

Abstract
Akt1 is frequently up-regulated in human tumors and has been shown to accelerate cell proliferation and to suppress programmed cell death; consequently, inhibition of the activity of Akt1 has been seen as an attractive target for therapeutic intervention. Paradoxically, hyperactivation of the Akt1 oncogene can also prevent the invasive behavior that underlies progression to metastasis. Here we show that overexpression of activated myr-Akt1 in human breast cancer cells phosphorylates and thereby targets the tumor suppressor tuberous sclerosis complex 2 (TSC2) for degradation, leading to reduced Rho-GTPase activity, decreased actin stress fibers and focal adhesions, and reduced motility and invasion. Overexpression of TSC2 rescues the migration phenotype of myr-Akt1-expressing tumor cells, and high levels of TSC2 in breast cancer patients correlate with increased metastasis and reduced survival. These data indicate that the functional properties of genes designated as oncogenes or tumor suppressor genes depend on the context of the cell type and the tissues studied, and suggest the need for caution in designing therapies targeting the function of individual genes in epithelial tissues.
Keywords: metastasis, oncogene, tumor suppressor
Metastasis occurs when cancer cells invade beyond the boundaries of the primary site and establish new tumors in distant organs; because metastases are responsible for most cancer deaths, attention has focused on the mechanisms by which cancer cells acquire invasive metastatic properties (1). Invasion by cancer cells into surrounding tissues involves alterations in cellular shape and stiffness that facilitate interaction with the new tissue microenvironment (2), and these modifications depend on reorganization of the actin cytoskeleton through the action of Rho-GTPases (3). Although 22 members of the Rho-GTPase family have been identified, it is the activation of the Rho subfamily that is principally responsible for the assembly of the contractile actomyosin machinery necessary for most types of cell motility (4). Inhibition of Rho or its effector Rho-kinase (ROCK) significantly compromises cellular migration and invasiveness (5–10), and increased expression/activation of Rho and its isoforms has been found in many types of cancers (11–14). A recently discovered mechanism for activating Rho in epithelial cells involves the tuberous sclerosis complex 2 gene product, TSC2 (15).
Tuberous sclerosis complex is a disorder characterized by hamartomas in the skin, kidney, heart, and lung and is caused by inactivating mutations in the tumor suppressor genes TSC1 and TSC2 (16). Although it has been best characterized as a controller of cell size through its regulation of ribosomal synthesis proteins including ribosomal S6 kinase (17, 18), TSC2 has been implicated in a number of related signaling pathways in a variety of cell types (19). Investigations of Madin–Darby canine kidney epithelial cells demonstrated that overexpression of TSC2 results in activation of Rho and increased cellular motility (15), and analysis of TSC2-deficient cells has shown reduced Rho activity (16), although these effects may depend on the cell type investigated (20). Although mutational inactivation of TSC2 contributes to tuberous sclerosis complex, regulation of intact TSC2 occurs principally through the action of the serine/threonine kinase Akt1/protein kinase B (21, 22). Phosphorylation of TSC2 by Akt1 stimulates association with 14-3-3 proteins that mediate subcellular translocation and proteolytic degradation of TSC2 (23–27).
Akt1 is one of the most frequently activated protein kinases in human cancer (28, 29). Activation of Akt1 is associated with resistance to apoptosis, as well as increased cell growth, proliferation, and energy metabolism (28, 30). Activated Akt1 is correlated with altered cell migration and invasion in several mammalian systems: constitutively active Akt1 or its isoforms can enhance the ability to invade in already invasive cultured cancer cells (31–33), but Akt1 can have the opposite effect on normal or less invasive cells (33). Moreover, a constitutively active allele of Akt1 blocks ErbB2/Neu-mediated invasion into surrounding tissue and lung metastasis in double transgenic mice (34). These results reinforce the idea that understanding cell- and tissue-specific signaling pathways is critical for evaluating the implications of activated upstream signaling molecules on complex phenotypic effects (35).
In a study investigating the mechanisms involved in the development of functional mammary epithelial cell structures, we found that signaling pathways activated by phosphatidyl inositol 3-kinase (PI3K) disrupted normal tissue structure by blocking polarity through increased Rac1 activity and stimulating cell proliferation through increased Akt1 activity (36). We found that cells expressing activated Akt1 showed the expected effects of increased proliferation and resistance to apoptosis but surprisingly also appeared to have substantially decreased invasiveness and motility. Here we show that inhibition of motility by activated Akt1 depends on down-regulation of Rho activity. We further show that TSC2 is a key intermediate in this mechanism, that activated Akt1 inhibits motility by stimulating the degradation of TSC2, and that increased expression of TSC2 can block the invasion-inhibitory effects of activated Akt1.
Results
Activated Akt1 Promotes Cell Proliferation, Survival, and Tumor Growth but Inhibits Cell Motility and Invasion.
