

Abstract
Mechanistic models of charge transfer (CT) in macromolecules often focus on CT energetics and distance as the chief parameters governing CT rates and efficiencies. However, in DNA, features unique to the DNA molecule, in particular, the structure and dynamics of the DNA base stack, also have a dramatic impact on CT. Here we probe the influence of subtle structural variations on base–base CT within a DNA duplex by examining photoinduced quenching of 2-aminopurine (Ap) as a result of hole transfer (HT) to guanine (G). Photoexcited Ap is used as a dual reporter of variations in base stacking and CT efficiency. Significantly, the unique features of DNA, including the strandedness and directional asymmetry of the double helix, play a defining role in CT efficiency. For an (AT)n bridge, the orientation of the base pairs is critical; the yield of intrastrand HT is markedly higher through (A)n compared with (T)n bridges, whereas HT via intrastrand pathways is more efficient than through interstrand pathways. Remarkably, for reactions through the same DNA bridge, over the same distance, and with the same driving force, HT from photoexcited Ap to G in the 5′ to 3′ direction is more efficient and less dependent on distance than HT from 3′ to 5′. We attribute these differences in HT efficiency to variations in base–base coupling within the DNA assemblies. Thus base–base coupling is a critical parameter in DNA CT and strongly depends on subtle structural nuances of duplex DNA.
The transport of electronic charge through double-helical DNA continues to fascinate and surprise us (1–5). A wealth of experimental evidence has established, undeniably, that DNA can act as a conduit for rapid and long-range charge transfer (CT), not only in DNA assemblies designed in the laboratory (6–24), but also in biologically significant environments (25, 26). DNA, remarkable for its role in the molecular basis of life, may also play a role as a mediator of CT in diverse areas of chemistry and biology. Indeed, the ability of DNA to mediate CT and the exquisite sensitivity of this chemistry to DNA structure has spawned the development of a completely new family of diagnostic tools (27, 28). Yet, while we continue to probe and exploit the distinctive ability of DNA to transport charge, fundamental questions concerning the mechanisms and features of DNA CT remain.
We investigate DNA CT by using a combination of spectroscopic, biochemical, and electrochemical tools to interrogate well-characterized assemblies that incorporate redox-active molecules from a large repertoire including metallointercalators, organic intercalators, and modified DNA bases (6–12, 25–28). Consistently, we have observed that DNA CT is remarkably sensitive to the structure and dynamics of the DNA bases. Experiments in which the DNA base stack is altered by, for instance, sequence variation (10), mismatches (9, 28), structural perturbations (11), or protein binding (12, 27) emphasize the importance of the integrity of the π-stack to DNA CT. In addition, time-resolved investigations of ethidium-modified DNA duplexes established that dynamic variations in stacking are crucial to DNA CT and suggest that molecular motions within DNA may serve to gate CT (8). This sensitivity of CT to DNA structure and dynamics attests to the fact that an effectively coupled π-stack, including the redox participants and the intervening DNA bases, is required for DNA CT.
Photoexcited 2-aminopurine (Ap*) is a sensitive probe both of structure and dynamics within DNA assemblies (29–31) and base–base CT (6, 7, 32). Using Ap*, we have directly observed ultrafast base–base CT and evaluated the dependence of CT on distance and driving force. Since Ap* acts as a dual reporter of DNA structural dynamics and CT, it is uniquely suited for probing base–base interactions and the influence of these interactions on CT through DNA. Such investigations also benefit from the fact that the probe, Ap*, is effectively a DNA base, thereby facilitating examination of structurally well-defined DNA assemblies unperturbed by auxiliary reagents. This study contrasts with most spectroscopic and biochemical investigations of DNA-mediated CT that use pendant redox reagents that are not integral components of DNA.
By analogy to well-described paradigms for CT in proteins (33), mechanistic studies of CT in DNA tend to focus on the same fundamental parameters, namely the energetics and distance dependence of CT. Features specific to DNA structure and dynamics that are undoubtedly significant to CT mechanisms have received much less attention. Yet countless studies of CT reactions in DNA over similar energetic and distance regimes have reported widely different rate constants and efficiencies, pointing to the significance of other parameters to DNA CT. Base–base coupling in DNA, resulting, qualitatively, from constructive orbital overlap, is expected to be crucial for CT through the dynamic stack of DNA bases. Certainly our experimental data emphasize the exquisite sensitivity of DNA CT to stacking interactions, indicating that for DNA, variations in base–base coupling may be as, if not more, important than energetics or distance in mechanistic descriptions of CT.
Here we test these ideas by probing the impact of the most subtle structural variations in the base stack on DNA CT. Our approach relies on sensitive spectroscopic probing of DNA structure and DNA-mediated CT with Ap*. In this way it is possible to characteristically modify DNA structure in assemblies where the CT distance and energetics are fixed, to discretely alter base–base coupling and assess how this influences DNA CT. These investigations reveal not only the remarkable dependence of DNA CT on base–base coupling, but also the sensitivity of base–base coupling to subtle structural nuances within DNA.
Materials and Methods
Oligonucleotides.
DNA oligonucleotides were synthesized on an Applied Biosystems DNA Synthesizer by using standard solid-phase techniques and twice purified on a Hewlett–Packard HPLC with a reverse-phase C-18 column with an acetonitrile/30 mM ammonium acetate gradient. DNA oligonucleotides were quantified by using UV-visible spectroscopy. Duplex solutions (100 mM sodium phosphate buffer, pH 7) were prepared by combining equimolar amounts of the desired DNA complements and annealing with regulated cooling from 90° to 4°C over a period of 3 h. Duplex formation was evaluated by examining the temperature-dependent absorbance of Ap at 325 nm. All duplexes displayed cooperative thermal denaturation profiles with melting temperatures ≥32°C and were therefore fully duplexed at the experimental temperatures of 5° or 10°C. The distances between Ap and I or G were obtained from molecular models of the DNA assemblies generated by using commercial software (insight ii) and standard B-DNA geometry.
Spectroscopy.
