

Abstract
Replacement of the p53 tumor suppressor gene is a rational approach to the management of malignant gliomas because p53 is frequently mutated or inactivated in these cancers. Major weaknesses of this approach are that malignant gliomas are mixtures of cells with wild-type and mutant p53 and that tumor cells exhibiting wild-type p53 are resistant to p53 gene transfer. An effective alternative is needed to overcome these difficulties. Recently, p53 upregulated modulator of apoptosis (PUMA) was identified as a p53-inducible pro-apoptotic molecule. Our purpose was to elucidate a role for PUMA in p53 gene therapy and to investigate whether PUMA is an efficient substitute for p53 in cancer therapy. We demonstrated that PUMA was upregulated in mutant p53 malignant glioma cells (U373-MG and T98G) undergoing apoptosis but was not upregulated in apoptosis-resistant wild-type p53 malignant glioma cells (U87-MG and D54) following adenoviral transfer of p53. Overexpression of PUMA resulted in massive apoptosis associated with mitochondrial damage and caspase-3 activation in all tumor cells tested. Use of the human telomerase reverse transcriptase (hTERT) promoter system induced apoptosis only in malignant glioma cells with telomerase activity, while sparing normal cells lacking telomerase. The ability of PUMA to induce apoptosis was greater than that of caspase-6 or caspase-8 transfer using the same system. Moreover, exogenous expression of PUMA under the hTERT promoter system significantly suppressed the growth of subcutaneous U87-MG tumors in nude mice and did not induce apoptosis in surrounding non-tumor tissues. These results indicate that PUMA, which is regulated under a tumor-specific expression system such as the hTERT promoter, may be better than p53 as a therapeutic tool for malignant gliomas.
Keywords: PUMA, p53, Apoptosis, hTERT, Glioma
OVERVIEW SUMMARY
Recently, p53 upregulated modulator of apoptosis (PUMA) was identified as a p53-inducible pro-apoptotic molecule. In the present study, we demonstrated that PUMA was up-regulated in mutant p53 malignant glioma cells undergoing apoptosis but was not up-regulated in apoptosis-resistant wild-type p53 malignant glioma cells following transfer of p53. Exogenous expression of PUMA resulted in massive apoptosis associated with mitochondrial damage and caspase-3 activation in both mutant and wild-type p53 malignant glioma cells. Using the human telomerase reverse transcriptase (hTERT) promoter system, PUMA caused apoptosis only in malignant glioma cells, but not in normal cells. Moreover, the in vivo therapeutic efficacy and absence of side effects by delivery of PUMA under the hTERT promoter system was demonstrated in subcutaneous tumors in nude mice. These findings suggest that PUMA under the hTERT promoter is substituted for p53 as a therapeutic tool for malignant gliomas.
INTRODUCTION
Apoptosis, also called programmed cell death, is a genetically encoded program that disposes of unwanted cells (Wyllie et al., 1980). Disruptions of this pathway have been implicated as a cause of cancer (Bold et al., 1997). Therefore, genetic restoration of the apoptotic pathway or introduction of pro-apoptotic molecules is an attractive approach for treating tumors, including malignant gliomas.
Treatment of cancers with exogenous expression of the wild-type p53 tumor suppressor gene through activation of an apoptotic pathway has been tested extensively in preclinical and clinical studies (Roth and Cristiano, 1997; Vousden and Lu, 2002). Numerous in vitro and in vivo studies have demonstrated the validity of using an adenovirus carrying wild-type p53 (Ad-p53) to induce apoptosis and thereby kill tumors. Several phase I/II clinical trials in patients with advanced cancers have demonstrated low toxicity, successful gene transfer, and evidence of tumor regression (Clayman et al., 1998; Swisher et al., 1999; Lang et al., 2003). Experiments using Ad-p53 as a means of cancer treatment, however, have revealed the limitation of this treatment: Tumor cells with wild-type p53 are highly resistant to the effect of Ad-p53 both in culture and in tumor xenograft models (Gomez-Manzano et al., 1996; Gomez-Manzano et al., 1997; Komata et al., 2000). These different responses to Ad-p53 would be problematic in the treatment of malignant gliomas in particular because the cells are a mixture of wild-type and mutant p53 (Lang et al., 1999). As a result, we examined a novel approach that might overcome this difficulty in using Ad-p53.
p53 up-regulated modulator of apoptosis (PUMA), a Bcl-2 homology 3 (BH3)-only pro-apoptotic Bcl-2 family member, was recently identified as a molecule that directly mediates p53-associated apoptosis (Yu et al., 2001; Nakano and Vousden, 2001; Han et al., 2001; Yu et al., 2003). PUMA protein associates with mitochondria and induces apoptosis much earlier than the apoptosis that results from exogenous expression of p53 when it is overexpressed in various cell lines. Furthermore, PUMA mediates not only p53-dependent apoptosis but also p53-independent apoptotic pathways (Jeffers et al., 2003; Villunger et al., 2003). These studies imply that PUMA is a principal mediator of apoptotic cell death.
On the basis of these findings, we speculated that PUMA might be a better therapeutic tool for cancer than is p53. To test our hypothesis, we treated mutant p53 and wild-type p53 malignant glioma cells with adenoviruses expressing p53 or PUMA (Ad-p53 or Ad-PUMA). We found that only mutant p53 tumor cells underwent apoptosis following Ad-p53 infection, whereas Ad-PUMA induced massive apoptosis in tumor cells regardless of their p53 status. However, our observation that overexpression of PUMA also induced apoptosis in normal cells suggested the possibility of PUMA-induced adverse effects.
The use of the human telomerase reverse transcriptase (hTERT) promoter as a tumor-targeting system (Koga et al., 2000; Komata et al., 2001) restricted the PUMA-induced apoptosis to tumor cells. Our findings from in vitro and in vivo models suggest that tumor-specific transfer of PUMA is a promising approach to the management of malignant gliomas and that it may be further applicable to a variety of cancers. To the best of our knowledge, this is the first report to demonstrate the therapeutic efficacy of PUMA in in vitro and in vivo models of cancer.
MATERIALS AND METHODS
Cells
Human malignant glioma U373-MG, U87-MG, and T98G cells and human normal fibroblasts MRC5 were purchased from American Tissue Culture Collection (Rockville, MD). Human malignant glioma D54 cells were a kind gift from Dr. Frederick Lang (M. D. Anderson Cancer Center). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen), 4 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Reagents
Hoechst 33258 was purchased from Sigma Chemical Co. (St. Louis, MO). Antibodies against caspase-6, caspase-8, Bax, hemagglutinin (HA), cytochrome c, and heat shock protein (HSP) 60 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against PUMA and actin were purchased from Abcam (Cambridge, MA) and Sigma.
Transient transfection
The preparation and infection of adenoviruses carrying p53 or green fluorescent protein (GFP) (Ad-p53 or Ad-GFP) has been described previously (Komata et al., 2000). Adenoviruses expressing wild-type PUMA (Ad-PUMA) or mutant PUMA with a deletion of the BH3 domain (Ad-PUMAΔBH3) fused to GFP (Yu et al., 2003) were generous gifts from Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD). HA-PUMA (Yu et al., 2001) also was a kind gift from Dr. Vogelstein. To construct the PUMA expression vector under the hTERT promoter, we used the HA-PUMA and the hTERT promoter-expression vector containing luciferase (pGL3-378) as described previously (Koga et al., 2000; Komata et al., 2001). First, the HA-PUMA region was removed from HA-PUMA vector using NcoI and SalI. The fragment was exchanged with luciferase region of pGL3-378 construct at NcoI/SalI site and designated as the hTERT-PUMA expression vector (hTERT/HA-PUMA). The cells were transiently transfected with HA-PUMA or hTERT/HA-PUMA by using FuGENE 6™ transfection reagent (Roche Applied Science, Indianapolis, IN) as described previously (Koga et al., 2000; Komata et al., 2001). The plasmid expressing luciferase under SV40 enhancer/promoter (PGL3-control) was used as a reporter gene when hTERT/HA-PUMA was transfected into telomerase-negative MRC5 cells.
