

Abstract
Cytosine methylation is critical for correct development and genome stability in mammals and plants. In order to elucidate the factors that control genomic DNA methylation patterning, a genetic screen for mutations that disrupt methylation-correlated silencing of the endogenous gene PAI2 was conducted in Arabidopsis. This screen yielded seven loss-of-function alleles in a SET domain protein with histone H3 Lys9 methyltransferase activity, SUVH4. The mutations conferred reduced cytosine methylation on PAI2, especially in non-CG sequence contexts, but did not affect methylation on another PAI locus carrying two genes arranged as an inverted repeat. Moreover, an unmethylated PAI2 gene could be methylated de novo in the suvh4 mutant background. These results suggest that SUVH4 is involved in maintenance but not establishment of methylation at particular genomic regions. In contrast, a heterochromatin protein 1 homolog, LHP1, had no effect on PAI methylation.
Keywords: DNA methylation/epigenetics/gene silencing/histone methylation
Introduction
Cytosine methylation is centrally important in regulating gene expression and genome stability in mammals, plants and some fungi. Changes in chromatin structure associated with methylated regions of the genome lead to transcriptional silencing. This silencing mechanism controls fundamental processes including imprinting and X chromo some inactivation in mammals, and suppression of transposon movement in plants. In mammalian genomes, the bulk of methylation is found in the symmetric dinucleotide context CG, whereas plant and fungal genomes carry methylation at both CG and non-CG cytosines.
Insights into the factors involved in DNA methylation have come from genetic approaches in model organisms. For example, three mammalian cytosine methyltransferases, Dnmt1, Dnmt3a and Dnmt3b, have been characterized by knock-out mutations in mice (Li et al., 1992; Okano et al., 1999). These studies revealed that each methyltransferase has a distinct role in genomic methylation patterning and in mouse development. Similarly, genetic studies of two Arabidopsis cytosine methyltransferases, MET1 and CMT3, have shown that these proteins have distinct functions. MET1, which is related to the mammalian DNMT1 gene, has been implicated in maintenance of methylation in the symmetric sequence context CG (Finnegan et al., 1996; Ronemus et al., 1996). CMT3, which combines a chromo-domain motif with the amino acid motifs characteristic of cytosine methyltransferases, has been implicated in maintenance of CNG and other non-CG methylation patterns (Bartee et al., 2001; Lindroth et al., 2001). A related chromomethylase gene from maize, ZMET1, has also been implicated in maintenance of CNG methylation (Papa et al., 2001).
Besides cytosine methyltransferases, relatively little is known about factors that control DNA methylation. Genetic screens in Arabidopsis showed that the DDM1 gene, which encodes a protein related to SWI2/SNF2 chromatin-remodeling factors, is important for maintenance of genomic methylation (Jeddeloh et al., 1999). Loss of DDM1 function confers globally reduced DNA methylation. Similar results have been obtained for a mouse mutant in the DDM1-orthologous gene Lsh (Dennis et al., 2001). DDM1 and Lsh thus indicate a relationship between chromatin structure and DNA methylation.
A genetic screen in the fungus Neurospora revealed that the dim-5 gene, which encodes a SET domain protein, is required for genomic methylation (Tamaru and Selker, 2001). Like the related SU(VAR)3-9 and CLR4 SET domain proteins that have been characterized from mammalian and yeast systems (Rea et al., 2000; Nakayama et al., 2001), the dim-5 gene product catalyzes the methylation of histone H3 at the lysine 9 (K9) position. This modification is associated with histones found in heterochromatic regions of eukaryotic genomes (Noma et al., 2001; Peters et al., 2001; Gendrel et al., 2002; Johnson et al., 2002). The finding that loss of DIM-5 function blocks both H3 methylation and genomic cytosine methylation provides another link between chromatin structure and DNA modification.
The Arabidopsis genome encodes nine SU(VAR)3-9-related genes, named SUVH1–SUVH9 [SU(VAR)3-9 homologs] (Baumbusch et al., 2001). In contrast to SU(VAR)3-9 and CLR4 proteins, the Arabidopsis SUVH proteins lack a chromo-domain at their N-terminus. Instead, the Arabidopsis SUVH proteins share a novel central sequence motif named the YDG domain, with divergent N-termini. Recently, mutations in the SUVH4 gene were isolated as suppressors of methylation and silencing of the SUPERMAN floral homeotic gene (Jackson et al., 2002). In this analysis, the heterologously expressed protein was shown to have histone H3 K9 methyltransferase activity in vitro, consistent with the predicted protein function.
Here we describe the isolation and characterization of seven new loss-of-function mutations in SUVH4. The suvh4 mutations were recovered in a forward genetic screen for trans-acting factors that control DNA methylation and silencing of the endogenous gene PAI2. This screen previously yielded 11 loss-of-function alleles in the CMT3 chromomethylase gene (Bartee et al., 2001).
In the Wassilewskija (WS) strain of Arabidopsis, PAI2 methylation is triggered by an unlinked inverted repeat PAI gene arrangement PAI1–PAI4 (Luff et al., 1999). Mutation of CMT3 reduces non-CG methylation on both PAI2 and the inverted repeat PAI genes, and blocks the ability of the inverted repeat to establish a new methylation imprint on a ‘naïve’ unmethylated singlet PAI2. In contrast, mutation of SUVH4 reduces non-CG methylation on PAI2 but not on the inverted repeat PAI genes, and does not block the ability to establish a new PAI2 methylation imprint. Taken together, our results lead to a model for the establishment and maintenance of methylation on unlinked repeated sequences in plant genomes: CMT3 is needed during both an initiation phase and a maintenance phase of methylation, whereas SUVH4 is only needed during the maintenance phase at targets where methylation is signaled in trans from an unlinked initiator locus. This system probably mirrors the mechanism of transposable element methylation as a means of genome defense (Yoder et al., 1997).
Results
Isolation of suvh4 mutants as suppressors of PAI2 silencing
An endogenous Arabidopsis gene that is subject to cytosine methylation and silencing, PAI2, serves as a facile reporter in genetic screens for methylation mutations. The PAI genes encode an intermediate enzyme in the tryptophan biosynthetic pathway. In some Arabidopsis strains including WS, these genes are arranged as an inverted repeat (PAI1–PAI4) at one locus, plus two singlet genes (PAI2 and PAI3) at unlinked loci, and are cytosine methylated at both CG and non-CG cytosines across their regions of sequence identity (Luff et al., 1999; Melquist et al., 1999). Only PAI1 and PAI2 encode a functional enzyme, and only PAI1 is expressed due to the presence of novel promoter sequences upstream of the methylated region (Melquist et al., 1999). The PAI2 gene is silenced by methylation of its proximal promoter sequences. To exploit PAI2 as a reporter locus, a pai1 missense mutant variant of WS (pai1C251Y) was isolated (Bartee and Bender, 2001). The pai1 strain displays PAI-deficient phenotypes, including blue fluorescence due to accumulation of a tryptophan pathway intermediate, yellow–green leaf pigmentation, reduced size, increased bushiness and reduced fertility, because the remaining functional gene, PAI2, is silenced by methylation. This fluorescent reporter strain was mutagenized and screened for isolates with suppression of the PAI-deficient fluorescent phenotype as a means of finding plants with reduced PAI2 silencing.