Steady-state fluorescence measurements on Ap-containing DNA duplexes were conducted with an ISS (Champaign, IL) K2 fluorimeter using cells with a 5-mm pathlength. Emission spectra (100 μM duplexes, 10°C) were obtained by exciting at 325 nm and monitoring the integrated emission between 340 and 500 nm. Fluorescence polarization measurements (100 μM duplexes, 10°C) examined the polarized emission at 370 nm after excitation with polarized light at 325 nm. Excitation spectra (5 μM duplexes, 5°C) were obtained by monitoring the emission at 370 nm while scanning excitation wavelengths between 240 and 350 nm. Evaluation of the yield of CT from emission spectra was accomplished by comparing the observed fluorescence intensity in redox-active G-containing duplexes to otherwise identical duplexes in which the G is replaced by inosine (I), an analog of G that is inactive toward CT quenching of Ap* (7). This control accounts for the subtle influences of oligonucleotide sequence and duplex environment on the fluorescence of Ap* and accurately quantifies fluorescence quenching caused by hole transfer (HT). The fraction of fluorescence quenching caused by HT from G to Ap* is thus defined by Fq, where Fq = 1 − ΦG/ΦI. Similarly, the dependence of HT yield on distance, r, can be described by the parameter γ, according to the equation, ln[(ΦI/ΦG) − 1] = −γr.
Results
Design and Characterization of DNA Assemblies.
Because of the unique photophysics of Ap relative to natural DNA bases (34–36), it is possible to selectively excite Ap and observe the fluorescence of Ap* within DNA duplexes. We use this fluorescence to probe interactions and reactions between Ap* and DNA bases. Our previous investigations of base–base HT demonstrate that guanine transfers an electron to Ap* in solution and DNA duplexes, resulting in fluorescence quenching (6, 7). The bases thymine (T), cytosine (C), and the base analog inosine (I) do not undergo this HT reaction with photoexcited Ap (7) (although T and C may react via reductive CT with Ap* in DNA; ref. 8). These observations are consistent with the driving force for HT that is ≈200 mV for G and thermodynamically unfavorable for the other nucleotides (7). Consequently, redox-active DNA duplexes for HT investigations contain Ap systematically separated from G with the remaining sequence being constructed by d(A)-d(T) or d(I)-d(C) base pairs that do not undergo significant HT with Ap*. Measurements of HT reactions in each DNA duplex are calibrated against an otherwise identical duplex in which the electron donor, G, has been replaced by the redox-inactive I. In addition to serving as references for CT reactions, these I-containing duplexes are used to probe nonredox interactions between Ap* and DNA bases.
The DNA duplexes used in this study possess DNA base analogs to tune the photophysics and redox characteristics of the duplexes with minimal structural impact (Fig. 1). Each DNA duplex contains Ap paired with T on the complementary strand. Investigations of the structure and stability of Ap-containing DNA duplexes by NMR, fluorescence, and calorimetry have shown that Ap undergoes normal Watson–Crick pairing with T and is well stacked within the DNA helix in a manner that is similar to adenine (37–40). The guanine analog, I, is base-paired with C. DNA duplexes substituted with I are not identical to their G-containing counterparts as the exchange of G for I results in the loss of the exocyclic amino group in the minor groove and the loss of one H bond in the base pair with C. However, the structural consequences of a single I substitution are relatively minor, based on crystal structures (41, 42) and theoretical investigations (43). In addition, we observe close similarities in the CD spectra, fluorescence excitation spectra, and fluorescence polarization between analogous G and I duplexes, which strongly suggests that differences in base stacking, structure, and dynamics are not significant.
Fig 1.

Idealized model (insight ii) of a B-DNA duplex (5′-TCTIApAGITCTATTCT-3′ and complement) and structures of the molecular constituents. The aromatic stack of DNA bases, shown in gray, blue (Ap), and red (guanine, G), is distinctly visible within the sugar phosphate backbone (green ribbons). The connection of deoxyribose sugar units via phosphate groups at distinct 5′ and 3′ positions imparts the DNA strand with directional asymmetry.
The specific sequences of the assemblies examined are presented in Fig. 2. Series I duplexes are designed to explore intrastrand versus interstrand HT reactions through A and T bridges. The duplexes of series II–V investigate the influence of the directional asymmetry of DNA on DNA HT. Each series examines photoinduced HT from Ap* to G in both the 5′ to 3′ direction (5′-3′ HT) and the 3′ to 5′ direction (3′-5′ HT). In the series II duplexes this is accomplished by simply exchanging the position of the hole donor, G, and the hole acceptor, Ap*, in the DNA duplexes. Consequently, the bases neighboring Ap and the position of Ap in the 5′-3′ HT assemblies (IIa) are not identical to the 3′-5′ HT assemblies (IIb). The series III duplexes are modified to ensure that the microenvironment surrounding Ap is constant for HT in both directions. In series IV, the position of Ap is fixed at the center of the duplexes, and the IVb assemblies are derived from the IVa assemblies simply by reversing the direction of the entire sequence. Finally, in the series V duplexes, the microenvironment and the position of Ap in the duplex are held constant and HT is considered in each direction, as well as both directions simultaneously.
Fig 2.

Sequences of DNA duplexes designed to examine the influence of base–base coupling on DNA CT. Intrastrand and interstrand coupling involving adenine and thymine bridges is investigated by using series I duplexes (X,X′ = A, T; Y,Y′ = I, G, C). The duplexes of series II—V (complements not shown) are designed to probe the influence of base-step direction on base–base coupling and CT in DNA (Y = I, G). The adenine analog, 2-Ap, is paired with thymine, whereas the guanine analog, inosine, is paired with cytosine.
To probe the possible consequences of our structural variations, the thermal denaturation profiles and fluorescence polarization of all duplexes were obtained. Thermal denaturation profiles (monitoring Ap absorbance at 325 nm) and melting temperatures (Tm) of analogous duplexes within each series are virtually identical (Tables 2 and 3, which are published as supporting information on the PNAS web site, www.pnas.org). In particular, no systematic differences in the Tm values were observed. The steady-state fluorescence polarization of Ap was found to be 0.3–0.4 in all DNA duplexes examined (Tables 2 and 3). These levels of steady-state fluorescence polarization are consistent with previous investigations of Ap-containing DNA duplexes (7, 39, 40) and indicate that Ap is indeed base-paired and stacked within all DNA duplexes. For instance, these polarization values can be contrasted with the very low fluorescence polarization of Ap in solution (<0.005) and the poorly stacked DNA base analog, 1,N6-ethenoadenine, within DNA duplexes (≈0.01) (7). The consistency in the fluorescence polarization for analogous duplexes within each series suggests that the microenvironment and flexibility of Ap is similar in all cases. Differences in the fluorescence polarization among the duplexes generally coincide with differences in relative quantum yield of Ap* (see below).
Fluorescence Investigations of DNA Assemblies.