Apoptosis assay
To determine whether tumor cells undergo apoptosis after treatment with Ad-p53, Ad-PUMA, HA-PUMA, or hTERT/HA-PUMA, we stained nuclei with Hoechst 33258 to detect chromatin condensation or nuclear fragmentation characteristic of apoptosis, as described previously (Kondo et al., 1995). Regarding the adenoviral infection, tumor cells were infected with Ad-p53, Ad-PUMA, or Ad-PUMAΔBH3 at a multiplicity of infection (MOI) of 5 to 50. To detect cells infected with Ad-p53, Ad-GFP was co-infected at the same MOI. After a 24-h incubation, cells were washed with PBS, fixed with 4% paraformaldehyde for 15 min, and stained with 1 μg/ml Hoechst 33258 for 15 min at room temperature. We visualized the nuclear morphology of the cells under a fluorescence microscope. The apoptotic index was determined as the percentage of apoptotic cells among 200 GFP-positive cells. After the tumor cells were transfected with the HA-PUMA or hTERT/HA-PUMA construct, we immunohistochemically stained them with anti-HA antibody prior to Hoechst staining. Again, the apoptotic index was determined as the percentage of apoptotic cells among 200 HA-positive cells. To compare the ability of PUMA and other apoptosis-inducing caspase genes to induce apoptosis, the expression vectors of constitutively active caspase-6 (rev-caspase-6) and caspase-8 under hTERT promoter (hTERT/rev-caspase-6 and hTERT/caspase-8) were transiently transfected as described previously (Komata et al., 2001; Komata et al., 2002). The apoptotic index was determined as the percentage of apoptotic cells among 200 caspase-6- or caspase-8-positive cells.
Luciferase assay
The transcriptional activities of PUMA in U373-MG and U87-MG cells infected with Ad-p53 were determined by using luciferase reporter plasmids as described previously (Komata et al., 2001). Tumor cells were plated at a density of 1.0 × 105 cells/ml 1 day prior to transfection of the luciferase reporter plasmids. Twenty-four h after transfection, the cells were infected with Ad-p53 and/or Ad-GFP at an MOI of 20. Luciferase activity was assessed 24 h after transfection with the luciferase kit (Promega, Madison, WI). The following plasmids were used: the SV40 enhancer/promoter (PGL3-control) for a positive control and 4×BS2WT-Luc and 4×BS2MUT-Luc (from Dr. Vogelstein) containing four copies of p53 binding sites in wild-type or mutant form, which are the major p53-responsive elements in the PUMA promoter (Yu et al., 2001). The luciferase activity of the PGL3-control plasmid in each cell line was considered as 100%.
Immunoblotting assay
Total proteins for PUMA detection were prepared as described previously (Kondo et al., 1995). To prepare cytosolic proteins for cytochrome c release, we washed treated or untreated cells (2 × 106) and incubated them for 30 min on ice in 300 μl lysis buffer (68 mM sucrose, 200 mM mannitol, 50 mM KCl, 1 mM EGTA, 1 mM EDTA, 1 mM DTT, and protease inhibitor). Cells were then lysed with dounce homogenizer and centrifuged at 4ºC (800 g). The supernatant was centrifuged at 13,000 g for 10 min. The supernatant was then stored at −80°C as cytosolic extract, and the concentration of clarified lysate was measured by the Bio-Rad Protein Assay (Bio-Rad Laboratories, Richmond, CA). Equal amounts of proteins were separated by electrophoresis on an 18% polyacrylamide gel in sodium dodecyl sulfate and thereafter subjected to electrotransfer to nitrocellulose membranes. Membranes were subjected to immunoblotting using the ECL detection system (Amersham, Arlington Heights, IL) according to the manufacturer’s instructions, as described previously (Kondo et al., 1995).
Cell viability assay
The cytotoxic effect of PUMA expression on tumor cells was determined by using Cell Proliferation Reagent WST-1 (Roche Applied Science, Indianapolis, IN). Cells were seeded at 1 ×104 cells/well in 96-well plate and incubated at 37 °C overnight. After infection with Ad-PUMA or Ad-PUMAΔBH3 at an MOI of 5 to 50 for 24 h, the cells were exposed to 10 μl of WST-1 reagent for 1 h at 37°C. The absorbance at 450 nm was measured in a microplate-reader. The viability of untreated cells was considered to be 100%.
Subcellular localization
Cells were grown overnight on chamber slides and after the indicated treatments, fixed in 4% paraformaldehyde for 15 min, and blocked with 3% goat serum for 30 min at room temperature. After the slides were washed, we incubated them with anti-Bax antibody diluted 1:1000 and anti-HSP60 diluted 1:500 with 3% goat serum in PBS at 4°C overnight. The slides were washed and then incubated with Alexa-conjugated anti–mouse antibody (A-21154; Molecular Probes, Inc., Eugene, OR) and Alexa-conjugated anti–rabbit antibody (A-11046; Molecular Probes) diluted 1:1000 in PBS for 30 min at room temperature. After three additional washes with PBS, the slides were mounted and analyzed by fluorescence microscopy. Cells with a punctate distribution of Bax and co-localization with HSP60 were scored as translocated. Bax-translocated cells were counted among 200 cells sufficiently stained with anti-Bax antibody.
Flow cytometric analysis of mitochondrial membrane potential
Rhodamine 123 (Molecular Probes), a cationic voltage-sensitive probe, was used to detect changes in mitochondrial membrane potential by using flow cytometry as described previously (Daido et al., 2004). Nonadherent and trypsinized adherent cells were collected, washed twice with PBS, and stained with 1 μM rhodamine 123 in PBS in the dark at 37 °C for 1 h. Samples were washed and re-suspended with PBS and then analyzed with the FACScan using CellQuest software.
Caspase-3 activity assay
Activity of caspase-3 was measured by using the Cleavalite™ caspase-3 activity assay kit (Chemicon International, Temecula, CA) as described previously (Kanzawa et al., 2004). Treated or untreated cells were trypsinized and washed with PBS. The cell pellets were incubated in the kit’s cell lysis buffer on ice for 10 min and centrifuged at 10,000 g for 5 min. Cell lysates were incubated at 37ºC for 2 h with a bioluminescent substrate for caspase-3, which has a Renilla luciferase containing the caspase-3 cleavage site, DEVD. Upon cleavage, it exhibits decreased bioluminescence.
Effect of PUMA expression in vivo
U87-MG cells (1.0 ×106 cells in 25 μl of serum-free DMEM) were inoculated subcutaneously into the right flank of 8 – 12-week-old female nude mice, as described previously (Koga et al., 2000; Komata et al., 2001). Tumor growth was monitored with calipers every other day. Tumor volume was calculated as (L ×W2)/2, where L is length (mm) and W is width (mm). When the tumors reached a mean volume of 30 – 50 mm3, we initiated treatment (day 1). 10 μg of the HA-PUMA, hTERT/HA-PUMA or hTERT/luciferase construct in the presence of 2 μl Lipofectamine 2000 (Invitrogen) dissolved in 20 μl sterile PBS was directly injected into the tumor every day for 5 days. To determine the effect of the treatment on non-tumor tissues, subcutaneous U87-MG tumors together with skin and surrounding tissues were collected from the mice next day (day 6) after the last treatment and frozen rapidly, and 15-μm cryosections were made for histological studies. Sections from the tumors with non-tumor tissues were used for double staining of the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) analysis using the ApopTag® Apoptosis Detection Kit (Invitrogen) and immunohistochemical assay using the anti-HA antibody. As additional experiments, we treated subcutaneous U87-MG tumors with HA-PUMA, hTERT/HA-PUMA or hTERT/luciferase for 5 days as described above and further examined if the tumor grew back during 2–3 additional weeks without additional treatment. Because some mice treated with hTERT/luciferase were moribund because of tumor on day 21, all mice were sacrificed then to compare the effectiveness of each treatment. All animal studies were performed in the veterinary facilities of the University of Texas M. D. Anderson Cancer Center in accordance with institutional, state, and federal laws and ethics guidelines for experimental animal care.