The PAI2 silencing suppressor screen yielded 11 loss-of-function mutations in the CMT3 cytosine methyltransferase gene as the predominant class of silencing suppressor mutations (Bartee et al., 2001). Here we describe the other major complementation group of mutations recovered from this screen: seven loss-of-function suvh4 mutations in the SUVH4 SET domain histone methyltransferase gene. In the pai1 reporter strain, suvh4 mutations conferred partial suppression of fluorescence (Figure 1). These phenotypes were similar to those conferred by cmt3 mutations, except that the suppression of fluorescence was weaker in suvh4 than in cmt3 mutant backgrounds. The suvh4 mutations did not confer any obvious morphological defects, even after several generations of inbreeding. There were also no obvious morphological defects for a suvh4 mutation crossed into the wild-type WS background.

Fig. 1. PAI2 silencing is suppressed by suvh4 mutations. Representative 2.5-week-old plants of the indicated strains are shown under visible (upper panel) and UV (lower panel) light. The suvh4302* and cmt3i11a alleles were used. The suppressed phenotype of suvh4R302* is similar to those of the other six suvh4 alleles.
suvh4 mutations reduce methylation at singlet PAI genes but not at the PAI1–PAI4 inverted repeat
In addition to suppression of silenced PAI2 phenotypes, the suvh4 mutations conferred reduced cytosine methylation on the PAI2 reporter gene. Methylation was monitored both by Southern blot assays and by sodium bisulfite genomic sequencing. For Southern blot analysis, genomic DNA was cleaved with the isoschizomers HpaII and MspI, which recognize 5′-CCGG-3′, and probed with a PAI probe. HpaII will not cleave if either the inner (CG) or the outer (CCG) cytosine of the recognition site is methylated, whereas MspI is only sensitive to methylation of the outer cytosine. Thus, if neither enzyme cleaves, the site carries outer CCG methylation, whereas if MspI but not HpaII cleaves, the site carries only inner CG methylation. These enzymes cleave once in each WS PAI locus at a site that is affected by methylation, plus cleaving at unmethylated flanking sites which lie different distances away for each locus (Figure 2A). These enzymes thus allow the methylation status of the site in each PAI locus to be monitored independently.

Fig. 2. suvh4 mutations confer reduced PAI2 methylation. (A) The HpaII (H) and MspI (M) restriction map of each WS PAI locus is shown, with the probed regions indicated by gray bars and the bisulfite sequenced regions indicated by hatched bars. (B) Genomic DNAs prepared from 4-week-old plants of the indicated genotypes were cleaved with either HpaII or MspI and used for Southern blot analysis with a PAI probe. P1-P4 is PAI1–PAI4, P2 is PAI2, and P3 is PAI3, with asterisks indicating the positions of species methylated at internal HpaII/MspI sites. Note that PAI3 is divergent from the probe sequence and thus gives a weaker signal than other PAI genes. (C) The blot shown in (B) was reprobed with a 180 bp CEN repeat probe. The phenotypes of the WS suvh4R302* allele shown are representative of the phenotypes observed with six other suhv4 alleles. The phenotypes of the WS pai1 suvh4R302* cmt3i11a double mutant line shown are representative of the phenotypes observed with three other independent double mutant lines.
HpaII/MspI Southern blot analysis of WS pai1 suvh4 DNA revealed that the recognition sites in the singlet PAI2 and PAI3 genes were partially demethylated and susceptible to increased cleavage by both enzymes relative to the parental strain; however, the recognition site in the PAI1–PAI4 inverted repeat retained methylation and resisted cleavage with both enzymes (Figure 2B). This pattern of demethylation was different from the pattern previously observed for cmt3 mutant DNA, where all three PAI loci are almost completely cleaved by MspI (Figure 2B; Bartee et al., 2001). The suvh4 pattern of reduced methylation for PAI2 but not PAI1–PAI4 versus the cmt3 pattern of reduced methylation on both PAI2 and PAI1–PAI4 was confirmed with a second Southern blot assay using the methylation-sensitive enzyme HincII, which monitors non-CG cytosines at the translational start codons of PAI1, PAI4 and PAI2 (Supplementary figure 1 available at The EMBO Journal Online). The PAI demethylation patterns characteristic of suvh4 persisted when the mutation was segregated away from pai1 in a back-cross to WS (data not shown). Detailed analysis of the effects of ddm1 and met1 mutations on PAI methylation and silencing were described previously (Bartee and Bender, 2001).
To score effects of suvh4 at the methylated 180 bp CEN repeats in Arabidopsis, the PAI HpaII/MspI Southern blot was reprobed with a CEN probe. This analysis revealed no significant enhancement of HpaII cleavage and only a slight enhancement of MspI cleavage relative to the parental control strain at CEN (Figure 2C). Other characterized methylation mutations, ddm1, met1 and cmt3, display different alterations in the patterns of CEN cleavage (Figure 2C; Vongs et al., 1993; Bartee and Bender, 2001). The ddm1 mutation confers increased cleavage by both HpaII and MspI, the met1 mutation confers increased cleavage by HpaII, and the cmt3 mutation confers increased cleavage by MspI.
The PAI1 and PAI2 genes were also subjected to sodium bisulfite genomic sequencing of methylation patterns (Frommer et al., 1992) in the regions just upstream of their translational start codons. These regions include distal heterologous sequences unique to each gene and proximal sequences that are nearly identical among the PAI genes (Figure 3). Sequencing of the same regions in wild-type WS showed that there is dense CG and non-CG methylation within the regions of PAI identity for each gene, but that there is very little methylation in the upstream heterologous regions (Luff et al., 1999). In a WS pai1 suvh4 DNA sample, methylation sequencing analysis showed that the PAI2 reporter locus had a reduction in overall methylation relative to wild-type WS, with most of the residual methylation retained at cytosines in the CG sequence context. This pattern is similar to that previously observed in a cmt3 mutant background (Bartee et al., 2001; Figure 3B; Supplementary figure 2). In contrast, for the PAI1 locus, there was no significant change in methylation relative to wild-type WS. This pattern is different from the PAI1 pattern observed in the cmt3 mutant background, where overall methylation is reduced and the residual methylation occurs primarily at cytosines in the sequence context CG.

