generate highly reactive intermediates, such as NO and CO
and CO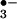 , some of which are inactivated by UA (4, 5). UA is an intermediate of purine metabolism in most mammals but, because of the unusual penetrance of a single point mutation in the urate oxidase gene, is the end product in higher-order primates and humans (19–21). When the normally low serum levels of UA in animals used to model neuroinflammatory disease are raised by repeated administration of UA, they become resistant to the induction of such diseases, which include experimental allergic encephalomyelitis (EAE) and Borna disease (8, 10–13, 22). Moreover, studies in EAE have demonstrated that treatment of an ongoing neuroinflammatory condition with UA has beneficial effects (10, 11).
, some of which are inactivated by UA (4, 5). UA is an intermediate of purine metabolism in most mammals but, because of the unusual penetrance of a single point mutation in the urate oxidase gene, is the end product in higher-order primates and humans (19–21). When the normally low serum levels of UA in animals used to model neuroinflammatory disease are raised by repeated administration of UA, they become resistant to the induction of such diseases, which include experimental allergic encephalomyelitis (EAE) and Borna disease (8, 10–13, 22). Moreover, studies in EAE have demonstrated that treatment of an ongoing neuroinflammatory condition with UA has beneficial effects (10, 11).
There are also indications that the elevated UA levels normally seen in humans may be beneficial. For example, MS patients generally have serum UA levels that are lower than those of controls, and gout (hyperuricemia) patients very rarely develop MS (10). Moreover, it has been demonstrated recently that serum UA levels are significantly lower in MS patients with a clinical remission and that this correlates with blood–brain barrier (BBB) dysfunction (17). We therefore have begun to examine the therapeutic potential of elevating UA levels in MS patients, first assessing whether oral administration of UA could be used to raise serum levels (23, 24). This proved ineffective likely because of the production of uricase by enteric bacteria (23). Our alternative approach was to administer a precursor of UA that is not susceptible to breakdown by uricase because metabolism of dietary purines is a major source of UA. Inosine is a purine nucleoside that has been used extensively in humans and marketed for a number of years as an energy supplement and performance enhancer, although there is little scientific evidence that it has these properties (25, 26). A well known side effect of inosine use is raised serum UA levels. We have found that the low serum UA levels of MS patients can be raised reliably and maintained at desired levels for extended periods of time by oral administration of inosine (24). Preliminary results suggest that there may be a therapeutic benefit to such treatment, and no signs of any deleterious effects have been noted (24). Inosinic acid, the 5′-monophosphate of inosine, is used as a flavor enhancer and, therefore, also is consumed extensively by humans, particularly in Japan (27).
Although little is known about the effects of inosinic acid on physiological processes, there is increasing evidence that inosine can modulate inflammatory processes (28–31) and neuronal growth properties (32, 33) that possibly are relevant to neurodegenerative disease. For example, administration of inosine in a mouse model of acute, lipopolysaccharide (LPS)-induced, lung inflammation had a broad range of antiinflammatory effects including the down-regulation of tumor necrosis factor-α expression (31). On the other hand, infusion of inosine directly into the cisterna magna or lateral ventricle of the rat brain after experimental stroke induction stimulated axonal outgrowth and rewiring in the intact side of the brain (33). In this study, we therefore have performed experiments to establish the nature of any inosine or inosinic acid effects on peroxynitrite-mediated chemical reactivities and the pathogenesis of EAE.
Experimental Procedures
Assessment of Peroxynitrite-Mediated Oxidation.
Peroxynitrite-mediated oxidation was measured in vitro by the conversion of dihydrorhodamine 123 (DHR123, Molecular Probes) to fluorescent rhodamine 123 (10, 12, 13). DHR123 (5 μM) and different concentrations of UA, inosine, and inosinic acid were added to cultures containing either 100 μM 3-morpholinosydnonimine⋅HCl (SIN-1, Alexis Biochemicals, San Diego) or LPS-stimulated RAW 264.7 cells, as sources of peroxynitrite, and incubated for 1 h at 37°C. Rhodamine 123 fluorescence then was measured in a microplate fluorometer (Cytofluor II, PerSeptive Biosystems, Framingham, MA), with an excitation wavelength of 485 nm and an emission wavelength of 530 nm, gain 70.
Western Analysis of ONOO−-Mediated Tyrosine Nitration.