Statistical analysis
All the experiments were repeated at least three times. The data were expressed as mean ± standard deviation (SD). Statistical analysis was performed with the Student’s t test (two-tailed). The criterion for statistical significance was taken as p<0.05.
RESULTS
Effect of Ad-p53 on tumor cells expressing mutant or wild-type p53
To ascertain the cell killing effect of Ad-p53 on malignant glioma cell lines with different p53 status, U373-MG (mutant p53) and U87-MG cells (wild-type p53) were infected with Ad-p53 and/or Ad-GFP at an MOI of 5 to 20 for 24 h. As shown in Fig. 1A, Hoechst 33258 staining revealed that most of the U373-MG cells treated with Ad-p53 plus Ad-GFP at 20 MOI underwent apoptosis, whereas no significant apoptosis was detectable in Ad-p53 plus Ad-GFP-infected U87-MG cells. To quantify the incidence of apoptosis in U373-MG or U87-MG cells infected with Ad-p53 and/or Ad-GFP, we determined the percentage of apoptotic cells out of 200 GFP-positive cells. As shown in Fig. 1B, in U373-MG cells expressing mutant p53, apoptosis was induced in 20.6%, 25.8%, and 47.4% of cells, which were infected with Ad-p53 plus Ad-GFP at 5, 10, or 20 MOI, respectively. In contrast, the induction of apoptosis was less than 10% in wild-type p53-expressing U87-MG cells infected with Ad-p53 plus Ad-GFP even at 20 MOI. The percentage of apoptosis in U373-MG and U87-MG cells induced by Ad-GFP alone was less than 5% (Fig. 1B).
Fig. 1.
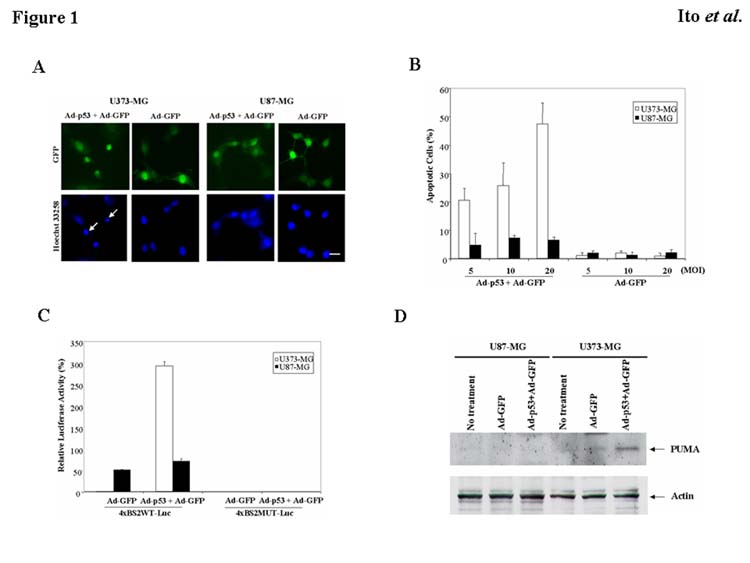
Induction of apoptosis in malignant glioma cells with different p53 status by Ad-p53. A: U373-MG (mutant p53) or U87-MG cells (wild-type p53) were infected with Ad-p53 and/or Ad-GFP at an MOI of 20. After 24 h, tumor cells were fixed and stained with Hoechst 33258. Arrows indicate representative apoptotic nuclei. Bars, 10 μm. B: Percentage of apoptotic cells in U373-MG or U87-MG cells infected with Ad-p53 and/or Ad-GFP at an MOI of 5, 10, or 20 for 24 h. Data shown are the mean ± SD of apoptotic cells among 200 GFP-positive cells chosen at random from three different areas. C: Transcriptional activity of the PUMA promoter. U373-MG or U87-MG cells were infected with Ad-p53 and/or Ad-GFP at an MOI of 20 for 24 h after reporters (4×BS2WT-Luc or 4×BS2MUT-Luc) were transfected. Luciferase activity in each cell line was normalized, with the represented value indicating a percentage of the positive control plasmid (PGL3-control) in each cell line. Data shown are the mean ± SD from three independent experiments. D: Increase in PUMA protein expression by Ad-p53. U373-MG or U87-MG cells were infected with Ad-GFP and/or Ad-p53 at an MOI of 20 for 24 h. Untreated cells were included as a control. Cell lysates were used for Western blots to assess the expression of PUMA. Actin serves as a loading control.
The different responses of U373-MG and U87-MG cells to Ad-p53 were consistent with those seen in previous investigations (Gomez-Manzano et al., 1996; Gomez-Manzano et al., 1997; Komata et al., 2000). Because PUMA is activated by p53 in the apoptotic pathway (Yu et al., 2001; Nakano and Vousden, 2001; Han et al., 2001; Yu et al., 2003), we speculated that PUMA might be activated in U373-MG cells but not in U87-MG cells after exposure to Ad-p53. To test this hypothesis, we transfected U373-MG and U87-MG cells with the wild-type or mutant PUMA promoter construct (4×BS2WT-Luc or 4×BS2MUT-Luc) prior to infection with Ad-GFP and/or Ad-p53. As shown in Fig. 1C, the transcriptional activity of PUMA was dramatically activated in U373-MG cells by an exogenous p53 expression compared to the control treated with Ad-GFP alone. On the other hand, U87-MG cells showed endogenous activity of PUMA transcription to a certain extent, which was little activated by Ad-p53 infection. Then we performed the Western blot analysis to determine whether PUMA protein expression was increased in U373-MG cells, but not in U87-MG cells by Ad-p53 infection. Fig. 1D revealed that treatment with Ad-p53 plus Ad-GFP increased the expression of PUMA in U373-MG cells, whereas PUMA expression was undetectable in U87-MG cells infected with Ad-GFP and/or Ad-p53. Similar responses to Ad-p53 were observed in T98G (mutant p53) and D54 cells (wild-type p53) (data not shown). These results indicate that Ad-p53 up-regulated PUMA in mutant p53 tumor cells, causing apoptosis, but not in tumor cells expressing wild-type p53.
Effect of PUMA overexpression on malignant glioma cells
To determine the antitumor effect of ectopic expression of PUMA on malignant glioma cell lines with different p53 status, we infected U373-MG and U87-MG cells with Ad-PUMA or Ad-PUMAΔBH3 at an MOI of 5 – 20 for 24 h and then measured the incidence of apoptosis using Hoechst 33258 staining. As shown in Fig. 2A, infection of Ad-PUMA at 20 MOI induced substantial apoptosis in both U373-MG and U87-MG cells, whereas little apoptotic cells were detectable in these cells infected with Ad-PUMAΔBH3. To quantify the incidence of apoptosis in tumor cells treated with Ad-PUMA or Ad-PUMAΔBH3, we determined the percentage of apoptotic cells out of 200 GFP-positive cells. As shown in Fig. 2B, apoptotic cells were observed in 80.5% of U373-MG cells and in 49.2% of U87-MG cells, all of which were infected at 20 MOI of Ad-PUMA. Apoptosis was induced in U373-MG and U87-MG cells by Ad-PUMA in a dose-dependent manner, although U373-MG cells were more sensitive to Ad-PUMA than were U87-MG cells. Interestingly, the incidence of apoptosis even in Ad-p53–sensitive U373-MG cells was significantly higher by Ad-PUMA than by Ad-p53 (p<0.01), suggesting that Ad-PUMA is superior to Ad-p53 in its ability to induce apoptosis.