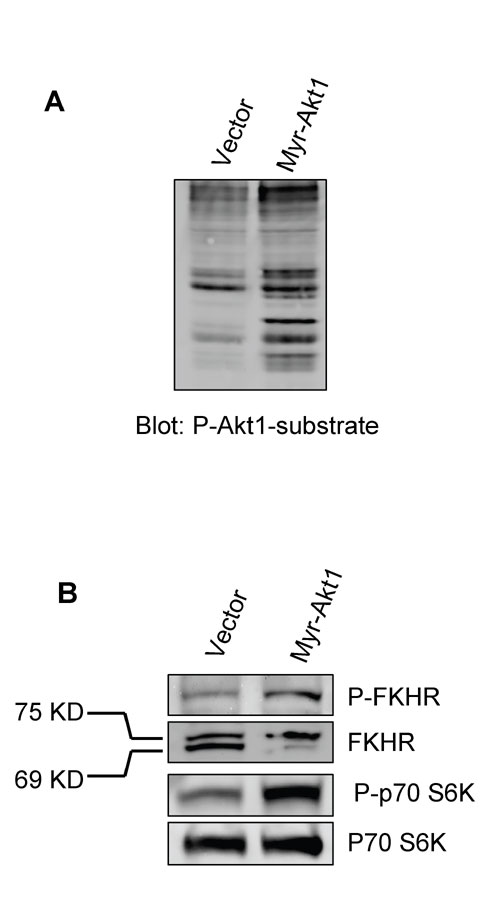
Akt1 has been implicated in multiple cellular processes, including cell survival, proliferation, growth, and apoptosis resistance (28, 37). The particular role it plays in a given tissue, however, depends on the model system investigated. We expressed constitutively activated, myristoylated Akt1 (myr-Akt1) (38) or empty vector in the human mammary epithelial cancer cell line HMT-3522 T4-2 (T4-2) (39, 40). In these pooled populations, increased Akt1 signaling induced by this construct was confirmed by phosphorylation of Akt1 substrates and specific downstream targets (Fig. 6, which is published as supporting information on the PNAS web site). Consistent with the results seen with the transgenic Akt1 mice (34), xenografts of myr-Akt1-expressing cells formed tumors that develop more rapidly and to a much larger size than control cells (P < 0.001; Fig. 1A and data not shown). Cells overexpressing myr-Akt1 showed significantly increased colony number and size in methylcellulose and soft agar assays (Fig. 1 B and C). When cultured in 3D laminin-rich extracellular matrix (lrECM) (Matrigel or collagen I+laminin1), cells expressing myr-Akt1 also showed larger colony size (36) (data not shown) because of increased cell proliferation and suppressed apoptosis as assessed by KI-67 and TUNEL immunostaining (Fig. 7, which is published as supporting information on the PNAS web site). These results demonstrate that Akt1 activation strongly stimulates cell survival and tumor growth in this experimental model. Opposite effects were seen in cell migration, motility, and invasion assays, however. Myr-Akt1 significantly inhibited cell motility in wound-healing assays (Fig. 1D), and by tracking cell location for 24 h, we found that the motility of isolated myr-Akt1 cells was significantly reduced relative to control cells (Fig. 1E), which suggests that the decreased migration in the scratch assay is not due to increased cell–cell adhesion. Consistent with these observations, we found that active Akt1 significantly reduced invasion of T4-2 cells through Matrigel-coated Boyden chambers (Fig. 1F). Previous studies have linked Akt activity to expression of matrix metalloproteinases (MMPs) (31), well known effectors of cell invasiveness, and although we found that cells expressing myr-Akt1 showed reduced expression of MMP-9, supplementation with much higher levels of MMP-9 in the media did not rescue the inhibition of motility by myr-Akt1 (data not shown).
Fig. 1.

Activated Akt1 promotes growth and anchorage independence in T4-2 cells but suppresses motility and invasion. Cells expressing myr-Akt1 showed increased tumor volume in nude mouse xenografts (A), increased colony number in methyl cellulose (B), and increased colony size in soft agar (C). However, myr-Akt1 inhibited cell migration (D) and motility (E), as well as invasion (F). All graphs display averages ± SEM. ∗, P < 0.05.
Cell Spreading and Formation of Focal Adhesions Are Inhibited by Activated Akt.
Cell motility is a dynamic process in which moving cells undergo dramatic changes in cell shape, associated with the creation of actin-rich protrusions, formation and disassembly of adhesive complexes, and establishment of migration polarity (2, 41). We found that cell spreading was greatly reduced in cells expressing myr-Akt1 (Fig. 2A); because actin polymerization is one of the major forces involved in cell spreading (42), we evaluated the actin cytoskeleton in myr-Akt1-expressing cells. We found that cells expressing active Akt1 had increased cortical actin staining and reduced stress fiber formation (Fig. 2B), and that initiation of cell movement in scratch assays was associated with fewer actin-rich projections toward the scratch wound in myr-Akt1 cells (data not shown). Reduced cell extensions in myr-Akt1 cells in the cell spreading and motility assays are consistent with the requirement for these structures during the onset and maintenance of cell motility (2).
Fig. 2.

Activated Akt1 alters cell-substratum adhesion. Cells expressing myr-Akt1 show reduced cell spreading (A), increased cortical distribution of actin (B), and fewer paxillin-containing focal adhesions (C). (Scale bars: A and C, 10 μm; B, 20 μm.)