BSA (Sigma) at 1 mg/ml in PBS was incubated with 1 mM SIN-1 for 2 h at 37°C in the presence and absence of UA, inosine, and inosinic acid (100–800 μM) and 10 mM HCO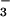 added as NaHCO3 (Sigma). Immediately after incubation, 5 μl of each sample was separated on a 12.5% SDS-polyacrylamide gel and transferred onto a poly(vinylidene difluoride) membrane (NEN; ref. 13). Nitrotyrosine-containing proteins were detected with rabbit polyclonal anti-nitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY) and developed with a diaminobenzidine substrate by using the Vectastain detection kit according to manufacturer recommendations (PK-6101, Vector Laboratories).
added as NaHCO3 (Sigma). Immediately after incubation, 5 μl of each sample was separated on a 12.5% SDS-polyacrylamide gel and transferred onto a poly(vinylidene difluoride) membrane (NEN; ref. 13). Nitrotyrosine-containing proteins were detected with rabbit polyclonal anti-nitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY) and developed with a diaminobenzidine substrate by using the Vectastain detection kit according to manufacturer recommendations (PK-6101, Vector Laboratories).
Activation of Monocytes and Assessment of iNOS Activity in Vitro.
Cells of the mouse monocyte–macrophage cell line RAW 264.7 (American Type Culture Collection) were grown to 80% confluence and activated with 1 μg/ml LPS (Escherichia coli serotype 055:B5, Sigma) in RPMI medium 1640 supplemented with 10% heat-inactivated FBS, 50 units of penicillin, 50 μg/ml streptomycin, and 5 mM l-glutamine in the presence or absence of UA, inosine, and inosinic acid (200 μM) overnight. Nitrite accumulation in the medium was assessed by using the Griess reaction as detailed (10). The activation of iNOS genes was assessed by real-time quantitative RT-PCR analysis of the expression of the specific mRNAs by using a Bio-Rad (Hercules, CA) iCycler iQ real-time detection system as detailed (22). Data were calculated based on a threshold cycle (Ct), determined as the cycle with a signal higher than that of the background (signal detected in cycles 2–10) plus 10 times its SD. Data are expressed as a fold increase in mRNA expression calculated by exp[Ct lowest expresser (e.g., unstimulated cells) − Ct test value] divided by the same value determined for the housekeeping gene GAPDH.
Induction of EAE.
Female 8- to 10-week-old PLSJL mice (The Jackson Laboratory) each were immunized s.c. at three sites along the back with 200 μl of an emulsion of 100 μg myelin basic protein (MBP) in complete Freund's adjuvant (1:1) containing 0.05% Mycobacterium butyricum plus an additional 4 mg/ml Mycobacterium tuberculosis H37 RA. Pertussis toxin (List Biological Laboratories, Campbell, CA), 400 ng, was given i.p. twice, on days 0 and 2. Mice were scored for clinical signs of EAE twice daily on the basis of the presence of the following symptoms: 0, normal mouse; 1, piloerection, tail weakness; 2, tail paralysis; 3, tail paralysis plus hindlimb weakness; 4, tail paralysis plus partial hindlimb paralysis; 5, total hindlimb paralysis; 6, hind- and forelimb paralysis; 7, moribund/dead.
Treatment of Mice.
Inosine and inosinic acid (5′-monophosphate, disodium salt, from yeast, Sigma) were administered twice daily either as i.p. doses of 500 mg/kg in 100 μl of saline or by gastric intubation of 1,500 mg/kg in 100 μl of saline with and without two daily i.p. injections of 250 mg/kg potassium oxonate (K-Ox) in 100 μl of saline to inhibit breakdown of UA by urate oxidase. To assess the effects on existing disease, treatment of mice began when clinical signs of EAE reached a score of at least 3.
HPLC Analysis of UA, Inosine, and Inosinic Acid Levels in Sera and CNS Tissue.
For sera, heparinized blood was collected and sera was isolated by centrifugation and deproteinized by using HClO4 and K2HPO4 as described (11). Spinal cord tissue was homogenized in 0.1 M perchloric acid, and the supernatant was deproteinized with K2HPO4 as detailed (11). HPLC analysis was performed by using a C18 reverse-phase column and a 30-min convex gradient of 100% buffer A (0.06 M K2HPO4 and 0.04 M KH2PO4 in H2O, pH 6.0) to 100% buffer B (0.06 M K2HPO4 and 0.04 M KH2PO4 in 25% methanol/75% H2O, pH 6.0) on a Beckman Coulter 125 solvent module and 168 diode array detector. UA was detected at 292 nm at ≈5 min, and inosinic acid and inosine were detected at 254 nm at ≈7 and 22 min, respectively. Concentrations were determined by comparison of the integral of peak areas with those of known controls.
Results
Chemical Reactivities Mediated Through Peroxynitrite Decomposition Are Inhibited in Vitro by UA but Not Inosine or Inosinic Acid.