Fig. 2.
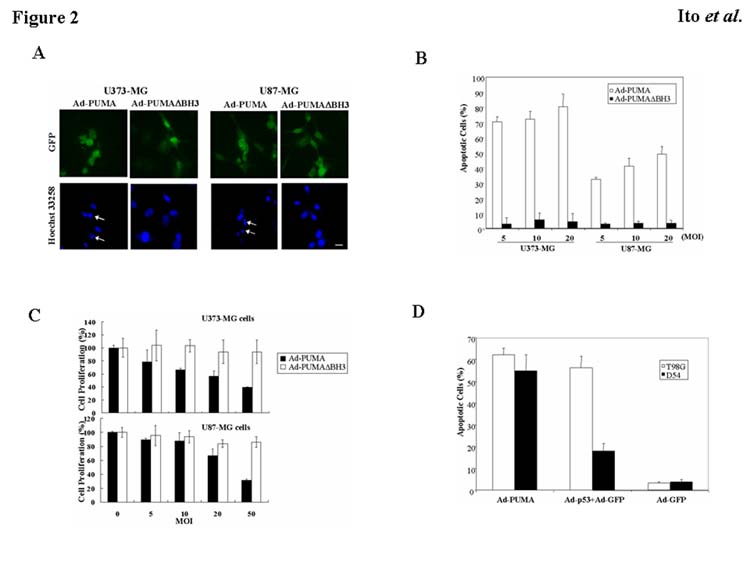
Antitumor effect of PUMA overexpression on malignant glioma cells with different p53 status. A: U373-MG (mutant p53) or U87-MG cells (wild-type p53) were infected with Ad-PUMA or Ad-PUMAΔBH3 at 20 MOI. After 24 h, tumor cells were fixed and stained with Hoechst 33258. Arrows indicate representative apoptotic nuclei. Bars, 10 μm. B: Percentage of apoptotic cells in U373-MG or U87-MG cells by Ad-PUMA or Ad-PUMAΔBH3 at an MOI of 5, 10, or 20 for 24 h. Data shown are the mean ± SD of apoptotic cells among 200 GFP-positive cells chosen at random from three different areas. C: Effect of PUMA overexpression on cell viability. U373-MG and U87-MG cells were seeded at 1 × 104 cells/well in 96-well plates and incubated overnight at 37°C. The cells were infected with Ad-PUMA or Ad-PUMAΔBH3 at an MOI of 5, 10, 20, or 50 for 24 h. Cell proliferation was assessed by a WST-1 assay. The proliferation of the uninfected cells was considered to be 100% for each cell line. Results shown are the mean ± SD of three independent experiments. D: Percentage of apoptotic cells in T98G (mutant p53) or D54 cells (wild-type p53) by Ad-PUMA, Ad-p53 and/or Ad-GFP at an MOI of 20 for 24 h. Data shown are the mean ± SD of apoptotic cells among 200 GFP-positive cells chosen at random from three different areas.
To test the effect of PUMA overexpression on cell viability, we infected U373-MG and U87-MG cells with Ad-PUMA or Ad-PUMAΔBH3 at an MOI of 5 to 50 for 24 h and performed a cell proliferation assay. As shown in Fig. 2C, the proliferation of U373-MG and U87-MG cells were inhibited by Ad-PUMA in a dose-dependent manner, whereas Ad-PUMAΔBH3 had little or no cytotoxic effect on tumor cells. To determine whether the results obtained from U373-MG and U87-MG cells can be generalized to other malignant glioma cells, we infected T98G (mutant p53) and D54 (wild-type p53) malignant glioma cells with Ad-PUMA for 24 h. A similar tendency in the induction of apoptosis by Ad-PUMA or Ad-p53 was observed in T98G and D54 cells (Fig. 2D). The incidence of apoptosis in cells infected with Ad-PUMA at 20 MOI was 62.3% of T98G cells and 54.8% of D54 cells. These results indicate that overexpression of PUMA caused apoptosis in both mutant p53 and wild-type p53 malignant glioma cells.
Mechanisms of PUMA-induced apoptosis
Bax translocation from cytosol to mitochondria is a critical step in the induction of apoptosis (Hsu et al., 1997; Wolter et al., 1997). Bax relates to apoptosis by interacting with anti-apoptotic molecules such as Bcl-2 and thus controlling the release of mitochondrial proteins such as cytochrome c (Rosse et al., 1998; Jurgensmeier et al., 1998). Using immunohistochemical double-staining of Bax and mitochondrial heat shock protein (HSP) 60, we investigated whether mitochondrial translocation of Bax is observed in Ad-PUMA–infected tumor cells. As shown in Fig. 3A, Bax was diffusely distributed throughout the cytosol in U373-MG cells infected with Ad-PUMAΔBH3. On the other hand, Bax moved to a punctate distribution, co-localizing with HSP60-positive mitochondria 24 h after exposure of U373-MG cells to Ad-PUMA (Fig. 3A). As shown in Fig. 3B, immunohistochemical staining revealed that treatment with Ad-PUMA induced Bax translocation in 38% of U373-MG cells and in 25% of U87-MG cells. These results indicate that overexpression of PUMA caused translocation of Bax from cytosol to mitochondria in malignant glioma cell lines regardless of their p53 status. This led to initiation of an apoptotic pathway, a development that is supported by recent studies (Yu et al., 2003; Melino et al., 2003).
Fig. 3.
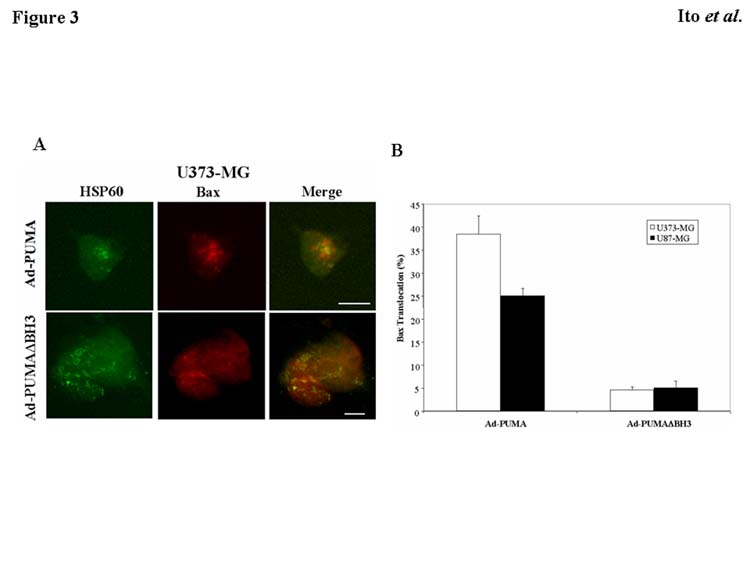
Bax translocation to the mitochondria by PUMA expression. A: Induction of Bax translocation from cytosol to mitochondria. U373-MG cells were infected with Ad-PUMA or Ad-PUMAΔBH3 at 20 MOI for 24 h. HSP60 and Bax protein were detected by immunofluorescence assay using anti-HSP60 antibody (green fluorescence) and anti-Bax antibody (red fluorescence), respectively. Co-localization of mitochondria and Bax is indicated as orange in the merged picture. Bars, 20 μm. B: Percentage of tumor cells exhibiting Bax translocation after exposure to Ad-PUMA or Ad-PUMAΔBH3 at an MOI of 20 for 24 h. Data shown are the mean ± SD of Bax-translocated cells among more than 100 cells chosen at random from three different areas.