Fig. 3. Bisulfite sequencing analysis of the PAI genes in a suvh4 mutant background. (A) Bisulfite genomic methylation sequencing was performed for the top strands of the PAI1 and PAI2 upstream regions in WS pai1 suvh4R302* or WS pai1 suvh4R302* cmt3i11a DNA. For each region, eight independent molecules were sequenced. Vertical lines indicate positions of cytosines, with the height of each line representing how many sequenced molecules had 5-methylcytosine (5-Me-C). Black indicates CG cytosines, blue indicates CNG cytosines, and red indicates other cytosines. Asterisks indicate sites with no methylation. The black horizontal line indicates the region of PAI identity, and the gray horizontal line indicates flanking upstream heterologous sequence unique to each gene. The exon and intron structures of PAI1 and PAI2 are shown as open boxes and dashed lines, respectively, under each sequence. These structures are based on full-length cDNA sequences for each gene (Melquist et al., 1999). (B) Within the region of PAI identity (black horizontal line in A), the percentages of available cytosines that are methylated in CG (black bars), CNG (white bars) or other (gray bars) sequence contexts are shown for either PAI1 (P1) or PAI2 (P2) in the indicated strains. Data for wild-type WS and WS pai1 cmt3 are from previous publications (Luff et al., 1999; Bartee et al., 2001). See also Supplementary figure 2.
Positional cloning of the suvh4 mutations
The suvh4 mutations were mapped as previously described for the cmt3 mutations isolated in the PAI2 silencing suppressor screen (Bartee et al., 2001; see Materials and methods). Mapping analysis showed linkage to a region on the upper arm of chromosome 5, between markers that lie ∼830 kb apart. We focused on the SUVH4 gene in this region as a candidate for the suppressor locus acting on information that suppressors of methylation and silencing of the floral homeotic gene SUPERMAN affect this locus (Jackson et al., 2002). The SUVH4 gene was confirmed as the mutant locus with two approaches. First, the gene was cloned and sequenced from the seven PAI2 silencing suppressor alleles. Each mutant isolate carried a single C:G to T:A transition mutation in the SUVH4 coding region predicted to abrogate protein function (Figure 4; Supplementary figure 3). The suvh4 alleles included two premature termination codons, two splice junction mutations and three missense mutations. To understand the consequences of the splice junction mutations, RNA prepared from mutant plants was used as a template for RT–PCR of the SUVH4 transcript, and the products were analyzed by cloning and sequencing. This analysis revealed that each mutation resulted in at least three different mis-spliced transcript species that created either frameshifts with premature termination of the protein-coding region, or in-frame internal deletions of the protein-coding region (Supplementary figure 4). Thus, both splice site mutations are likely to be null alleles. The S200F and R260H missense mutations alter residues that lie in the YDG domain, and the E341K missense mutation alters a residue in the pre-SET domain of the Arabidopsis SUVH group of proteins (Baumbusch et al., 2001). These residues are conserved in all nine members of the SUVH family, with the exception of SUVH7, which carries a cysteine rather than a serine at the position analogous to S200 in SUVH4. The conserved nature of the mutated residues and the observation that the missense mutations have phenotypes of similar severity to the nonsense and splice mutations suggest that the missense alleles are strong loss-of-function alleles.

Fig. 4. Positions of seven suvh4 alleles recovered as PAI2 silencing suppressors. The predicted amino acid sequence of WS SUVH4 is shown, with the positions of introns marked with black arrowheads. Missense mutations are indicated by the new amino acid above the affected codon. Nonsense mutations are indicated by an asterisk above the affected codon. Splice mutations are indicated by an x at the appropriate junction of the affected intron. The YDG, pre-SET, SET and post-SET domains as previously defined for SUVH4 relative to related proteins (Baumbusch et al., 2001; Jackson et al., 2002) are underlined. The WS genomic sequence of SUVH4 is available as DDBJ/EMBL/GenBank accession No. AF538715.
As a second approach to verify the SUVH4 gene as the site of the mutations, the pai1 suvh4 isolates were transformed with a genomic clone of the wild-type SUVH4 gene. Strongly fluorescent transformants with phenotypes similar to that of the parental pai1 SUVH4 strain were recovered, showing that the cloned SUVH4 gene can complement the silencing defect (Figure 5). Transformant lines assayed by Southern blotting in the T2 generation showed remethylation of PAI2 to the levels observed in WS and WS pai1, although in some cases PAI3 was refractory to remethylation, as previously observed (Luff et al., 1999). In contrast, when the pai1 suvh4 mutant was transformed with a CMT3 genomic clone previously shown to complement the cmt3 mutation (Bartee et al., 2001), all of the transformed progeny retained the suppressed pai1 suvh4 phenotype. This result suggests that extra copies of CMT3 in suvh4 are not sufficient to bypass the mutant defect.

Fig. 5. A SUVH4 transgene can complement the suvh4 mutant defect. (A) Representative T2 2-week-old seedlings of the suvh4R302* mutant transformed with either a SUVH4 genomic clone or a CMT3 genomic clone (two independent lines each) are shown under visible (upper panel) or UV (lower panel) light. (B) Genomic DNAs prepared from single leaves of 4-week-old T2 plants of the same lines shown in (A) were cleaved with either HpaII (H) or MspI (M) and used for Southern blot analysis with a PAI probe. P1-P4 is PAI1–PAI4, P2 is PAI2, and P3 is PAI3, with asterisks indicating the positions of species methylated at internal HpaII/MspI sites.
The suvh4 cmt3 double mutant retains partial PAI2 silencing and methylation
The genetic screen for suppressors of PAI2 silencing revealed that both the histone methyltransferase SUVH4 and the DNA methyltransferase CMT3 are important for maintenance of DNA methylation at this locus, particularly in CNG and other non-CG sequence contexts. A simple interpretation of these results is that SUVH4-mediated histone modifications at the PAI2 locus facilitate CMT3 action at PAI2. To examine the relationship between SUVH4 and CMT3 functions further, we constructed a double suvh4 cmt3 mutant in the WS pai1 background and examined its cytosine methylation and silencing phenotypes relative to each of the parental single mutants.