Peroxynitrite decomposition mediates several chemical reactions that are highly sensitive to inhibition by UA, including oxidation of DHR123 to rhodamine 123 and tyrosine nitration (13). Neither inosine nor inosinic acid had any effect on the oxidation of DHR123, whereas UA at physiological levels completely inhibited the reaction, whether mediated by a peroxynitrite donor or activated monocytes (Fig. 1). A similar result was observed for the nitration of serum proteins by peroxynitrite, where UA strongly prevented the reaction, whereas comparable concentrations of inosine and inosinic acid had no noticeable effect (Fig. 2).
Fig 1.

DHR123 oxidation by the peroxynitrite donor SIN-1 and activated monocytes is inhibited by UA but not by inosine or inosinic acid. UA, inosine, and inosinic acid were titrated into cultures containing 100 μM SIN-1 as a source of peroxynitrite (A) or added at a final concentration of 200 μM to cultures containing RAW 264.7 cells activated with LPS (B) as described in Experimental Procedures. The conversion of 5 μM DHR123 to rhodamine 123 was measured with a fluorometer (excitation, 485 nm; emission, 530 nm; gain 70). The conversion of DHR123 to rhodamine 123 was used as an indicator of oxidation. Data are expressed as DHR123 oxidation and are presented as means ± SD.
Fig 2.

UA but not inosine or inosinic acid inhibits tyrosine nitration by SIN-1. BSA (1 mg/ml) in PBS was incubated with 1 mM SIN-1 for 2 h at 37°C in the presence of 10 mM HCO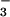 , with and without decreasing concentrations of UA, inosine, and inosinic acid. Immediately after incubation, 5 μl of each sample was separated on a 12.5% SDS/PAGE gel and transferred onto a poly(vinylidene difluoride) membrane. Nitrotyrosine-containing proteins were detected by using a rabbit polyclonal anti-nitrotyrosine antibody as detailed in Experimental Procedures. 1, Molecular mass markers; 2, SIN-1 and HCO
, with and without decreasing concentrations of UA, inosine, and inosinic acid. Immediately after incubation, 5 μl of each sample was separated on a 12.5% SDS/PAGE gel and transferred onto a poly(vinylidene difluoride) membrane. Nitrotyrosine-containing proteins were detected by using a rabbit polyclonal anti-nitrotyrosine antibody as detailed in Experimental Procedures. 1, Molecular mass markers; 2, SIN-1 and HCO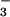 ; 3, SIN-1, HCO
; 3, SIN-1, HCO , and 800 μM UA; 4, SIN-1, HCO
, and 800 μM UA; 4, SIN-1, HCO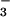 , and 400 μM UA; 5, SIN-1, HCO
, and 400 μM UA; 5, SIN-1, HCO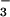 , and 200 μM UA; 6, SIN-1, HCO
, and 200 μM UA; 6, SIN-1, HCO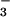 , and 100 μM UA; 7, SIN-1, HCO
, and 100 μM UA; 7, SIN-1, HCO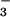 , and 800 μM inosine; 8, SIN-1, HCO
, and 800 μM inosine; 8, SIN-1, HCO , and 400 μM inosine; 9, SIN-1, HCO
, and 400 μM inosine; 9, SIN-1, HCO , and 200 μM inosine; 10, SIN-1, HCO
, and 200 μM inosine; 10, SIN-1, HCO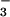 , and 100 μM inosine; 11, SIN-1, HCO
, and 100 μM inosine; 11, SIN-1, HCO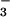 , and 800 μM inosinic acid; 12, SIN-1, HCO
, and 800 μM inosinic acid; 12, SIN-1, HCO , and 400 μM inosinic acid; 13, SIN-1, HCO
, and 400 μM inosinic acid; 13, SIN-1, HCO , and 200 μM inosinic acid; 14, SIN-1, HCO
, and 200 μM inosinic acid; 14, SIN-1, HCO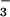 , and 100 μM inosinic acid; and 15, molecular mass markers.
, and 100 μM inosinic acid; and 15, molecular mass markers.
iNOS Gene Expression by Monocytes in Vitro Is Unaffected by UA, Inosine, or Inosinic Acid.
Our previous studies have not detected any changes in monocyte or lymphocyte activation associated with UA treatment (12, 13). However, there is evidence that inosine may have inhibitory effects on pathways related to immune function (28–31). iNOS is an enzyme that catalyzes the production of NO by monocytes during an inflammatory response and has been implicated in the pathogenesis of both MS and EAE (6–9, 11–13). Therefore, we assessed whether, unlike UA, inosine or inosinic acid could inhibit the activation of iNOS in cultures of mouse RAW monocytes stimulated by LPS. Fig. 3 shows that neither the levels of iNOS mRNA expression nor the accumulation of the NO metabolite, nitrite, in the cultures is altered by the addition of UA, inosine, or inosinic acid. This indicates that none of these compounds has a direct inhibitory effect on the induction of iNOS.