Focal adhesions are required for cell attachment and serve as points of contact for cell spreading and traction during migration (43); we examined the effect on focal adhesion formation in cells expressing myr-Akt1 by immunostaining for paxillin. We determined that activated Akt1 decreased the formation of focal adhesions without affecting the level of endogenous paxillin expression (Fig. 2C and data not shown). These results suggest that reduced motility in myr-Akt1 cells is due to alterations in the assembly and function of the actin cytoskeleton.
Active Akt1 Inhibits Rho.
Assembly and organization of the actin cytoskeleton are controlled by Rho GTPases, and the formation of stress fibers and focal adhesions is a characteristic effect of activation of Rho GTPases (44). Using a Rhotekin-GST fusion protein pull-down assay, we found that active Rho levels were significantly lower in myr-Akt1-expressing cells, starting 2 h after cell plating (Fig. 3A). To determine the effect of the decreased Rho activity on actin organization, cells plated on coverslips were stained with phalloidin at the same time points as pull-down assays. At all time points, myr-Akt1-expressing cells spread more slowly and formed fewer stress fibers than vector control cells (Fig. 2 A and B and data not shown). We found no substantial difference in Rac1 or Cdc42 activities (Fig. 3B). Consistent with the implication that compromised cell motility and invasiveness in myr-Akt1-expressing cells was due to inhibition of Rho activity, treatment with exoenzyme C3 transferase (C3 toxin), which inhibits Rho when added to the media (45, 46), was as effective at blocking cell migration as was expression of myr-Akt1 (Fig. 3C).
Fig. 3.

Rho GTPases are selectively inhibited by Akt1. Cells were plated for the indicated times, then lysates were analyzed for activity by pull-down with GST-Rhotekin (A) or GST-PAK-CD (B). (C) Inhibition of Rho by pretreatment with recombinant C3 transferase (10 μg/ml) reduced migration of vector control cells in scratch assay to levels of myr-Akt1-expressing cells. The graph displays the average ± SEM. ∗, P < 0.05.
Active Akt1 Controls Migration and Invasion by Modulating Levels of the Tumor Suppressor TSC2.
In many systems, Akt1 has been shown to phosphorylate TSC2, triggering 14-3-3-dependent proteolytic degradation (16, 19). Moreover, elevated TSC2 and TSC1 have been shown to increase Rho activity in epithelial Madin–Darby canine kidney cells (15) and in fibroblasts (47), respectively. Accordingly, we hypothesized that Akt1-mediated inhibition of Rho activity was the consequence of phosphorylation and down-modulation of TSC2. Consistent with this possibility, we found increased phosphorylation and decreased total levels of TSC2 in cells expressing myr-Akt1 (Fig. 4A), and decreased TSC2 expression in myr-Akt1 tumor xenografts (Fig. 4B). Using immunoprecipitation experiments, we found significantly increased association of TSC2 with 14-3-3 in cells expressing myr-Akt1 (Fig. 4C).
Fig. 4.

TSC2 mediates the effects of Akt1 on motility and invasion. (A) TSC2 shows increased phosphorylation but decreased total levels in cells expressing myr-Akt1. (B) Decreased levels of TSC2 in tumor xenografts from myr-Akt1 cells. (Scale bar: 50 μm.) (C) TSC2 shows increased association with 14-3-3 in cells expressing myr-Akt1. (D) Expression of functional TSC2 in myr-Akt1 cells as shown by reduced phosphorylation of p70S6K. (E) Expression of TSC2 rescues cell invasiveness and motility in myr-Akt1 cells. The graphs display averages ± SEM. ∗, P < 0.05; ∗∗, P < 0.02; ∗∗∗, P < 0.001.
To determine whether exogenous expression of TSC2 could compensate for the inhibition of cell motility and invasion induced by active Akt1, wild-type TSC2 was stably expressed in vector control and myr-Akt1 cells (Fig. 4E). Functional expression of TSC2 was demonstrated by decreased phosphorylation of p70S6K (Fig. 4D) (48). We found that myr-Akt1 cells expressing TSC2 showed enhanced migration and invasion (Fig. 4E), as well as increased cell spreading, formation of stress fibers, and focal adhesions (Fig. 8, which is published as supporting information on the PNAS web site).
Relative Expression of Akt1 and TSC2 Predicts Progression to Metastasis in Human Breast Cancer Patients.
These results suggested a model (Fig. 5A) in which active Akt1 reduces the levels of TSC2, which causes a reduction in the TSC2-dependent activation of Rho, causing in turn decreased cell migration, motility, and invasiveness. This system presents a striking example of the importance of cell-type specificity for the function of key signaling molecules, inasmuch as the expression of the oncogene Akt1 is inhibiting the invasive behavior that underlies progression to metastasis, whereas the tumor suppressor TSC2 is stimulating this same tumor progression step. Moreover, because the inhibition of Rho activity and consequent effects on invasiveness depend on the relative activities of Akt1 and TSC2, this model suggests that the effect of TSC2 to stimulate the prometastatic invasive phenotype would be most pronounced in cells expressing lower levels of Akt1.
Fig. 5.