Because Bax translocation to the mitochondria correlates with mitochondrial membrane depolarization and the release of cytochrome c (Dewson et al., 2003), we sought to determine whether Ad-PUMA damages mitochondrial membrane integrity and releases cytochrome c into the cytosol. Fig. 4A shows that treatment with Ad-PUMA for 24 h induced loss of membrane potential in U373-MG and U87-MG cells, whereas no significant change was seen in these tumor cells with no treatment and with Ad-PUMAΔBH3 infection. Furthermore, the amount of cytochrome c in the cytoplasm increased in both U373-MG and U87-MG cell lines after exposure to Ad-PUMA (Fig. 4B), indicating that cytochrome c was released into the cytosol by Ad-PUMA. Then we examined whether PUMA expression activates an apoptosis-executioner, caspase-3, by using a caspase-3 activity assay. Infection of Ad-PUMA at an MOI of 20 decreased bioluminescence to 58% and 49% of the control in U373-MG and U87-MG, respectively (Fig. 4C). This finding demonstrates that caspase-3 is activated in both types of tumor cells after exposure to Ad-PUMA. These results indicate that mitochondrial translocation of Bax by PUMA expression causes damage to mitochondrial membrane integrity, release of cytochrome c, and activation of caspase-3. This series of events results in the execution of apoptosis in both mutant p53 and wild-type p53 malignant glioma cells.
Fig. 4.
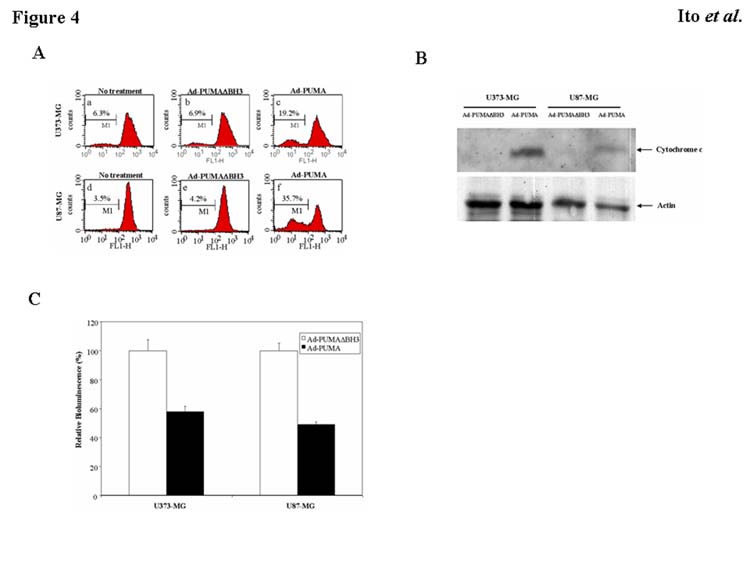
Mechanisms of PUMA-induced apoptosis. A: Disruption of mitochondrial membrane potential in malignant glioma cells by overexpression of PUMA. Rhodamine 123 was used to determine changes in mitochondrial membrane potential using FACS analysis. After treatment with Ad-PUMA or Ad-PUMAΔBH3 at an MOI of 20 for 24 h, attached and detached U373-MG and U87-MG cells were collected and stained with rhodamine 123. B: Release of cytochrome c into cytosol by Ad-PUMA. U373-MG and U87-MG cells were infected with Ad-PUMA or Ad-PUMAΔBH3 at 20 MOI for 24 h. Cytosolic extracts were used for Western blots to assess release of cytochrome c. Actin serves as a loading control. C: Activation of caspase-3 by Ad-PUMA. Activity of caspase-3 in U373-MG and U87-MG cells infected with Ad-PUMA or Ad-PUMAΔBH3 at an MOI of 20 for 24 h was measured with the CleavaLite™ Caspase-3 activity assay kit. Results shown are the mean ± SD of three independent experiments.
Tumor-specific apoptosis by the hTERT/HA-PUMA construct
We recognized that exogenous expression of PUMA might induce apoptosis not only in cancer cells, but also in normal cells. To investigate this possibility, we infected normal fibroblasts MRC5 with Ad-PUMA. Approximately 50% of the cells underwent apoptosis (data not shown), suggesting that PUMA might have a significant adverse effect when used for cancer therapy. Therefore, we decided to adapt the tumor-targeting system that we had recently demonstrated by using the hTERT promoter system (Koga et al., 2000; Komata et al., 2001) to construct the expression vector of PUMA under the hTERT promoter (hTERT/HA-PUMA). To determine whether hTERT/HA-PUMA–transfected cells undergo apoptosis, we performed immunohistochemical staining using anti-HA antibody followed by Hoechst 33258 staining. As predicted from the results with Ad-PUMA, transfection with the HA-PUMA construct induced apoptosis in both normal fibroblast MRC5 cells and malignant glioma U373-MG and U87-MG cells (Fig. 5A). In contrast, U373-MG and U87-MG cells transfected with the hTERT/HA-PUMA construct underwent apoptosis, whereas little apoptotic cells were detected in luciferase-positive MRC5 cells co-transfected with the hTERT/HA-PUMA and the plasmid expressing luciferase under SV40 enhancer/promoter (PGL3-control) constructs. To quantify the incidence of apoptosis in cells transfected with the HA-PUMA or hTERT/HA-PUMA construct, we determined the percentage of apoptotic cells out of 200 HA- or luciferase-positive cells. As shown in Fig. 5B, the HA-PUMA construct induced apoptosis in 75% of U373-MG cells, 66% of U87-MG cells, and 51% of MRC5 cells, respectively. However, the hTERT/HA-PUMA construct induced apoptosis selectively in tumor cells (59% of U373-MG cells and 64% of U87-MG cells). Only 9% of MRC5 cells expressing luciferase by pGL3-control underwent apoptosis. To compare the ability of PUMA and other apoptosis-inducing molecules caspases to induce apoptosis, cells were transiently transfected with hTERT/rev-caspase-6 or hTERT/caspase-8 construct as describe previously (Komata et al., 2001; Komata et al., 2002). As shown in Fig. 5B, the ability of PUMA to induce apoptosis in malignant glioma cells was significantly greater than that of rev-caspase-6 or caspase-8 (P<0.001, respectively). These results indicate that under the hTERT promoter, PUMA-induced apoptosis was restricted to malignant glioma cells, and normal cells were spared. Additionally, PUMA was superior to caspases as well as p53 regarding the ability to induce apoptosis.
Fig. 5.
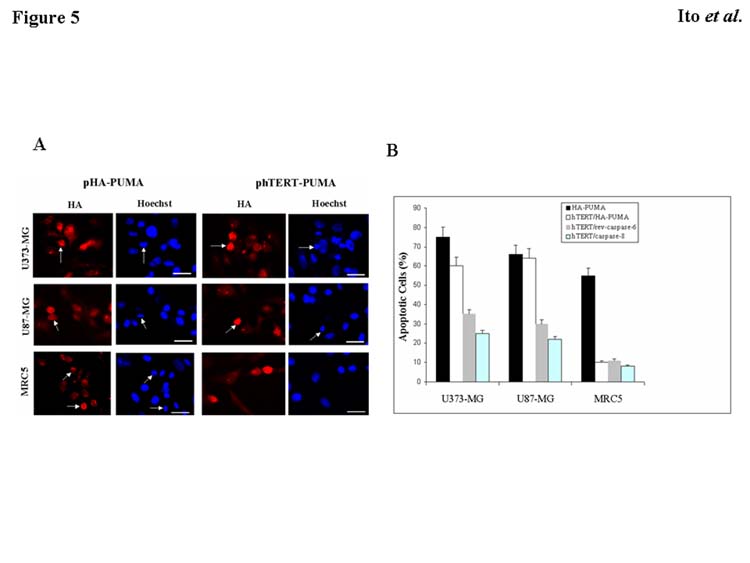
Induction of apoptosis in malignant glioma cells or normal cells by HA-PUMA or hTERT/HA-PUMA. A: Malignant glioma cells (U373-MG and U87-MG) and normal fibroblasts MRC5 were transfected with the HA-PUMA or hTERT/HA-PUMA vector. The PGL3-control was used as a reporter gene when the hTERT/HA-PUMA construct was transfected into MRC5 cells. After 48 h, immunohistochemical staining using anti-HA or anti-luciferase antibody was performed, followed by Hoechst 33258 staining. Arrows indicate representative apoptotic nuclei. Bars, 10 μm. B: Percentage of apoptotic cells in U373-MG, U87-MG, and MRC5 cells transfected with the HA-PUMA, hTERT/HA-PUMA, hTERT/rev-caspase-6, or hTERT/caspase-8 construct for 48 h. Data shown are the mean ± SD of apoptotic cells among 200 HA-, luciferase-, caspase-6, or caspase-8-positive cells chosen at random from three different areas.