The emission and excitation spectra of Ap* in redox-inactive DNA duplexes report on the microenvironment surrounding Ap and interactions of Ap with other DNA bases. In the excitation spectra, both the long wavelength band (≈320 nm), corresponding to direct excitation of Ap, and the short wavelength band (≈270 nm), corresponding to energy transfer from the natural DNA bases, can serve as valuable diagnostic tools (44, 45). Representative excitation spectra are presented in Fig. 3. Incorporation of Ap into oligonucleotide duplexes induces a red-shift in the long wavelength band of ≈10 nm that is attributed to reduced solvent exposure associated with the duplex environment. The Ap-containing duplexes used here exhibit identical long wavelength maxima in the excitation spectra (Fig. 3), corresponding to a red-shift of ≈7 nm from free, neutral Ap. The relative intensity of the short wavelength band is associated with the efficiency of energy transfer and has also been used as a measure of base-stacking interactions (44, 45). The variation in the relative intensity of the short wavelength band among different DNA assemblies is revealed by Fig. 3. The absence of the short wavelength band for 5′-IApI(A)nI assemblies (series III) is attributed to inefficient energy transfer from the neighboring inosines. Previous investigations have demonstrated that adenine is the most efficient energy donor and that stacks of adenines behave as funnels for energy transfer to Ap in DNA duplexes (44). More subtle distinctions in the short wavelength excitation band are detected between the 5′-IAp(A)nI-3′ (series IIa) and 5′-I(A)nApI-3′ (series IIb) assemblies. These differences suggest that energy transfer occurs more efficiently in the series IIa assemblies, an observation that may be rationalized in terms of differences in base stacking within the 5′-Ap(A)n-3′ and 5′(A)nAp-3′ motifs.
Fig 3.

Excitation spectra of Ap in DNA duplexes demonstrating the influence of sequence and base-step direction on base–base energy transfer and stacking interactions. The following duplexes (5 μM duplex in 100 mM sodium phosphate buffer, pH 7, at 5°C) are shown: series IIa, 5′-TCTIApAAAIITCTTCT-3′ (•); series IIb, 5′-TCTIIAAAApTCTTCT-3′ (○); and series IIIa, 5′-TCTCIApIAAIITCTTCT (▪).
Table 1 compiles the relative steady-state emission intensities of Ap (Φr) in redox-inactive DNA duplexes. These data reveal how Ap* fluorescence sensitively probes the DNA environment at the base level. Consider, for instance, the dramatic differences in the values of Φr for DNA assemblies that have A as compared with T adjacent to Ap. The values of Φr are considerably higher when Ap has neighboring Ts on the same strand. This effect is further exacerbated as the number of consecutive Ts adjacent to Ap increases. These relatively high values of Φr suggest that Ap is less tightly stacked with consecutive thymines than with consecutive adenines along the same strand. It is remarkable that base-pair orientation so dramatically influences the quantum yield of Ap* fluorescence. Indeed base-stacking interactions in B-DNA do exhibit a distinct strandedness.
Table 1.
Relative fluorescence quantum yields, Φr, for Ap in redox-inactive (Y = I) DNA duplexes, and the fractional quenching (Fq = 1 − (ΦG/ΦI)) for HT from Ap* to G as a function of the base-pair orientation and directional asymmetry of DNA
| Duplex
|
Series
|
Φr(Y = I) | Fq | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n = 0 | n = 1 | n = 2 | n = 3 | n = 0 | n = 1 | n = 2 | n = 3 | ||
| Base-pair orientation | |||||||||
| 5′-T C T I Ap(A)nY I T C T A T T C T-3′ | I/IIa | 0.048 (8) | 0.037 (6) | 0.031 (8) | 0.025 (6) | 0.92 (2) | 0.69 (3) | 0.38 (3) | 0.09 (1) |
| 3′-A I A C T (T)nC C A I A T A A I A-5′ | Intrastrand (A)n | ||||||||
| 5′-T C T I Ap(A)n C I T C T A T T C T-3′ | I | 0.040 (6) | 0.023 (3) | 0.029 (3) | 0.028 (1) | 0.11 (6) | 0.30 (5) | 0.22 (9) | 0.12 (3) |
| 3′-A I A C T (T)n Y C A I A T A A I A-5′ | Interstrand (A)n | ||||||||
| 5′-T C T I Ap(T)nY I T C T A T T C T-3′ | I | 0.042 (8) | 0.090 (3) | 0.113 (1) | 0.115 (1) | 0.89 (1) | nq | nq | nq |
| 3′-A I A C T (A)nY C A I A T A A I A-5′ | Intrastrand (T)n | ||||||||
| 5′-T C T I Ap(T)n C I T C T A T T C T-3′ | I | 0.046 (6) | 0.116 (2) | 0.180 (4) | 0.21 (2) | 0.12 (1) | nq | nq | nq |
| 3′-A I A C T (A)n Y C A I A T A A I A-5′ | Interstrand (T)n | ||||||||
| 5′-T C T I Ap A T Y I T C T A T C T-3′ | I | 0.027 (1) | 0.23 (1) | ||||||
| 3′-A I A C T T A C C A I A T A I A-5′ | Intrastrand (AT) | ||||||||
| 5′-T C T I Ap A T C I T C T A T C T-3′ | I | 0.029 (1) | 0.11 (3) | ||||||
| 3′-A I A C T T A Y C A I A T A I A-5′ | Interstrand (AT) | ||||||||
| 5′-T C T I Ap A T A Y I T C T T C T-3′ | I | 0.026 (1) | 0.12 (1) | ||||||
| 3′-A I A C T T A T C C A I A A I A-5′ | Intrastrand (ATA) | ||||||||
| 5′-3′ orientation | |||||||||
| 5′-T C T I I(A)nAp Y T C T A T T C T-3′ | IIb | 0.063 (2) | 0.036 (2) | 0.047 (1) | 0.040 (1) | 0.93 (1) | 0.50 (1) | 0.06 (1) | nq |
| 3′-A I A C C(T)n T C A I A T A A I A-5′ | (A)n 3′–5′ HT | ||||||||
| m = 0 | m = 1 | m = 1 | m = 1 | m = 0 | m = 1 | m = 1 | m = 1 | ||
| n = 0 | n = 0 | n = 1 | n = 2 | n = 0 | n = 0 | n = 1 | n = 2 | ||
| Ap flanked by inosines | |||||||||
| 5′-T C T I Ap(I)m(A)n Y T C T A T T C T-3′ | IIIa | 0.038 (7) | 0.044 (2) | 0.044 (2) | 0.044 (2) | 0.91 (1) | 0.34 (1) | 0.18 (4) | 0.04 (2) |
| 3′-A I A C T (C)m(T)n C A I A T A A I A-5′ | 5′–3′ HT | ||||||||
| 5′-T C T I Y (A)n(I)mAp I T C T A T C T-3′ | IIIb | 0.064 (1) | 0.062 (2) | 0.066 (7) | 0.067 (2) | 0.95 (2) | 0.34 (3) | 0.03 (1) | nq |
| 3′-A I A C C (T)n(C)m T C A I A T A I A-5′ | 3′–5′ HT | ||||||||
| Ap in center of duplex | |||||||||
| 5′-A T C A T A T I Ap A Y I T C T A T-3′ | IVa | 0.018 (1) | 0.64 (1) | ||||||
| 3′-T A I T A T A C T T C C A I A T A-5′ | 5′–3′ HT | ||||||||
| 5′-T A T C T I Y A Ap I T C T A C T A-3′ | IVb | 0.047 (1) | 0.36 (3) | ||||||
| 3′-A T A I A C C T T C A I A T I A T-5′ | 3′–5′ HT | Y1 = G | 0.31 (3) | ||||||
| 5′-A C T A T A Y2I Ap I Y1 A T A T C A-3′ | V | 0.046 (1) | Y2 = G | 0.15 (2) | |||||
| 3′-T I A T A T C C T C C T A T A I T-5′ | 5′–3′ + 3′–5′ HT | Y1,2 = G | 0.43 (2) | ||||||
Measurements were made at 10°C using 100 μM duplexes in 100 mM sodium phosphate buffer, pH 7. The numbers in brackets are the SDs for three to six experiments, each with different duplex samples. No HT quenching of Ap* fluorescence is indicated by nq.