Overall, the pai1 suvh4 cmt3 double mutant displayed suppressed PAI2 silencing and methylation phenotypes similar to those of the cmt3 single mutant. Double mutant plants had a weakly fluorescent phenotype like that of the pai1 cmt3 single mutant (Figure 1). In Southern blot assays for methylation, double mutant DNA displayed increased cleavage of PAI2 with HpaII relative to either single mutant parent, whereas patterns of PAI1–PAI4, PAI3 and CEN cleavage were similar to those displayed by cmt3 single mutant DNA (Figure 2). HincII Southern blot analysis showed no difference between double mutant and cmt3 single mutant patterns of cleavage in the PAI1–PAI4 and PAI2 loci (Supplementary figure 1). Sodium bisulfite genomic sequencing of the PAI1 and PAI2 upstream regions in double mutant DNA revealed patterns similar to those observed previously for the cmt3 single mutant, with the bulk of residual methylation retained at cytosines in the context CG (Figure 3). However, the density of methylation was slightly lower in the PAI2 upstream region of double mutant DNA than in cmt3 DNA. These results are consistent with the model that SUVH4 is involved primarily in facilitating CMT3 action, with little effect on other factors that contribute to maintenance of PAI or CEN methylation.
An Arabidopsis HP1 homolog is not required for PAI methylation
Heterochromatin protein 1 (HP1) has been implicated as a component of heterochromatin in animal and fungal systems (reviewed in Jenuwein and Allis, 2001; Richards and Elgin, 2002). This protein contains a chromo-domain which binds to histone H3 methylated at K9. The Arabidopsis genome encodes a single protein with HP1 homology, called like HP1 (LHP1) (Gaudin et al., 2001). The LHP1 protein was shown to interact with the CMT3 protein in vitro, leading to the proposal that LHP1 serves as a bridge between the SUVH4-catalyzed histone modification and the targeting of CMT3 (Jackson et al., 2002). This model predicts that loss of LHP1 function should lead to loss of methylation from CMT3 target loci. To test this prediction, we assayed an lhp1 mutant isolated in the WS strain (Gaudin et al., 2001) for changes in PAI and CEN methylation using Southern blot analysis with HpaII and MspI (Figure 6), or for changes in PAI methylation with HincII (Supplementary figure 1). These analyses revealed no differences between parental WS and WS lhp1 for PAI or CEN methylation, suggesting that LHP1 protein is not involved in maintaining DNA methylation at these loci.
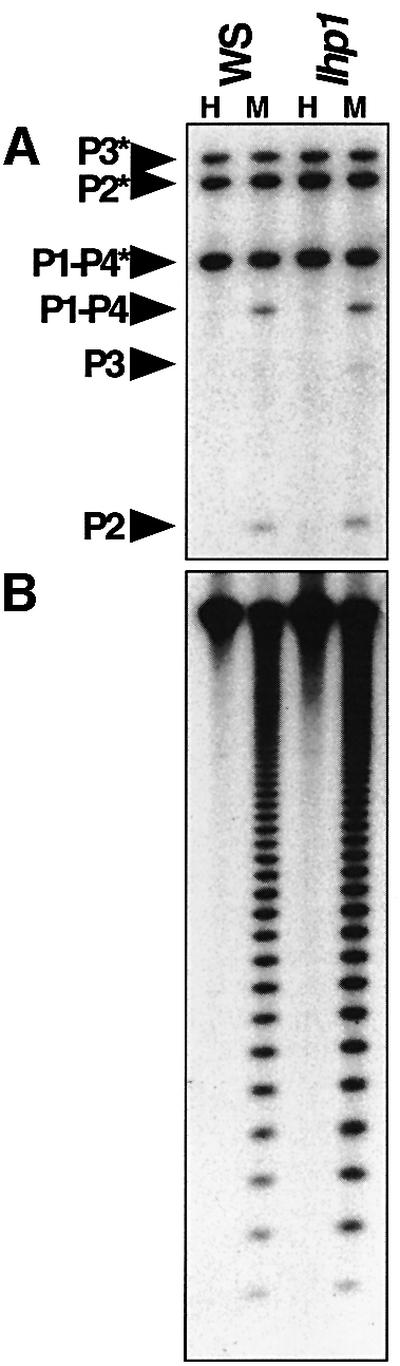
Fig. 6. The lhp1 mutation does not affect PAI or CEN methylation in WS. (A) Genomic DNAs prepared from 4-week-old plants of the indicated genotypes were cleaved with either HpaII (H) or MspI (M) and used for Southern blot analysis with a PAI probe. P1-P4 is PAI1–PAI4, P2 is PAI2, and P3 is PAI3, with asterisks indicating the positions of species methylated at internal HpaII/MspI sites. (B) The blot shown in (A) was reprobed with a 180 bp CEN repeat probe. The lhp1-1 allele, which displays characteristic developmental defects, was used for this analysis (Gaudin et al., 2001).
CMT3, but not SUVH4, is required for establishment of methylation of PAI2
In previous work, we showed that the PAI1–PAI4 inverted repeat locus provides a signal for methylation of PAI sequences. In the genomes of most commonly used strains of Arabidopsis, including Columbia (Col) and Landsberg erecta (Ler), there are three singlet PAI genes (PAI1, PAI2 and PAI3), and these genes have no detectable cytosine methylation (Melquist et al., 1999). Only strains such as WS that carry a PAI1–PAI4 inverted repeat gene structure at the PAI1 locus display dense methylation of their PAI genes. If the PAI1–PAI4 inverted repeat from WS is combined with an unmethylated PAI2 locus from Col via genetic crosses, the PAI2 locus becomes methylated de novo (Luff et al., 1999). This methylation increases progressively upon inbreeding: the Col PAI2 methylation reaches a density similar to that found on WS PAI2 by the F4 generation in such WS × Col hybrid lines.
We wanted to determine whether inverted repeat-triggered PAI2 methylation can occur in strains that are deficient in either SUVH4 or CMT3. To address this question, we performed crosses between a WS pai1 suvh4 mutant and a Ler suvh4 mutant, or between a WS pai1 cmt3 mutant and a Ler cmt3 mutant, and examined whether the Ler PAI2 could acquire silencing and cytosine methylation when combined in a hybrid genome with the WS pai1–PAI4 inverted repeat. As a control, we performed the analogous wild-type cross between WS pai1 and Ler. The use of the WS pai1–PAI4 locus with a missense mutation in the PAI1 gene allowed us to monitor silencing of Ler PAI2 via acquisition of a blue fluorescent PAI-deficient phenotype.
There is almost no methylation on PAI2 sequences in wild-type Ler, as determined both by HpaII/MspI Southern blot analysis (Figure 7; Melquist et al., 1999) and by bisulfite genomic sequencing of the PAI2 upstream region (two methylated sites out of 584 total cytosines monitored). Similarly, HpaII/MspI Southern blot analysis shows that the PAI genes are not methylated in SUVH4- or CMT3-deficient Ler backgrounds (Figure 7).