In vivo effect of PUMA expression on malignant glioma cells
To determine the in vivo antitumor effect of PUMA expression, we inoculated U87-MG cells subcutaneously in nude mice. After the subcutaneous tumors were established, we injected the hTERT/luciferase, HA-PUMA, or hTERT/HA-PUMA construct (10 μg each) in the presence of Lipofectamin 2000 directly into tumors every day for 5 days. To determine whether antitumor effect persists or whether there is a rapid resumption of tumor growth after cessation of the treatment, we observed the tumor growth up to day 21. As shown in Fig. 6A, treatment with the hTERT/HA-PUMA or HA-PUMA construct significantly suppressed the growth of U87-MG subcutaneous tumors when compared with the control treatment using the hTERT/luciferase (P<0.001). As predicted from the in vitro experiments, the inhibitory effect of hTERT/HA-PUMA on subcutaneous tumors was not significantly different from that of HA-PUMA. The mean volume of the tumors treated with the hTERT/luciferase construct reached 191 ± 32 % of the initial tumor size 21 days after the first injection. The mean volume of the tumors treated with the hTERT/HA-PUMA or HA-PUMA construct was 87 ± 13 % or 72 ± 10 % of the initial tumor size on day 21, respectively. These results indicate that treatment with the hTERT/HA-PUMA construct produced a significant antitumor effect on subcutaneous tumors.
Fig. 6.
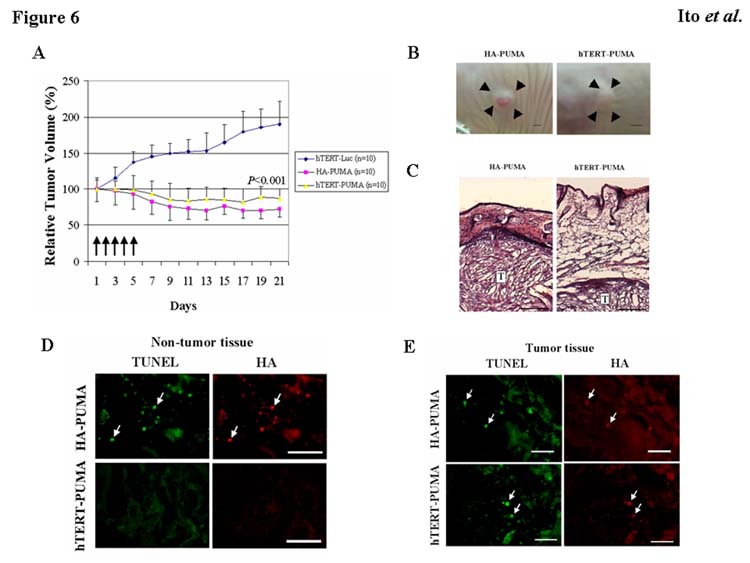
Effect of PUMA expression on subcutaneous tumors in nude mice. A: Tumors from U87-MG cells (1 × 106 cells) were established subcutaneously in nude mice. When the tumors reached a mean volume of 30–50 mm3, the hTERT/HA-PUMA, HA-PUMA or hTERT/luciferase construct (10 μg/20 μl PBS) mixed with Lipofectamine 2000 (2 μl) was administered directly into the tumors daily for 5 days. Tumor volume was determined using calipers up to day 21. 10 mice were used in each treatment group. Five arrows indicate when the mice were treated. Results shown are the mean ± SD of the percentage of change in tumor volume. *P<0.001 (on day 21). B: Photographs of subcutaneous U87-MG tumors next day (day 6) after the last injection of HA-PUMA or hTERT/HA-PUMA construct as described above. Arrowheads indicate the border of subcutaneous tumors. Bars, 2 mm. C: Hematoxylin and eosin (HE) staining of subcutaneous U87-MG tumors together with non-tumor tissues collected from the mice treated as described in B. T indicates subcutaneous tumors. Bars, 250 μm. D: HA expression and TUNEL analysis in non-tumor tissues collected from the mice treated as described in B. Arrows indicate representative TUNEL- and HA-positive cells. Bars, 50 μm. E: HA expression and TUNEL analysis in subcutaneous tumors collected from the mice treated as described in B. Arrows indicate representative TUNEL- and HA-positive cells. Bars, 50 μm.
7 of 10 mice treated with the HA-PUMA vector showed redness in their skin where the HA-PUMA plasmid was repeatedly injected (Fig. 6B). These were spontaneously cured without additional treatment after 5 times injections. To further examine the tissue damage observed only in HA-PUMA treatment group, we prepared additional animals and sacrificed them next day (day 6) after the last injection of HA-PUMA or hTERT/HA-PUMA to collect tumors together with skin and surrounding non-tumor tissues. As shown in Fig. 6C, non-tumor subcutaneous tissues were remarkably degenerated and reduced in thickness in HA-PUMA treatment group compared with mice treated with hTERT/HA-PUMA. Furthermore, some cells positive for TUNEL and HA were observed in non-tumor tissues from the mice treated with HA-PUMA, whereas such double-positive cells indicating apoptosis induced by PUMA were scarcely observed in hTERT/HA-PUMA-treated non-tumor tissues (Fig. 6D). In contrast, most of HA-positive tumor cells in HA-PUMA and hTERT/HA-PUMA treatment group were TUNEL-positive (Fig. 6E). These results indicate that PUMA was expressed selectively in cancer cells by using the hTERT promoter. The absence of apoptosis in non-tumor tissues demonstrates the benefit of using the hTERT/HA-PUMA construct instead of the HA-PUMA.
DISCUSSION
Accumulating evidence from preclinical and clinical studies indicates that transfer of p53 is an effective therapy for malignant gliomas (Gomez-Manzano et al., 1996; Gomez-Manzano et al., 1997; Li et al., 1999; Lang et al., 2003). However, the fact that exogenous expression of p53 has little effect on malignant glioma cells harboring wild-type p53 (Gomez-Manzano et al., 1996; Gomez-Manzano et al., 1997; Komata et al., 2000) and the fact that malignant gliomas are a mixture of cells with mutant and wild-type p53 (Lang et al., 1999) prompted us to explore an alternative approach to overcome those difficulties. Our data presented here demonstrate that the ability of PUMA to induce apoptotic cell death in malignant glioma cells is significantly greater than that of p53 and that the antitumor effect of PUMA is independent of the p53 status of tumors. Moreover, our investigations demonstrate that using the hTERT promoter system enables PUMA to induce tumor-specific apoptosis in in vitro and in vivo models. In addition, PUMA was superior to caspase-6 and caspase-8 with regard to the ability to induce apoptosis. These data strongly suggest that the transfer of the PUMA gene under the hTERT promoter is a promising strategy for treating malignant gliomas.