We have recently reported the fractional quenching due to HT in these duplexes (49). The low interstrand quenching observed for HT from Ap* to G in the 5′-ApC-3′ duplex is attributed to a competitive electron transfer reaction from Ap* to C (6, 49). This value of fractional quenching was therefore not included in the evaluation of γ for the Ap(A)nC duplexes.
The sensitivity of Ap* to subtle structural perturbations is also revealed by the consistently larger Φr of assemblies IIb, IIIb, and IVb as compared with IIa, IIIa, and IVa, respectively. These duplexes differ only in the relative position (5′ or 3′) of identical bases neighboring Ap. In some cases (e.g., series III) this effect is propagated to bases beyond the bases immediately adjacent to Ap. The similarity of these duplexes is in sharp contrast with the markedly different Φr. This directional asymmetry in Ap* Φr attests to the differences in base-stacking interactions between 5′-3′ and 3′-5′ base steps.
Intrastrand and Interstrand Base–Base CT Involving Adenine and Thymine.
The yields and distance dependence of HT between Ap* and G in each DNA assembly are presented in Table 1. Two significant facts concerning photoinduced base–base HT involving A and T are revealed by these data: Ts are inferior to As as bridges for intrastrand HT from Ap* and G, and HT through intrastrand pathways is notably more efficient than HT through interstrand pathways. The difference between A and T bridges is dramatic. For instance, where A facilitates intrastrand photoinduced HT over at least 14 Å the presence of intervening Ts essentially shuts off the reaction. The absence of intrastrand HT between Ap* and G through T bridges is consistent with our time-resolved investigations of base–base CT in DNA duplexes, which suggested that an intervening pyrimidine does not facilitate HT between Ap* and G (6). This finding may be related, in part, to an electron transfer to T from Ap* (6). However, the notion that T may be an ineffective bridge for HT is suggested also by our studies of Ap* fluorescence in redox-inactive duplexes (see above). The unusually high values of Φr for Ap* in DNA assemblies with adjacent Ts is indicative of rather poor base stacking within the Ap(T)n motif. Notably these data also indicate that a pathway involving strand crossing and hole migration through the (A)n bridge is not operative here.
As a further test of the consequences of T bridges and strand crossings, intrastrand HT through ApATG and ApATAG as well as interstrand HT through ApATC duplexes was examined. In these assemblies T is not adjacent to Ap, so any quenching of Ap* by electron transfer to T will be eliminated. We have shown that photoinduced interstrand HT is less efficient and may be 3 orders of magnitude slower than intrastrand HT (7). The generality of this trend is confirmed with these duplexes that possess HT pathways involving interstrand purine/purine coupling (Ap/A, A/A, and A/G). As shown in Table 1, in contrast to (T)n bridges, HT between Ap* and G is observed through the mixed AT sequences. However, the yields of HT through ApATG and ApATC are relatively poor. Clearly the efficiency of intrastrand HT is lower through AT than AA bridges, and the efficiency of interstrand HT is reduced relative to intrastrand HT. These data underscore the notion that HT involving T bridges and strand crossing is less efficacious than intrastrand HT via As, even when interstrand purine-purine (including A/A) steps exist. The preference for intrastrand CT has also recently been observed for guanine oxidation in DNA duplexes after high energy ionization (46) and in photoinduced CT reactions through DNA hairpins (13).
HT Is Sensitive to the Directional Asymmetry of Double-Helical DNA.
Donor-acceptor distance and CT energetics are fundamental parameters thought primarily to control the rate constants and yields of DNA CT. Within the series I assemblies, these distances and energetics of HT are essentially constant (see below). In fact, for this series even the (A-T)n base-pair sequence of the bridge is maintained. Only the orientation of the bridge base pairs relative to the donor and acceptor is altered. Yet, the yield of HT varies dramatically.
Striking illustration of how subtle variations in DNA structure can affect CT is provided by comparing the yields of photoinduced HT in assemblies IIa, IIIa, IVa, and Va with IIb, IIIb, IVb, and Vb, respectively (Table 1). In these assemblies, the HT distance and energetics, as well as base-pair orientation within the bridge, are identical. Furthermore, in the series III and V duplexes, the base environment surrounding Ap is constant. Nonetheless, dramatic differences in HT yields and distance dependence are observed (Fig. 4). We also considered that the position of Ap within the duplex might be an important variable. Hence in the series IV and V duplexes the position of Ap within the duplex is fixed. Again, significant and consistent differences in quenching to the 5′ versus 3′ side are observed.
Fig 4.

Variation in the yield of intrastrand CT between Ap* and G as a function of distance determined from steady-state fluorescence measurements of redox-active (ΦG) and redox-inactive (ΦI) duplexes (ln[(ΦI/Φg − 1)] = −γr): (a) series II duplexes, IIa (•), γ = 0.46(2) and IIb (○), γ = 0.77(1); (b) series III duplexes, IIIa (•), γ = 0.56(1) and IIIb (○), γ = 0.95(9).