Analysis of pathway implication on human breast cancer metastasis. (A) Pathway model. a, Active Akt1 phosphorylates TSC2, stimulating down-modulation of TSC2 levels; b, TSC2 activates Rho; c, Active Rho promotes cytoskeletal rearrangements permissive for cell invasion. (B) Metastasis-free survival of human breast tumors stratified by expression of Akt1 and TSC2. Higher expression of TSC2 together with low expression of Akt1 is predictive of decreased time to metastasis in breast tumors.
To evaluate the applicability of these findings, we interrogated a comprehensive RNA expression microarray data set of 295 primary human breast cancers (49). To analyze the contribution of TSC2 and Akt1 to tumor progression and metastasis, tumors were divided in quartiles by expression level of each marker, and metastasis-free survival was calculated for the upper and lower quartiles (n = 74 samples for each single gene). Although elevated expression of TSC2 did appear to correlate with decreased time to metastasis (data not shown), this result fell short of the standard for statistical significance (P = 0.053). However, consideration of Akt1 expression levels in combination with TSC2 expression produced a different result (Fig. 5B). We found that tumors in both the highest quartile for TSC2 expression and the lowest quartile for Akt1 (blue line, n = 11) showed significantly decreased time to metastasis relative to tumors in the lowest quartile of TSC2 and Akt1 expression (black line, n = 28; P < 0.005). Comparison of tumors with high TSC2 and high Akt1 (red line, n = 26) to tumors with low TSC2 and high Akt1 (green line, n = 14), by contrast, revealed no statistically significant difference.
Discussion
Akt1 has been classified as an oncogene on the basis of its frequent up-regulation in a wide variety of human tumors (reviewed in refs. 50 and 51), but increased expression of Akt1 does not always correlate with invasiveness (52, 53), the defining stage of malignancy. Recent experimental studies have confirmed these observations: comparisons of ErbB2/Akt1 double transgenic mice with ErbB2 single transgenics have shown that expression of activated Akt1 accelerates tumorigenicity but blocks metastasis (34), and in articles appearing after this work was completed, expression of activated Akt1 was shown to inhibit motility and invasiveness of MDA-MB-435 (54) and MCF-10A (55) cells. These observations are corroborated by other studies of cultured cancer cells, because almost without exception, increased activation of Akt1 correlates with increased proliferation and anchorage-independent growth, but the effects of activated Akt1 on cell migration and invasiveness depend on the cell type that is investigated (31–33, 54–57). Here, we present a mechanism to account for this apparent paradox: overexpression of an activated isoform of Akt1 leads to phosphorylation and degradation of TSC2 and decreased Rho activity, resulting in a reduction in stress fiber and focal adhesion formation, reduced random and persistent migration, and invasion (Fig. 5A).
Such observations suggest the existence of a dichotomy between proliferation and cell motility. Although often regulated by the same extracellular cues and intracellular signal transduction pathways, cell migration and proliferation rarely occur simultaneously. During wound healing, keratinocytes migrate into the wound site before proliferating (58), and during embryonic development, neural crest cells can migrate across the distance of the embryo without entering the cell cycle (59). Additional examples can be found during development of the lung and vasculature, where buds and sprouts have been seen to form before initiation of cell proliferation (60, 61). Similarly, a switch between proliferation and migration has been observed in gliomas, other brain tumors, and bladder carcinoma (62–64). There are several possible explanations for this phenomenon. Much of the cytoskeletal machinery required for migration is also required for cell-cycle progression and cytokinesis. In addition to regulating the actin cytoskeleton during locomotion, RhoA accumulates at, and recruits myosin to, the cleavage furrow during cell division (65). It is conceivable that maintaining the cell in a nonmotile state during cell-cycle progression allows for the proper distribution of RhoA and other resources to facilitate cytokinesis, and this organization may constitute part of the checkpoint machinery. Our data implicate Akt1/TSC2 as a crucial signaling node in this decision-making process; a similar role has been reported for the adaptor protein Shc (66). If so, a breakdown of the separation of proliferative and invasive behaviors, as is observed in advanced stages of tumor progression, could contribute to improper apportioning of chromosomes during cell division, leading to genomic amplifications and deletions, thereby accelerating cancer progression.
Much of the current research into cancer etiology, progression, and potential therapies is centered on understanding and circumventing the activation of protooncogenes and for reactivating or substituting for the loss of protective tumor suppressor genes. However, classification of a gene as an oncogene or tumor suppressor may result from limited observations of specific cell lines, transgenic mouse tissues, or types of human tumors. Given that cancer is not a single disease, evaluating the status of a tumor simply by the presence of certain activated “oncogenes” or inactivated “tumor suppressors” is incomplete and potentially misleading. Indeed, a similar effect has been proposed for KLF4, which acts as a tumor suppressor in gastrointestinal cancer but has oncogenic properties in breast cancer cells (67); similarly, cell context-dependent tumor-suppressive and oncogenic effects have been found for TGF-β (68) and oncostatin (69). These findings suggest the importance of a more nuanced approach, because the functional outcome of activating or inactivating a gene depends largely on the cell type, its preexisting genetic background, and the local microenvironment (70). Here, the overexpression of Akt1 (a classical oncogene) leads to larger tumors that are less invasive, and that (given that the majority of mortality resulting from breast cancer is a result of metastatic disease) could potentially lead to a more favorable outcome clinically if occurring in the absence of other tumor-specific modifications (34). Conversely, increased expression of TSC2 (a classical tumor suppressor) stimulates invasiveness and correlates with a less favorable clinical outcome (Fig. 5B). Ongoing efforts to therapeutically target the Akt1 pathway in breast cancer patients (71) might benefit from consideration of TSC2 expression status in the target population, inasmuch as reducing Akt1 activity in tumors expressing high levels of TSC2 could potentially enhance progression to a more malignant phenotype.