Fig. 7. The WS pai1–PAI4 inverted repeat locus triggers de novo methylation and silencing of an unmethylated Ler PAI2 gene in a SUVH4-deficient but not a CMT3-deficient background. (A) To the left, representative WxL wild-type (F4), WxL suvh4 (F5) or WxL cmt3 (F5) 2.5-week-old seedlings are shown under visible (left panel) or UV (right panel) light. To the right, diagrams show the PAI genotypes and methylation phenotypes in these lines. PAI genes inherited from WS are indicated as black arrows. Note that none of the three WS PAI genes encodes functional PAI enzyme. The WS PAI1–PAI4 and PAI3 loci both lie on the upper arm of chromosome 1, indicated by a slash separating the two loci. The Ler PAI2 gene is indicated by a red arrow. Methylation is indicated by boxes around the affected genes, with a solid line representing dense methylation and a dashed line representing partial methylation. (B) Genomic DNAs prepared from 4-week-old plants of the indicated strains were cleaved with either HpaII (H) or MspI (M) and used for Southern blot analysis with a PAI probe. P1-P4 is WS PAI1–PAI4, P1 is Ler PAI1, P2 is PAI2, and P3 is PAI3, with asterisks indicating the positions of species methylated at internal HpaII/MspI sites. WxL wild-type, WxL suvh4 and WxL cmt3 DNAs were prepared from F4, F5 and F5 generation plants, respectively.
In the wild-type, the SUVH4-deficient and the CMT3-deficient WS pai1 × Ler experiments, populations of F2 progeny plants were scored for their genotypes at the PAI1 and PAI2 loci as previously described (Luff et al., 1999) to identify individuals homozygous for both the trigger WS pai1–PAI4 inverted repeat and the target Ler PAI2 gene. We examined six independent ‘WxL’ wild-type control lines, two independent ‘WxL suvh4’ lines and four independent ‘WxL cmt3’ lines.
In the WxL wild-type control experiment, three lines were non-fluorescent and three lines were fluorescent in the F2 generation. All six lines segregated all or almost all fluorescent progeny in the F3 generation. By the F4 generation, all six lines were strongly fluorescent, similar to the parental WS pai1 strain (Figures 1 and 7). The fluorescent phenotype correlated with partial methylation of the Ler PAI2 gene, as determined by HpaII/MspI Southern blotting of DNA prepared from F4 generation plants (Figure 7). These WS pai1 × Ler PAI2 de novo methylation patterns show that the single base missense mutation in the pai1 gene does not impair the ability of the inverted repeat to trigger methylation. However, these strains did not display complete remethylation of PAI2 to WS levels by the F4 generation, as previously observed in the analogous WS × Col experiment (Luff et al., 1999). This difference might represent a selection against the most strongly silenced PAI-deficient female gametes in the pai1 background (Bender and Fink, 1995).
In the SUVH4-deficient experiment, neither of the two WxL suvh4 lines was detectably fluorescent in the F2 generation. However, both lines segregated ∼30% F3 progeny that were fluorescent, suggesting that there was progressive partial methylation of PAI2 during the F3 generation. Two individual non-fluorescent F3 plants of each line gave rise to F4 progeny that segregated all or almost all fluorescent plants, consistent with a progressive trend towards increased methylation and silencing. In fact, DNA prepared from progeny F5 populations showed slight PAI2 methylation by HpaII/MspI Southern blot analysis, similar to the patterns observed in the WS pai1 suvh4 strain (Figures 2 and 7). WxL suvh4 F5 plants also displayed an intermediate fluorescence phenotype similar to the WS pai1 suvh4 strain (Figures 1 and 7). These results indicate that the Ler PAI2 gene can acquire and maintain methylation and silencing despite a SUVH4 deficiency.
In the CMT3-deficient experiment, none of four WxL cmt3 lines was detectably fluorescent in the F2 generation. In contrast to the SUVH4-deficient experiment, none of these lines segregated fluorescent progeny plants in the F3 generation. Furthermore, two individual F3 plants from each line yielded F4 progeny populations that were completely non-fluorescent. Similarly, two individual F4 plants from each line yielded F5 progeny populations that were completely non-fluorescent. DNA prepared from these F5 populations showed no significant PAI2 methylation by HpaII/MspI Southern blot analysis (Figure 7); in comparison, WS pai1 cmt3 strains display intermediate fluorescence and a readily detectable methylated PAI2 species in a HpaII digest (Figures 1 and 2; Bartee et al., 2001). The pai1–PAI4 locus and the PAI3 locus were inherited from the WS pai1 cmt3 parent in all four WxL cmt3 lines, and these loci maintained CG methylation to the same levels as observed in the parent strain (Figures 2 and 7). These results suggest that although existing methylation imprints can be maintained in the absence of CMT3 function, new PAI2 methylation imprints cannot be propagated to the point where they result in methylation or silencing phenotypes like those observed for the parental pai1 cmt3 strain.
Discussion
Genomic DNA methylation is catalyzed by cytosine methyltransferase enzymes, but the factors that control the targeting and activity of these enzymes at different genomic sites are not well understood. Here we show that a SET domain histone methyltransferase protein, SUVH4, is important for maintenance of particular cytosine methylation patterns in the Arabidopsis genome. SUVH4 is involved mainly in maintenance of non-CG methylation at a subset of the sites controlled by the predicted cytosine methyltransferase CMT3.
In Arabidopsis, two cytosine methyltransferases, MET1 and CMT3, have been characterized by genetic analysis. Loss of MET1 function reduces methylation primarily in the sequence context CG, which is the predominant methylation context in the Arabidopsis genome, and leads to developmental pleiotropy (Finnegan et al., 1996; Ronemus et al., 1996). In contrast, loss of CMT3 function reduces methylation primarily in CNG and other non-CG contexts and does not confer obvious developmental alterations (Bartee et al., 2001; Lindroth et al., 2001).