Previous experiments using acridine intercalators as photooxidants have considered the possibility that HT in DNA may depend on the directional asymmetry of the double helix (18, 19, 47). These experiments found either no distinct trend in the rate constants for HT as a function of directionality (47) or HT that was faster in the 3′-5′ than 5′-3′ direction (18, 19). In the latter case, the possibility that differences in electronic coupling involving the acridine intercalator were responsible for the observed distinctions in HT rate constant could not be ruled out. Higher yields of HT between guanine doublets in the 5′-3′ direction versus the 3′-5′ direction is suggested by the data of Nakatani et al. (24). Again, however, the structural impact of the photooxidant, a cyanobenzophenone-substituted uridine, is difficult to assess. The data presented here represent an experimental demonstration of the impact of directionality on CT between DNA bases.‡ Here the directional properties of the DNA double helix are shown to influence both the yield and distance dependence of CT.
Discussion
Ap as a Tool in Probing DNA Structure and Stacking.
Ap is one of the most widely used probes of DNA structure at the base level, and particular emphasis has been placed on the ability of Ap* to report on base-stacking interactions. Many such investigations rely on the notion that base stacking is solely responsible for quenching Ap* fluorescence. Yet, various mechanisms and/or conditions, including stacking (39, 48), hydrogen bonding (34, 38), base–base collisions (48), and CT reactions (6, 7, 32) have been proposed to explain the quenching of Ap* fluorescence in DNA. Furthermore, the dynamic exchange of Ap between conformational states in DNA modulates stacking, H bonding, and other base–base interactions and reactions on the fluorescence time scale, and thereby regulates all quenching reactions.
We distinguish CT from other modes of fluorescence quenching by examining the fluorescence in analogous redox-active and redox-inactive duplexes. We use the other modes of fluorescence quenching, observed in redox-inactive duplexes, to develop a picture of the structure and stacking of Ap and nearby bases in duplex DNA. Generally speaking, the tighter the association of Ap within the duplex, the greater the degree of fluorescence quenching. Thus although stacking alone may not be responsible for Ap* fluorescence quenching, when comparing Ap fluorescence in similar DNA duplexes it is likely that a greater degree of quenching reflects more time in a stacked DNA-like environment. To strengthen conclusions based on fluorescence quenching, fluorescence excitation spectra are also used to probe stacking interactions between Ap and neighboring bases. This approach has revealed valuable information about the differences in structure and dynamics between DNA and DNA:RNA hybrids and how these differences are correlated to CT reactions (49).
The data presented here emphasize the unique ability of Ap* to probe the environment within the DNA base stack. The emission intensities for redox-inactive duplexes shown in Table 1 reveal that even the most subtle structural variations are readily sensed by Ap*. Exchanging the bases within a single base pair between DNA strands (series I) exchanging the bases on the 5′ and 3′ side of Ap (series II and III), or indeed simply reversing the direction of the entire DNA strand (series IV), induces dramatic changes in the relative emission quantum yield of Ap*. Furthermore, the excitation spectra (Fig. 3) suggest that the environmental changes caused by these minor structural perturbations are most likely changes in base stacking around Ap*. Remarkably, Ap* easily senses these perturbations even when they occur well beyond the nearest-neighbor base, for instance, up to at least 4 bp away.
Correlating HT Yields with Base–Base Coupling.
Following Marcus theory (50), the rate constant for nonadiabatic CT from a donor (d) to an acceptor (a) separated by a distance (r) is defined by the free energy change (ΔG), the reorganization energy (λ), and the effective electronic coupling, Hda:
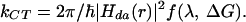 |
The effective electronic coupling for a CT reaction in DNA (Hda) depends on the coupling of donor and acceptor with the bases in the DNA bridge, as well as the energy gap, or energy mismatch, between the donor and the DNA bridge (51). We are interested in probing the importance of coupling in mechanistic descriptions of DNA CT. In the simplest description, electronic coupling for HT between DNA bases is a consequence of overlap between the highest occupied molecular orbitals (HOMOs) of adjacent bases and therefore depends on the atomic distribution and symmetry of each HOMO. Where constructive overlap exists, coupling between the orbitals and delocalization can occur. Here we refer to this situation, qualitatively, as base–base coupling. Constructive overlap among HOMOs has been correlated with calculated values of electronic coupling between DNA bases (52).
The DNA assemblies used (Fig. 2) have been designed to modulate base–base coupling with minimal impact on other parameters that influence CT rate constants and yields. Certainly the CT distance in analogous assemblies (e.g., series IIa and IIb, n = 0) is the same. Likewise, the reorganization energy in analogous assemblies is also expected to be essentially constant. The driving force for HT between Ap* and G is estimated to be ≈200 mV based on the redox potentials of the free base (Ap) and nucleotide (G) in solution (7, 53, 54). Within DNA it is likely that the driving force differs somewhat, and it is also possible that the precise HT energetics depend on the bases adjacent to the redox participants. To eliminate the possibility that energetic perturbations are responsible for our differences in HT yield, we examined assemblies where the microenvironment surrounding Ap is constant (series III and IV). These experiments indicate that sequence effects on the redox potential of Ap are not the source of our varied HT efficiencies.
The bases neighboring G on the 5′ and 3′ side are necessarily reversed in many of the assemblies. The resultant change in HT driving force, if any, cannot account for the dramatic differences in HT yield observed here. Theoretical calculations of the influence of flanking sequence on base ionization potential have found that the 3′ base exerts the dominant impact, and that the influence depends on the oxidation potential of the flanking bases (55). In our assemblies G is flanked by A (or Ap) and I (e.g., 5′-AGI-3′ versus 5′-IGA-3′). Since the oxidation potential of I (7) and A (53, 54) are relatively close, little difference [<100 mV (55)] in the oxidation potential of G is expected between these assemblies. The differences in HT yield observed here are greater than those observed for HT between Ap* and G versus 7-deazaguanine (Z), where the HT driving force differs by 300 mV (7). Furthermore, the data in Table 1 reveal that the variations in HT yield are not consistent with perturbations in driving force. For instance, in series IV the yield of HT is larger for the 5′-AGI-3′ assembly (IVa) than for the 5′-IGA-3′ assembly (IVb). As the oxidation potential of A is less than I, the opposite trend would be expected based on differences in driving force. Alternatively, in the series V duplexes, the yield of HT in the 5′-IGA-3′ assembly is higher than in the 5′-IGA-3′ assembly.