Materials and Methods
Cell Line, Constructs, and Antibodies.
HMT-3522 mammary epithelial cells were cultured as described in refs. 40 and 72. The myr-Akt1 pWZL retroviral construct myrΔ4–129 and kinase-dead Akt1 (T308A/S473A) constructs were kindly provided by Richard Roth (Stanford University). TSC2/pMSCVneo was a gift from Elizabeth P. Henske (Fox Chase Cancer Center, Philadelphia). Transfection of Phoenix packaging cells (gift from Garry P. Nolan, Stanford University), production of retroviral stock, and viral infection were according to standard protocol. The stably expressing cells were selected in the presence of neomycin (500 μg/ml) or hygromycin B (50 μg/ml), and surviving clones were pooled. Antibodies were E-cadherin, paxillin (clone 165), hamartin (BD Biosciences), total and phospho-p70 S6 kinase (threonine 389), phospho-(Ser/Thr) Akt1 substrate, total and phospho-FKHR (serine 256), phospho-tuberin (threonine 1462) (Cell Signaling Technology), Akt1 and tuberin (C-20; Santa Cruz Biotechnology), 14-3-3 pan Ab-4 (clone CG15; NeoMarkers). Rho antibody was clone 55 from BD Biosciences, which does not distinguish between RhoA, RhoB, and RhoC.
Immunoblot Analysis and Immunoprecipitation.
Immunoblot analysis was performed described in ref. 72. For immunoprecipitation, cells were lysed in IP buffer [1% Triton X-100/150 mM NaCl/10 mM Tris·HCl (pH 7.4)/1 mM EDTA/1 mM EGTA, containing 2 mM NaF, 1 mM sodium orthovanadate, 10 μg/ml leupeptin, 10 μg/ml pepstatin, 10 μg/ml aprotinin, 10 μg/ml E 64, and 1 mM Pefabloc]. Equal amounts of protein lysates were precleared by 50 μl of Protein G Plus-conjugated agarose beads (Santa Cruz Biotechnology) before the addition of primary antibody. Samples were incubated at 4°C for 1 h. Subsequently, samples were incubated with Protein G Plus-conjugated beads for 1 h at 4°C. The beads were washed three times with IP buffer before being heated with sample buffer and analyzed by SDS/PAGE and Western blotting.
Immunofluorescence and Immunohistochemistry.
Cells were fixed either in 2% paraformaldehyde at room temperature for 20 min or in 1:1 methanol/acetone at −20°C for 10 min. The samples were blocked with blocking buffer (130 mM NaCl/15 mM Na2HPO4/3.5 mM NaH2PO4/0.1% BSA/0.2% Triton X-100/0.05% Tween 20) containing 10% goat serum and then incubated with primary antibodies followed directly by either FITC- or Texas red-conjugated secondary antibodies for 45 min. Nuclei were counterstained with 4′,6-diamidino-2 phenylindole (DAPI) (Sigma). The images were collected with a Zeiss 410 LSM confocal microscope (Zeiss Pluar ×40 oil objective lenses) or a SPOT RT Slider digital camera (spot v.3.2 software; Diagnostic Instruments, Sterling Heights, MI) attached to a Zeiss Photomicroscope III (Zeiss Plan Neofluar ×40 oil objective lenses).
Invasion and Migration Assays.
For invasion assay, cell-culture inserts (8 μm, 24-well format; BD Discovery Labware) were evenly coated with diluted Matrigel. Cells (1 × 105) were added to the upper chamber, and the lower chamber was filled with 300 μl of medium. The culture was maintained for 48 h. The number of cells was counted under ×20 objective in each of five randomly chosen fields. Alternatively, the membrane of the insert was cut, and the dye was eluted from the membrane with 200 μl of 10% acetic acid. The OD reading was obtained at a wavelength of 590 nm. The stained insert without adding cells was used as background reading. Each experiment was duplicated. The cell migration assay is similar to the invasion assay except that inserts were not coated with Matrigel and that 10% FBS was added to lower chamber and the culture was maintained for 24 h. For cell-migration wound-healing assays, cells were plated into 24-well plates (Nalge Nunc) at the density of 4 × 104 cells per cm2. After cells had grown to confluence, a wound was made across the well with blue tips under the guide of a ruler. The gap of the wound was marked and imaged immediately after wounding and 24 h later. The gap was measured and presented as net closure per 24 h. Each experiment was performed in duplicate or triplicate.
Anchorage-Independence Growth Assays.
For soft agar assay, 5,000 cells were plated in 1 ml of DMEM/F12 containing 0.3% agarose, overlaid with 1 ml of 1% agarose. Cultures were maintained for 15 days. Colonies from duplicated wells were measured and scored positive when the colony sizes exceeded a diameter of 50 μm. For methyl cellulose assay, 100,000 cells were seeded per 60-mm dish in 5 ml of DMEM/F12 containing 1.5% methyl cellulose (Fisher Scientific). Colonies were scored after 2 weeks.