Fig 3.

LPS-induced iNOS expression and nitrite formation by monocytes in vitro are unaffected by the addition of UA, inosine, and inosinic acid. RAW 264.7 cells were activated by LPS as described in Experimental Procedures in the presence and absence of 200 μM UA, inosine, or inosinic acid. iNOS expression was determined by real-time quantitative RT-PCR (A), and nitrite production in culture medium was measured by the Griess reaction (B) as detailed in Experimental Procedures. Results are expressed as mean ± SD of a minimum of two independent samples assayed in duplicate.
Oral Administration of Inosine and Inosinic Acid Primarily Raises Serum UA Levels in Mice.
The oral administration of inosine to MS patients has proven to effectively raise their normally low serum UA levels (23, 24). Because, unlike humans, mice metabolize UA to allantoin, we first determined whether their serum UA levels could be elevated similarly. In addition to inosine, we also assessed the capacity of its derivative, inosinic acid, to raise serum UA levels in mice. Inosinic acid is particularly interesting in this regard because it has been consumed regularly by a large number of individuals as a flavor enhancer, without reported side effects (27). The i.p. administration of 500 mg/kg of either inosine or inosinic acid in 100 μl of saline to adult female PLSJL mice caused a transient, minor increase in serum inosine levels (peak, ≈30 μM at 15 min postinjection) as well as a strong elevation in serum UA (data not shown). Serum UA levels were maximal at 15 min postinjection (≈400 μM) but declined rapidly to between 100 and 150 μM, where it was maintained for 2–4 h (data not shown).
As is evident from Fig. 4, serum UA levels can be raised only transiently in mice likely because of the rapid metabolism of UA by urate oxidase. To maintain elevated serum UA levels over a more extended period, we have examined the utility of several urate oxidase inhibitors including K-Ox, 8-azaxanthine, and 9-methyl-UA. K-Ox has been used more extensively in vivo (e.g., ref. 34) and therefore has become our urate oxidase inhibitor of choice. By titrating K-Ox in mice treated with UA, we have determined that serum UA levels can be raised more effectively and maintained at higher levels for over 8 h when using a single i.p. dose of 10 mg of K-Ox (Fig. 4).
Fig 4.

Inhibition of urate oxidase with potassium oxonate delays UA metabolism in mice. Groups of mice (n = 3–4) were immunized with MBP as detailed in Experimental Procedures and injected i.p with UA (1, 5, or 10 mg) and K-Ox (1, 5, or 10 mg) as indicated. Serum samples were collected 30 min, 1 h, 4 h, and 8 h postinjection. Serum UA levels were assessed by HPLC as described in Experimental Procedures. Data are presented as mean ± SD.
Using K-Ox to inhibit the metabolism of UA, we compared the efficacy of inosinic acid vs. inosine in raising serum UA levels after oral administration. After a single i.p. dose of K-Ox, serum UA levels were elevated strongly for periods of up to 8 h by the administration of inosinic acid, whereas inosine, initially, was less effective (Fig. 5). As shown in Fig. 5, oral administration of both inosine and inosinic acid caused a substantial elevation in serum UA levels but only a minor increase in serum inosine. Inosinic acid ingestion resulted in peak serum UA levels at 15 min that were roughly twice those caused by inosine, but these declined within 2 h such that the elevated UA levels caused by both reagents were approximately the same (Fig. 5). In both cases, serum inosine levels dropped to background between 30 and 60 min after ingestion, whereas UA levels remained elevated for more than 4 h (Fig. 5). Inosinic acid was not detected consistently in serum at any time point (Fig. 5).
Fig 5.

Effect of oral administration of inosinic acid and inosine on serum UA, inosine, and inosinic acid levels. Groups of MBP-immunized mice (n = 5) were treated once with 5 mg of K-Ox i.p. and 30 mg of either inosinic acid or inosine in 100 μl of saline by gastric intubation. Serum samples were collected at 15 min, 30 min, 1 h, 2 h, and 4 h posttreatment. Serum levels of UA, inosine, and inosinic acid were measured by HPLC as described in Experimental Procedures. Results are expressed as mean ± SD.
Oral Administration of Inosine and Inosinic Acid Raises UA Levels in the CNS Tissues of Mice with EAE.