PUMA has recently been identified as a gene that is activated by p53 in cells undergoing p53-induced apoptosis (Yu et al., 2001; Nakano and Vousden, 2001; Han et al., 2001; Yu et al., 2003). PUMA shares homology with Bcl-2 family proteins within a short stretch of amino acids termed the BH3 domain, a region that allows interaction among the family members. Proteins with similar homology, called BH3-only proteins, constitute the third subgroup of the Bcl-2 family and include Bad, Bid, Bik, and Bim (Kelekar and Thompson, 1998). These proteins play an important role in apoptosis induction.
PUMA is required for p53-dependent apoptosis induced by DNA damage, hypoxia, and oncogenes (Yu et al., 2001; Nakano and Vousden, 2001; Han et al., 2001; Yu et al., 2003). Intriguingly, exogenous expression of PUMA protein resulted in a more rapid and profound apoptosis than did p53. Indeed, our study also demonstrated the superiority of PUMA to p53 in its ability to induce apoptosis in malignant glioma cells. Additionally, overexpression of PUMA induces activation of caspases and cytochrome c release (Yu et al., 2001; Nakano and Vousden, 2001), and Bax plays a critical role in inducing these events (Yu et al., 2001; Yu et al., 2003). Bax, a pro-apoptotic Bcl-2 family member, is a p53 target gene (Oltvai et al., 1993). Under normal conditions, Bax protein exists as an inactive monomer in the cytosol and is induced to homo-oligomerize and translocate to mitochondria upon death stimuli, thus leading to cytochrome c release and caspase activation (Hsu et al., 1997; Wolter et al., 1997; Rosse et al., 1998; Jurgensmeier et al., 1998). In the present study, we demonstrated that the mitochondrial translocation of Bax, followed by damage to mitochondrial membrane integrity, cytochrome c release, and activation of caspase-3, was induced by PUMA expression. These findings are consistent with those of previous investigations (Yu et al., 2003; Melino et al., 2003). More importantly, genetic disruption of Bax confers resistance to the apoptosis resulting from PUMA expression (Yu et al., 2003). In other words, it is likely that cells without Bax are resistant to the transfer of PUMA, and the more Bax that cells express, the more sensitive they are to PUMA-based therapy. Because Bax is expressed in the vast majority of malignant gliomas (Rieger et al., 1998), these tumors are considered to be among the best candidates for PUMA gene therapy.
More recently, it has been shown that knockout of PUMA can result in apoptotic deficiencies similar to those observed in p53 knockout mice (Jeffers et al., 2003; Villunger et al., 2003). Thymocyte apoptosis induced by ionizing radiation, deregulated c-Myc expression, and cytokine withdrawal was blocked in the PUMA knockout mice as in p53 knockout mice. PUMA also is necessary for the radiation-induced apoptosis that occurs in the developing nervous system. These findings establish PUMA as a principal mediator of p53-dependent and -independent apoptotic cell death. In our study, PUMA induced apoptosis in malignant glioma cells regardless of the p53 genotype. Genetic restoration of the apoptotic molecules is an attractive approach to the treatment of cancer. We and other investigators have recently demonstrated that transfer of apoptosis-inducible genes, such as members of the caspase family, successfully inhibits the growth of malignant glioma cells in vitro and in vivo through induction of apoptosis (Yu et al., 1996; Kondo et al., 1998; Shinoura et al., 2000). These genes act for downstream in the apoptotic pathway, whereas PUMA acts as the initial mediator in the pathway. In the present study, we demonstrated the superiority of PUMA to initiator caspase-8 and executioner caspase-6 in inducing apoptosis in U373-MG and U87-MG cells. These findings provide strong support for the idea that PUMA can be used as a new target for anticancer therapy more effectively than p53 or caspases.
In the present study, we adopted the hTERT promoter system that we recently developed (Koga et al., 2000; Komata et al., 2001) to restrict expression of PUMA protein to malignant glioma cells. The hTERT is one of three major subunits of telomerase, a ribonucleoprotein enzyme (Meyerson et al., 1997; Nakamura et al., 1997). Although hTERT and another subunit, the RNA component (hTR) (Feng et al., 1995), are necessary for telomerase activity, their expression patterns are different (Weinrich et al., 1997; Nakayama et al., 1998). The expression of hTERT is highly associated with telomerase activity, whereas the expression of hTR is also detected in telomerase-negative cells (Weinrich et al., 1997; Nakayama et al., 1998). In other words, hTERT is the major determinant of telomerase activity, indicating that control of telomerase expression is predominantly regulated at the transcriptional level of hTERT. Telomerase is a particularly attractive target for the malignant glioma–specific expression system of a therapeutic gene such as caspase or PUMA. That is because telomerase is detected in the vast majority of malignant gliomas, but not in normal brain tissues (Langford et al., 1995; DeMasters et al., 1997; Le et al., 1998). Moreover, among patients with glioblastoma multiforme (GBM)—the most malignant type of glioma—those whose cells lack telomerase have better survival than do those with telomerase-positive GBM (Hakin-Smith et al., 2003).
In summary, PUMA was activated in mutant p53 malignant glioma cells that were sensitive to Ad-p53 but not in wild-type p53 malignant glioma cells that were insensitive to Ad-p53. Both mutant p53 and wild-type p53 malignant glioma cells underwent apoptosis following treatment with Ad-PUMA, and the incidence of apoptosis was significantly higher in Ad-PUMA than in Ad-p53. Utilizing the hTERT promoter system restricted PUMA-induced cell death to malignant glioma cells and kept normal cells from undergoing apoptosis. Our findings using in vitro and in vivo models suggest that transfer of PUMA regulated by the hTERT promoter is a promising strategy for treating malignant gliomas without inducing apoptosis in non-tumor cells. Because approximately 90% of a variety of other tumors have telomerase activity, the strategy of using the hTERT/HA-PUMA construct is applicable to these cancers.
Acknowledgments
We thank Dr. Bert Vogelstein and Dr. Frederick Lang for their kind gifts of reagents. We also thank Marianne Doran and Emporia Faith Hollingsworth for editing manuscript and technical assistance. This study was supported in part by the USPHS Grants CA088936 and CA108558 awarded by the National Cancer Institute, in part by a start-up fund from The University of Texas M. D. Anderson Cancer Center, in part by a generous donation from the Anthony D. Bullock III Foundation (all to S. Ko.), and in part by the Cancer Center Support Grant (CCSG)/Shared Resources.