Xenografts.
Nude mice, 4–5 weeks of age, were purchased from Simonsen Laboratories (Santa Clara, CA). T4-2 cells expressing myr-Akt1 or vector alone were propagated as monolayers, trypsinized, and resuspended in PBS at the concentration of 1 × 108 cells per ml. Then 0.1 ml of cell suspension (1 × 107 cells) was injected s.c. into rear flanks of nude mice, and tumor size was monitored twice per week by measuring tumor size (length × width × height). All of the mice were killed by CO2 at day 17 because of tumor burden. Tumors were dissected, fixed in 2% formaldehyde, paraffin-embedded, and sectioned (5 μm).
GST Fusion Protein Pull-Down Assay.
The cells were lysed in GST-Fish buffer [10% glycerol/50 mM Tris (pH 7.4)/100 mM NaCl/1% Nonidet P-40/2 mM MgCl2, containing 10 μg/ml leupeptin, 10 μg/ml pepstatin, 10 μg/ml aprotinin, 10 μg/ml E 64, and 1 mM Pefabloc) and centrifuged at 16,000 × g at 4°C. Equal amounts of protein supernatants were incubated with recombinant GST-Rhotekin or GST-Pak-CD fusion protein-coupled Sepharose beads at 4°C for 45 min. The beads were washed with an excess of lysis buffer, eluted in sample buffer, and analyzed by SDS/PAGE and Western blotting with antibody against Rho, Rac1, or Cdc42.
Statistics.
All data analysis was performed by using prism (GraphPad, San Diego). Bar graphs represent means ± SEM. The database consisting of the microarray profiles of the 295 human breast tumors and associated clinical data (49) was obtained from Rosetta Inpharmatics. Patients were stratified into quartiles for expression of TSC2 or Akt1, and metastasis-free survival was computed by using the Kaplan–Meier method; statistical significance was assessed by using the log-rank test.
Supplementary Material
Acknowledgments
We thank Paraic Kenny and Mark LaBarge for insightful discussions and assistance with statistical analysis. This work was supported by a grant from the U.S. Department of Energy, Office of Biological and Environmental Research (DE-AC03-SF0098), an innovator award from the Department of Defense Breast Cancer Research Program (DOD BCRP) (BC012005) (to M.J.B.), a DOD BCRP predoctoral fellowship (to H.L.), DOD BCRP postdoctoral fellowships (to C.M.N. and J.E.F.), and an American Cancer Society fellowship (to D.C.R.).
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Chambers A. F., Groom A. C., MacDonald I. C. Nat. Rev. Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 2.Friedl P., Wolf K. Nat. Rev. Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 3.Yamazaki D., Kurisu S., Takenawa T. Cancer Sci. 2005;96:379–386. doi: 10.1111/j.1349-7006.2005.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaffe A. B., Hall A. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 5.Gildea J. J., Harding M. A., Seraj M. J., Gulding K. M., Theodorescu D. Cancer Res. 2002;62:982–985. [PubMed] [Google Scholar]
- 6.Jo M., Thomas K. S., Somlyo A. V., Somlyo A. P., Gonias S. L. J. Biol. Chem. 2002;277:12479–12485. doi: 10.1074/jbc.M111147200. [DOI] [PubMed] [Google Scholar]
- 7.Kusama T., Mukai M., Iwasaki T., Tatsuta M., Matsumoto Y., Akedo H., Nakamura H. Cancer Res. 2001;61:4885–4891. [PubMed] [Google Scholar]
- 8.Somlyo A. V., Bradshaw D., Ramos S., Murphy C., Myers C. E., Somlyo A. P. Biochem. Biophys. Res. Commun. 2000;269:652–659. doi: 10.1006/bbrc.2000.2343. [DOI] [PubMed] [Google Scholar]
- 9.Kaneko K., Satoh K., Masamune A., Satoh A., Shimosegawa T. Pancreas. 2002;24:34–41. doi: 10.1097/00006676-200201000-00005. [DOI] [PubMed] [Google Scholar]
- 10.Bourguignon L. Y., Zhu H., Shao L., Zhu D., Chen Y. W. Cell Motil. Cytoskeleton. 1999;43:269–287. doi: 10.1002/(SICI)1097-0169(1999)43:4<269::AID-CM1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 11.Fritz G., Just I., Kaina B. Int. J. Cancer. 1999;81:682–687. doi: 10.1002/(sici)1097-0215(19990531)81:5<682::aid-ijc2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 12.Abraham M. T., Kuriakose M. A., Sacks P. G., Yee H., Chiriboga L., Bearer E. L., Delacure M. D. Laryngoscope. 2001;111:1285–1289. doi: 10.1097/00005537-200107000-00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ridley A. J. Breast Cancer Res. Treat. 2004;84:13–19. doi: 10.1023/B:BREA.0000018423.47497.c6. [DOI] [PubMed] [Google Scholar]