Experimental evidence indicating that base–base coupling, and not other CT parameters, is chiefly responsible for the observed differences in HT yield is obtained from fluorescence investigations of redox-inactive duplexes. Here the fluorescence characteristics of Ap* illustrate the consequences of our structural perturbations in terms of the stacking and association of Ap and nearby bases in the DNA duplex. The differences in Φr among the redox-inactive duplexes (Table 1) demonstrate that these minor structural variations do exert an impact on base stacking. Furthermore, the variations in base stacking observed in redox-inactive duplexes generally parallel the trends in HT yield observed in redox-active duplexes. For instance, we find lower emission quantum yields and, concurrently, higher yields of HT and more shallow distance dependencies for the 5′-3′ HT assemblies, series IIa, IIIa, IVa, and Va, than for the analogous 3′-5′ HT assemblies, series IIb, IIIb, IVb, and Vb. This finding is consistent with the notion that factors that regulate quenching mechanisms in redox-inactive duplexes are equally and similarly important to quenching via CT. This is true because both redox and nonredox mechanisms of quenching are intimately related to base–base stacking.
Experimental and Theoretical Considerations of Interstrand Versus Intrastrand Base–Base Coupling.
Theoretical calculations of the electronic coupling between bases and the effective coupling in DNA bridges (51, 52, 56–60) may be compared with experimentally determined rates and yields of CT in DNA assemblies. Such comparisons are approximations at best, given the current need of theoretical treatments to neglect, for instance, solvent, counterions, cooperativity, and structural dynamics, and the difficulty of experiments to extract differences in effective coupling from other variables. Furthermore, theoretical calculations have thus far generally been restricted to coupling for thermal HT between guanine bases and therefore are not readily extrapolated to experimental investigations of, for instance, photoinduced processes involving auxiliary redox participants. With these distinctions in mind, however, such comparisons can reveal important considerations that must be made in both theoretical and experimental investigations of base–base coupling.
Theoretical calculations of the efficacy of A versus T as a bridge for intrastrand HT between G bases have generated mixed conclusions. Generally, individual TT couplings are determined to be among the highest, although the effective coupling through (A)n bridges has been found to be greater due to the more favorable energy gap (56, 58). Other studies suggest, however, that T is the more effective bridge (59), the distance dependence may be steeper through A rather than T bridges (58, 59), and (A)n is a more effective bridge only for n ≤ 3 (60). Experimentally, we observe that T is substantially inferior to A as a bridge for photoinduced intrastrand HT between Ap* and G. In most studies using pendant redox reagents, the auxiliary reagent may interact with both DNA strands, making it difficult to evaluate HT efficiency through T versus A bridges. However, the results reported here are consistent with experimental investigations of thermal HT between Ap radical cation and G (17), and with thermal HT between Gs over both short (one base) (13) and long (4–10 bases) range (10). The preference for HT through (A)n bridges is therefore not specifically related to our photoexcited hole donor, or HT distance. More likely the general trend of slower rate constants and lower efficiencies for HT through T bridges is a consequence of the reduced effective base–base coupling. Here, the reduced coupling afforded by T for HT between Ap* and G is attributed to a combination of both electronic and structural effects. For instance, the high oxidation potential of T is expected to diminish the electronic coupling for HT. In fact, we have previously proposed that neighboring Ts might react with Ap* by electron transfer, rather than HT (6). We also know that the stacking interactions of T with Ap are weakened relative to A, as evidenced by, for instance, the differences in the yield of Ap* fluorescence. This structural feature will likewise contribute to diminished coupling between Ap* and T.
Regardless of the efficacy of T or A bridges for intrastrand HT, theoretical calculations of electronic coupling generally conclude that purine–purine couplings define the pathway of CT. In other words, hole motion will proceed through the purine path, be it intrastrand or interstrand. This notion has been bolstered by the fact that large interstrand couplings between purine bases, particular As, have been calculated theoretically (51, 52, 56, 58, 59). Such values suggest that intrastrand and interstrand CT involving As should occur with similar rates and yields, A-T base pair orientation should not significantly affect HT, and a “zig-zag” mechanism should prevail. Support for these large interstrand couplings has been suggested (51, 61) to be experimental verification of a zig-zag mechanism by Giese and coworkers (20, 62). However, a zig-zag mechanism for HT between DNA bases need not be invoked to rationalize the results of Giese et al. as the strand crossing could occur between the initially generated sugar radical cation and the neighboring G on the opposite strand (46).
We have demonstrated that photoinduced interstrand HT is substantially slower and less efficient than intrastrand HT (7). Here our investigations of photoinduced HT between DNA bases confirm this feature of DNA CT and clearly dispute a zig-zag mechanism, even when purine/purine interstrand pathways are accessible. We find no experimental verification that interstrand coupling between purine bases, including As, is as large as intrastrand coupling. The notion that interstrand coupling between purines is significantly less than intrastrand coupling is supported by experimental investigations of HT between G bases (13, 46), and between Ap radical cation and G (17). It is therefore not restricted to photoinduced reactions or reactions involving Ap. Furthermore, the low yield for HT in the ApATG and ApATC assemblies unambiguously demonstrates that a zig-zag pathway does not prevail, even when A/A interstrand pathways are available, and the possibility that interaction between Ap* and T is hindering HT through A is removed.
Sensitivity of HT to Directional Asymmetry Emphasizes the Importance of Base–Base Coupling.
The idea that the directional asymmetry of DNA should be manifested at the base level is not novel. Certainly the structure and dynamics of, for instance, a 5′-AG-3′ step, are not expected to be the same as a 5′-GA-3′ step. Indeed, even simple models will illustrate that the base–base overlap within these steps differs. Here we have directly observed the impact of DNA directional asymmetry at the base level. The dramatic influence of directional asymmetry on the values of Φr for Ap* clearly reflects differences in stacking to the 3′ versus the 5′ direction. These differently stacked motifs are characterized by different base–base coupling, and, correspondingly, variations in the efficiency and distance dependence of DNA HT are observed. Significantly, base steps characterized by relatively poor stacking interactions, as evident from fluorescence quantum yields and excitation spectra, exhibit lower yields and steeper distance dependencies for HT. This finding is likewise consistent with a qualitative picture of HOMO distribution and overlap in 5′-ApG-3′ versus 5′-GAp-3′ or 5′-ApA-3′ versus 5′-AAp-3′ base steps (Fig. 5).
Fig 5.