Normally, the BBB is relatively impervious to UA and UA levels in CNS tissues are low (11). When the BBB becomes compromised during the course of EAE or MS, UA accumulates in CNS tissue (11, 35, 36). Presumably, ingested inosine or inosinic acid also could accumulate in CNS tissue under similar circumstances, provided that they can cross the BBB and are metabolized slowly in CNS tissues. To determine whether this may be the case, mice with clinical signs of EAE were fed inosine or inosinic acid and their CNS tissues were assessed for levels of these and UA. As shown in Fig. 6, UA levels in the spinal cord tissues of mice with EAE became elevated but there was no significant effect on endogenous inosine levels and inosinic acid remained essentially undetectable.
Fig 6.

Effects of inosine and inosinic acid administration on the levels of inosine, inosinic acid, and UA in the CNS tissues of PLSJL mice with active EAE. Groups of three to five PLSJL mice with active EAE (mean clinical score, 3.8 ± 0.2) were treated once with 5 mg of K-Ox i.p. and 30 mg of either inosinic acid or inosine in 100 μl saline by gastric intubation. Untreated, healthy PLSJL mice served as donors of normal tissue. At 90 min, 4 h, and 8 h posttreatment, animals were killed and spinal cord tissues were collected for analysis. The levels of inosinic acid, inosine, and UA in the spinal cord tissues were determined by HPLC as described in Experimental Procedures. Data are expressed as the fold increase in tissue levels, determined by dividing the levels detected in test animals by the average normal level. Results are presented as mean ± SD. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 compared with normal levels by one-way ANOVA with post hoc Dunnett's test.
Oral Administration of Inosinic Acid Inhibits the Development of EAE and Promotes Recovery from Clinical Signs of the Disease.
Based on its efficacy in raising serum UA levels in mice, and apparent safety in humans, we have examined the effects of inosinic acid on the development of EAE. As an initial test, we administered inosinic acid i.p. to PLSJL mice immunized with MBP, based on our prior experiments with UA (10–13). With two daily doses of inosinic acid, the onset of clinical signs of EAE was delayed and the severity of the disease was reduced (Fig. 7). Moreover, the 30% mortality seen in vehicle-treated controls also was prevented by inosinic acid treatment (Fig. 7). Our previous studies established that mice with clinical signs of EAE recover rapidly when given UA (10, 11). To assess whether raising serum UA levels by oral administration of a precursor has similar effects, we fed inosinic acid to mice with existing signs of active EAE while prolonging raised UA levels by i.p. administration of K-Ox. As can be seen readily from Fig. 8, such treatment promoted early recovery from clinical signs of EAE, whereas disease severity in mice that received K-Ox alone remained relatively constant. Like K-Ox treatment alone, sham treatment with saline also had no effect on the clinical course of the disease (data not shown).
Fig 7.

Effects of inosinic acid administration on the clinical course of EAE in PLSJL mice. Groups of PLSJL mice (n = 10) were immunized with MBP as detailed in Experimental Procedures. From day 7 postimmunization (arrow in A), animals received two daily i.p. doses of either 10 mg of inosinic acid in saline (INA) or saline alone (Vehicle). Mice were scored twice daily for clinical signs of EAE by independent investigators as detailed (10). Results are presented as percent incidence of disease (A), mean disease score ± SE (B), and percent mortality (C). The clinical scores between inosinic acid and vehicle-treated mice are significantly different (P < 0.001) by the Wilcoxon signed-rank test.
Fig 8.

Administration of inosinic acid plus potassium oxonate promoted recovery from clinical signs of active EAE. PLSJL mice were immunized with MBP as detailed in Experimental Procedures. When mice developed a clinical EAE score between 3 and 4, they were randomized into two groups of six mice each, one group receiving 5 mg of K-Ox in 100 μl of saline i.p. three times daily plus 100 μl of saline by gastric intubation twice a day (K-Ox + Saline), and the second receiving the same regimen of K-Ox plus 30 mg of inosinic acid in 100 μl of saline by gastric intubation twice a day (K-Ox + INA). Mice were scored twice daily for clinical signs of EAE by independent investigators. *, P < 0.05; **, P < 0.01 compared with K-OX + saline group by the Mann–Whitney U test.