The WS PAI genes display a high level of CG and non-CG methylation across their regions of shared sequence identity (Luff et al., 1999), suggesting that they are preferred substrates for both MET1 and CMT3. Indeed, loss of function in either cytosine methyltransferase gene leads to partially reduced methylation on the PAI1–PAI4 inverted repeat methylation trigger locus and on two unlinked singlet genes PAI2 and PAI3 (Figure 2; Bartee and Bender, 2001; Bartee et al., 2001). In the case of cmt3 mutations, the three PAI loci display a weak reduction in CG methylation and a strong reduction in non-CG methylation relative to parental WS. Our finding that suvh4 mutations cause a similar loss of mostly non-CG methylation from the singlet PAI2 gene (Figures 2 and 3) suggests that the SUVH4 histone-modifying activity is necessary for CMT3 to act at this region. Previous in vitro studies showed that heterologously expressed CMT3 does not interact directly with K9-methylated H3 peptides (Jackson et al., 2002), and the proposal that the Arabidopsis HP1 homolog LHP1 is a bridging factor for this interaction is not supported by our observation that an lhp1 mutation has no effect on PAI or CEN methylation (Figure 6). Therefore, SUVH4 might facilitate CMT3 action at PAI2 through an indirect effect on overall chromatin structure.
Strikingly, in contrast to cmt3 mutations, suvh4 mutations do not affect maintenance of dense CG and non-CG methylation on the PAI1–PAI4 inverted repeat locus (Figures 2 and 3). This result suggests that SUVH4-mediated histone methylation is not needed for CMT3 or other cytosine methyltransferases to act at PAI1–PAI4. Similarly, suvh4 mutations only partially reduce CCG methylation at CEN repeats relative to cmt3 mutations (Figure 2; Jackson et al., 2002), suggesting only a partial requirement for SUVH4 at these repeats to facilitate CMT3 action. One explanation for these differences in suvh4 and cmt3 DNA methylation phenotypes is that other SUVH proteins (Baumbusch et al., 2001) contribute to histone modifications in the PAI1–PAI4 and CEN regions of the genome. However, recent chromatin immunoprecipitation experiments with antibodies against histone H3 methylated at K9 showed that suvh4 mutant chromatin is depleted for K9 methylation at CEN and other DNA methylated genomic sites, arguing that SUVH4 is the major H3 K9-modifying activity for heterochromatin in Arabidopsis (Johnson et al., 2002). An alternative view is that repeated sequences like PAI1–PAI4 and CEN might have chromatin structure features that attract CMT3 independently of K9 methylation status.
Like our screen for suppressors of hypermethylated PAI2 silencing, a screen for suppressors of hypermethylated SUPERMAN silencing yielded loss-of-function mutations in CMT3 and SUVH4 (Lindroth et al., 2001; Jackson et al., 2002). Analysis of the effects of these mutations at methylated transposon sequences and the CEN repeats revealed the general pattern that cmt3 mutations confer stronger effects than suvh4 mutations on CNG methylation, consistent with our findings. The parallel results from the two different systems indicate that CMT3 and SUVH4 are major factors in the control of non-CG methylation genome-wide. However, a notable difference between the two systems is the effect of suvh4 mutation on inverted repeat methylation. In the SUPERMAN screen, an inverted repeat transgene carrying SUPERMAN genomic sequences was used to stabilize hypermethylation of the endogenous singlet SUPERMAN gene. Collective bisulfite genomic sequencing of these three SUPERMAN copies revealed strong demethylation by mutation of SUVH4 (Jackson et al., 2002), implying that the inverted repeat copies and the singlet gene are affected uniformly. This demethylation of the SUPERMAN inverted repeat by suvh4 could reflect its lower CG content or its transgenic nature versus the endogenous PAI1–PAI4 inverted repeat.
The maintenance of CG methylation in suvh4 mutants suggests that the MET1 DNA methyltransferase does not require SUVH4-mediated histone methylation. It will be interesting to determine whether the MET1-related mammalian methyltransferase Dnmt1 might be similarly independent of H3 K9 methylation. In Neurospora, loss of H3 K9 methylation confers complete loss of CG and non-CG methylation (Tamaru and Selker, 2001). This finding suggests that the mechanistic relationship between histone modification and DNA methylation has evolved differently in Neurospora from that in plants. In this regard, DNA methylation in Neurospora is controlled by the DIM-2 methyltransferase, which is structurally distinct from both MET1- and CMT3-related enzymes (Kouzminova and Selker, 2001).
In plants, certain aberrant RNA species such as double-stranded RNAs and RNA virus replication intermediates can direct DNA methylation of identical sequences (reviewed in Wassenegger, 2000; Bender, 2001). Because RNA-directed DNA methylation is typically densely patterned with a high proportion of non-CG methylation, it has been proposed that CMT3 is the methyltransferase that acts in response to an RNA signal (Martienssen and Colot, 2001; Matzke et al., 2001). In the WS PAI system, read-through RNA species from the PAI1–PAI4 inverted repeat could provide a trigger for PAI-directed dense DNA methylation. Our finding that loss of CMT3 function blocks the ability of the PAI1–PAI4 inverted repeat to promote methylation on an unmethylated PAI2 target (Figure 7) is consistent with a central role for CMT3 in this potentially RNA-directed process.
One possibility is that CMT3 could act as both a de novo and a maintenance methyltransferase for PAI methylation. During the initiation of methylation, CMT3 would be required for a primary methylation imprint. The primary imprint would then be propagated and maintained at CG cytosines by MET1 and at non-CG cytosines by CMT3. In this model, SUVH4 would be needed to facilitate CMT3 action during the maintenance phase. However, another class of putative cytosine methyltransferases, the DRMs, has recently been shown to be required for de novo methylation of other Arabidopsis methylation target loci (Cao and Jacobsen, 2002). Therefore, an alternative possibility is that the DRMs provide the primary methylation imprint, but that CMT3 is required to propagate this imprint up to the point where MET1 will effectively maintain CG methylation. A third possibility is that the PAI1–PAI4 methylation trigger locus must itself be densely methylated at CG and non-CG cytosines in order to produce a methylation signal that can act in trans on PAI2; the absence of this dense methylation in a cmt3 background would block production of the signal. This third possibility is consistent with the observation that PAI2 de novo methylation can be triggered in a suvh4 mutant background (Figure 7), where dense methylation on PAI1–PAI4 is retained (Figures 2 and 3).
Mammalian genomes contain mostly CG DNA methylation patterning, and this limited methylation sequence context is sufficient to mediate effective gene silencing. The CG methylation context is also the predominant pattern found in the Arabidopsis genome. The question then arises of why Arabidopsis and other plants have evolved the capacity to maintain additional non-CG methylation patterns via CMT3-related cytosine methyltransferases. Our results suggest that non-CG methylation acts as a reinforcement to CG methylation in promoting an additional level of gene silencing. In the WS pai1 strain where PAI2 is densely methylated at both CG and non-CG residues, PAI2 is strongly silenced and the plant has a severely PAI-deficient phenotype; when non-CG methylation is lost from PAI2 via suvh4 or cmt3 mutation, PAI2 silencing persists but at a reduced level (Figure 1). Because dense non-CG patterning is associated with RNA-directed DNA methylation, an attractive hypothesis is that DNA sequences perceived as being related to RNA viruses or mobile elements due to production of aberrant RNAs are targeted for an extra level of methylation and silencing as an extra level of genome defense. In fact, mutation of CMT3 reactivates transcription of some retroelement-related sequences (Lindroth et al., 2001), and CMT3-mediated methylation has been shown to be targeted preferentially to some transposable element sequences (Tompa et al., 2002). Thus, CMT3 and SUVH4 are likely to be necessary for the long-term fitness of the plant genome in the face of invasive virus and transposon sequences.