Qualitative representation of HOMOs of 5′ApG-3′ versus 5′-GAp-3′ (a) and 5′ApA-3′ versus 5′-AAp-3′ (b) steps. The extent of orbital overlap and delocalization is notably greater for the 5′ApG-3′ and 5′-ApA-3′ steps. The HOMOs were calculated by using HYPERCHEM 5.1 (restricted Hartree–Fock level with 3-21G* basis set). The atomic coordinates of the bases were obtained from insight ii based on standard B-DNA geometry and the sugar-phosphate backbones were replaced with a methyl group.
The sensitivity of base–base coupling to directional asymmetry provides experimental evidence for an emerging theme in theoretical studies (51, 52, 56–60, 63, 64). This theme emphasizes, in a general sense, the striking sensitivity of electronic coupling in DNA to subtle structural perturbations, including directional asymmetry. Interestingly, theory indicates that the coupling between individual base steps is higher for 5′-GA-3′ than 5′-AG-3′ (52, 56–59). The opposite trend is found here, although we are observing differences in effective base–base coupling between Ap* and G. Importantly, a current challenge is to achieve coincidence between experiment and theory regarding specific trends in coupling as a function of structural features.
Implications.
The remarkable sensitivity of HT to the base-pair orientation and directional asymmetry of DNA is a powerful illustration of the fundamental role of base–base coupling in DNA CT. These data emphasize how CT through DNA, unlike proteins, depends strongly on specific base–base coupling, not just distance and energetics. Consequently, DNA CT reports on coupling, dynamics, and structure.
Supplementary Material
Acknowledgments
We gratefully acknowledge the National Institutes of Health for generous financial support of this research (Grant GM49216). We also thank the Natural Sciences and Engineering Research Council of Canada for a postdoctoral fellowship (to M.A.O.).
Abbreviations
CT, charge transfer
HT, hole transfer
Ap, 2-aminopurine
Ap*, photoexcited Ap
HOMO, highest occupied molecular orbital
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 30, 2002.
A previously reported “side selectivity” for HT reactions between GG sites in anthraquinone-modified DNA duplexes was attributed to structural perturbations within the DNA conjugates (21).
References
- 1.Dekker C. & Ratner, M. A. (2001) Phys. World 14, 29-33. [Google Scholar]
- 2.Boon E. M. & Barton, J. K. (2002) Curr. Opin. Struct. Biol. 12, 320-329. [DOI] [PubMed] [Google Scholar]
- 3.Giese B. (2002) Annu. Rev. Biochem. 71, 51-70. [DOI] [PubMed] [Google Scholar]
- 4.Schuster G. B. (2000) Acc. Chem. Res. 33, 253-260. [DOI] [PubMed] [Google Scholar]
- 5.Lewis F. D., Letsinger, R. L. & Wasielewski, M. R. (2001) Acc. Chem. Res. 34, 159-170. [DOI] [PubMed] [Google Scholar]
- 6.Wan C. Z., Fiebig, T., Schiemann, O., Barton, J. K. & Zewail, A. H. (2000) Proc. Natl. Acad. Sci. USA 97, 14052-14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelley S. O. & Barton, J. K. (1999) Science 283, 375-381. [DOI] [PubMed] [Google Scholar]
- 8.Wan C. Z., Fiebig, T., Kelley, S. O., Treadway, C. R., Barton, J. K. & Zewail, A. H. (1999) Proc. Natl. Acad. Sci. USA 96, 6014-6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhattacharya P. K. & Barton, J. K. (2001) J. Am. Chem. Soc. 123, 8649-8656. [DOI] [PubMed] [Google Scholar]
- 10.Williams T. T., Odom, D. T. & Barton, J. K. (2000) J. Am. Chem. Soc. 122, 9048-9049. [Google Scholar]
- 11.Hall D. B. & Barton, J. K. (1997) J. Am. Chem. Soc. 119, 5045-5046. [Google Scholar]
- 12.Rajski S. R. & Barton, J. K. (2001) Biochemistry 40, 5556-5564. [DOI] [PubMed] [Google Scholar]
- 13.Lewis F. D., Zuo, X., Liu, J., Hayes, R. T. & Wasielewski, M. R. (2002) J. Am. Chem. Soc. 124, 4568-4569. [DOI] [PubMed] [Google Scholar]
- 14.Lewis F. D., Kalgutkar, R. S., Wu, Y. S., Liu, X. Y., Liu, J. Q., Hayes, R. T., Miller, S. E. & Wasielewski, M. R. (2000) J. Am. Chem. Soc. 122, 12346-12351. [Google Scholar]
- 15.Lewis F. D., Wu, T. F., Liu, X. Y., Letsinger, R. L., Greenfield, S. R., Miller, S. E. & Wasielewski, M. R. (2000) J. Am. Chem. Soc. 122, 2889-2902. [Google Scholar]
- 16.Shafirovich V., Dourandin, A. & Geacintov, N. E. (2001) J. Phys. Chem. B 105, 8431-8435. [Google Scholar]
- 17.Shafirovich V., Dourandin, A., Huang, W. D., Luneva, N. P. & Geacintov, N. E. (2000) Phys. Chem. Chem. Phys. 2, 4399-4408. [Google Scholar]
- 18.Hess S., Gotz, M., Davis, W. B. & Michel-Beyerle, M. E. (2001) J. Am. Chem. Soc. 123, 10046-10055. [DOI] [PubMed] [Google Scholar]
- 19.Davis W. B., Naydenova, I., Haselsberger, R., Ogrodnik, A., Giese, B. & Michel-Beyerle, M. E. (2000) Angew. Chem. Int. Ed. 39, 3649-3652. [DOI] [PubMed] [Google Scholar]
- 20.Meggers E., Michel-Beyerle, M. E. & Giese, B. (1998) J. Am. Chem. Soc. 120, 12950-12955. [Google Scholar]
- 21.Ly D., Sanii, L. & Schuster, G. B. (1999) J. Am. Chem. Soc. 121, 9400-9410. [Google Scholar]
- 22.Henderson P. T., Jones, D., Hampikian, G., Kan, Y. Z. & Schuster, G. B. (1999) Proc. Natl. Acad. Sci. USA 96, 8353-8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakatani K., Dohno, C. & Saito, I. (2000) J. Am. Chem. Soc. 122, 5893-5894. [Google Scholar]