Discussion
Raising their normally low serum UA levels protects mice from the development of EAE (10). Moreover, UA treatment of mice with EAE promotes their recovery (10, 11). The therapeutic effects of UA in EAE and other animal models of CNS inflammation (10–13, 22), together with evidence of an inverse correlation between serum UA levels and the incidence of MS, have renewed interest in the specificity of this purine metabolite. Although UA has long been recognized as an antioxidant (2), recent observations suggest that it is not a general antioxidant but a specific inhibitor of radicals generated by the decomposition of peroxynitrite, the product of NO and superoxide, in a biological milieu (4, 5). These UA-sensitive, peroxynitrite-decomposition radicals, possibly NO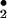 and CO
and CO , therefore are likely to be responsible for the elements of a CNS inflammatory response that have been associated with peroxynitrite (4, 5). Using UA administration to probe the effects of peroxynitrite in CNS inflammation, we have discovered that it contributes not only to CNS tissue damage but also to enhanced BBB permeability and immune cell invasion into the CNS (11–13, 22). Peripheral aspects of the immune response that leads to CNS inflammation, such as the up-regulation of iNOS expression by monocytes, are unchanged by UA treatment (12, 13). We, therefore, have concluded that the peroxynitrite-based radicals inactivated by UA not only cause tissue damage by tyrosine nitration and other chemical reactions but also trigger changes in BBB function that promote cell infiltration into CNS tissues (11–13, 22). Ultimately, these processes result in clinical signs of disease that can be prevented by the administration of UA or a precursor such as inosinic acid.
, therefore are likely to be responsible for the elements of a CNS inflammatory response that have been associated with peroxynitrite (4, 5). Using UA administration to probe the effects of peroxynitrite in CNS inflammation, we have discovered that it contributes not only to CNS tissue damage but also to enhanced BBB permeability and immune cell invasion into the CNS (11–13, 22). Peripheral aspects of the immune response that leads to CNS inflammation, such as the up-regulation of iNOS expression by monocytes, are unchanged by UA treatment (12, 13). We, therefore, have concluded that the peroxynitrite-based radicals inactivated by UA not only cause tissue damage by tyrosine nitration and other chemical reactions but also trigger changes in BBB function that promote cell infiltration into CNS tissues (11–13, 22). Ultimately, these processes result in clinical signs of disease that can be prevented by the administration of UA or a precursor such as inosinic acid.
Inosine administration to humans clearly can be used to raise serum UA levels in MS patients (23, 24). The current findings suggest that inosinic acid is adsorbed in the mouse gut more rapidly/efficiently than inosine but that both can effectively raise serum UA levels. That serum UA levels become elevated for extended periods of time after inosine or inosinic acid administration when K-Ox is present, but inosine levels are increased only transiently, attests to the fact that inosine is metabolized rapidly to UA. As is the case with UA (10–13), inosinic acid treatment not only inhibits the development of EAE but also promotes recovery from existing disease.
Is it possible that increased inosine levels are directly responsible for the therapeutic effects seen in EAE? To significantly reduce the production of several cytokines by LPS-stimulated peritoneal macrophages in vitro, 30-min pretreatment with 10–100 μM inosine followed by a 24-h incubation was necessary (28). Pretreatment for 30 min with 100 mg/kg inosine applied directly into the peritoneum was required to transiently inhibit cytokine production in mice in response to LPS that also was administered i.p. (28). Moreover, the continued presence of inosine evidently was necessary to stimulate axonal growth in vivo (33). In our studies, the activation of monocytes to express iNOS or produce NO in vitro was unaffected by the presence of inosine, inosinic acid, or UA. In addition, unlike UA, neither inosinic acid nor inosine inhibits the peroxynitrite-mediated nitration of tyrosine or oxidation of DHR, reactivities that have been associated with the pathogenesis of EAE (10–13). Because serum inosine levels were elevated only to a peak of ≈100 μM for ≈30 min and no increase in CNS levels were seen in our experiments, we consider it unlikely that inosine is directly therapeutic in EAE. As expected if UA is the functional molecule after the administration of inosinic acid to mice with EAE, serum UA levels were maintained for extended periods of time and elevated levels of UA, but not inosine or inosinic acid, were found in diseased spinal cord tissue. We therefore conclude that the effects of inosinic acid in EAE are likely to be mediated through its metabolism to UA, culminating in the inactivation of peroxynitrite-decomposition radicals at the BBB and in diseased spinal cord tissue.
Perhaps the best direct evidence of peroxynitrite's involvement in the pathogenesis of MS is that active plaques often contain accumulations of iNOS-positive cells and nitrotyrosine (6–9). However, the inverse relationship between serum UA levels and the incidence of MS also attests to the possibility of an association between peroxynitrite and the disease process. Several studies have confirmed our original observation that serum UA levels tend to be lower in MS patients than controls (14–17). We recently have found that this difference extends to identical twins, where individuals with MS have lower serum UA levels than their healthy siblings (16). Analysis of the 1995 Medicare/Medicaid database revealed that the incidence of MS among gout patients was exceptionally low (10). Even if most of the gout patients were male, which is not the case for the predominant age group in the database, and two-thirds of the MS patients were female, the combined incidence of MS and gout should have been ≈10-fold higher than observed. The implication from these observations is that UA may protect against the development of MS, a possibility made more distinct by the demonstration in animal models that UA administration, in addition to preventing a particular element of CNS tissue damage, can inhibit CNS inflammation through protecting against BBB-permeability changes (11, 13, 22). That the therapeutic effects of inosinic acid treatment of EAE are associated with increased serum and tissue levels of UA, rather than inosine, leads us to conclude that the mechanism of action is through inactivation of peroxynitrite decomposition products by UA. We expect that this also will be the case if inosine administration to MS patients proves therapeutic.