- 14.Clark E. A., Golub T. R., Lander E. S., Hynes R. O. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 15.Astrinidis A., Cash T. P., Hunter D. S., Walker C. L., Chernoff J., Henske E. P. Oncogene. 2002;21:8470–8476. doi: 10.1038/sj.onc.1205962. [DOI] [PubMed] [Google Scholar]
- 16.Mak B. C., Yeung R. S. Cancer Invest. 2004;22:588–603. doi: 10.1081/cnv-200027144. [DOI] [PubMed] [Google Scholar]
- 17.Krymskaya V. P. Cell. Signalling. 2003;15:729–739. doi: 10.1016/s0898-6568(03)00040-8. [DOI] [PubMed] [Google Scholar]
- 18.McManus E. J., Alessi D. R. Nat. Cell Biol. 2002;4:E214–E216. doi: 10.1038/ncb0902-e214. [DOI] [PubMed] [Google Scholar]
- 19.Li Y., Corradetti M. N., Inoki K., Guan K. L. Trends Biochem. Sci. 2004;29:32–38. doi: 10.1016/j.tibs.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 20.Goncharova E., Goncharov D., Noonan D., Krymskaya V. P. J. Cell Biol. 2004;167:1171–1182. doi: 10.1083/jcb.200405130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inoki K., Li Y., Zhu T., Wu J., Guan K. L. Nat. Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 22.Potter C. J., Pedraza L. G., Xu T. Nat. Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 23.Li Y., Inoki K., Vacratsis P., Guan K. L. J. Biol. Chem. 2003;278:13663–13671. doi: 10.1074/jbc.M300862200. [DOI] [PubMed] [Google Scholar]
- 24.Dan H. C., Sun M., Yang L., Feldman R. I., Sui X. M., Ou C. C., Nellist M., Yeung R. S., Halley D. J., Nicosia S. V., et al. J. Biol. Chem. 2002;277:35364–35370. doi: 10.1074/jbc.M205838200. [DOI] [PubMed] [Google Scholar]
- 25.Li Y., Inoki K., Yeung R., Guan K. L. J. Biol. Chem. 2002;277:44593–44596. doi: 10.1074/jbc.C200510200. [DOI] [PubMed] [Google Scholar]
- 26.Liu M. Y., Cai S., Espejo A., Bedford M. T., Walker C. L. Cancer Res. 2002;62:6475–6480. [PubMed] [Google Scholar]
- 27.Nellist M., Goedbloed M. A., de Winter C., Verhaaf B., Jankie A., Reuser A. J., van den Ouweland A. M., van der Sluijs P., Halley D. J. J. Biol. Chem. 2002;277:39417–39424. doi: 10.1074/jbc.M204802200. [DOI] [PubMed] [Google Scholar]
- 28.Vivanco I., Sawyers C. L. Nat. Rev. Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 29.Scheid M. P., Woodgett J. R. Nat. Rev. Mol. Cell Biol. 2001;2:760–768. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- 30.Blume-Jensen P., Hunter T. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 31.Kim D., Kim S., Koh H., Yoon S. O., Chung A. S., Cho K. S., Chung J. FASEB J. 2001;15:1953–1962. doi: 10.1096/fj.01-0198com. [DOI] [PubMed] [Google Scholar]
- 32.Grille S. J., Bellacosa A., Upson J., Klein-Szanto A. J., van Roy F., Lee-Kwon W., Donowitz M., Tsichlis P. N., Larue L. Cancer Res. 2003;63:2172–2178. [PubMed] [Google Scholar]
- 33.Arboleda M. J., Lyons J. F., Kabbinavar F. F., Bray M. R., Snow B. E., Ayala R., Danino M., Karlan B. Y., Slamon D. J. Cancer Res. 2003;63:196–206. [PubMed] [Google Scholar]
- 34.Hutchinson J. N., Jin J., Cardiff R. D., Woodgett J. R., Muller W. J. Cancer Res. 2004;64:3171–3178. doi: 10.1158/0008-5472.can-03-3465. [DOI] [PubMed] [Google Scholar]
- 35.Bissell M. J., Kenny P. A., Radisky D. C. Cold Spring Harbor Symp. Quant. Biol. 2005;70:1–14. doi: 10.1101/sqb.2005.70.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu H., Radisky D. C., Wang F., Bissell M. J. J. Cell Biol. 2004;164:603–612. doi: 10.1083/jcb.200306090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanhaesebroeck B., Leevers S. J., Ahmadi K., Timms J., Katso R., Driscoll P. C., Woscholski R., Parker P. J., Waterfield M. D. Annu. Rev. Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 38.Barthel A., Kohn A. D., Luo Y., Roth R. A. Endocrinology. 1997;138:3559–3562. doi: 10.1210/endo.138.8.5263. [DOI] [PubMed] [Google Scholar]
- 39.Briand P., Nielsen K. V., Madsen M. W., Petersen O. W. Cancer Res. 1996;56:2039–2044. [PubMed] [Google Scholar]
- 40.Weaver V. M., Petersen O. W., Wang F., Larabell C. A., Briand P., Damsky C., Bissell M. J. J. Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 42.Pollard T. D., Borisy G. G. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 43.Horwitz A. R., Parsons J. T. Science. 1999;286:1102–1103. doi: 10.1126/science.286.5442.1102. [DOI] [PubMed] [Google Scholar]