Materials and methods
Mutant isolation and sequencing
Seeds of WS pai1C251Y were mutagenized with ethylmethane sulfonate (EMS), and M2 progeny seedlings were grown on agar medium and scored at 2 weeks for reduced fluorescence as previously described (Bartee et al., 2001). Putative mutants were transplanted to soil, and genomic DNA prepared from a single leaf was used for HpaII/MspI Southern blot analysis of methylation patterns. The PAI and CEN Southern blot probes were as previously described (Bartee and Bender, 2001). From the screen of 20 000 M2 seedlings, seven independent isolates with the Southern blot pattern characteristic of suvh4 mutants (Figure 2) were recovered, and these isolates proved to be suvh4 mutants by mapping, sequencing and complementation analysis with a SUVH4 transgene.
The suvh4 suppressor isolates were mapped by crossing with the polymorphic strain Niederzenz (Nd-0), which has an arrangement of densely methylated PAI genes similar to that found in WS (Melquist et al., 1999). F2 progeny of the cross with a weak fluorescent phenotype indicative of homozygosity for both the pai1 mutation and the suvh4 mutation were selected, and confirmed as having the suppressor mutation by an MspI Southern blot methylation assay. A mapping population of confirmed pai1 suvh4 F2 plants was then scored with standard cleaved amplified polymorphic sequence (CAPS) (Konieczny and Ausubel, 1993) and simple sequence length polymorphism (SSLP) (Bell and Ecker, 1994) markers that are polymorphic between WS and Nd-0. This analysis showed linkage to a single locus on the upper arm of chromosome 5. The locus was narrowed down to an ∼830 kb region between the Arabidopsis BAC clones MSH12 and T21H19. Information about the markers used at these loci is provided in the Supplementary data.
Mutant alleles of SUVH4 were amplified by PCR from genomic DNA. Products from two independent PCRs were cloned and sequenced for each allele. To facilitate genetic analysis, a PCR-based marker was designed where the base change created by the suvh4R302* mutation is combined with a mismatch at the end of a nearby PCR primer to create a restriction site polymorphism (see Supplementary data).
The WS pai1 suvh4 cmt3 double mutant strain was made by crossing pai1 suvh4R302* with a pai1 cmt3 allele (intron 11 acceptor site, cmt3i11a) previously isolated as a PAI2 silencing suppressor (Bartee et al., 2001), and screening the progeny plants for genotype by PCR-based assays. The suvh4R302* mutation was scored as described above, and the cmt3i11a mutation was scored by a restriction enzyme site change created by the mutation (see Supplementary data). The cmt3i11a mutation is a G to A transition at the splice acceptor site of the intron immediately upstream of the active site motif of CMT3 that creates an MseI site. The splice mutation causes mis-splicing to a cryptic acceptor site 1 bp downstream of the normal splice junction, causing a frameshift that disrupts the active site sequence and results in premature termination at codon 476. This cmt3 allele is thus likely to be a null mutation. Four independent double mutant lines were pursued by inbreeding and Southern blot methylation assays, and each gave identical phenotypes. A representative line was used for the methylation analyses shown in Figures 2 and 3.
For the Ler PAI2 de novo methylation experiment shown in Figure 7, the WS pai1 suvh4R302*, WS pai1 cmt3i11a, Ler kyp-2 (Jackson et al., 2002) and Ler cmt3-7 (Lindroth et al., 2001) alleles were used.
Sodium bisulfite genomic sequencing of methylation patterns
Mutant effects on PAI methylation were monitored by sequencing the top strands of the regions upstream of the start codons of the PAI1 and PAI2 genes in bisulfite-modified genomic DNA. The DNA samples used were prepared from the representative pai1 suvh4R302* allele that had been backcrossed to WS once and inbred for three generations, and from a pai1 suvh4R302* cmt3i11a double mutant that had been inbred for two generations. The same DNA samples were used for Southern blot analysis of DNA methylation patterns (Figure 2). Bisulfite treatment was performed as previously described (Jeddeloh et al., 1998; Luff et al., 1999), except that denaturation of genomic DNA was performed in 0.3 M NaOH for 20 min at 37°C. Products were amplified by PCR from the modified genomic DNA template and cloned into the pGEM T-EASY (Promega) vector for sequencing.
Plant transformation with the SUVH4 genomic clone
The SUVH4 transgene is an 11.8 kb Col genomic fragment extending from a SalI site that lies 6.4 kb upstream of the start codon to a SalI site that lies 1.0 kb downstream of the stop codon subcloned into the SalI site of the pBIN19 transformation vector (Bevan, 1984). This clone was isolated by hybridization from a Col genomic λ plaque library (Bender and Fink, 1998). The CMT3 transgene was described previously (Bartee et al., 2001). Transgenes were introduced into the WS pai1C251Y suvh4R302* strain by an in planta transformation method (Clough and Bent, 1998).
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Steve Jacobsen (University of California, Los Angeles) for pre-publication information about positional cloning of suvh4 mutations, and for the gift of SUVH4 sequencing primers, Ler cmt3-7 seeds and Ler kyp-2 seeds. We would also like to thank Valerie Gaudin (INRA, Versailles) for the gift of WS lhp1-1 seeds. This work was supported by grants from the Searle Scholars Foundation (97-E-103), March of Dimes Birth Defects Foundation (FY00-418) and NIH (1R01GM61148) to J.B. F.M. was supported by a post-doctoral fellowship from the European Molecular Biology Organization (ALTF 443-2000). L.B. was supported by NCI training grant 5-T32-CA09110.