Acknowledgments
We thank C. M. Brimer, W. K. Hooper, and K. M. Hooper for technical assistance. This work was supported by Award Number RG 2896A8/5 to D.C.H. from the National Multiple Sclerosis Society and by a grant to the Biotechnology Foundation Laboratories from the Commonwealth of Pennsylvania.
Abbreviations
UA, uric acid/urate
BBB, blood–brain barrier
EAE, experimental allergic encephalomyelitis
iNOS, inducible NO synthase/NOS2
K-Ox, potassium oxonate
LPS, lipopolysaccharide
MS, multiple sclerosis
MBP, myelin basic protein
DHR, dihydrorhodamine
SIN-1, 3-morpholinosydnonimine⋅HCl
References
- 1.Aruoma O. I. & Halliwell, B. (1989) FEBS Lett. 244, 6-80. [DOI] [PubMed] [Google Scholar]
- 2.Whiteman M. & Halliwell, B. (1996) Free Radical Res. 25, 275-283. [DOI] [PubMed] [Google Scholar]
- 3.Santos C. X. C., Anjos, E. I. & Augusto, O. (1999) Arch. Biochem. Biophys. 372, 285-294. [DOI] [PubMed] [Google Scholar]
- 4.Squadrito S. L., Cueto, R., Splenser, A. E., Valavanidis, A., Zhang, H., Uppu, R. M. & Pryor, W. A. (2000) Arch. Biochem. Biophys. 376, 333-337. [DOI] [PubMed] [Google Scholar]
- 5.Whiteman M., Ketsawatsakul, U. & Halliwell, B. (2002) Ann. N.Y. Acad. Sci. 962, 242-259. [DOI] [PubMed] [Google Scholar]
- 6.Bo L., Dawson, T. M., Wesselingh, S., Mork, S., Choi, S., Kong, P. A., Hanley, D. & Trapp, B. D. (1994) Ann. Neurol. 36, 778-786. [DOI] [PubMed] [Google Scholar]
- 7.Bagasra O., Michaels, F. H., Zheng, Y. M., Bobroski, L. E., Spitsin, S. V., Fu, Z. F., Tawadros, R. & Koprowski, H. (1995) Proc. Natl. Acad. Sci. USA 92, 12041-12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hooper D. C., Bagasra, O., Marini, J. C., Zborek, A., Ohnishi, S. T., Kean, R., Champion, J. M., Sarker, A. B., Bobroski, L., Farber, J. L., et al. (1997) Proc. Natl. Acad. Sci. USA 94, 2528-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cross A. H., Maning, P. T., Keeling, R. M., Schmidt, R. E. & Misko, T. P. (1998) J. Neuroimmunol. 88, 45-56. [DOI] [PubMed] [Google Scholar]
- 10.Hooper D. C., Spitsin, S., Kean, R. B., Champion, J. M., Dickson, G. M., Chaudhry, I. & Koprowski, H. (1998) Proc. Natl. Acad. Sci. USA 95, 675-680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hooper D. C., Scott, G. S., Zborek, A., Mikheeva, T., Kean, R. B., Koprowski, H. & Spitsin, S. V. (2000) FASEB J. 14, 691-698. [DOI] [PubMed] [Google Scholar]
- 12.Spitsin S. V., Scott, G. S., Kean, R. B., Mikheeva, T. & Hooper, D. C. (2000) Neurosci. Lett. 292, 137-141. [DOI] [PubMed] [Google Scholar]
- 13.Kean R. B., Spitsin, S. V., Mikheeva, T., Scott, G. S. & Hooper, D. C. (2000) J. Immunol. 165, 6511-6518. [DOI] [PubMed] [Google Scholar]
- 14.Sotgiu S., Vanoli, M., Pugliatti, G., Fresu, V., Agnetti, I. & Rostati, A. G. (1999) J. Neuroimmunol. 101, A53. [Google Scholar]
- 15.Drulovic J., Dujmovic, I., Stojsavljevic, N., Mesaros, S., Miljkovic, D., Peric, V., Dragutinovic, G., Marinkoviv, J., Levic, Z. & Stojkovic, M. M. (2001) J. Neurol. 248, 121-126. [DOI] [PubMed] [Google Scholar]
- 16.Spitsin S. V., Hooper, D. C., Mikheeva, T. & Koprowski, H. (2001) Multiple Sclerosis 7, 165-166. [DOI] [PubMed] [Google Scholar]