References
- Bold RJ, Termuhlen PM, McConkey DJ. Apoptosis, cancer and cancer therapy. Surg Oncol. 1997;6:133–142. doi: 10.1016/s0960-7404(97)00015-7. [DOI] [PubMed] [Google Scholar]
- Clayman GL, El-Naggar AK, Lippman SM, Henderson YC, Frederick M, Merritt JA, Zumstein LA, Timmons TM, Liu TJ, Ginsberg L, Roth JA, Hong WK, Bruso P, Goepfert H. Adenovirus-mediated p53 gene transfer in patients with advanced recurrent head and neck squamous cell carcinoma. J Clin Oncol. 1998;16:2221–2232. doi: 10.1200/JCO.1998.16.6.2221. [DOI] [PubMed] [Google Scholar]
- Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- DeMasters BK, Markham N, Lillehei KO, Shroyer KR. Differential telomerase expression in human primary intracranial tumors. Am J Clin Pathol. 1997;107:548–554. doi: 10.1093/ajcp/107.5.548. [DOI] [PubMed] [Google Scholar]
- Dewson G, Snowden RT, Almond JB, Dyer MJ, Cohen GM. Conformational change and mitochondrial translocation of Bax accompany proteasome inhibitor–induced apoptosis of chronic lymphocytic leukemic cells. Oncogene. 2003;22:2643–2654. doi: 10.1038/sj.onc.1206326. [DOI] [PubMed] [Google Scholar]
- Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, Yu J, Le S, West MD, Harlery CB, Andrews WH, Greider CW, Villeponteau B. The RNA component of human telomerase. Science. 1995;269:1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- Gomez-Manzano C, Fueyo J, Kyritsis AP, Steck PA, Roth JA, McDonnell TJ, Steck KD, Levin VA, Yung WK. Adenovirus-mediated transfer of the p53 gene produces rapid and generalized death of human glioma cells via apoptosis. Cancer Res. 1996;56:694–699. [PubMed] [Google Scholar]
- Gome-Manzano C, Fueyo J, Kyritsis AP, McDonnell TJ, Steck PA, Levin VA, Yung WK. Characterization of p53 and p21 functional interactions in glioma cells en route to apoptosis. J. Natl. Cancer Inst. 1997;89:1036–1044. doi: 10.1093/jnci/89.14.1036. [DOI] [PubMed] [Google Scholar]
- Hakin-Smith V, Jellinek DA, Levy D, Carroll T, Teo M, Timperley WR, McKay MJ, Reddel RR, Royds JA. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet. 2003;361:836–838. doi: 10.1016/s0140-6736(03)12681-5. [DOI] [PubMed] [Google Scholar]
- Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, Zhu L, Chittenden T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci USA. 2001;98:11318–11323. doi: 10.1073/pnas.201208798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP. PUMA is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- Kelekar A, Thompson CB. Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol. 1998;8:324–330. doi: 10.1016/s0962-8924(98)01321-x. [DOI] [PubMed] [Google Scholar]
- Koga S, Hirohata S, Kondo Y, Komata T, Takakura M, Inoue M, Kyo S, Kondo S. A novel telomerase-specific gene therapy: gene transfer of caspase-8 utilizing the human telomerase catalytic subunit gene promoter. Hum Gene Ther. 2000;11:1397–1406. doi: 10.1089/10430340050057477. [DOI] [PubMed] [Google Scholar]
- Komata T, Kondo Y, Koga S, Ko SC, Chung LW, Kondo S. Combination therapy of malignant glioma cells with 2-5A-antisense telomerase RNA and recombinant adenovirus p53. Gene Ther. 2000;7:2071–2079. doi: 10.1038/sj.gt.3301327. [DOI] [PubMed] [Google Scholar]
- Komata T, Kondo Y, Kanzawa T, Hirohata S, Koga S, Sumiyoshi H, Srinivasula SM, Barna BP, Germano IM, Takakura M, Inoue M, Alnemri ES, Shay JW, Kyo S, Kondo S. Treatment of malignant glioma cells with the transfer of constitutively active caspase-6 using the human telomerase catalytic subunit (human telomerase reverse transcriptase) gene promoter. Cancer Res. 2001;61:5796–5802. [PubMed] [Google Scholar]
- Kondo S, Barna BP, Morimura T, Takeuchi J, Yuan J, Akbasak A, Barnett GH. Interleukin-1 beta-converting enzyme mediates cisplatin-induced apoptosis in malignant glioma cells. Cancer Res. 1995;55:6166–6171. [PubMed] [Google Scholar]
- Kondo S, Tanaka Y, Kondo Y, Ishizaka Y, Hitomi M, Haqqi T, Liu J, Barnett GH, Alnemri ES, Barna BP. Retroviral transfer of CPP32beta gene into malignant gliomas in vitro and in vivo. Cancer Res. 1998;58:962–967. [PubMed] [Google Scholar]
- Lang FF, Yung WK, Sawaya R, Tofilon PJ. Adenovirus-mediated p53 gene therapy for human gliomas. Neurosurgery. 1999;45:1093–1104. doi: 10.1097/00006123-199911000-00016. [DOI] [PubMed] [Google Scholar]
- Lang FF, Bruner JM, Fuller GN, Aldape K, Prados MD, Chang S, Berger MS, McDermott MW, Kunwar SM, Junck LR, Chandler W, Zwiebel JA, Kaplan RS, Yung WK. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21:2508–2518. doi: 10.1200/JCO.2003.21.13.2508. [DOI] [PubMed] [Google Scholar]
- Langford LA, Piatyszek MA, Xu R, Schold SC, Jr, Shay JW. Telomerase activity in human brain tumors. Lancet. 1995;346:1267–1268. doi: 10.1016/s0140-6736(95)91865-5. [DOI] [PubMed] [Google Scholar]
- Le S, Zhu JJ, Anthony DC, Greider CW, Black PM. Telomerase activity in human gliomas. Neurosurgery. 1998;42:1120–1125. doi: 10.1097/00006123-199805000-00099. [DOI] [PubMed] [Google Scholar]
- Li H, Alonso-Vanegas M, Colicos MA, Jung SS, Lochmuller H, Sadikot AF, Snipes GJ, Seth P, Karpati G, Nalbantoglu J. Intracerebral adenovirus-mediated p53 tumor suppressor gene therapy for experimental human glioma. Clin Cancer Res. 1999;5:637–642. [PubMed] [Google Scholar]
- Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C, Vousden KH. p73 induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2003;279:8076–8083. doi: 10.1074/jbc.M307469200. [DOI] [PubMed] [Google Scholar]
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff ML, Liu Q, Bacchetti S, Haber DA, Weinberg RA. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Tahara H, Tahara E, Saito M, Ito K, Nakamura H, Nakanishi T, Ide T, Ishikawa F. Telomerase activation by hTR in human normal fibroblasts and hepatocellualr carcinomas. Nat Genet. 1998;18:65–68. doi: 10.1038/ng0198-65. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Rieger L, Weller M, Bornemann A, Schabet M, Dichgans J, Meyermann R. BCL-2 family protein expression in human malignant glioma: a clinical-pathological correlative study. J Neurol Sci. 1998;155:68–75. doi: 10.1016/s0022-510x(97)00277-3. [DOI] [PubMed] [Google Scholar]
- Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Roth JA, Cristiano RJ. Gene therapy for cancer: what have we done and where are we going? J Natl Cancer Inst. 1997;89:21–39. doi: 10.1093/jnci/89.1.21. [DOI] [PubMed] [Google Scholar]
- Shinoura N, Koike H, Furitu T, Hashimoto M, Asai A, Kirino T, Hamada H. Adenovirus-mediated transfer of caspase-8 augments cell death in gliomas: implication for gene therapy. Hum Gene Ther. 2000;11:1123–1137. doi: 10.1089/10430340050015185. [DOI] [PubMed] [Google Scholar]
- Swisher SG, Roth JA, Nemunaitis J, Lawrence DD, Kemp BL, Carrasco CH, Connors DG, El-Naggar AK, Fossella F, Glisson BS, Hong WK, Khuri FR, Kurie JM, Lee JJ, Lee JS, Mack M, Merritt JA, Nguyen DM, Nesbitt JC, Perez-Soler R, Pisters KM, Putnam JB, Jr, Richli WR, Savin M, Waugh MK, et al. Adenovirus-mediated p53 gene transfer in advanced non-small-cell lung cancer. J Natl Cancer Inst. 1999;91:763–771. doi: 10.1093/jnci/91.9.763. [DOI] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- Weinrich SL, Pruzan R, Ma L, Ouellette M, Tesmer VM, Holt SE, Bodnar AG, Lichtsteiner S, Kim NW, Trager JB, Taylor RD, Carlos R, Andrews WH, Wright WE, Shay JW, Harley CB, Mrin GB. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTR. Nat Genet. 1997;17:498–502. doi: 10.1038/ng1297-498. [DOI] [PubMed] [Google Scholar]
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA. 2003;100:1931–1936. doi: 10.1073/pnas.2627984100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JS, Sena-Esteves M, Paulus W, Breakefield XO, Reeves SA. Retroviral delivery and tetracycline-dependent expression of IL-1beta-converting enzyme (ICE) in a rat glioma model provides controlled induction of apoptotic death in tumor cells. Cancer Res. 1996;56:5423–5427. [PubMed] [Google Scholar]