- 24.Nakatani K., Dohno, C. & Saito, I. (1999) J. Am. Chem. Soc. 121, 10854-10855. [Google Scholar]
- 25.Núñez M. E., Holmquist, G. P. & Barton, J. K. (2001) Biochemistry 40, 12465-12471. [DOI] [PubMed] [Google Scholar]
- 26.Núñez M. E., Noyes, K. T. & Barton, J. K. (2002) Chem. Biol. 9, 403-415. [DOI] [PubMed] [Google Scholar]
- 27.Boon E. M., Salas, J. E. & Barton, J. K. (2002) Nat. Biotechnol. 20, 282-286. [DOI] [PubMed] [Google Scholar]
- 28.Boon E. M., Ceres, D. M., Drummond, T. G., Hill, M. G. & Barton, J. K. (2000) Nat. Biotechnol. 18, 1096-1100. [DOI] [PubMed] [Google Scholar]
- 29.Rachofsky E. L., Seibert, E., Stivers, J. T., Osman, R. & Ross, J. B. A. (2001) Biochemistry 40, 957-967. [DOI] [PubMed] [Google Scholar]
- 30.Mandal S. S., Fidalgo da Silva, E. & Reha-Krantz, L. J. (2002) Biochemistry 41, 4399-4406. [DOI] [PubMed] [Google Scholar]
- 31.Hochstrasser R. A., Carver, T. E., Sowers, L. C. & Millar, D. P. (1994) Biochemistry 33, 11971-11979. [DOI] [PubMed] [Google Scholar]
- 32.Larsen O. F. A., van Stokkum, I. H. M., Gobets, B., van Grondelle, R. & van Amerongen, H. (2001) Biophys. J. 81, 1115-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gray H. B. & Winkler, J. R. (1996) Annu. Rev. Biochem. 65, 537-561. [DOI] [PubMed] [Google Scholar]
- 34.Ward D. C., Reich, E. & Stryer, L. (1969) J. Biol. Chem. 244, 1228-1237. [PubMed] [Google Scholar]
- 35.Smagowicz J. & Wierzchowski, K. L. (1974) J. Lumin. 8, 210-232. [Google Scholar]
- 36.Nir E., Kleinermanns, K., Grace, L. & de Vries, M. S. (2001) J. Phys. Chem. A 105, 5106-5110. [Google Scholar]
- 37.Eritja R., Kaplan, B. E., Mhaskar, D., Sowers, L. C., Petruska, J. & Goodman, M. F. (1986) Nucleic Acids Res. 14, 5869-5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu D. G., Evans, K. O. & Nordlund, T. M. (1994) Biochemistry 33, 9592-9599. [DOI] [PubMed] [Google Scholar]
- 39.Nordlund T. M., Andersson, S., Nilsson, L., Rigler, R., Gräslund, A. & McLaughlin, L. W. (1989) Biochemistry 28, 9095-9103. [DOI] [PubMed] [Google Scholar]
- 40.Guest C. R., Hochstrasser, R. A., Sowers, L. C. & Millar, D. P. (1991) Biochemistry 30, 3271-3279. [DOI] [PubMed] [Google Scholar]
- 41.Xuan J.-C. & Weber, I. T. (1992) Nucleic Acids Res. 20, 5457-5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lipanov A., Kopka, M. L., Kaczor-Grzeskowiak, M., Quintana, J. & Dickerson, R. E. (1993) Biochemistry 32, 1373-1389. [DOI] [PubMed] [Google Scholar]
- 43.Cuberno E., Güimil-García, R., Luque, F. J., Eritja, R. & Orozco, M. (2001) Nucleic Acids Res. 29, 2522-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu D. G. & Nordlund, T. M. (2000) Biophys. J. 78, 1042-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stivers J. T. (1998) Nucleic Acids Res. 26, 3837-3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Neill P., Parker, A. W., Plumb, M. A. & Siebbeles, L. D. A. (2001) J. Phys. Chem. B 105, 5283-5290. [Google Scholar]
- 47.Fukui K., Tanaka, K., Fujitsuka, M., Watanabe, A. & Ito, O. (1999) J. Photochem. Photobiol. B 50, 18-27. [Google Scholar]
- 48.Rachofsky E. L., Osman, R. & Ross, J. B. A. (2001) Biochemistry 40, 946-956. [DOI] [PubMed] [Google Scholar]
- 49.O'Neill M. A. & Barton, J. K. (2002) J. Am. Chem. Soc. 124, 13053-13066. [DOI] [PubMed] [Google Scholar]
- 50.Marcus R. A. & Sutin, N. (1985) Biochim. Biophys. Acta 811, 265-322. [Google Scholar]
- 51.Voityuk A. A., Rösch, N., Bixon, M. & Jortner, J. (2000) J. Phys. Chem. B 104, 9740-9745. [Google Scholar]
- 52.Brunaud G., Castet, F., Fritsch, A., Kreisser, M. & Ducasse, L. (2001) J. Phys. Chem. B 105, 12665-12673. [Google Scholar]
- 53.Steenken S. & Jovanovic, S. V. (1997) J. Am. Chem. Soc. 119, 617-618. [Google Scholar]
- 54.Seidel C. A. M., Schultz, A. & Sauer, M. H. (1996) J. Phys. Chem. 100, 5541-5553. [Google Scholar]
- 55.Voityuk A. A., Jortner, J., Bixon, M. & Rösch, N. (2000) Chem. Phys. Lett. 324, 430-434. [Google Scholar]
- 56.Voityuk A. A., Jortner, J., Bixon, M. & Rösch, N. (2001) J. Chem. Phys. 114, 5614-5620. [Google Scholar]
- 57.Rak J., Voityuk, A. A., Marquez, A. & Rösch, N. (2002) J. Phys. Chem. B 106, 7919-7926. [Google Scholar]
- 58.Troisi A. & Orlandi, G. (2001) Chem. Phys. Lett. 344, 509-518. [Google Scholar]
- 59.Olofsson J. & Larsson, S. (2001) J. Phys. Chem. B 105, 10398-10406. [Google Scholar]
- 60.Voityuk A. A. & Rösch, N. (2002) J. Phys. Chem. B 106, 3013-3018. [Google Scholar]
- 61.Jortner J., Bixon, M., Langenbacher, T. & Michel-Beyerle, M. E. (1998) Proc. Natl. Acad. Sci. USA 95, 12759-12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bixon M., Giese, B., Wessely, S., Langenbacher, T., Michel-Beyerle, M. E. & Jortner, J. (1999) Proc. Natl. Acad. Sci. USA 96, 11713-11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Voityuk A. A., Siriwong, K. & Rösch, N. (2001) Phys. Chem. Chem. Phys. 3, 5421-5425. [Google Scholar]
- 64.Troisi A. & Orlandi, G. (2002) J. Phys. Chem. B 106, 2093-2101. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}