- 17.Toncev G., Milicic, B., Toncev, S. & Samardzic, G. (2002) Eur. J. Neurol. 9, 221-226. [DOI] [PubMed] [Google Scholar]
- 18.Ischiropoulos H., Zhu, L. & Beckman, J. S. (1992) Arch. Biochem. Biophys. 298, 446-451. [DOI] [PubMed] [Google Scholar]
- 19.Varela-Eschavarria A., Montes de Oca-Luna, R. & Barrera-Saldana, H. A. (1988) FASEB J. 2, 3092-3096. [DOI] [PubMed] [Google Scholar]
- 20.Wu X. W., Muzny, D. M., Lee, C. C. & Catskey, C. T. (1992) J. Mol. Evol. 34, 78-84. [DOI] [PubMed] [Google Scholar]
- 21.Yeldandi A. V., Yeldandi, V., Kumar, S., Murthy, C. V., Wang, X. D., Alvares, K., Rao, M. S. & Reddy, J. K. (1991) Gene 109, 281-284. [DOI] [PubMed] [Google Scholar]
- 22.Hooper D. C., Kean, R. B., Scott, G. S., Spitsin, S. V., Morimoto, K., Bette, M., Röhrenbeck, A. M., Dietzschold, B. & Weihe, E. (2001) J. Immunol. 167, 3470-3477. [DOI] [PubMed] [Google Scholar]
- 23.Koprowski H., Spitsin, S. V. & Hooper, D. C. (2001) Ann. Neurol. 49, 139. [DOI] [PubMed] [Google Scholar]
- 24.Spitsin S., Hooper, D. C., Leist, T., Streletz, L. J., Mikheeva, T. & Koprowski, H. (2001) Multiple Sclerosis 7, 313-319. [DOI] [PubMed] [Google Scholar]
- 25.Williams M. H., Kreider, R. B., Hunter, D. W., Somma, C. T., Shall, L. M., Woodhouse, M. L. & Rokitski, L. (1990) Med. Sci. Sports Exercise 22, 517-522. [PubMed] [Google Scholar]
- 26.Starling R. D., Trappe, T. A., Short, K. R., Sheffield-Moore, M., Jozsi, A. C., Fink, W. J. & Costill, D. L. (1996) Med. Sci. Sports Exercise 28, 1193-1198. [DOI] [PubMed] [Google Scholar]
- 27.Fuke S. & Konosu, S. (1991) Physiol. Behav. 49, 863-868. [DOI] [PubMed] [Google Scholar]
- 28.Haskó G., Kuhel, D. G., Németh, Z. H., Mabley, J. G., Stachlewitz, R. F., Virág, L., Lohinai, Z., Southan, G. J., Salzman, A. L. & Szabó, C. (2000) J. Immunol. 164, 1013-1019. [DOI] [PubMed] [Google Scholar]
- 29.Liaudet L., Mabley, J. G., Soriano, F. G., Pacher, P., Marton, A., Haskó, G. & Szabó, C. (2001) Am. J. Respir. Crit. Care Med. 164, 1213-1220. [DOI] [PubMed] [Google Scholar]
- 30.Marton A., Pacher, P., Murthy, K. G., Nemeth, Z. H., Haskó, G. & Szabó, C. (2001) Int. J. Mol. Med. 8, 617-621. [PubMed] [Google Scholar]
- 31.Liaudet L., Mabley, J. G., Pacher, P., Virag, L., Soriano, F. G., Marton, A., Haskó, G., Deitch, E. A. & Szabó, C. (2002) Ann. Surg. 235, 568-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benowitz L. I., Goldberg, D. E., Madsen, J. R., Soni, D. & Irwin, N. (1999) Proc. Natl. Acad. Sci. USA 96, 113486-113490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen P., Goldberg, D. E., Kolb, B., Lanser, M. & Benowitz, L. I. (2002) Proc. Natl. Acad. Sci. USA 99, 9031-9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mangoff S. C. & Milner, J. A. (1978) Proc. Soc. Exp. Biol. Med. 157, 110-115. [DOI] [PubMed] [Google Scholar]
- 35.Honegger C. G., Krenger, W. & Langemann, H. (1986) Neurosci. Lett. 69, 109-114. [DOI] [PubMed] [Google Scholar]
- 36.Langemann H., Kabiersch, A. & Newcombe, J. (1992) Eur. Neurol. 32, 248-252. [DOI] [PubMed] [Google Scholar]