References
- Bartee L. and Bender,J. (2001) Two Arabidopsis methylation-deficiency mutations confer only partial effects on a methylated endogenous gene family. Nucleic Acids Res., 29, 2127–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartee L., Malagnac,F. and Bender,J. (2001) Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev., 15, 1753–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbusch L.O., Thorstensen,T., Krauss,V., Fischer,A., Naumann,K., Assalkhou,R., Schulz,I., Reuter,G. and Aalen,R.B. (2001) The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res., 29, 4319–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C.J. and Ecker,J.R. (1994) Assignment of 30 microsatellite loci to the linkage map of Arabidopsis. Genomics, 19, 137–144. [DOI] [PubMed] [Google Scholar]
- Bender J. (2001) A vicious cycle: RNA silencing and DNA methylation in plants. Cell, 106, 129–132. [DOI] [PubMed] [Google Scholar]
- Bender J. and Fink,G.R. (1995) Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell, 83, 725–734. [DOI] [PubMed] [Google Scholar]
- Bender J. and Fink,G.R. (1998) A Myb homologue, ATR1, activates tryptophan gene expression in Arabidopsis. Proc. Natl Acad. Sci. USA, 95, 5655–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan M. (1984) Binary Agrobacterium vectors for plant transformation. Nucleic Acids Res., 12, 8711–8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X. and Jacobsen,S.E. (2002) Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol., 12, 1138–1144. [DOI] [PubMed] [Google Scholar]
- Clough S.J. and Bent,A.F. (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J., 16, 735–743. [DOI] [PubMed] [Google Scholar]
- Dennis K., Fan,T., Geiman,T., Yan,Q. and Muegge,K. (2001) Lsh, a member of the SNF2 family, is required for genome-wide methylation. Genes Dev., 15, 2940–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan E.J., Peacock,W.J. and Dennis,E.S. (1996) Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl Acad. Sci. USA, 93, 8449–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M., McDonald,L.E., Millar,D.S., Collis,C.M., Watt,F., Grigg, G.W., Molloy,P.L. and Paul,C.L. (1992) A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA, 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin V., Libault,M., Pouteau,S., Juul,T., Zhao,G., Lefebvre,D. and Grandjean,O. (2001) Mutations in like heterochromatin protein 1 affect flowering time and plant architecture in Arabidopsis. Development, 128, 4847–4858. [DOI] [PubMed] [Google Scholar]
- Gendrel A.V., Lippman,Z., Yordan,C., Colot,V. and Martienssen,R. (2002) Dependence of heterochromatic histone H3 methylation patterns on the Arabidopsis gene DDM1. Science, 297, 1871–1873. [DOI] [PubMed] [Google Scholar]
- Jackson J.P., Lindroth,A.M., Cao,X. and Jacobsen,S.E. (2002) Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature, 416, 556–560. [DOI] [PubMed] [Google Scholar]
- Jeddeloh J.A., Bender,J. and Richards,E.J. (1998) The DNA methylation locus DDM1 is required for maintenance of gene silencing in Arabidopsis. Genes Dev., 12, 1714–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeddeloh J.A., Stokes,T.L. and Richards,E.J. (1999) Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat. Genet., 22, 94–97. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. and Allis,C.D. (2001) Translating the histone code. Science, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Johnson L.M., Cao,X. and Jacobsen,S.E. (2002) Interplay between two epigenetic marks: DNA methylation and histone H3 lysine 9 methylation. Curr. Biol., 12, 1360–1367. [DOI] [PubMed] [Google Scholar]
- Konieczny A. and Ausubel,F.M. (1993) A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J., 4, 403–410. [DOI] [PubMed] [Google Scholar]
- Kouzminova E. and Selker,E.U. (2001) dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J., 20, 4309–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E., Bestor,T.H. and Jaenisch,R. (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell, 69, 915–926. [DOI] [PubMed] [Google Scholar]
- Lindroth A.M., Cao,X., Jackson,J.P., Zilberman,D., McCallum,C.M., Henikoff,S. and Jacobsen,S.E. (2001) Requirement of Chromo methylase3 is required for maintenance of CpXpG methylation. Science, 292, 2077–2080. [DOI] [PubMed] [Google Scholar]
- Luff B., Pawlowski,L. and Bender,J. (1999) An inverted repeat triggers cytosine methylation of identical sequences in Arabidopsis. Mol. Cell, 3, 505–511. [DOI] [PubMed] [Google Scholar]
- Martienssen R.A. and Colot,V. (2001) DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science, 293, 1070–1074. [DOI] [PubMed] [Google Scholar]
- Matzke M., Matzke,A.J.M. and Kooter,J.M. (2001) RNA: guiding gene silencing. Science, 293, 1080–1083. [DOI] [PubMed] [Google Scholar]
- Melquist S., Luff,B. and Bender,J. (1999) Arabidopsis PAI gene arrangements, cytosine methylation and expression. Genetics, 153, 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J., Rice,J.C., Strahl,B.D., Allis,C.D. and Grewal,S.I.S. (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science, 292, 110–113. [DOI] [PubMed] [Google Scholar]
- Noma K., Allis,C.D. and Grewal,S. (2001) Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science, 293, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Okano M., Bell,D.W., Haber,D.A. and Li,E. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Papa C.M., Springer,N.M., Muszynski,M.G., Meeley,R. and Kaeppler, S.M. (2001) Maize chromomethylase Zea methyltransferase2 is required for CpNpG methylation. Plant Cell, 13, 1919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A.H.F.M. et al. (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell, 107, 323–337. [DOI] [PubMed] [Google Scholar]
- Rea S. et al. (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature, 406, 593–599. [DOI] [PubMed] [Google Scholar]
- Richards E.J. and Elgin,S.C.R. (2002) Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell, 108, 489–500. [DOI] [PubMed] [Google Scholar]
- Ronemus M.J., Galbiati,M., Ticknor,C., Chen,J. and Dellaporta,S.L. (1996) Demethylation-induced developmental pleiotropy in Arabidopsis. Science, 273, 654–657. [DOI] [PubMed] [Google Scholar]
- Tamaru H. and Selker,E.U. (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature, 414, 277–283. [DOI] [PubMed] [Google Scholar]
- Tompa R., McCallum,C.M., Delrow,J., Henikoff,J.G., van Steensel,B. and Henikoff,S. (2002) Genome-wide profiling of DNA methylation reveals transposon targets of Chromomethylase3. Curr. Biol., 12, 65–68. [DOI] [PubMed] [Google Scholar]
- Vongs A., Kakutani,T., Martienssen,R.A. and Richards,E.J. (1993) Arabidopsis thaliana DNA methylation mutants. Science, 260, 1926–1928. [DOI] [PubMed] [Google Scholar]
- Yoder J.A., Walsh,C.P. and Bestor,T.H. (1997) Cytosine methylation and the ecology of intragenomic parasites. Trends Genet., 13, 335–340. [DOI] [PubMed] [Google Scholar]
- Wassenegger M. (2000) RNA-directed DNA methylation. Plant Mol. Biol., 43, 203–220. [DOI] [PubMed] [Google Scholar]