- 44.Hall A. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 45.Valderrama F., Luna A., Babia T., Martinez-Menarguez J. A., Ballesta J., Barth H., Chaponnier C., Renau-Piqueras J., Egea G. Proc. Natl. Acad. Sci. USA. 2000;97:1560–1565. doi: 10.1073/pnas.97.4.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wozniak M. A., Kwong L., Chodniewicz D., Klemke R. L., Keely P. J. Mol. Biol. Cell. 2005;16:84–96. doi: 10.1091/mbc.E04-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamb R. F., Roy C., Diefenbach T. J., Vinters H. V., Johnson M. W., Jay D. G., Hall A. Nat. Cell Biol. 2000;2:281–287. doi: 10.1038/35010550. [DOI] [PubMed] [Google Scholar]
- 48.Inoki K., Corradetti M. N., Guan K. L. Nat. Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 49.van de Vijver M. J., He Y. D., van’t Veer L. J., Dai H., Hart A. A., Voskuil D. W., Schreiber G. J., Peterse J. L., Roberts C., Marton M. J., et al. N. Engl. J. Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 50.Bellacosa A., Kumar C. C., Di Cristofano A., Testa J. R. Adv. Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 51.Altomare D. A., Testa J. R. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 52.Balsara B. R., Pei J., Mitsuuchi Y., Page R., Klein-Szanto A., Wang H., Unger M., Testa J. R. Carcinogenesis. 2004;25:2053–2059. doi: 10.1093/carcin/bgh226. [DOI] [PubMed] [Google Scholar]
- 53.Ruggeri B. A., Huang L., Wood M., Cheng J. Q., Testa J. R. Mol. Carcinog. 1998;21:81–86. [PubMed] [Google Scholar]
- 54.Yoeli-Lerner M., Yiu G. K., Rabinovitz I., Erhardt P., Jauliac S., Toker A. Mol. Cell. 2005;20:539–550. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 55.Irie H. Y., Pearline R. V., Grueneberg D., Hsia M., Ravichandran P., Kothari N., Natesan S., Brugge J. S. J. Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanno S., Mitsuuchi Y., Altomare D. A., Xiao G. H., Testa J. R. Cancer Res. 2001;61:589–593. [PubMed] [Google Scholar]
- 57.Enomoto A., Murakami H., Asai N., Morone N., Watanabe T., Kawai K., Murakumo Y., Usukura J., Kaibuchi K., Takahashi M. Dev. Cell. 2005;9:389–402. doi: 10.1016/j.devcel.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 58.Martin P. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 59.Perris R. Trends Neurosci. 1997;20:23–31. doi: 10.1016/S0166-2236(96)10063-1. [DOI] [PubMed] [Google Scholar]
- 60.Nogawa H., Morita K., Cardoso W. V. Dev. Dyn. 1998;213:228–235. doi: 10.1002/(SICI)1097-0177(199810)213:2<228::AID-AJA8>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 61.Ausprunk D. H., Folkman J. Microvasc. Res. 1977;14:53–65. doi: 10.1016/0026-2862(77)90141-8. [DOI] [PubMed] [Google Scholar]
- 62.Giese A., Loo M. A., Tran N., Haskett D., Coons S. W., Berens M. E. Int. J. Cancer. 1996;67:275–282. doi: 10.1002/(SICI)1097-0215(19960717)67:2<275::AID-IJC20>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 63.Khoshyomn S., Lew S., DeMattia J., Singer E. B., Penar P. L. J. Neurooncol. 1999;45:111–116. doi: 10.1023/a:1006375316331. [DOI] [PubMed] [Google Scholar]
- 64.Boyer B., Valles A. M., Tucker G. C., Delouvee A., Thiery J. P. Symp. Soc. Exp. Biol. 1993;47:183–195. [PubMed] [Google Scholar]
- 65.Bement W. M., Benink H. A., von Dassow G. J. Cell Biol. 2005;170:91–101. doi: 10.1083/jcb.200501131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Collins L. R., Ricketts W. A., Yeh L., Cheresh D. J. Cell Biol. 1999;147:1561–1568. doi: 10.1083/jcb.147.7.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rowland B. D., Bernards R., Peeper D. S. Nat. Cell Biol. 2005;7:1074–1082. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 68.Sieweke M. H., Thompson N. L., Sporn M. B., Bissell M. J. Science. 1990;248:1656–1660. doi: 10.1126/science.2163544. [DOI] [PubMed] [Google Scholar]
- 69.Porter A. C., Vaillancourt R. R. Oncogene. 1998;17:1343–1352. doi: 10.1038/sj.onc.1202171. [DOI] [PubMed] [Google Scholar]
- 70.Dolberg D. S., Bissell M. J. Nature. 1984;309:552–556. doi: 10.1038/309552a0. [DOI] [PubMed] [Google Scholar]
- 71.Cheng J. Q., Lindsley C. W., Cheng G. Z., Yang H., Nicosia S. V. Oncogene. 2005;24:7482–7492. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- 72.Wang F., Weaver V. M., Petersen O. W., Larabell C. A., Dedhar S., Briand P., Lupu R., Bissell M. J. Proc. Natl. Acad. Sci. USA. 1998;95:14821–14826. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}